Improvement of Biocatalysts for Industrial and Environmental Purposes by Saturation Mutagenesis


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
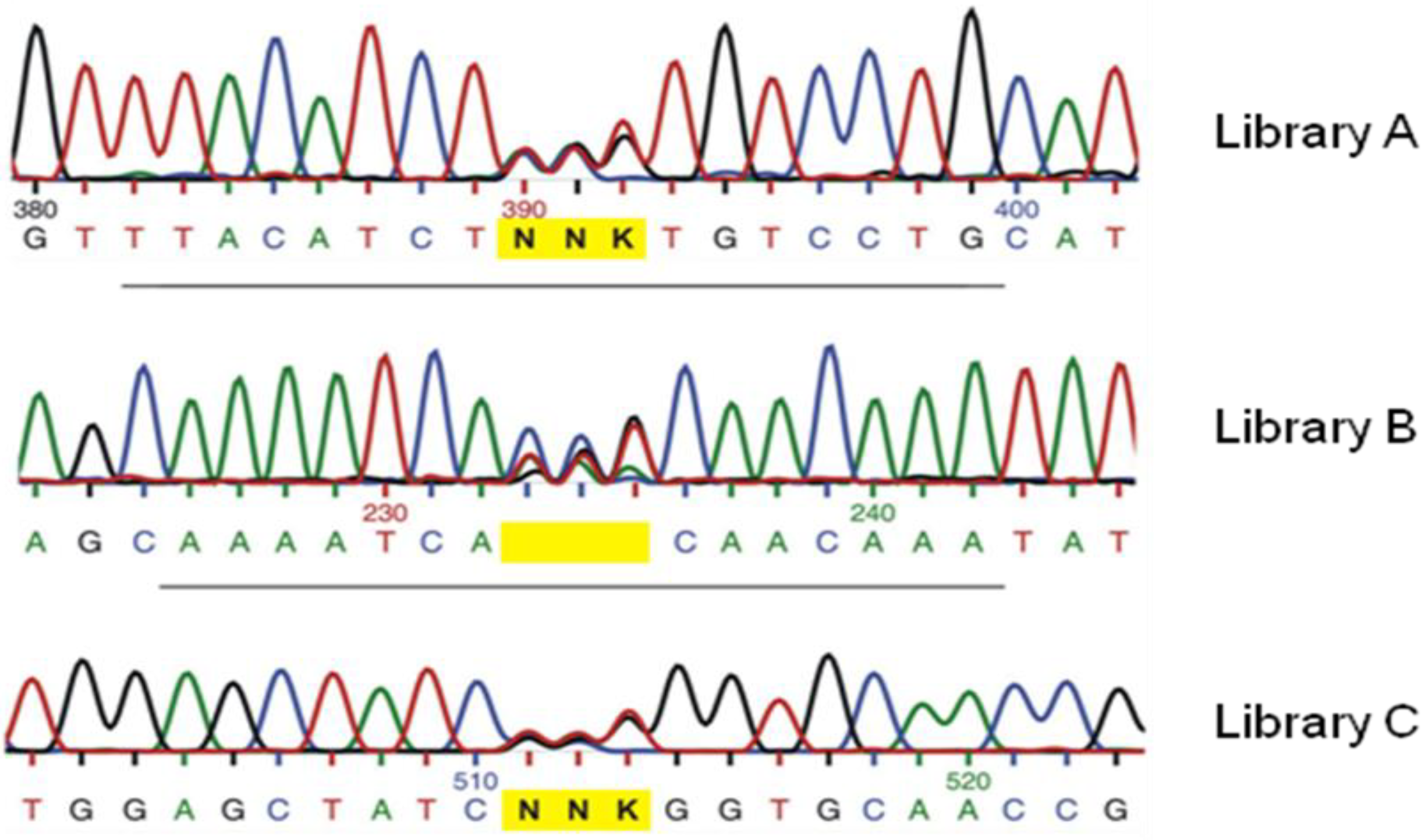{kind=link}
{kind=link}
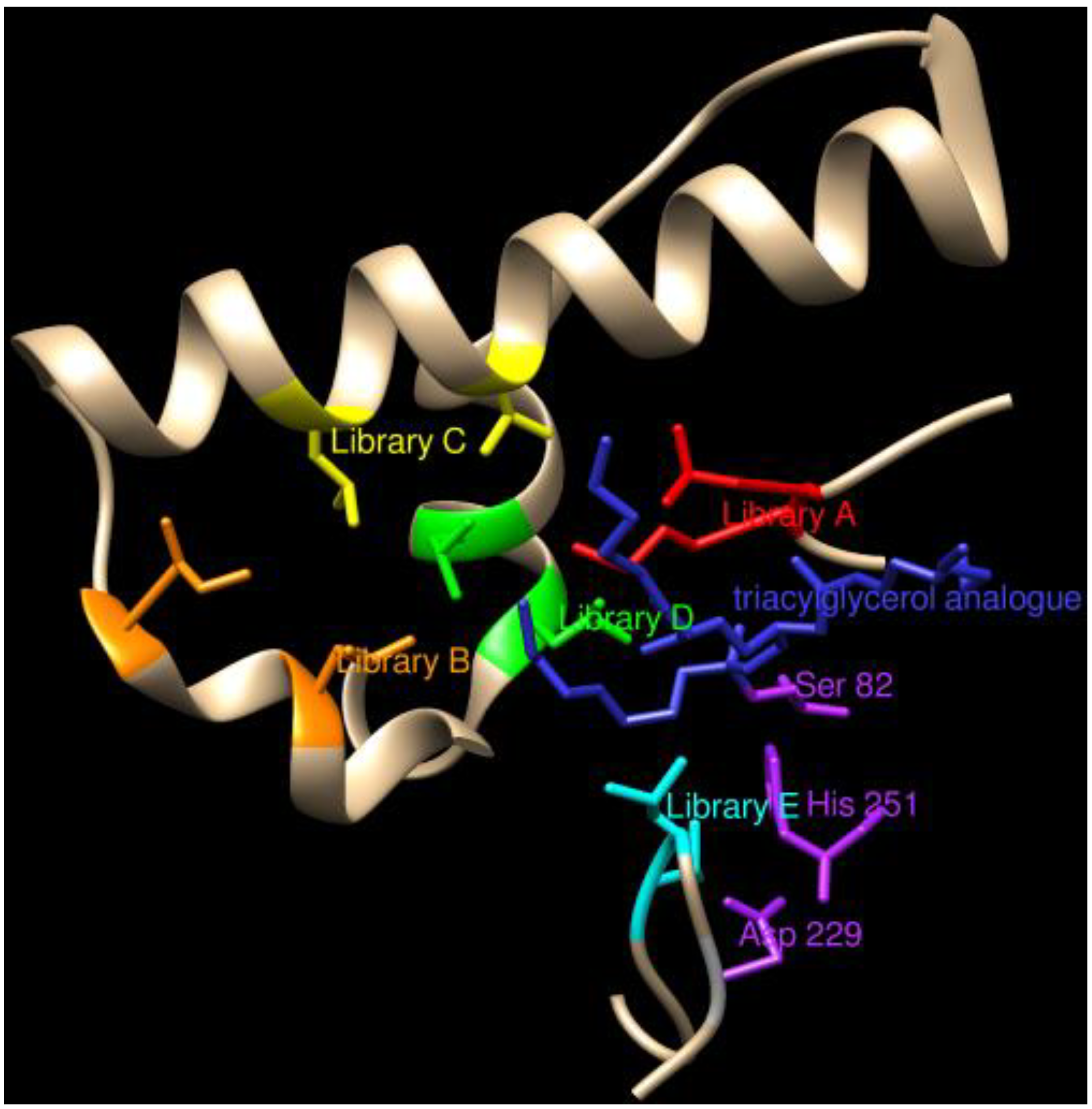{kind=link}
{kind=link}
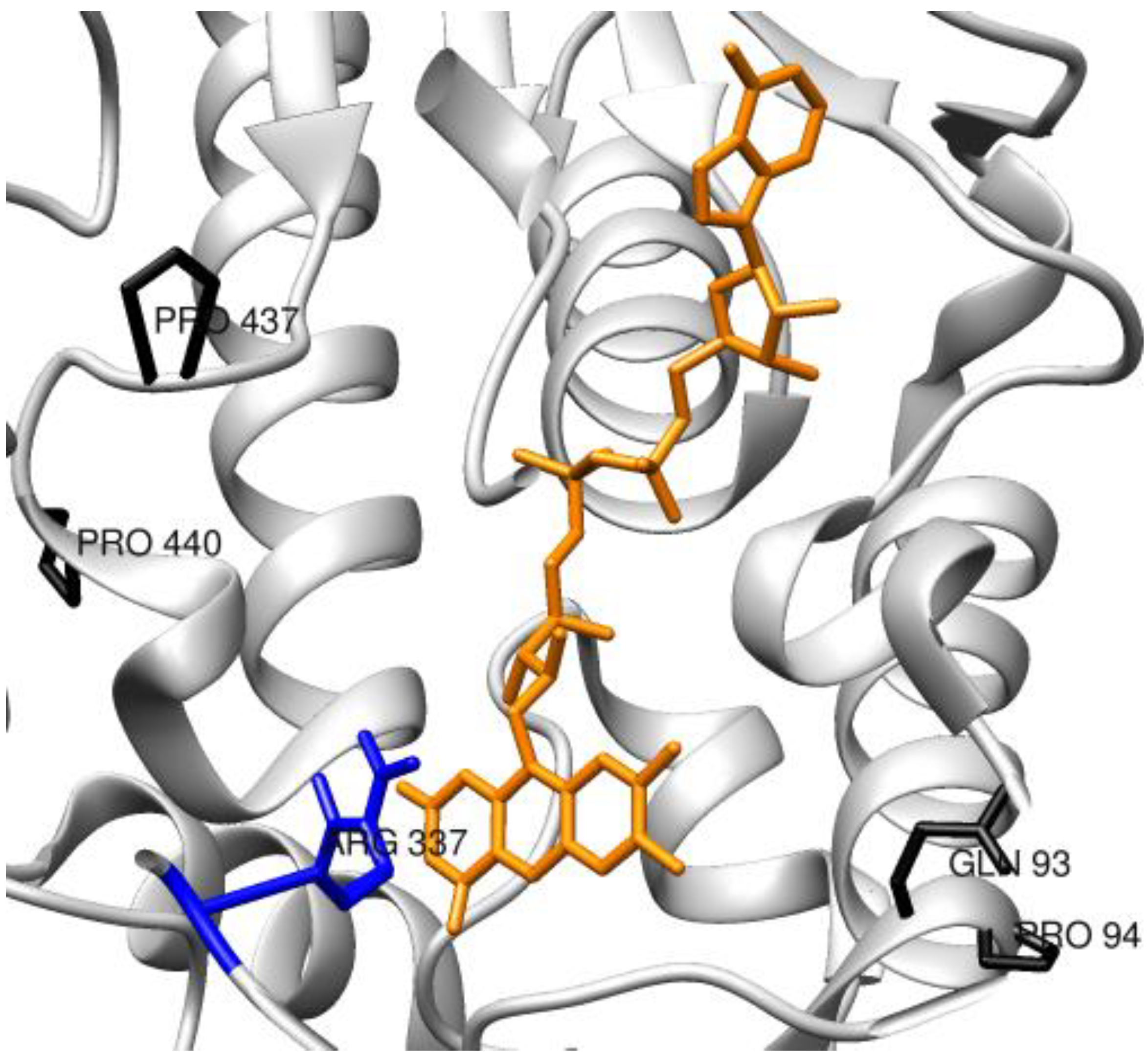{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental
2.1. Strategies for the Generation of Libraries of Mutants
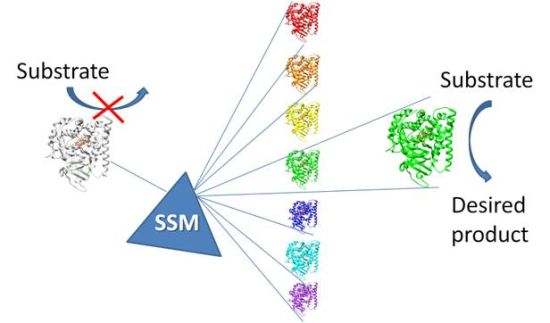
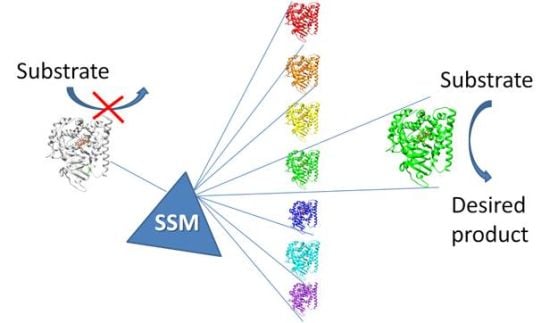
2.1.1. Site Saturation Mutagenesis (SSM)
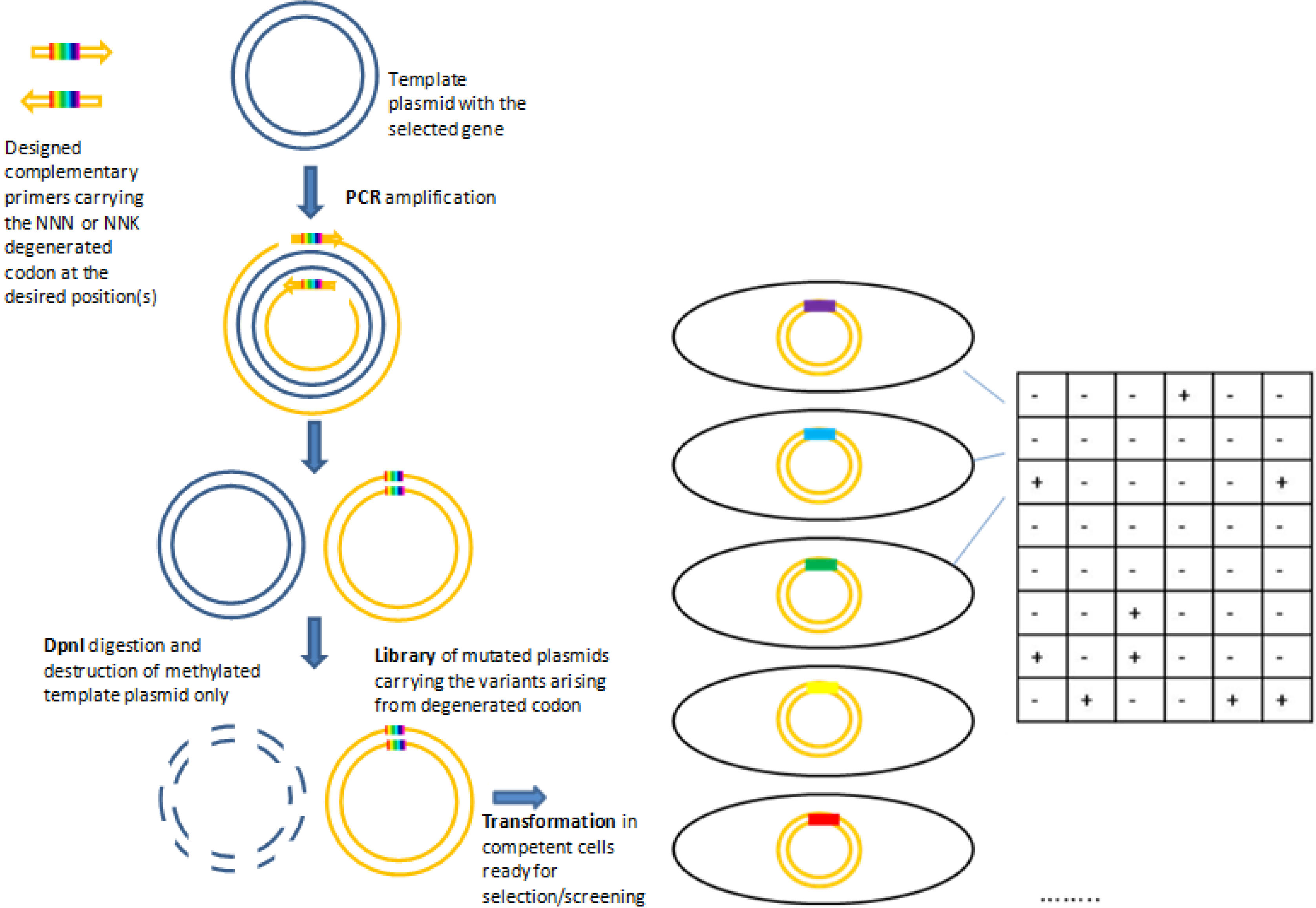
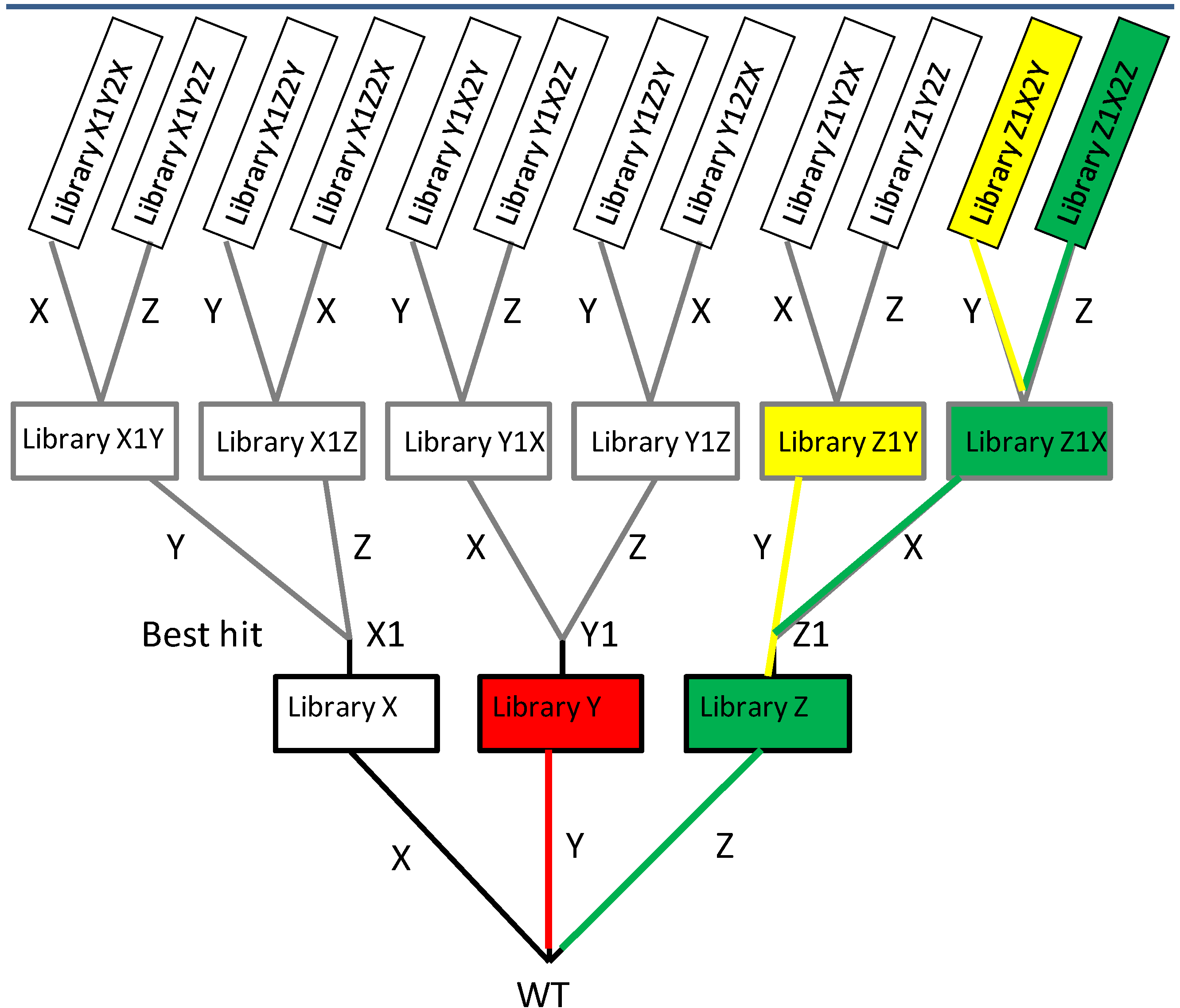
2.1.2. Iterative Saturation Mutagenesis (ISM)
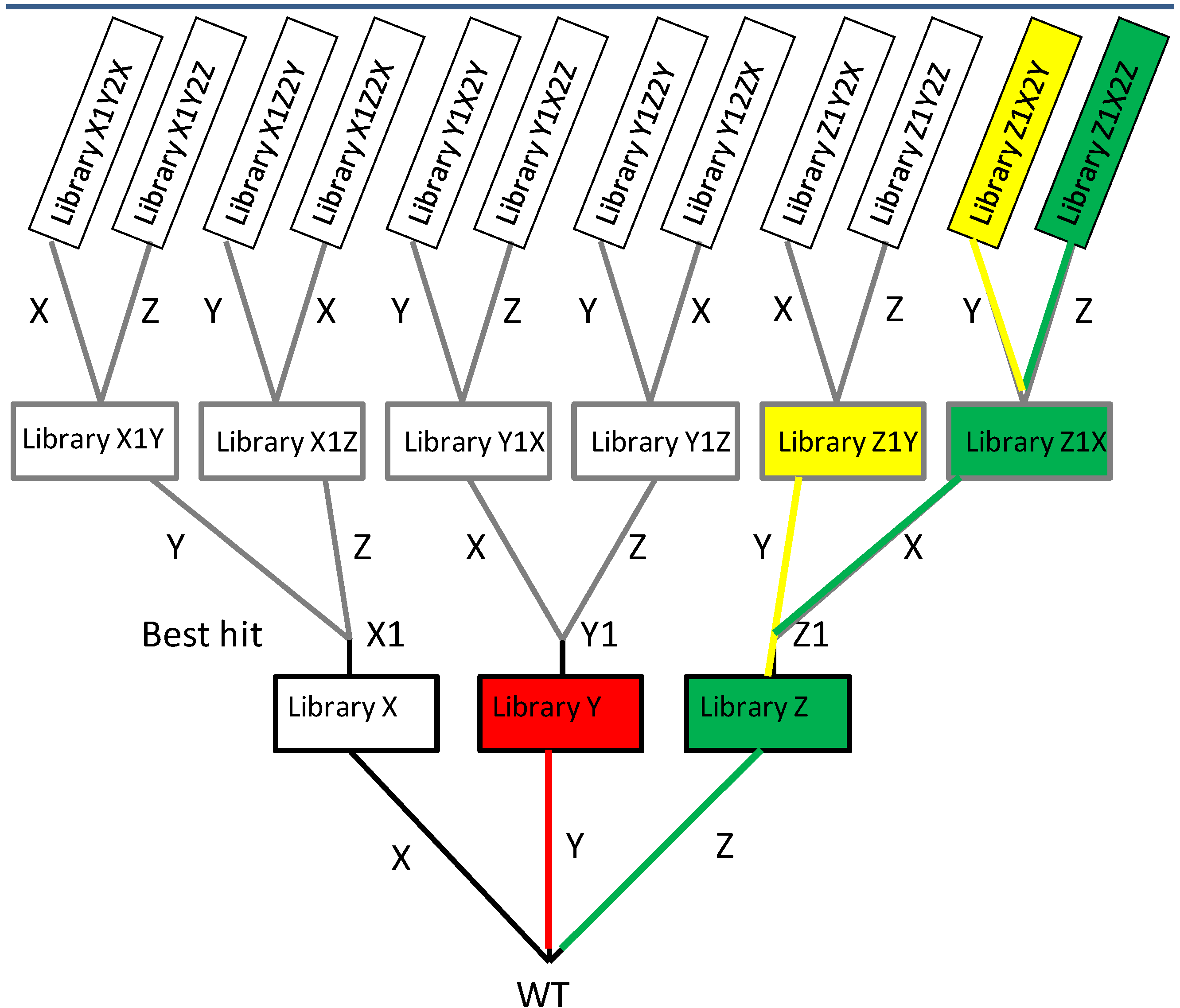
2.1.3. Combinatorial Active-Site Saturation Test (CAST)
2.1.4. B-Factor Iterative Test (B-FIT)
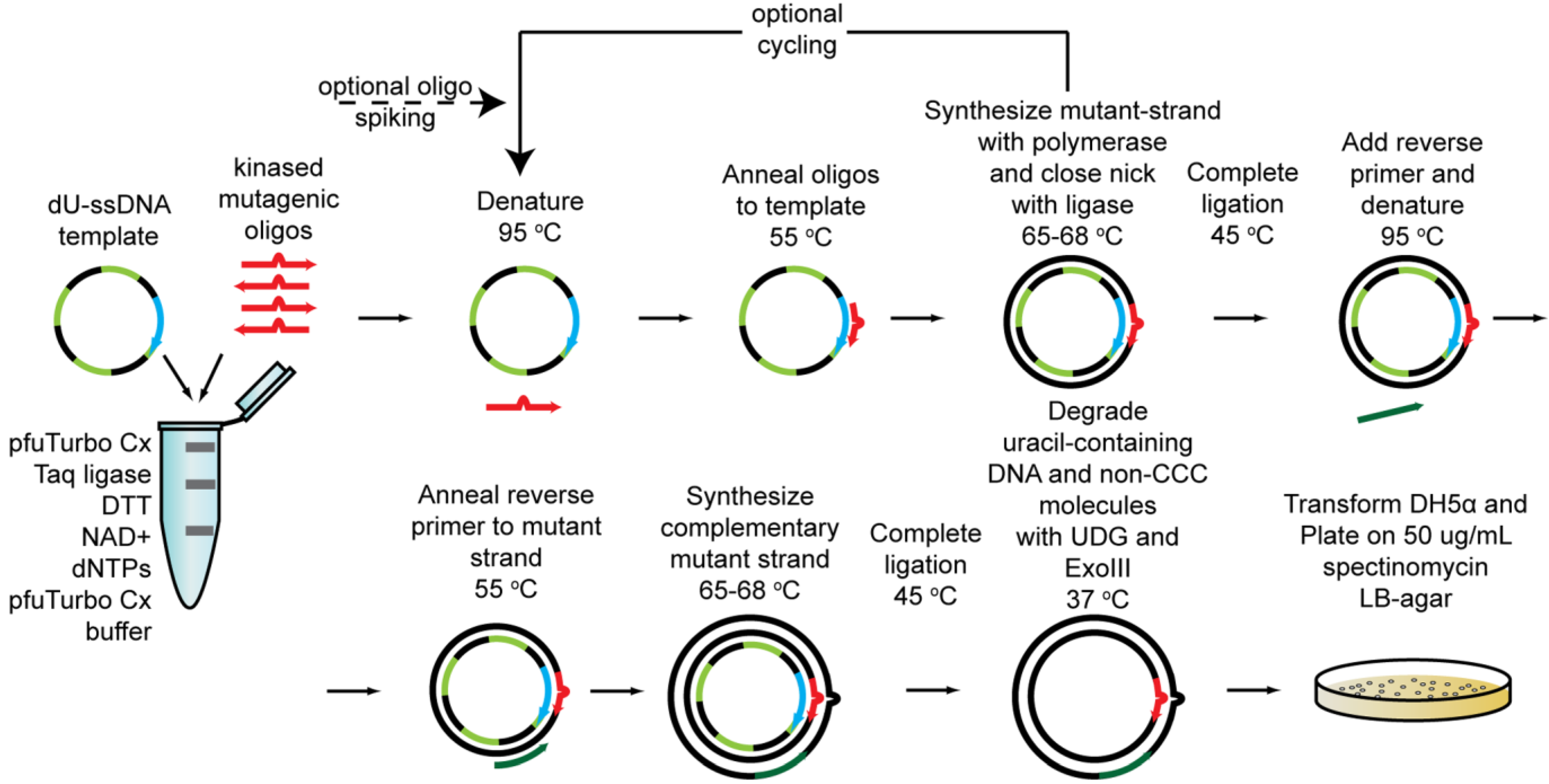
2.1.5. Cassette Mutagenesis and Other Approaches for Multisite Saturation Mutagenesis
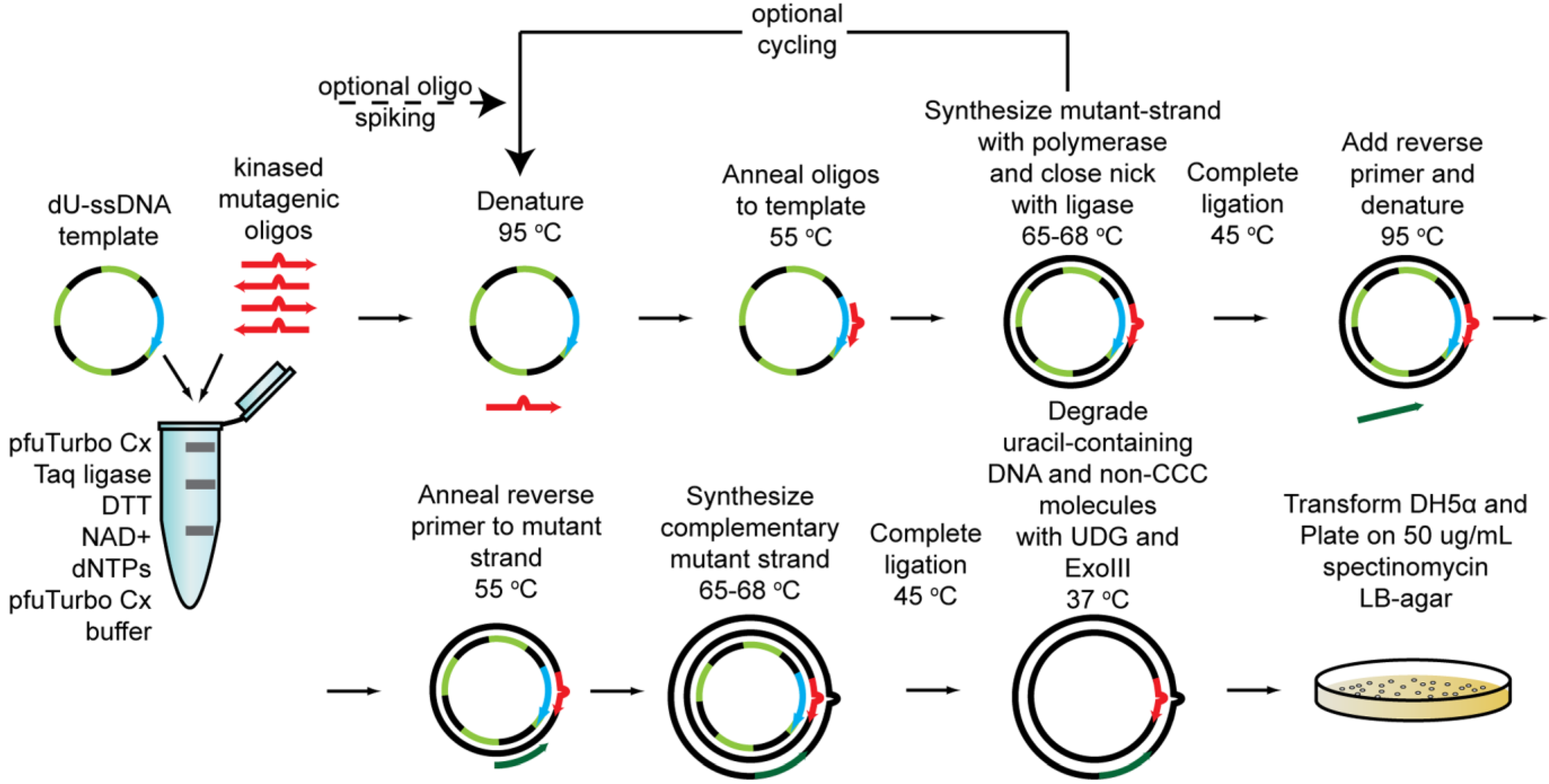
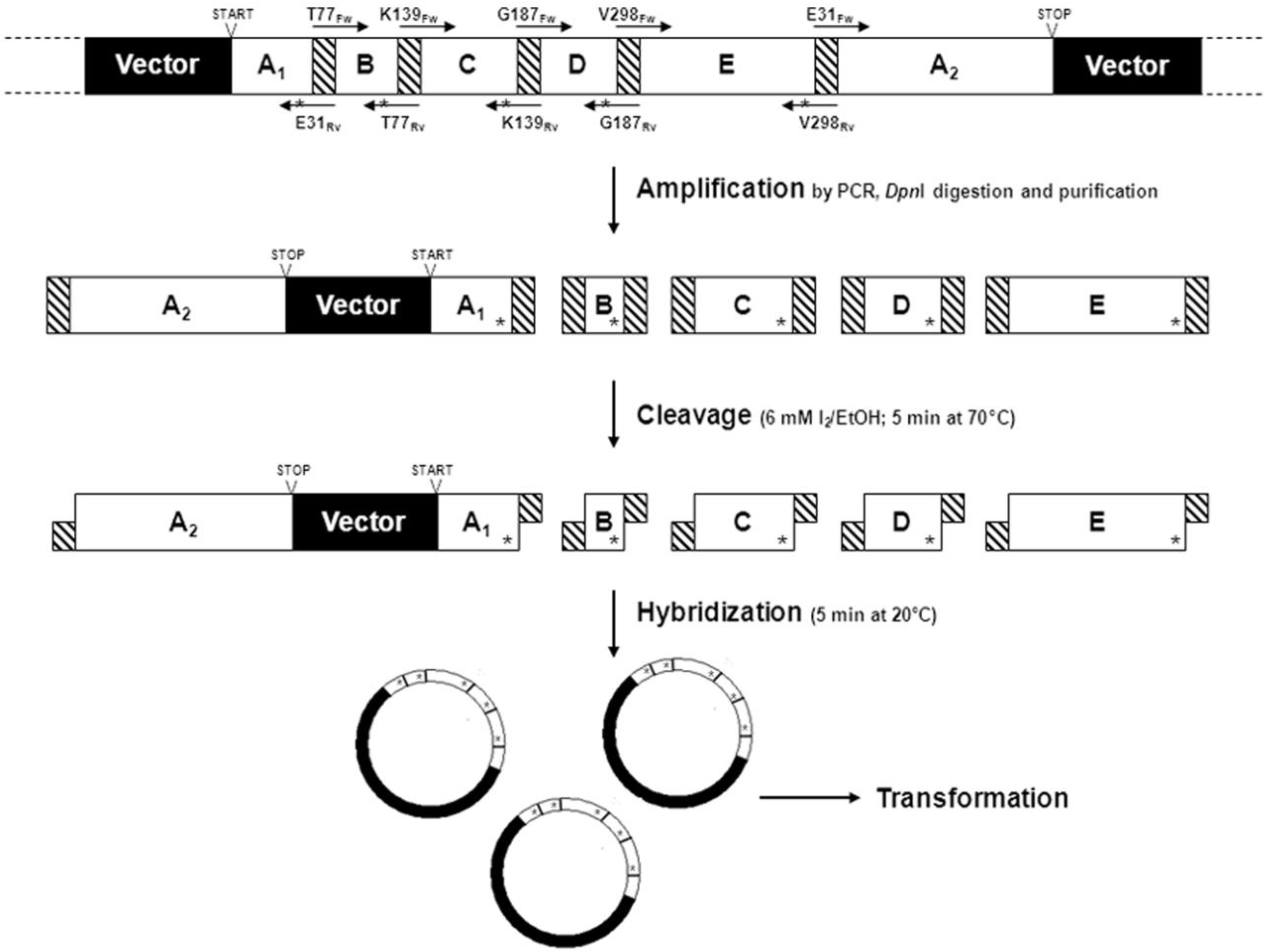
2.1.6. Reducing Amino Acid Alphabet
2.2. Statistical Robustness of the Method and Requirements for Library Screenings
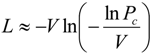
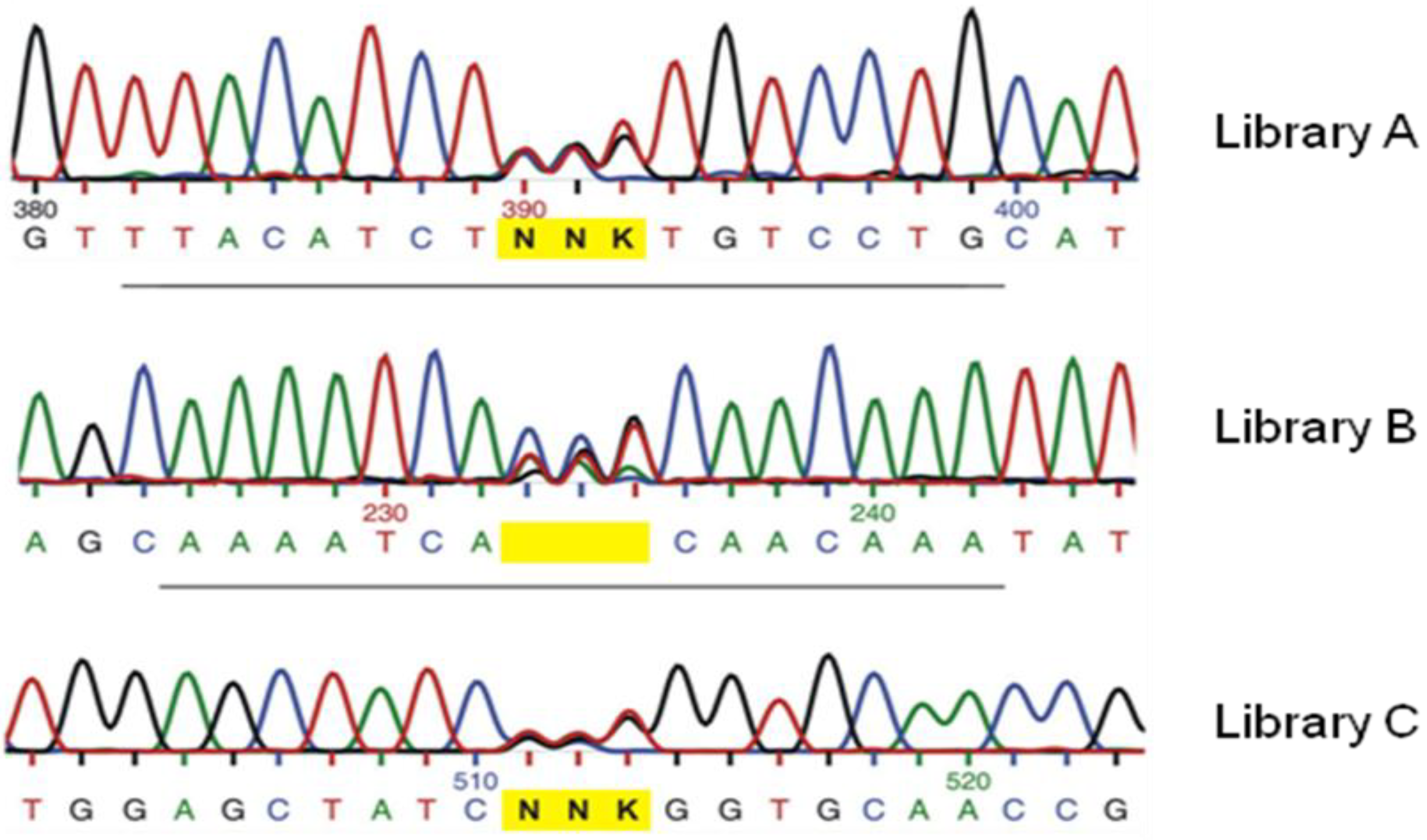
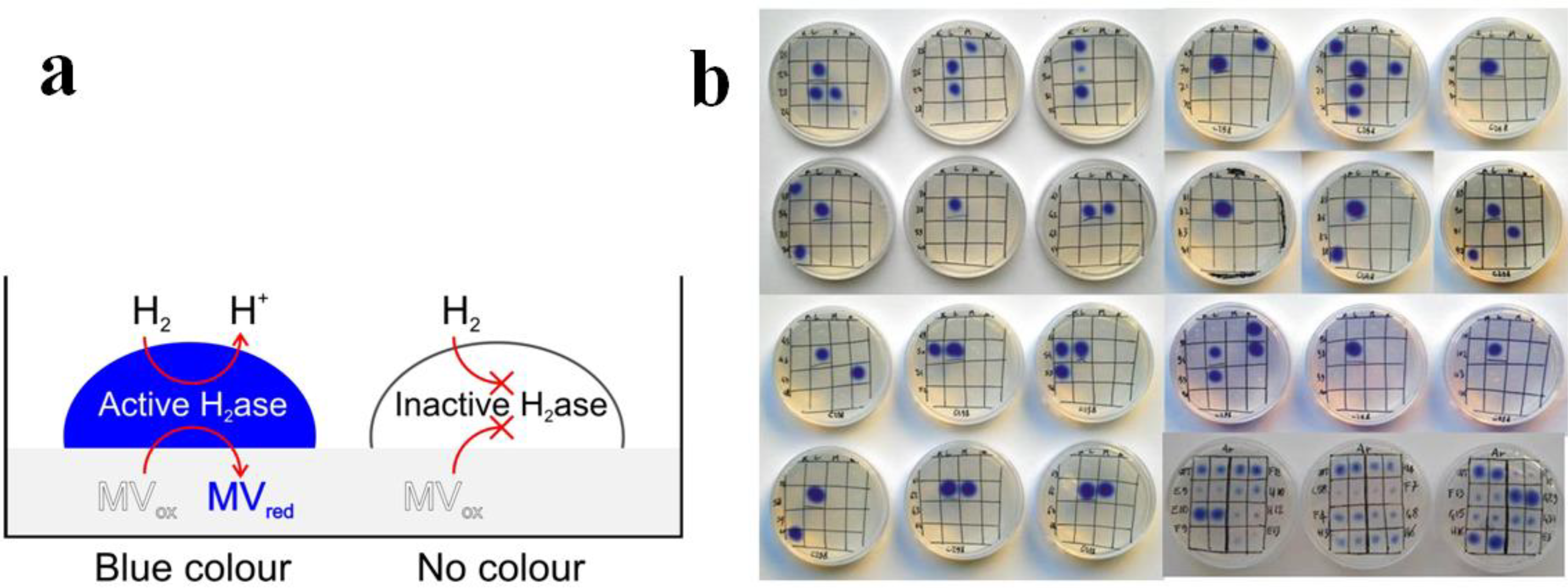
3. Recent Successful Applications
3.1. Enzymes Relevant for Industry
3.1.1. Lipases
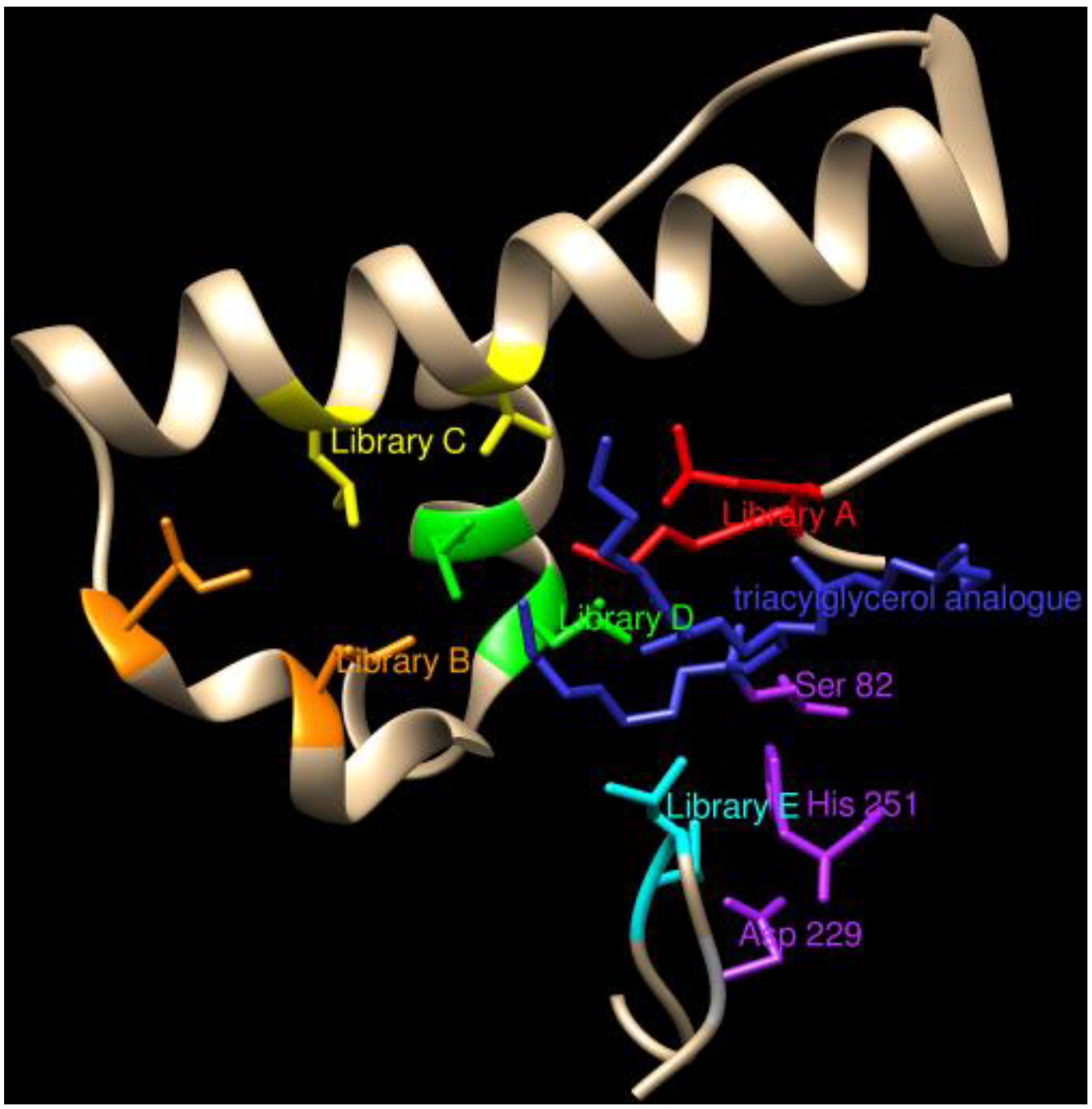
3.1.2. Esterases and Other Hydrolases
3.1.3. Oxygenases and Other Redox Enzymes
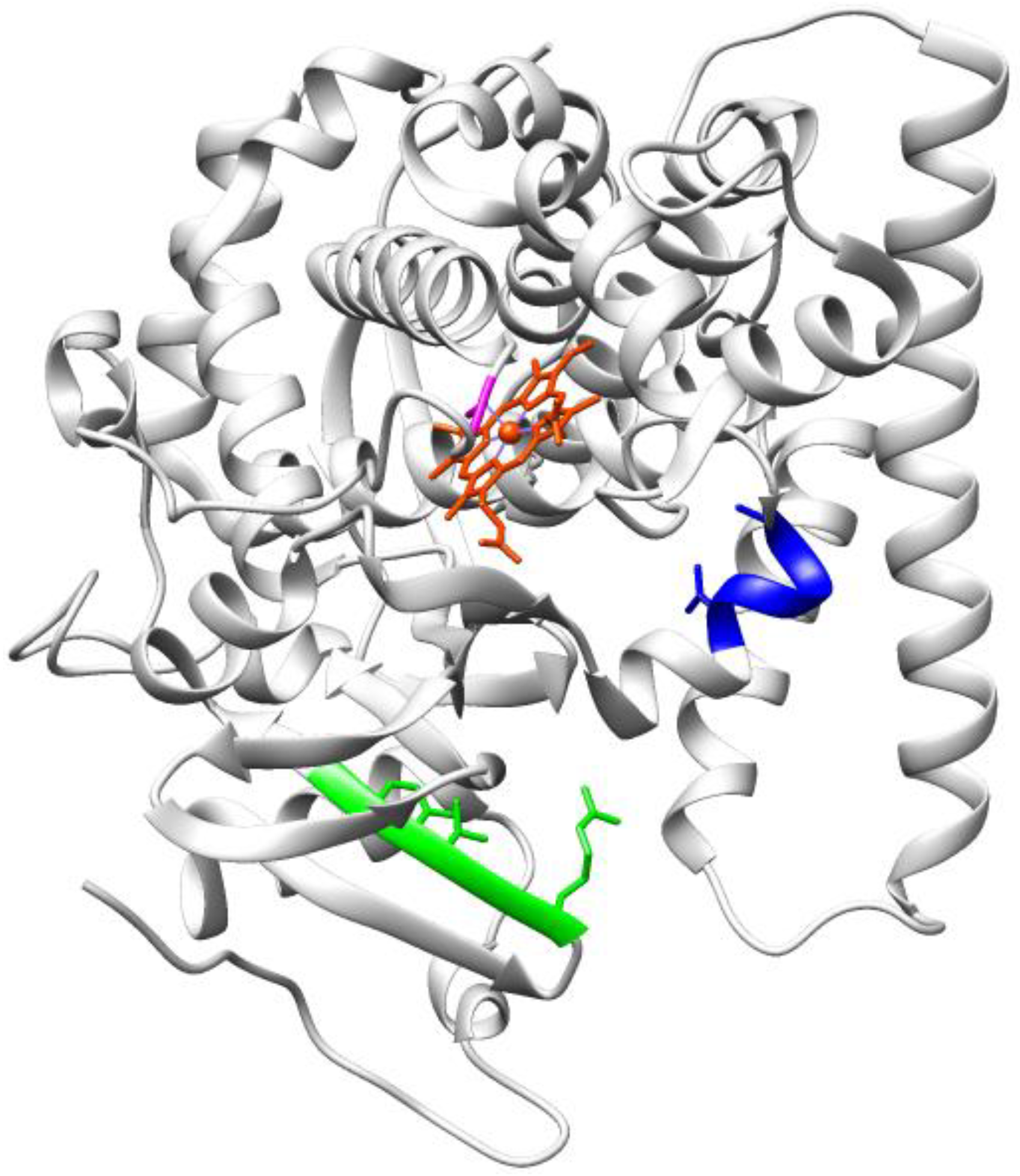
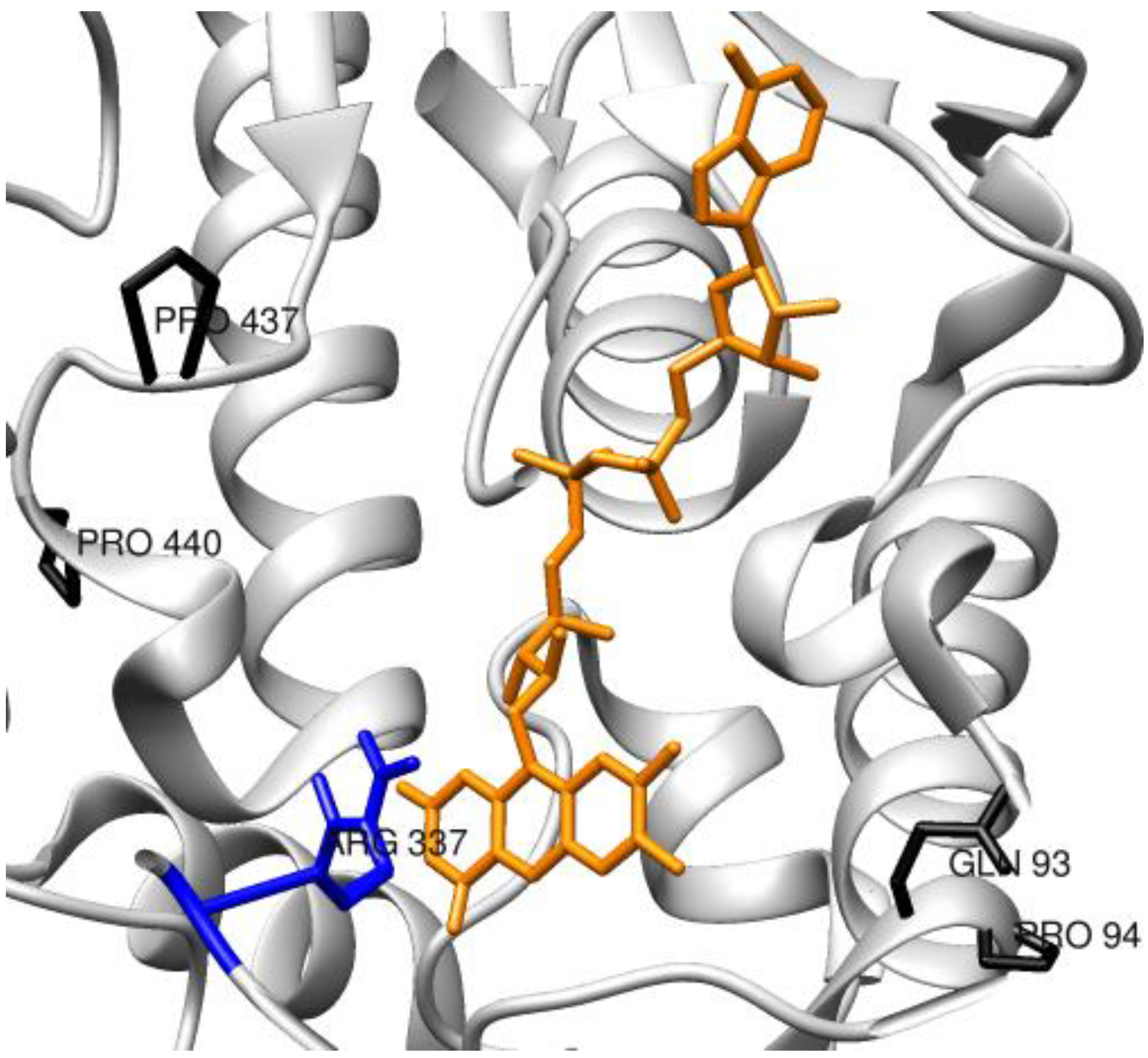
3.2. Enzymes Relevant to Environmental and Clean Energy Approaches
3.2.1. Oxygenases and Other Oxidoreductases for Bioremediation
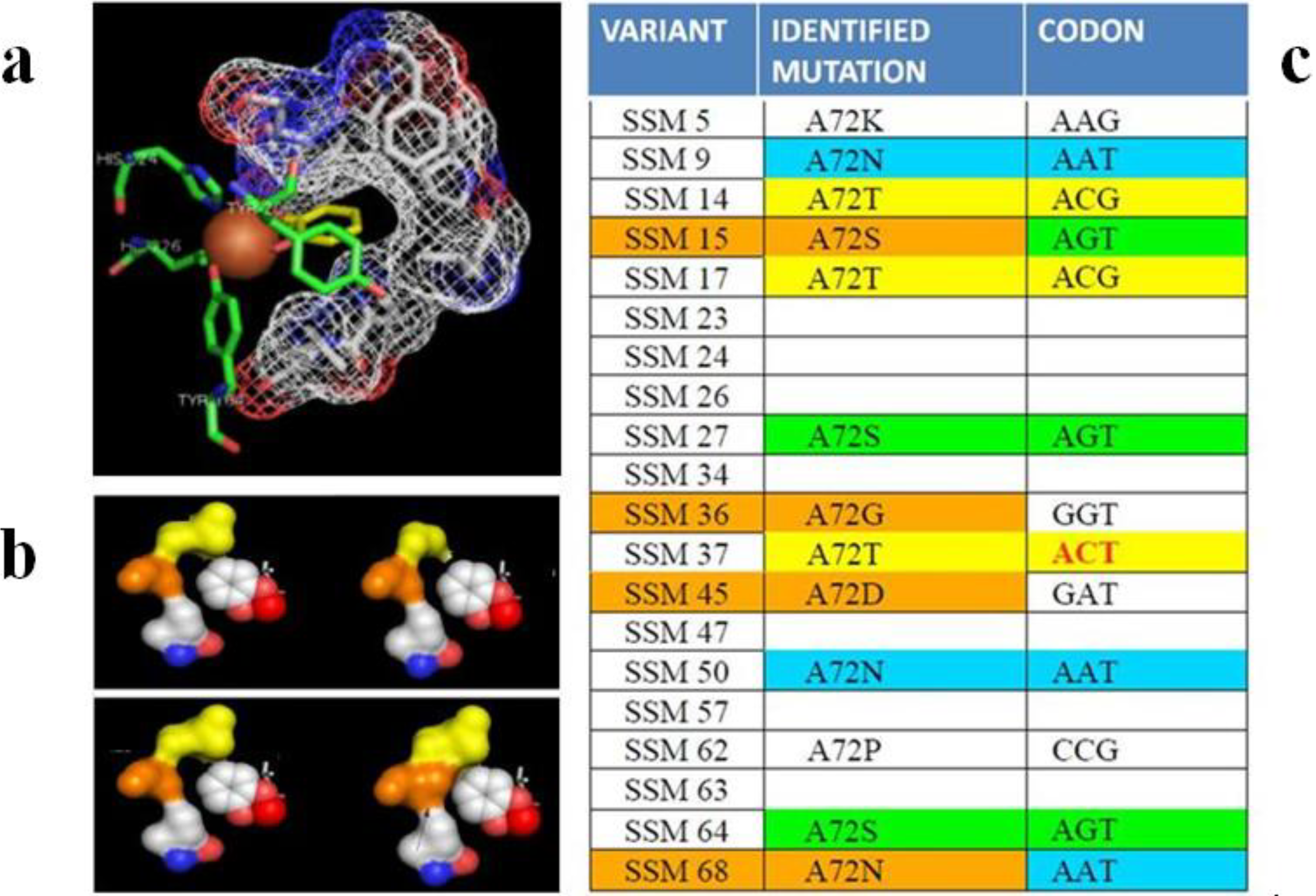
3.2.2. Cellulases, Haloalkane Dehalogenase and Other Hydrolases for Waste Degradation
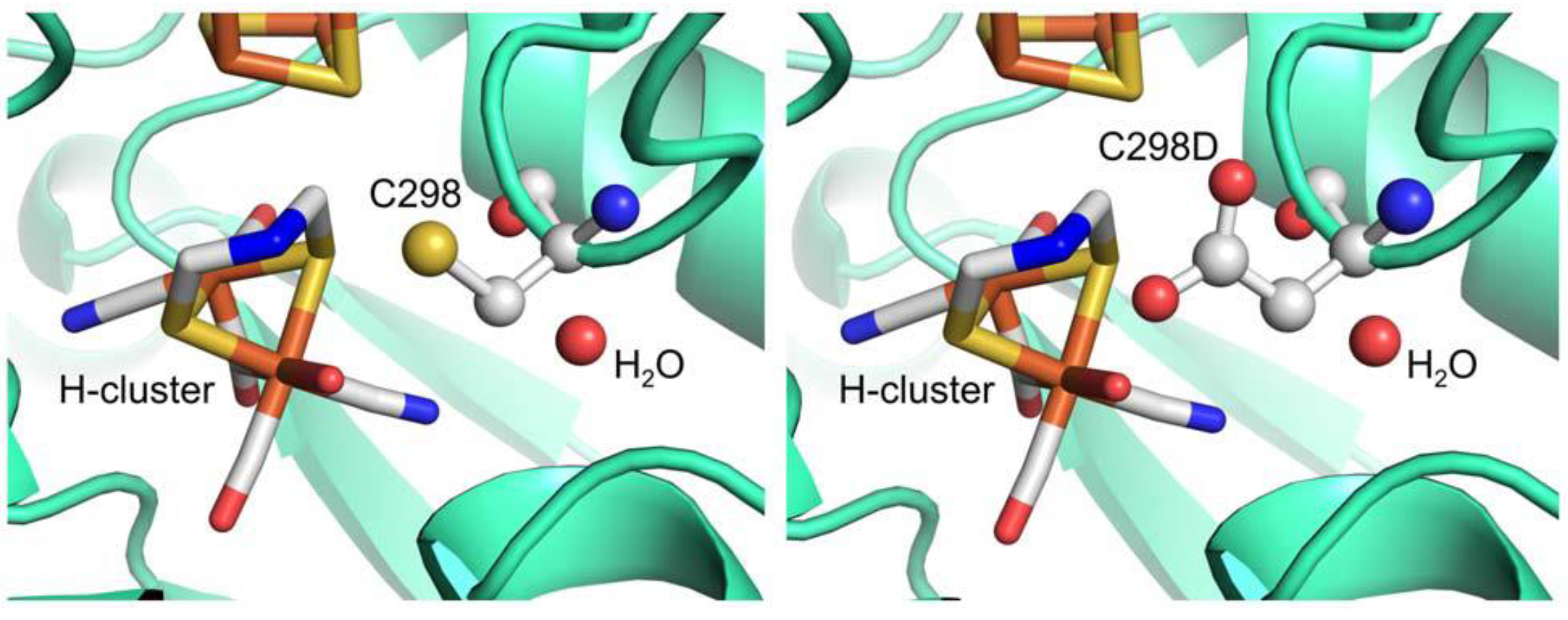
3.2.3. Hydrogenases and Other Enzymes Relevant to Clean Energy Production
4. Conclusions
Conflicts of Interest
References
- Peters, M.W.; Meinhold, P.; Glieder, A.; Arnold, F.H. Regio- and enantioselective alkane hydroxylation with engineered cytochromes P450 BM-3. J. Am. Chem. Soc. 2003, 125, 13442–13450. [Google Scholar] [CrossRef]
- Bocola, M.; Otte, N.; Jaeger, K.E.; Reetz, M.T.; Thiel, W. Learning from directed evolution: Theoretical investigations into cooperative mutations in lipase enantioselectivity. Chembiochem 2004, 5, 214–223. [Google Scholar] [CrossRef]
- Bartsch, S.; Kourist, R.; Bornscheuer, U.T. Complete inversion of enantioselectivity towards acetylated tertiary alcohols by a double mutant of a Bacillus subtilis esterase. Angew. Chem. Int. Ed. 2008, 47, 1508–1511. [Google Scholar] [CrossRef]
- Glieder, A.; Farinas, E.T.; Arnold, F.H. Laboratory evolution of a soluble, self-sufficient, highly active alkane hydroxylase. Nat. Biotechnol. 2002, 20, 1135–1139. [Google Scholar] [CrossRef]
- Schmidt, D.M.Z.; Mundorff, E.C.; Dojka, M.; Bermudez, E.; Ness, J.E.; Govindarajan, S.; Babbitt, P.C.; Minshull, J.; Gerlt, J.A. Evolutionary potential of (β/α)(8)-barrels: Functional promiscuity produced by single substitutions in the enolase superfamily. Biochemistry 2003, 42, 8387–8393. [Google Scholar] [CrossRef]
- Bosma, T.; Danborsky, J.; Stucki, G.; Janssen, D.B. Biodegradation of 1,2,3-trichloropropane through directed evolution and heterologous expression of a haloalkane dehalogenase gene. Appl. Environ. Microbiol. 2002, 68, 3582–3587. [Google Scholar] [CrossRef]
- Reetz, M.T.; Soni, P.; Acevedo, J.P.; Sanchis, J. Creation of an amino acid network of structurally coupled residues in the directed evolution of a thermostable enzyme. Angew. Chem. Int. Ed. Engl. 2009, 48, 8268–8272. [Google Scholar] [CrossRef]
- Zumarraga, M.; Bulter, T.; Shleev, S.; Polaina, J.; Martinez-Arias, A.; Plou, F.J.; Ballesteros, A.; Alcalde, M. In vitro evolution of a fungal laccase in high concentrations of organic cosolvents. Chem. Biol. 2007, 14, 1052–1064. [Google Scholar] [CrossRef]
- Siegel, J.B.; Zanghellini, A.; Lovick, H.M.; Kiss, G.; Lambert, A.R.; St. Clair, J.L.; Gallaher, J.L.; Hilvert, D.; Gelb, M.H.; Stoddard, B.L.; et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science 2010, 329, 309–313. [Google Scholar] [CrossRef]
- Khersonsky, O.; Kiss, G.; Röthlisberger, D.; Dym, O.; Albeck, S.; Houk, K.N.; Baker, D.; Tawfik, D.S. Bridging the gaps in design methodologies by evolutionary optimization of the stability and proficiency of designed Kemp eliminase KE59. Proc. Natl. Acad. Sci. USA 2012, 109, 10358–10363. [Google Scholar] [CrossRef]
- Merski, M.; Shoichet, B.K. Engineering a model protein cavity to catalyze the Kemp elimination. Proc. Natl. Acad. Sci. USA 2012, 109, 16179–16183. [Google Scholar] [CrossRef]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef]
- Janssen, D.B. Evolving haloalkane dehalogenases. Curr. Opin. Chem. Biol. 2004, 8, 150–159. [Google Scholar] [CrossRef]
- Pavlova, M.; Klvana, M.; Prokop, Z.; Chaloupkova, R.; Banas, P.; Otyepka, M.; Wade, R.C.; Tsuda, M.; Nagata, Y.; Damborsky, J. Redesigning dehalogenase access tunnels as a strategy for degrading an anthropogenic substrate. Nat. Chem. Biol. 2009, 5, 727–733. [Google Scholar] [CrossRef]
- Fasan, R.; Meharenna, Y.T.; Snow, C.D.; Poulos, T.L.; Arnold, F.H. Evolutionary history of a specialized p450 propane monooxygenase. J. Mol. Biol. 2008, 383, 1069–1080. [Google Scholar] [CrossRef]
- Stemmer, W.P. Rapid evolution of a protein in vitro by DNA shuffling. Nature 1994, 370, 389–391. [Google Scholar] [CrossRef]
- Ostermeier, M.; Shim, J.H.; Benkovic, S.J. A combinatorial approach to hybrid enzymes independent of DNA homology. Nat. Biotechnol. 1999, 17, 1205–1209. [Google Scholar] [CrossRef]
- Pelletier, J.N. A RACHITT for our toolbox. Nat. Biotechnol. 2001, 19, 314–315. [Google Scholar] [CrossRef]
- Sieber, V.; Martinez, C.A.; Arnold, F.H. Libraries of hybrid proteins from distantly related sequences. Nat. Biotechnol. 2001, 19, 456–460. [Google Scholar] [CrossRef]
- Wang, M.; Si, T.; Zhao, H. Biocatalyst development by directed evolution. Bioresour. Technol. 2012, 115, 117–125. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef]
- Yuan, L.; Kurek, I.; English, J.; Keenan, R. Laboratory-directed protein evolution. Microbiol. Mol. Biol. Rev. 2005, 69, 373–392. [Google Scholar] [CrossRef]
- Bloom, J.D.; Meyer, M.M.; Meinhold, P.; Otey, C.R.; MacMillan, D.; Arnold, F.H. Evolving strategies for enzyme engineering. Curr. Opin. Struct. Biol. 2005, 15, 447–452. [Google Scholar] [CrossRef]
- Valetti, F.; Gilardi, G. Directed evolution of enzymes for product chemistry. Nat. Prod. Rep. 2004, 21, 490–511. [Google Scholar] [CrossRef]
- Farinas, E.T.; Bulter, T.; Arnold, F.H. Directed enzyme evolution. Curr. Opin. Biotechnol. 2001, 12, 545–551. [Google Scholar] [CrossRef]
- Romero, P.A.; Arnold, F.H. Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell. Biol. 2009, 10, 866–876. [Google Scholar] [CrossRef]
- Turner, N.J. Directed evolution drives the next generation of biocatalysts. Nat. Chem. Biol. 2009, 5, 567–573. [Google Scholar] [CrossRef]
- Guo, F.; Xu, H.; Xu, H.; Yu, H. Compensation of the enantioselectivity-activity trade-off in the directed evolution of an esterase from Rhodobacter sphaeroides by site-directed saturation mutagenesis. Appl. Microbiol. Biotechnol. 2013, 97, 3355–3362. [Google Scholar] [CrossRef]
- Reetz, M.T.; Bocola, M.; Carballeira, J.D.; Zha, D.; Vogel, A. Expanding the range of substrate acceptance of enzymes: Combinatorial active-site saturation test. Angew. Chem. Int. Ed. Engl. 2005, 44, 4192–4196. [Google Scholar] [CrossRef]
- Reetz, M.T.; Carballeira, J.D. Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat. Protoc. 2007, 2, 891–903. [Google Scholar] [CrossRef]
- Loke, P.; Sim, T.S. A comparison of three site-directed mutagenesis kits. Z. Naturforsch. C 2001, 56, 810–813. [Google Scholar]
- Reetz, M.T.; Prasad, S.; Carballeira, J.D.; Gumulya, Y.; Bocola, M. Iterative saturation mutagenesis accelerates laboratory evolution of enzyme stereoselectivity: Rigorous comparison with traditional methods. J. Am. Chem. Soc. 2010, 132, 9144–9152. [Google Scholar] [CrossRef]
- Gumulya, Y.; Sanchis, J.; Reetz, M.T. Many pathways in laboratory evolution can lead to improved enzymes: How to escape from local minima. Chembiochem 2012, 13, 1060–1066. [Google Scholar] [CrossRef]
- Prasad, S.; Bocola, M.; Reetz, M.T. Revisiting the lipase from Pseudomonas aeruginosa: Directed evolution of substrate acceptance and enantioselectivity using iterative saturation mutagenesis. Chemphyschem 2011, 12, 1550–1557. [Google Scholar] [CrossRef]
- Available online: http://www.reportlinker.com/p0747897-summary/World-Enzymes-Industry.html.
- Engström, K.; Nyhlén, J.; Sandström, A.G.; Bäckvall, J.E. Directed evolution of an enantioselective lipase with broad substrate scope for hydrolysis of alpha-substituted esters. J. Am. Chem. Soc. 2010, 132, 7038–7042. [Google Scholar] [CrossRef]
- Wen, S.; Tan, T.; Zhao, H. Improving the thermostability of lipase Lip2 from Yarrowia lipolytica. J. Biotechnol. 2012, 164, 248–253. [Google Scholar] [CrossRef]
- Gumulya, Y.; Reetz, M.T. Enhancing the thermal robustness of an enzyme by directed evolution: Least favorable starting points and inferior mutants can map superior evolutionary pathways. Chembiochem 2011, 12, 2502–2510. [Google Scholar] [CrossRef]
- Reetz, M.T.; Soni, P.; Fernández, L.; Gumulya, Y.; Carballeira, J.D. Increasing the stability of an enzyme toward hostile organic solvents by directed evolution based on iterative saturation mutagenesis using the B-FIT method. Chem. Commun. (Camb.) 2010, 46, 8657–8658. [Google Scholar] [CrossRef]
- Reetz, M.T.; Soni, P.; Fernández, L. Knowledge-guided laboratory evolution of protein thermolability. Biotechnol. Bioeng. 2009, 102, 1712–1717. [Google Scholar] [CrossRef]
- Wang, C.; Huang, R.; He, B.; Du, Q. Improving the thermostability of alpha-amylase by combinatorial coevolving-site saturation mutagenesis. BMC Bioinformatics 2012, 13, e263. [Google Scholar] [CrossRef]
- Gouveia-Oliveira, R.; Pedersen, A.G. Finding coevolving amino acid residues using row and column weighting of mutual information and multi-dimensional amino acid representation. Algorithms Mol. Biol. 2007, 2, 12. [Google Scholar] [CrossRef]
- Derbyshire, K.M.; Salvo, J.J.; Grindley, N.D. A simple and efficient procedure for saturation mutagenesis using mixed oligodeoxynucleotides. Gene 1986, 46, 145–152. [Google Scholar] [CrossRef]
- Firnberg, E.; Ostermeier, M. PFunkel: Efficient, expansive, user-defined mutagenesis. PLoS One 2012, 7, e52031. [Google Scholar] [CrossRef]
- Dennig, A.; Shivange, A.V.; Marienhagen, J.; Schwaneberg, U. OmniChange: The sequence independent method for simultaneous site-saturation of five codons. PLoS One 2011, 6, e26222. [Google Scholar]
- Walter, K.U.; Vamvaca, K.; Hilvert, D. An active enzyme constructed from a 9-amino acid alphabet. J. Biol. Chem. 2005, 280, 37742–37746. [Google Scholar] [CrossRef]
- Reetz, M.T.; Kahakeaw, D.; Sanchis, J. Shedding light on the efficacy of laboratory evolution based on iterative saturation mutagenesis. Mol. Biosyst. 2009, 5, 115–122. [Google Scholar] [CrossRef]
- Reetz, M.T.; Kahakeaw, D.; Lohmer, R. Addressing the numbers problem in directed evolution. Chembiochem 2008, 9, 1797–1804. [Google Scholar] [CrossRef]
- Reetz, M.T.; Wu, S. Greatly reduced amino acid alphabets in directed evolution: Making the right choice for saturation mutagenesis at homologous enzyme positions. Chem. Commun. (Camb.) 2008, 5499–5501. [Google Scholar] [CrossRef]
- Kille, S.; Acevedo-Rocha, C.G.; Parra, L.P.; Zhang, Z.G.; Opperman, D.J.; Reetz, M.T.; Acevedo, J.P. Reducing codon redundancy and screening effort of combinatorial protein libraries created by saturation mutagenesis. ACS Synth. Biol. 2013, 2, 83–92. [Google Scholar] [CrossRef]
- Bosley, A.D.; Ostermeier, M. Mathematical expressions useful in the construction, description and evaluation of protein libraries. Biomol. Eng. 2005, 22, 57–61. [Google Scholar] [CrossRef]
- Mena, M.A.; Daugherty, P.S. Automated design of degenerate codon libraries. Protein Eng. Des. Sel. 2005, 18, 559–561. [Google Scholar] [CrossRef]
- Firth, A.E.; Patrick, W.M. Statistics of protein library construction. Bioinformatics 2005, 21, 3314–3315. [Google Scholar] [CrossRef]
- Patrick, W.M.; Firth, A.E.; Blackburn, J.M. User-friendly algorithms for estimating completeness and diversity in randomized protein-encoding libraries. Protein Eng. 2003, 16, 451–457. [Google Scholar] [CrossRef]
- Morra, S.; Giraudo, A.; di Nardo, G.; King, P.W.; Gilardi, G.; Valetti, F. Site saturation mutagenesis demonstrates a central role for cysteine 298 as proton donor to the catalytic site in CaHydA [FeFe]-hydrogenase. PLoS One 2012, 7, e48400. [Google Scholar]
- Koga, Y.; Kato, K.; Nakano, H.; Yamane, T. Inverting enantioselectivity of Burkholderia cepacia KWI-56lipase by combinatorial mutation and high-throughput screening using single-molecule PCR and in vitro expression. J. Mol. Biol. 2003, 331, 585–592. [Google Scholar] [CrossRef]
- Levin, A.M.; Weiss, G.A. Optimizing the affinity and specificity of proteins with molecular display. Mol. Biosyst. 2006, 2, 49–57. [Google Scholar] [CrossRef]
- Granieri, L.; Baret, J.C.; Griffiths, A.D.; Merten, C.A. High-throughput screening of enzymes by retroviral display using droplet-based microfluidics. Chem. Biol. 2010, 17, 229–235. [Google Scholar] [CrossRef]
- Tsotsou, G.E.; Cass, A.E.G.; Gilardi, G. High throughput assay for cytochrome P450 BM3 for screening libraries of substrates and combinatorial mutants. Biosens. Bioelectron. 2002, 17, 119–131. [Google Scholar] [CrossRef]
- Despotovic, D.; Vojcic, L.; Prodanovic, R.; Martinez, R.; Maurer, K.H.; Schwaneberg, U. Fluorescent assay for directed evolution of perhydrolases. J. Biomol. Screen. 2012, 17, 796–805. [Google Scholar] [CrossRef]
- Sass, S.; Kadow, M.; Geitner, K.; Thompson, M.L.; Talmann, L.; Bottcher, D.; Schmidt, M.; Bornscheuer, U.T. A high-throughput assay method to quantify Baeyer-Villiger monooxygenase activity. Tetrahedron 2012, 68, 7575–7580. [Google Scholar] [CrossRef]
- Sideri, A.; Goyal, A.; di Nardo, G.; Tsotsou, G.E.; Gilardi, G. Hydroxylation of non-substituted polycyclic aromatic hydrocarbons by cytochrome P450 BM3 engineered by directed evolution. J. Inorg. Biochem. 2013, 120, 1–7. [Google Scholar] [CrossRef]
- Tsotsou, G.E.; Sideri, A.; Goyal, A.; di Nardo, G.; Gilardi, G. Identification of mutant Asp251Gly/Gln307His of cytochrome P450 BM3 for the generation of metabolites of diclofenac, ibuprofen and tolbutamide. Chemistry 2012, 18, 3582–3588. [Google Scholar] [CrossRef]
- Di Nardo, G.; Gilardi, G. Optimization of the Bacterial Cytochrome P450 BM3 System for the production of human drug metabolites. Int. J. Mol. Sci. 2012, 13, 15901–15924. [Google Scholar] [CrossRef]
- Tsotsou, G.E.; di Nardo, G.; Sadeghi, S.J.; Fruttero, R.; Lazzarato, L.; Bertinaria, M.; Gilardi, G. A rapid screening for cytochrome P450 catalysis on new chemical entities: Cytochrome P450 BM3 and 1,2,5-oxadiazole derivatives. J. Biomol. Screen. 2013, 18, 211–218. [Google Scholar] [CrossRef]
- Stapleton, J.A.; Swartz, J.R. A cell-free microtiter plate screen for improved [FeFe] hydrogenases. PLoS One 2010, 5, e10554. [Google Scholar] [CrossRef]
- Chuah, J.A.; Tomizawa, S.; Yamada, M.; Tsuge, T.; Doi, Y.; Sudesh, K.; Numata, K. Characterization of site-specific mutations in a short-chain-length/medium-chain-length polyhydroxyalkanoate synthase: In vivo and in vitro studies of enzymatic activity and substrate specificity. Appl. Environ. Microbiol. 2013, 79, 3813–3821. [Google Scholar] [CrossRef]
- Jakoblinnert, A.; van den Wittenboer, A.; Shivange, A.V.; Bocola, M.; Heffele, L.; Ansorge-Schumacher, M.; Schwaneberg, U. Design of an activity and stability improved carbonyl reductase from Candida parapsilosis. J. Biotechnol. 2013, 165, 52–62. [Google Scholar] [CrossRef]
- Vojcic, L.; Despotovic, D.; Maurer, K.H.; Zacharias, M.; Bocola, M.; Martinez, R.; Schwaneberg, U. Reengineering of subtilisin Carlsberg for oxidative resistance. Biol. Chem. 2013, 394, 79–87. [Google Scholar]
- Wu, Q.; Soni, P.; Reetz, M.T. Laboratory evolution of enantiocomplementary Candida antarctica lipase B mutants with broad substrate scope. J. Am. Chem. Soc. 2013, 135, 1872–1881. [Google Scholar] [CrossRef]
- Nallaseth, F.S.; Anderson, S. A screen for over-secretion of proteins by yeast based on a dual component cellular phosphatase and immuno-chromogenic stain for exported bacterial alkaline phosphatase reporter. Microb. Cell Fact. 2013, 12, e36. [Google Scholar] [CrossRef]
- Zheng, H.; Wang, X.; Yomano, L.P.; Geddes, R.D.; Shanmugam, K.T.; Ingram, L.O. Improving Escherichia coli FucO for furfural tolerance by saturation mutagenesis of individual amino acid positions. Appl. Environ. Microbiol. 2013, 79, 3202–3208. [Google Scholar] [CrossRef]
- Zhou, H.; Qu, Y.; Kong, C.; Shen, E.; Wang, J.; Zhang, X.; Ma, Q.; Zhou, J. The key role of a non-active-site residue Met148 on the catalytic efficiency of meta-cleavage product hydrolase BphD. Appl. Microbiol. Biotechnol. 2013. [Google Scholar] [CrossRef]
- Phelan, R.M.; Townsend, C.A. Mechanistic insights into the bifunctional non-heme iron oxygenase carbapenem synthase by active site saturation mutagenesis. J. Am. Chem. Soc. 2013, 135, 7496–7502. [Google Scholar] [CrossRef]
- Geier, M.; Braun, A.; Fladischer, P.; Stepniak, P.; Rudroff, F.; Hametner, C.; Mihovilovic, M.D.; Glieder, A. Double site saturation mutagenesis of the human cytochrome P450 2D6 results in regioselective steroid hydroxylation. FEBS J. 2013, 280, 3094–3108. [Google Scholar] [CrossRef]
- Molloy, E.M.; Field, D.; O’Connor, P.M.; Cotter, P.D.; Hill, C.; Ross, R.P. Saturation mutagenesis of lysine 12 leads to the identification of derivatives of nisin A with enhanced antimicrobial activity. PLoS One 2013, 8, e58530. [Google Scholar]
- Shainsky, J.; Bernath-Levin, K.; Isaschar-Ovdat, S.; Glaser, F.; Fishman, A. Protein engineering of nitrobenzene dioxygenase for enantioselective synthesis of chiral sulfoxides. Protein Eng. Des. Sel. 2013, 26, 335–345. [Google Scholar] [CrossRef]
- Agudo, R.; Roiban, G.D.; Reetz, M.T. Induced axial chirality in biocatalytic asymmetric ketone reduction. J. Am. Chem. Soc. 2013, 135, 1665–1668. [Google Scholar] [CrossRef]
- Jakoblinnert, A.; Wachtmeister, J.; Schukur, L.; Shivange, A.V.; Bocola, M.; Ansorge-Schumacher, M.B.; Schwaneberg, U. Reengineered carbonyl reductase for reducing methyl-substituted cyclohexanones. Protein Eng. Des. Sel. 2013, 26, 291–298. [Google Scholar] [CrossRef]
- Sandström, A.G.; Wikmark, Y.; Engström, K.; Nyhlén, J.; Bäckvall, J.E. Combinatorial reshaping of the Candida antarctica lipase A substrate pocket for enantioselectivity using an extremely condensed library. Proc. Natl. Acad. Sci. USA 2012, 109, 78–83. [Google Scholar]
- Korman, T.P.; Sahachartsiri, B.; Charbonneau, D.M.; Huang, G.L.; Beauregard, M.; Bowie, J.U. Dieselzymes: Development of a stable and methanol tolerant lipase for biodiesel production by directed evolution. Biotechnol. Biofuels 2013, 6, e70. [Google Scholar] [CrossRef]
- Anbar, M.; Bayer, E.A. Approaches for improving thermostability characteristics in cellulases. Methods Enzymol. 2012, 510, 261–271. [Google Scholar] [CrossRef]
- Sygmund, C.; Santner, P.; Krondorfer, I.; Peterbauer, C.K.; Alcalde, M.; Nyanhongo, G.S.; Guebitz, G.M.; Ludwig, R. Semi-rational engineering of cellobiose dehydrogenase for improved hydrogen peroxide production. Microb. Cell Fact. 2013, 12, e38. [Google Scholar] [CrossRef] [Green Version]
- Yi, Z.L.; Pei, X.Q.; Wu, Z.L. Introduction of glycine and proline residues onto protein surface increases the thermostability of endoglucanase CelA from Clostridium thermocellum. Bioresour. Technol. 2011, 102, 3636–3638. [Google Scholar] [CrossRef]
- Yi, Z.L.; Zhang, S.B.; Pei, X.Q.; Wu, Z.L. Design of mutants for enhanced thermostability of β-glycosidase BglY from Thermus thermophilus. Bioresour. Technol. 2013, 129, 629–633. [Google Scholar] [CrossRef]
- Reetz, M.T.; Wilensek, S.; Zha, D.; Jaeger, K.E. Directed evolution of an enantioselective enzyme through combinatorial multiple-cassette mutagenesis. Angew. Chem. Int. Ed. Engl. 2001, 40, 3589–3591. [Google Scholar] [CrossRef]
- Jochens, H.; Bornscheuer, U.T. Natural diversity to guide focused directed evolution. Chembiochem 2010, 11, 1861–1866. [Google Scholar] [CrossRef]
- Jochens, H.; Aerts, D.; Bornscheuer, U.T. Thermostabilization of an esterase by alignment-guided focussed directed evolution. Protein Eng. Des. Sel. 2010, 23, 903–909. [Google Scholar] [CrossRef]
- Zheng, H.; Reetz, M.T. Manipulating the stereoselectivity of limonene epoxide hydrolase by directed evolution based on iterative saturation mutagenesis. J. Am. Chem. Soc. 2010, 132, 15744–15751. [Google Scholar] [CrossRef]
- Ye, L.J.; Wang, L.; Pan, Y.; Cao, Y. Changing the specificity of α-amino acid ester hydrolase toward para-hydroxyl cephalosporins synthesis by site-directed saturation mutagenesis. Biotechnol. Lett. 2012, 34, 1719–1724. [Google Scholar] [CrossRef]
- Garrett, J.B.; Kretz, K.A.; O’Donoghue, E.; Kerovuo, J.; Kim, W.; Barton, N.R.; Hazlewood, G.P.; Short, J.M.; Robertson, D.E.; Gray, K.A. Enhancing the thermal tolerance and gastric performance of a microbial phytase for use as a phosphate-mobilizing monogastric-feed supplement. Appl. Environ. Microbiol. 2004, 70, 3041–3046. [Google Scholar] [CrossRef]
- Kille, S.; Zilly, F.E.; Acevedo, J.P.; Reetz, M.T. Regio- and stereoselectivity of P450-catalysed hydroxylation of steroids controlled by laboratory evolution. Nat. Chem. 2011, 3, 738–743. [Google Scholar] [CrossRef]
- Li, H.M.; Mei, L.H.; Urlacher, V.B.; Schmid, R.D. Cytochrome P450 BM-3 evolved by random and saturation mutagenesis as an effective indole-hydroxylating catalyst. Appl. Biochem. Biotechnol. 2008, 144, 27–36. [Google Scholar] [CrossRef]
- Ba, L.; Li, P.; Zhang, H.; Duan, Y.; Lin, Z. Semi-rational engineering of cytochrome P450sca-2 in a hybrid system for enhanced catalytic activity: Insights into the important role of electron transfer. Biotechnol. Bioeng 2013. [Google Scholar] [CrossRef]
- Wu, S.; Acevedo, J.P.; Reetz, M.T. Induced allostery in the directed evolution of an enantioselective Baeyer-Villiger monooxygenase. Proc. Natl. Acad. Sci. USA 2010, 107, 2775–2780. [Google Scholar] [CrossRef]
- Reetz, M.T.; Wu, S. Laboratory evolution of robust and enantioselective Baeyer-Villiger monooxygenases for asymmetric catalysis. J. Am. Chem. Soc. 2009, 131, 15424–15432. [Google Scholar] [CrossRef]
- Willetts, A.; Joint, I.; Gilbert, J.A.; Trimble, W.; Mühling, M. Isolation and initial characterization of a novel type of Baeyer-Villiger monooxygenase activity from a marine microorganism. Microb. Biotechnol. 2012, 5, 549–559. [Google Scholar] [CrossRef]
- Minerdi, D.; Zgrablic, I.; Sadeghi, S.J.; Gilardi, G. Identification of a novel Baeyer-Villiger monooxygenase from Acinetobacter radioresistens: Close relationship to the Mycobacterium tuberculosis prodrug activator EtaA. Microb. Biotechnol. 2012, 5, 700–716. [Google Scholar] [CrossRef]
- Mascotti, M.L.; Juri Ayub, M.; Dudek, H.; Sanz, M.K.; Fraaije, M.W. Cloning, overexpression and biocatalytic exploration of a novel Baeyer-Villiger monooxygenase from Aspergillus fumigatus Af293. AMB Express 2013, 3, e33. [Google Scholar] [CrossRef]
- Gao, X.; Huang, F.; Feng, J.; Chen, X.; Zhang, H.; Wang, Z.; Wu, Q.; Zhu, D. Engineering the meso-diaminopimelate dehydrogenase from Symbiobacterium thermophilum by site-saturation mutagenesis for D-phenylalanine synthesis. Appl. Environ. Microbiol. 2013, 79, 5078–5081. [Google Scholar]
- Paul, D.; Pandey, G.; Pandey, J.; Jain, R.K. Accessing microbial diversity for bioremediation and environmental restoration. Trends Biotechnol. 2005, 23, 135–142. [Google Scholar] [CrossRef]
- Goldsmith, M.; Ashani, Y.; Simo, Y.; Ben-David, M.; Leader, H.; Silman, I.; Sussman, J.L.; Tawfik, D.S. Evolved stereoselective hydrolases for broad-spectrum G-type nerve agent detoxification. Chem. Biol. 2012, 19, 456–466. [Google Scholar] [CrossRef]
- Du, W.; Li, W.; Sun, T.; Chen, X.; Liu, D. Perspectives for biotechnological production of biodiesel and impacts. Appl. Microbiol. Biotechnol. 2008, 79, 331–337. [Google Scholar] [CrossRef]
- Parawira, W. Enzyme research and applications in biotechnological intensification of biogas production. Crit. Rev. Biotechnol. 2012, 32, 172–186. [Google Scholar] [CrossRef]
- King, P.W. Designing interfaces of hydrogenase-nanomaterial hybrids for efficient solar conversion. Biochim. Biophys. Acta 2013, 1827, 949–957. [Google Scholar] [CrossRef]
- Morra, S.; Valetti, F.; Sadeghi, S.J.; King, P.W.; Meyer, T.; Gilardi, G. Direct electrochemistry of an [FeFe]-hydrogenase on a TiO2 electrode. Chem. Commun. (Camb.) 2011, 47, 10566–10568. [Google Scholar]
- Vardar, G.; Wood, T.K. Protein engineering of toluene-o-xylene monooxygenase from Pseudomonas stutzeri OX1 for synthesizing 4-methylresorcinol, methylhydroquinone, and pyrogallol. Appl. Environ. Microbiol. 2004, 70, 3253–3562. [Google Scholar] [CrossRef]
- Tao, Y.; Fishman, A.; Bentley, W.E.; Wood, T.K. Altering toluene 4-monooxygenase by active-site engineering for the synthesis of 3-methoxycatechol, methoxyhydroquinone, and methylhydroquinone. J. Bacteriol. 2004, 186, 4705–4713. [Google Scholar] [CrossRef]
- Canada, K.A.; Iwashita, S.; Shim, H.; Wood, T.K. Directed evolution of toluene ortho-monooxygenase for enhanced 1-naphthol synthesis and chlorinated ethene degradation. J. Bacteriol. 2002, 184, 344–349. [Google Scholar] [CrossRef]
- Fortin, P.D.; MacPherson, I.; Neau, D.B.; Bolin, J.T.; Eltis, L.D. Directed evolution of a ring-cleaving dioxygenase for polychlorinated biphenyl degradation. J. Biol. Chem. 2005, 280, 42307–42314. [Google Scholar]
- Ang, E.L.; Obbard, J.P.; Zhao, H. Directed evolution of aniline dioxygenase for enhanced bioremediation of aromatic amines. Appl. Microbiol. Biotechnol. 2009, 81, 1063–1070. [Google Scholar] [CrossRef]
- Ang, E.L.; Obbard, J.P.; Zhao, H. Probing the molecular determinants of aniline dioxygenase substrate specificity by saturation mutagenesis. FEBS J. 2007, 274, 928–939. [Google Scholar] [CrossRef]
- Leungsakul, T.; Keenan, B.G.; Yin, H.; Smets, B.F.; Wood, T.K. Saturation mutagenesis of 2,4-DNT dioxygenase of Burkholderia sp. strain DNT for enhanced dinitrotoluene degradation. Biotechnol. Bioeng. 2005, 92, 416–426. [Google Scholar] [CrossRef]
- Caglio, R.; Valetti, F.; Caposio, P.; Gribaudo, G.; Pessione, E.; Giunta, C. Fine-tuning of catalytic properties of catechol 1,2-dioxygenase by active site tailoring. Chembiochem 2009, 10, 1015–1024. [Google Scholar] [CrossRef]
- Di Nardo, G.; Roggero, C.; Campolongo, S.; Valetti, F.; Trotta, F.; Gilardi, G. Catalytic properties of catechol 1,2-dioxygenase from Acinetobacter radioresistens S13 immobilized on nanosponges. Dalton Trans. 2009, 7, 6507–6512. [Google Scholar]
- Caglio, R.; Pessione, E.; Valetti, F.; Giunta, C.; Ghibaudi, E. An EPR, thermostability and pH-dependence study of wild-type and mutant forms of catechol 1,2-dioxygenase from Acinetobacter radioresistens S13. Biometals 2013, 26, 75–84. [Google Scholar] [CrossRef]
- Micalella, C.; Martignon, S.; Bruno, S.; Pioselli, B.; Caglio, R.; Valetti, F.; Pessione, E.; Giunta, C.; Rizzi, M. X-ray crystallography, mass spectrometry and single crystal microspectrophotometry: A multidisciplinary characterization of catechol 1,2 dioxygenase. Biochim. Biophys. Acta 2011, 1814, 817–823. [Google Scholar] [CrossRef]
- Chen, M.M.; Snow, C.D.; Vizcarra, C.L.; Mayo, S.L.; Arnold, F.H. Comparison of random mutagenesis and semi-rational designed libraries for improved cytochrome P450 BM3-catalyzed hydroxylation of small alkanes. Protein Eng. Des. Sel. 2012, 25, 171–178. [Google Scholar] [CrossRef]
- Jordan, D.B.; Wagschal, K.; Fan, Z.; Yuan, L.; Braker, J.D.; Heng, C. Engineering lower inhibitor affinities in β-D-xylosidase of Selenomonas ruminantium by site-directed mutagenesis of Trp145. J. Ind. Microbiol. Biotechnol. 2011, 38, 1821–1835. [Google Scholar] [CrossRef]
- Van Leeuwen, J.G.; Wijma, H.J.; Floor, R.J.; van der Laan, J.M.; Janssen, D.B. Directed evolution strategies for enantiocomplementary haloalkane dehalogenases: From chemical waste to enantiopure building blocks. Chembiochem 2012, 13, 137–148. [Google Scholar] [CrossRef]
- Frey, M. Hydrogenases: Hydrogen-activating enzymes. Chembiochem 2002, 3, 153–160. [Google Scholar] [CrossRef]
- Maeda, T.; Sanchez-Torres, V.; Wood, T.K. Protein engineering of hydrogenase 3 to enhance hydrogen production. Appl. Microbiol. Biotechnol. 2008, 79, 77–86. [Google Scholar] [CrossRef]
- Buhrke, T.; Lenz, O.; Krauss, N.; Friedrich, B.J. Oxygen tolerance of the H2-sensing [NiFe] hydrogenase from Ralstonia eutropha H16 is based on limited access of oxygen to the active site. Biol. Chem. 2005, 280, 23791–23796. [Google Scholar] [CrossRef]
- Cornish, A.J.; Gartner, K.; Yang, H.; Peters, J.W.; Hegg, E.L. Mechanism of proton transfer in [FeFe]-hydrogenase from Clostridium pasteurianum. J. Biol. Chem. 2011, 286, 38341–38347. [Google Scholar] [CrossRef]
- Knorzer, P.; Silakov, A.; Foster, C.E.; Armstrong, F.A.; Lubitz, W.; Happe, T. Importance of the protein framework for catalytic activity of [FeFe]-hydrogenases. J. Biol. Chem. 2012, 286, 38341–38347. [Google Scholar]
- Lautier, T.; Ezanno, P.; Baffert, C.; Fourmond, V.; Cournac, L.; Fontecilla-Camps, J.C.; Soucaille, P.; Bertrand, P.; Meynial-Salles, I.; Léger, C. The quest for a functional substrate access tunnel in FeFe hydrogenase. Faraday Discuss 2011, 148, 385–407. [Google Scholar] [CrossRef]
- Stapleton, J.A.; Swartz, J.R. Development of an in vitro compartmentalization screen for high-throughput directed evolution of [FeFe] hydrogenases. PLoS One 2010, 5, e15275. [Google Scholar] [CrossRef]
- Bingham, A.S.; Smith, P.R.; Swartz, J.R. Evolution of an [FeFe] hydrogenase with decreased oxygen sensitivity. Int. J. Hydrogen Energy 2012, 37, 2965–2976. [Google Scholar] [CrossRef]
- Winkler, M.; Esselborn, J.; Happe, T. Molecular basis of [FeFe]-hydrogenase function: An insight into the complex interplay between protein and catalytic cofactor. Biochim. Biophys. Acta 2013, 1827, 974–985. [Google Scholar] [CrossRef]
- Reda, T.; Plugge, C.M.; Abram, N.J.; Hirst, J. Reversible interconversion of carbon dioxide and formate by an electroactive enzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 10654–10658. [Google Scholar]
- Andreadeli, A.; Platis, D.; Tishkov, V.; Popov, V.; Labrou, N.E. Structure-guided alteration of coenzyme specificity of formate dehydrogenase by saturation mutagenesis to enable efficient utilization of NADP+. FEBS J. 2008, 275, 3859–3869. [Google Scholar] [CrossRef]
- Andrews, F.H.; McLeish, M.J. Using site-saturation mutagenesis to explore mechanism and substrate specificity in thiamin diphosphate-dependent enzymes. FEBS J. 2013. [Google Scholar] [CrossRef]
- Goldsmith, M.; Tawfik, D.S. Directed enzyme evolution: Beyond the low-hanging fruit. Curr. Opin. Struct. Biol. 2012, 22, 406–412. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Valetti, F.; Gilardi, G. Improvement of Biocatalysts for Industrial and Environmental Purposes by Saturation Mutagenesis. Biomolecules 2013, 3, 778-811. https://doi.org/10.3390/biom3040778
Valetti F, Gilardi G. Improvement of Biocatalysts for Industrial and Environmental Purposes by Saturation Mutagenesis. Biomolecules. 2013; 3(4):778-811. https://doi.org/10.3390/biom3040778
Chicago/Turabian StyleValetti, Francesca, and Gianfranco Gilardi. 2013. "Improvement of Biocatalysts for Industrial and Environmental Purposes by Saturation Mutagenesis" Biomolecules 3, no. 4: 778-811. https://doi.org/10.3390/biom3040778