Biocatalytic Synthesis of Chiral Alcohols and Amino Acids for Development of Pharmaceuticals

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Enzymatic Preparation of Chiral Alcohols
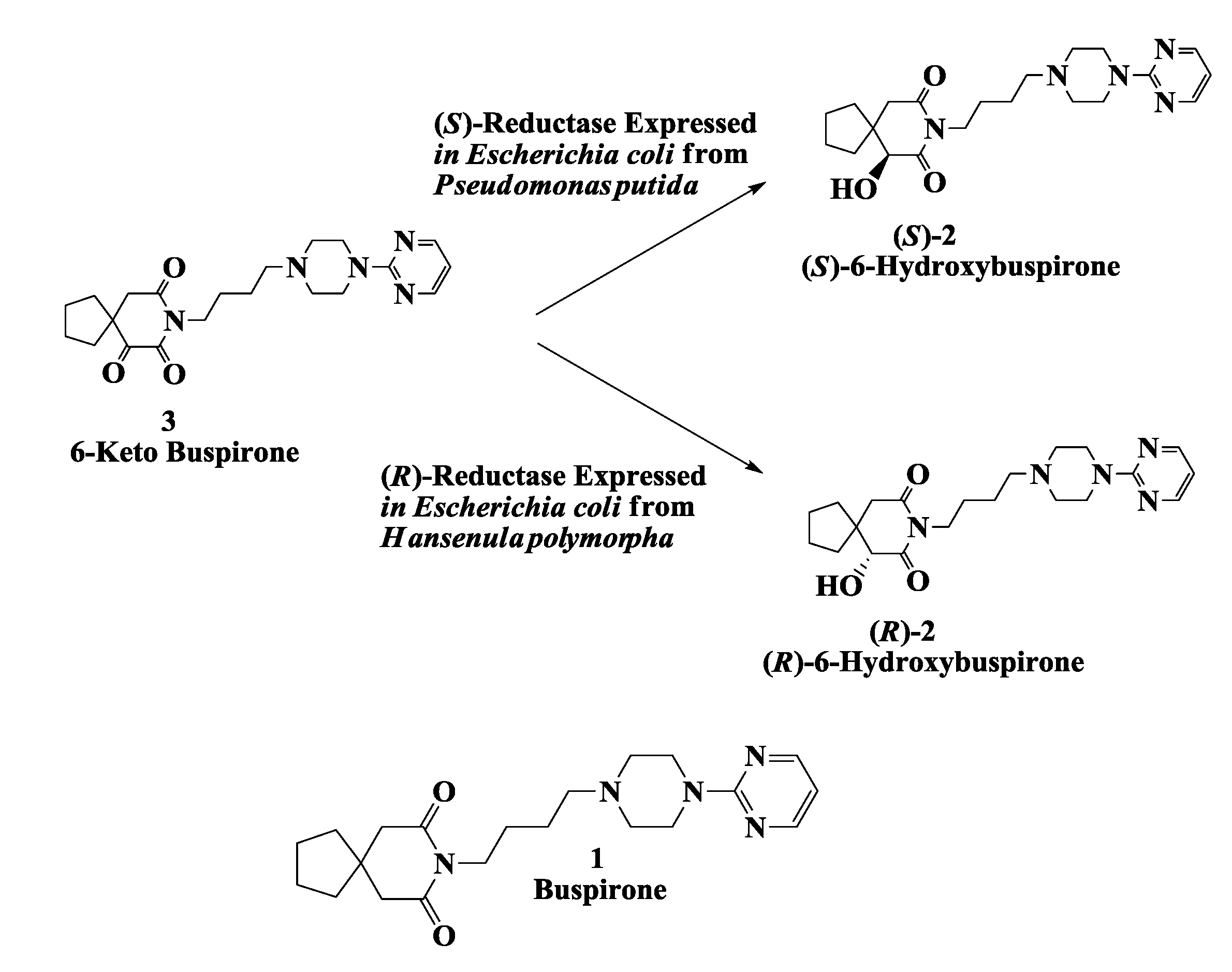
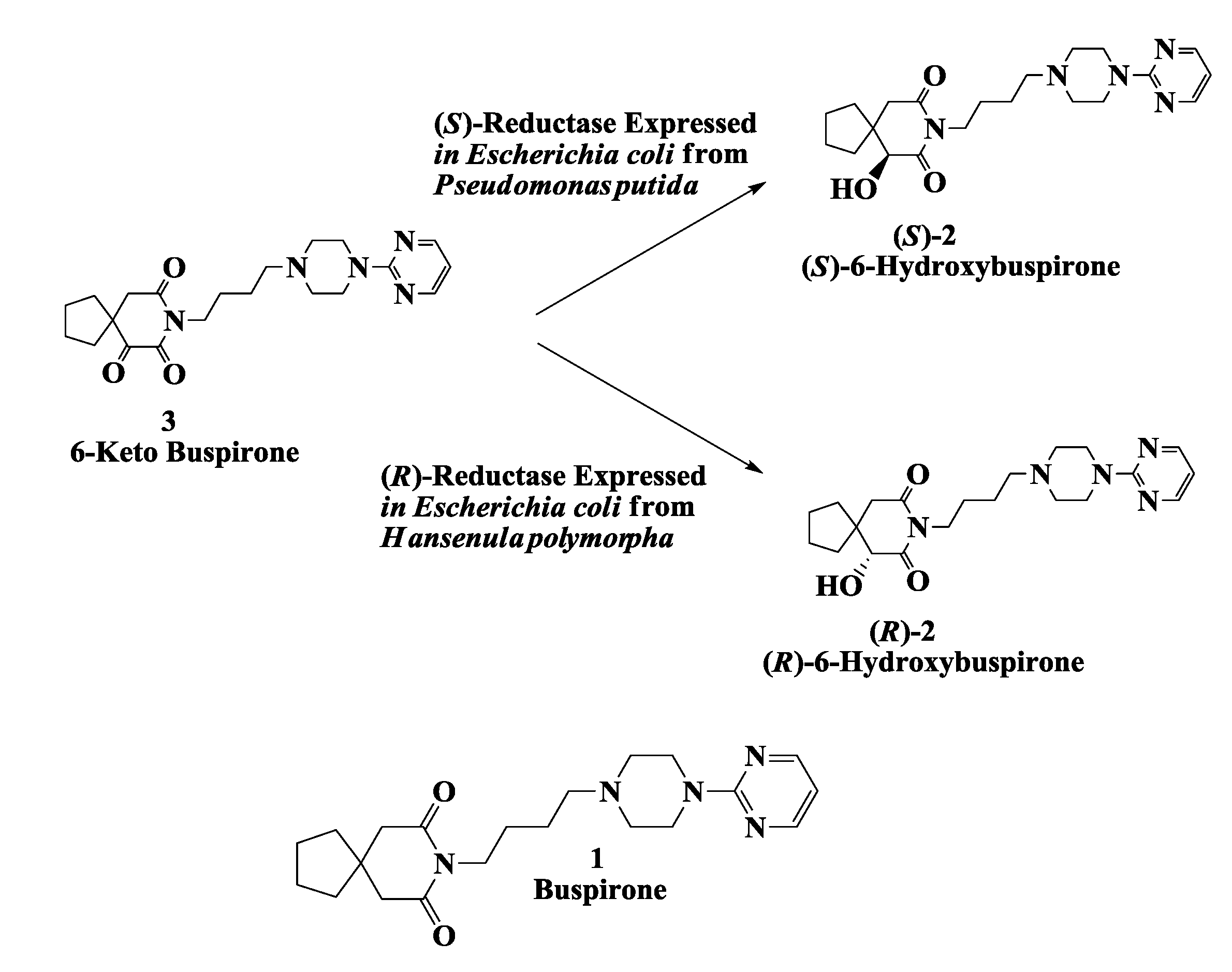
2.1. Hydroxy Buspirone (Antianxiety Drug): Enzymatic Preparation of 6-Hydroxybuspirone
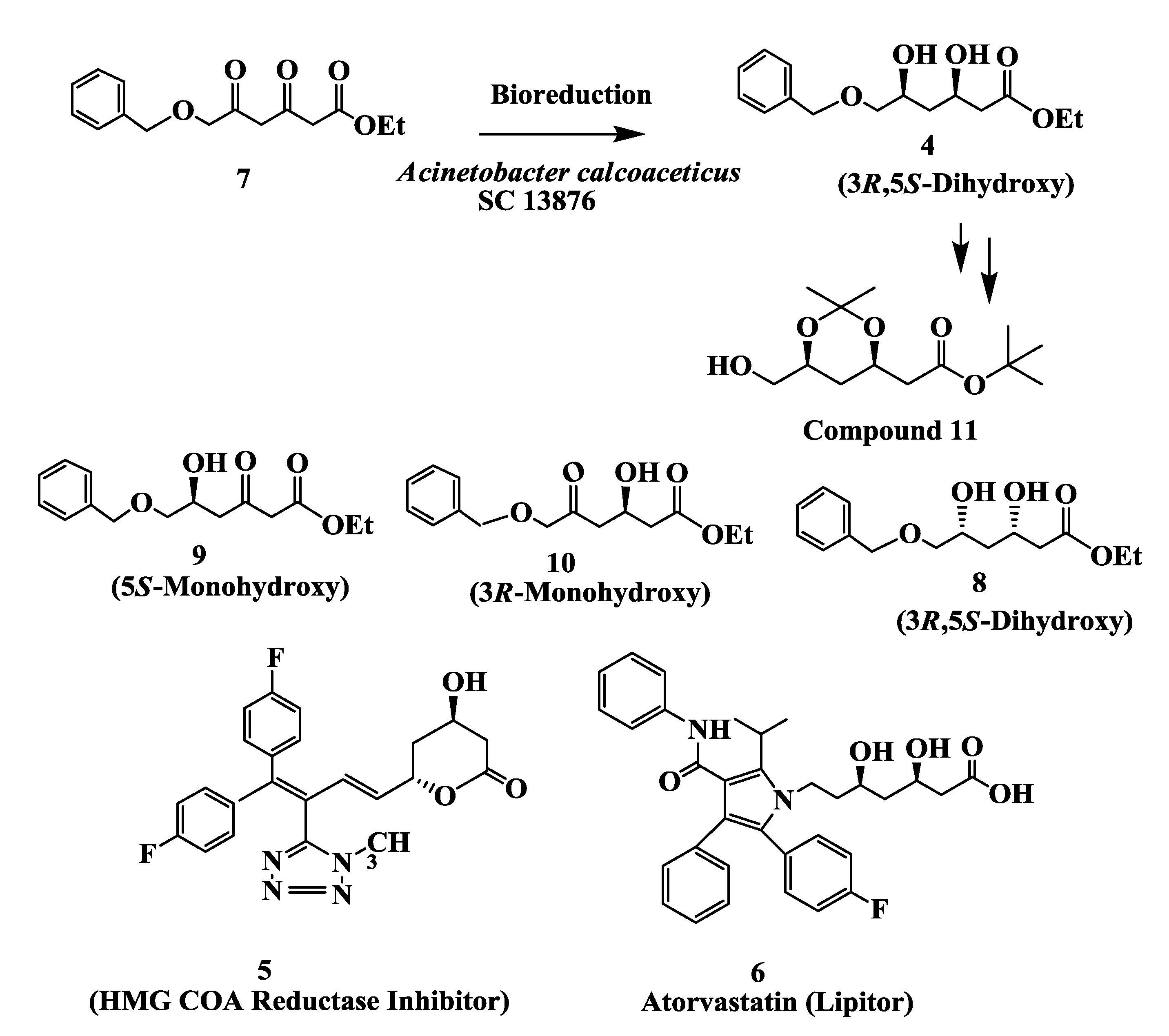
2.2. Cholesterol Lowering Agents: Enzymatic Preparation of (3S,5R)-Dihydroxy-6-(Benzyloxy) Hexanoic Acid, Ethyl Ester 4
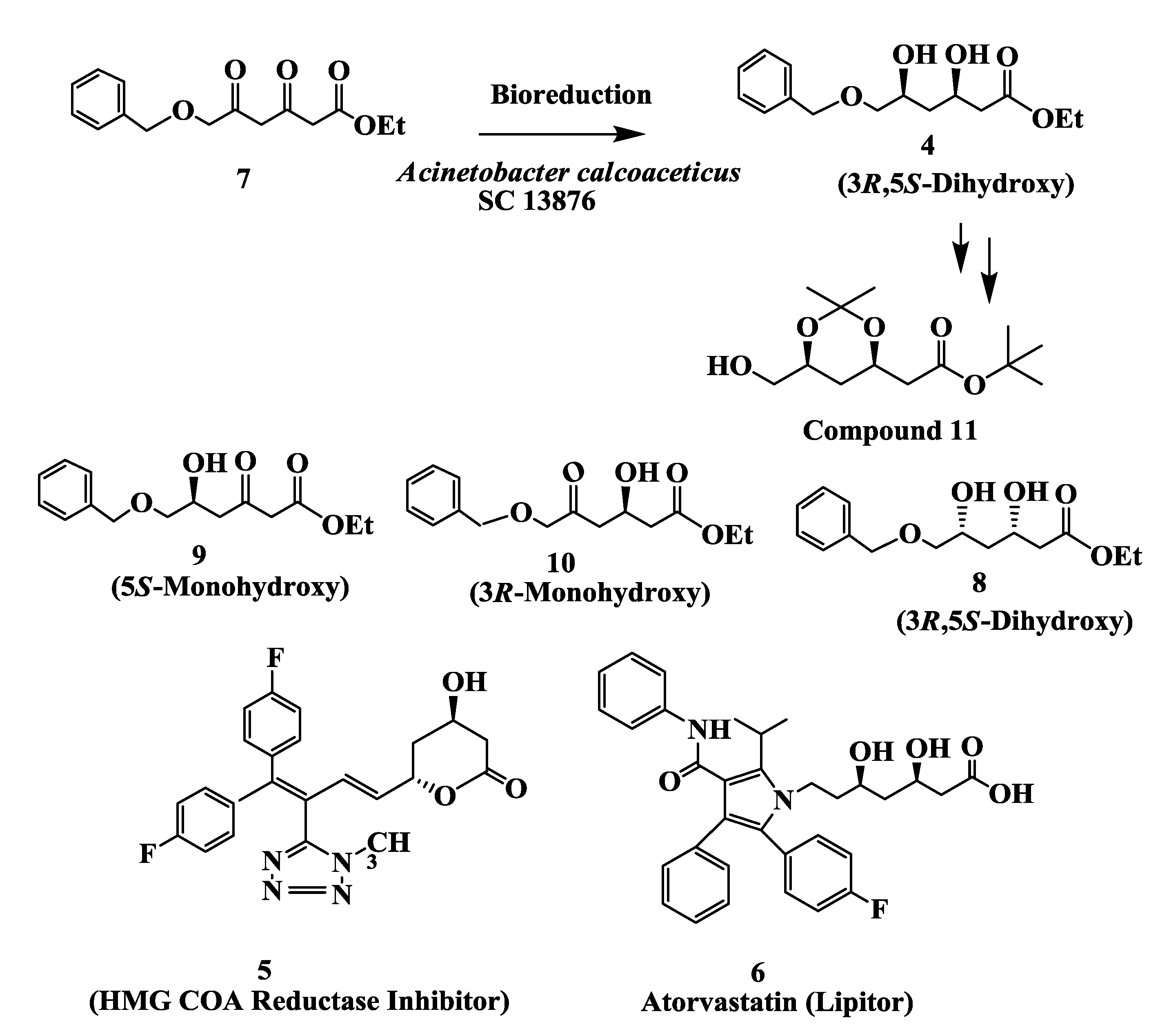
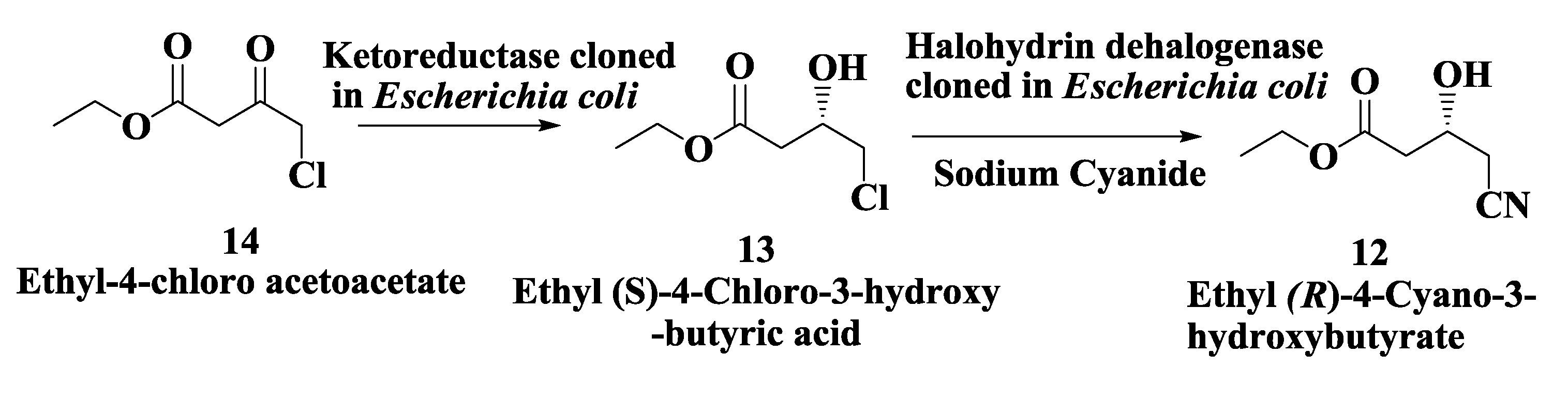
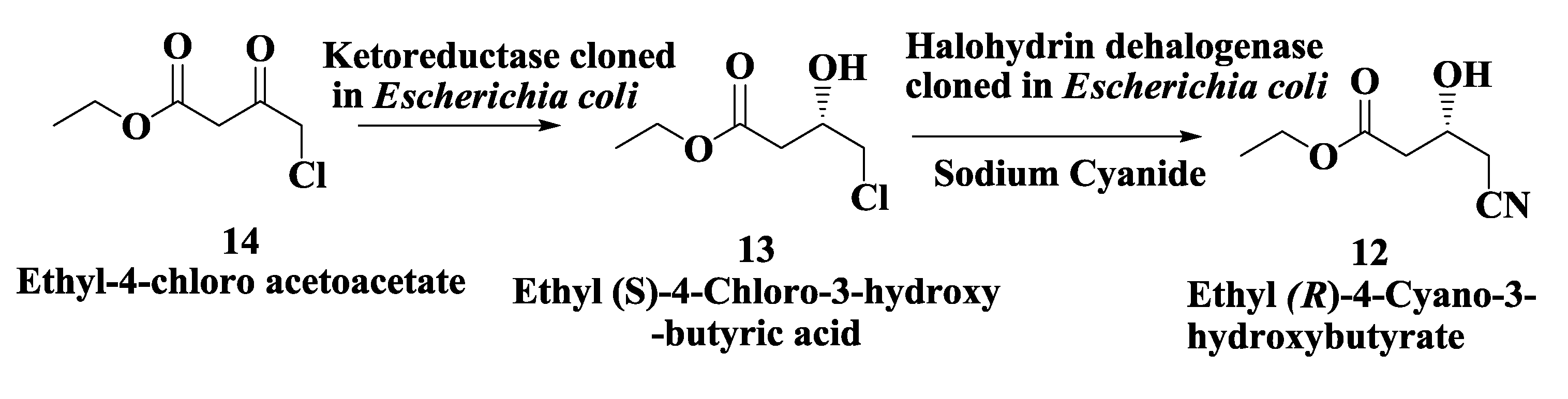
2.3. Atorvastatin: Enzymatic Preparation of (R)-4-Cyano-3-Hydroxybutyrate
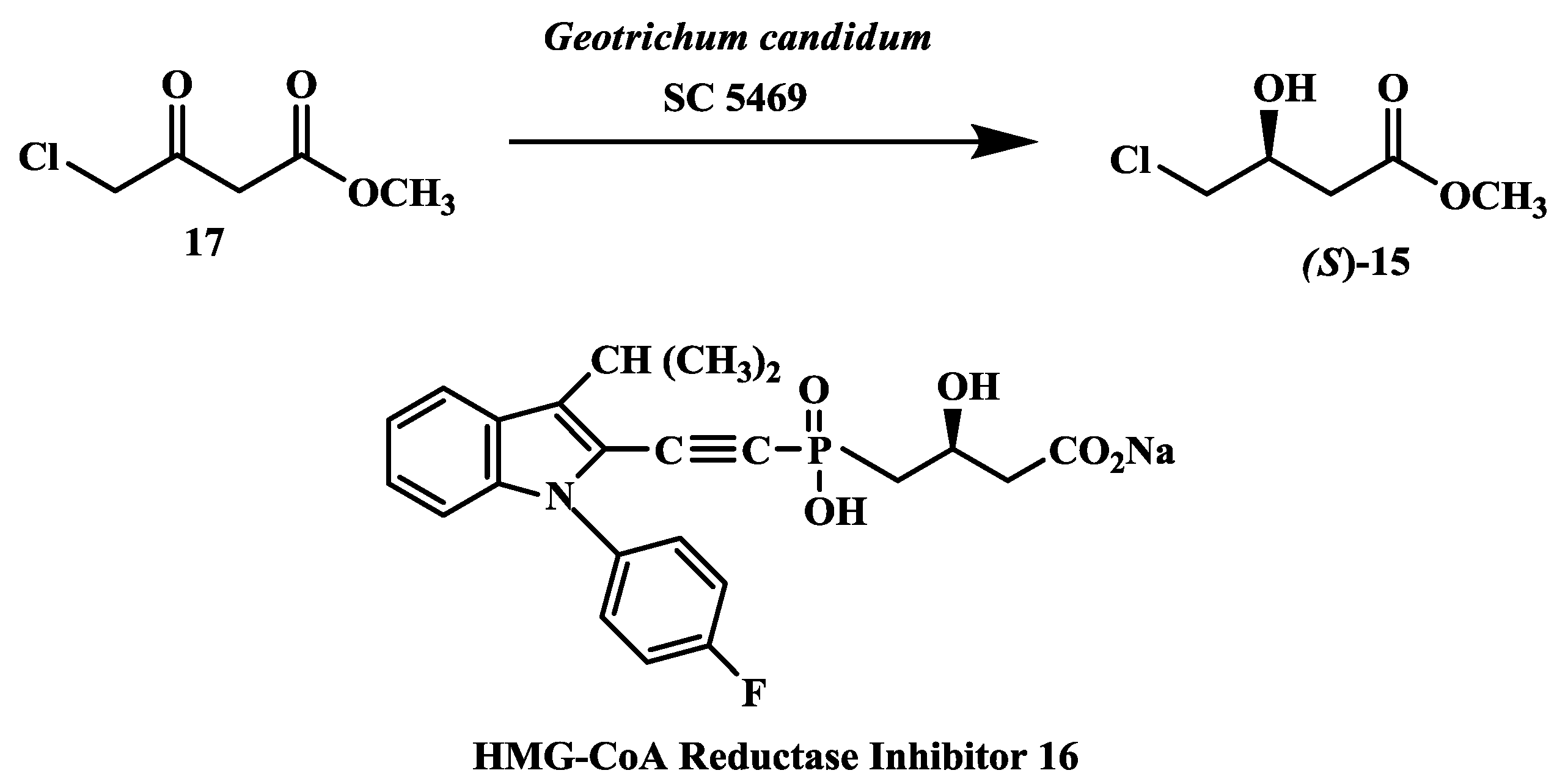
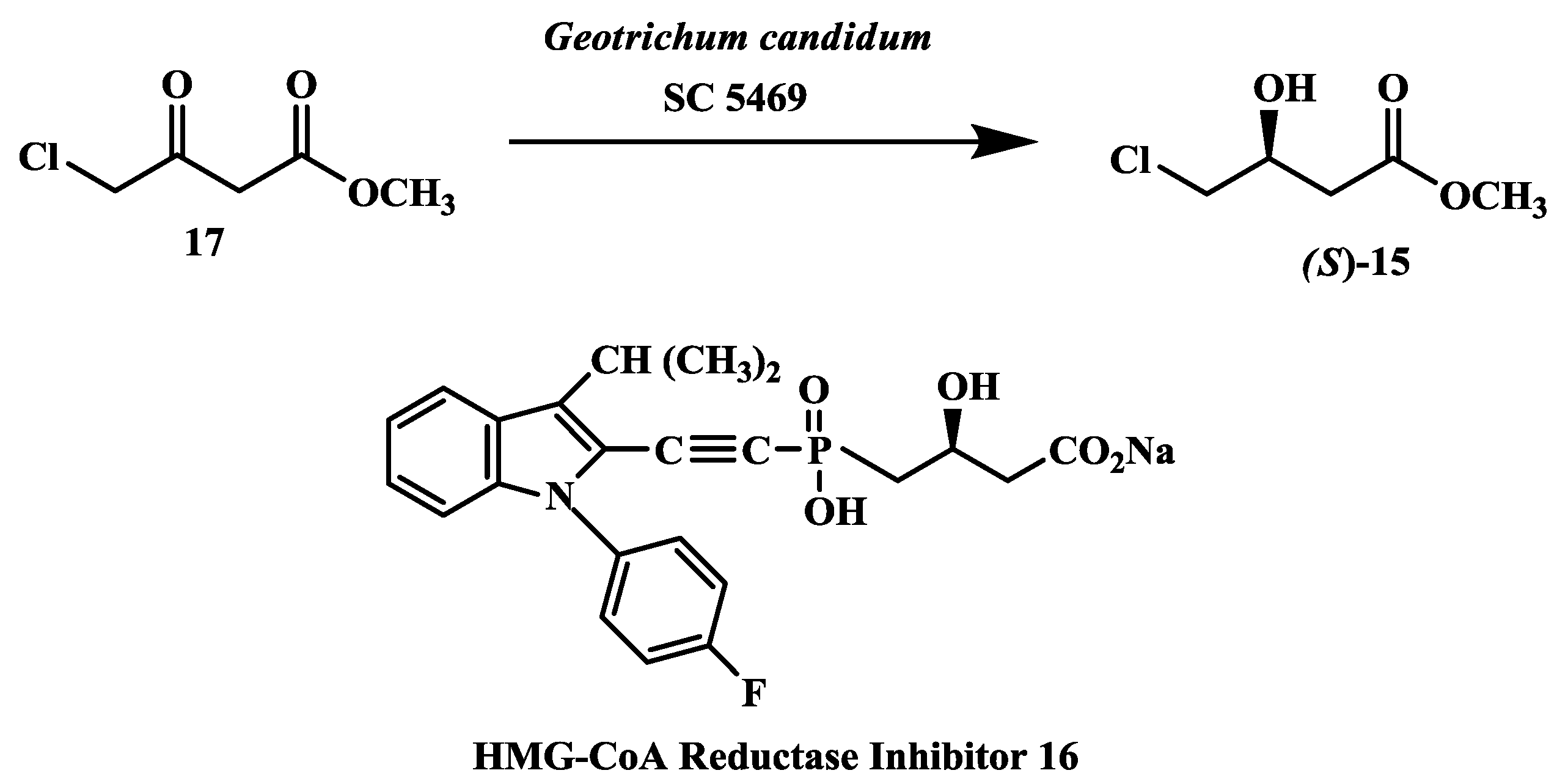
2.4. Preparation of (S)-4-Chloro-3-Hydroxybutanoic Acid Methyl Ester
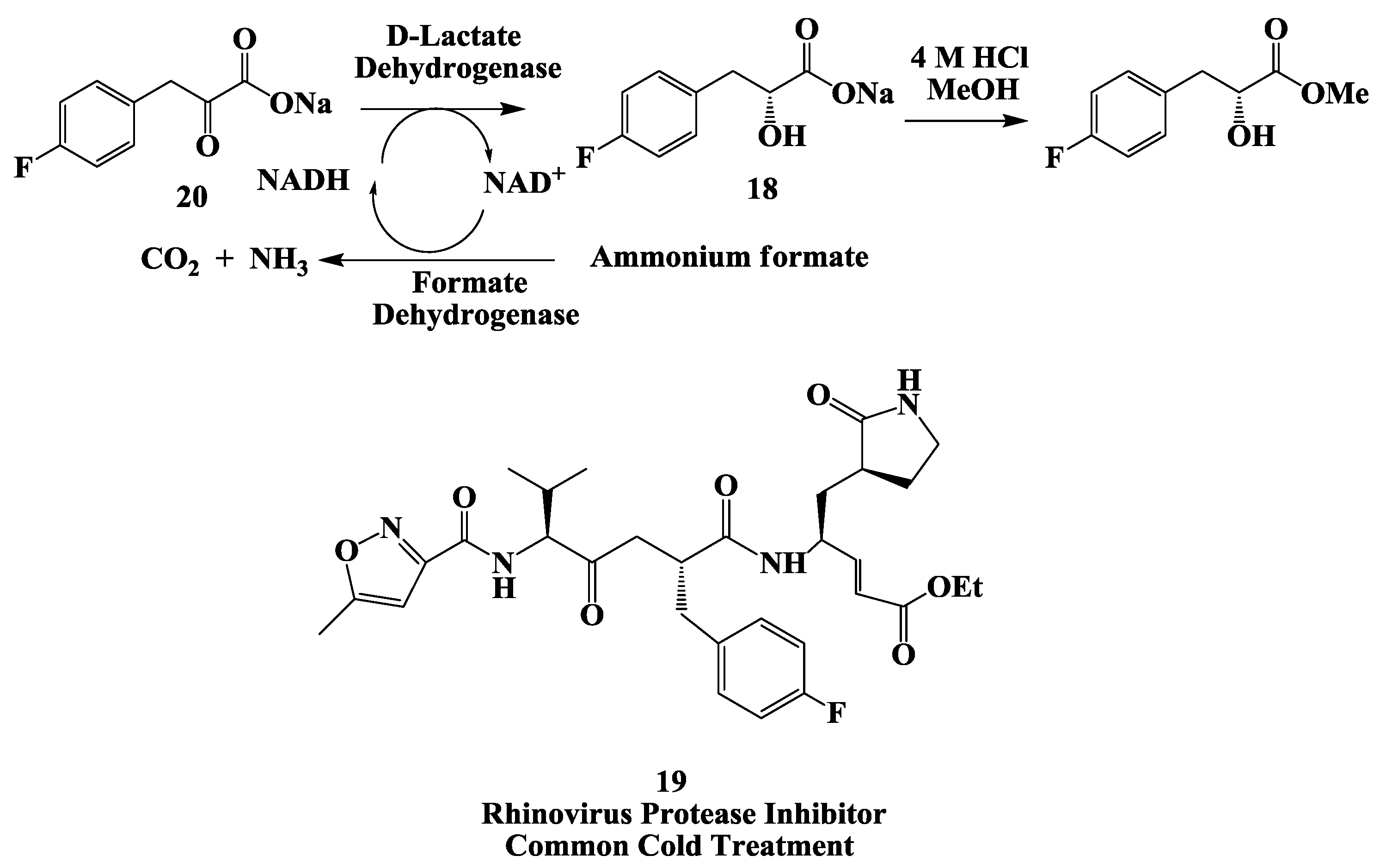
2.5. Rhinovirus Protease Inhibitor: Enzymatic Process for the Preparation of (R)-3-(4-Fluorophenyl)-2-Hydroxy Propionic Acid

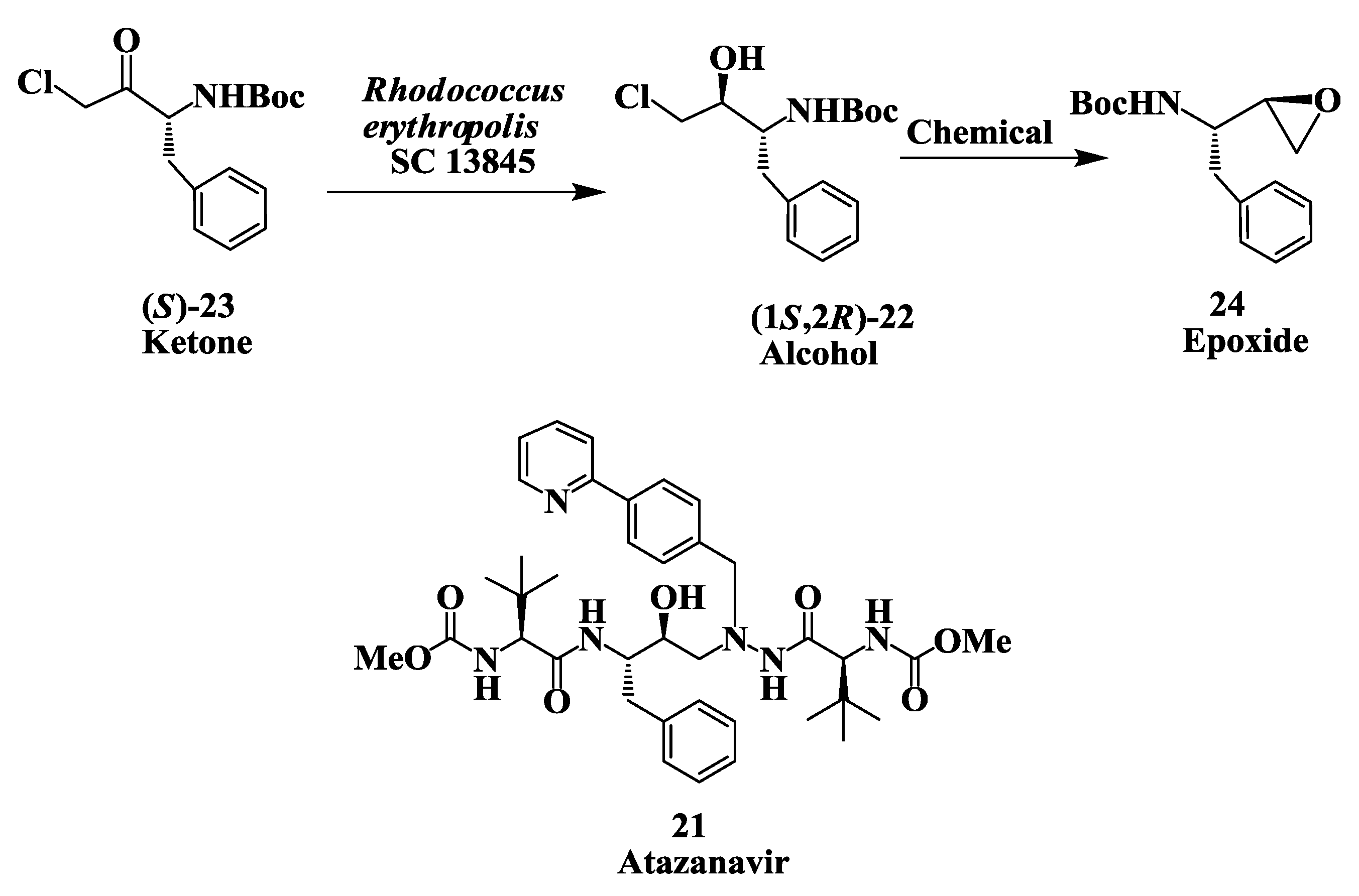
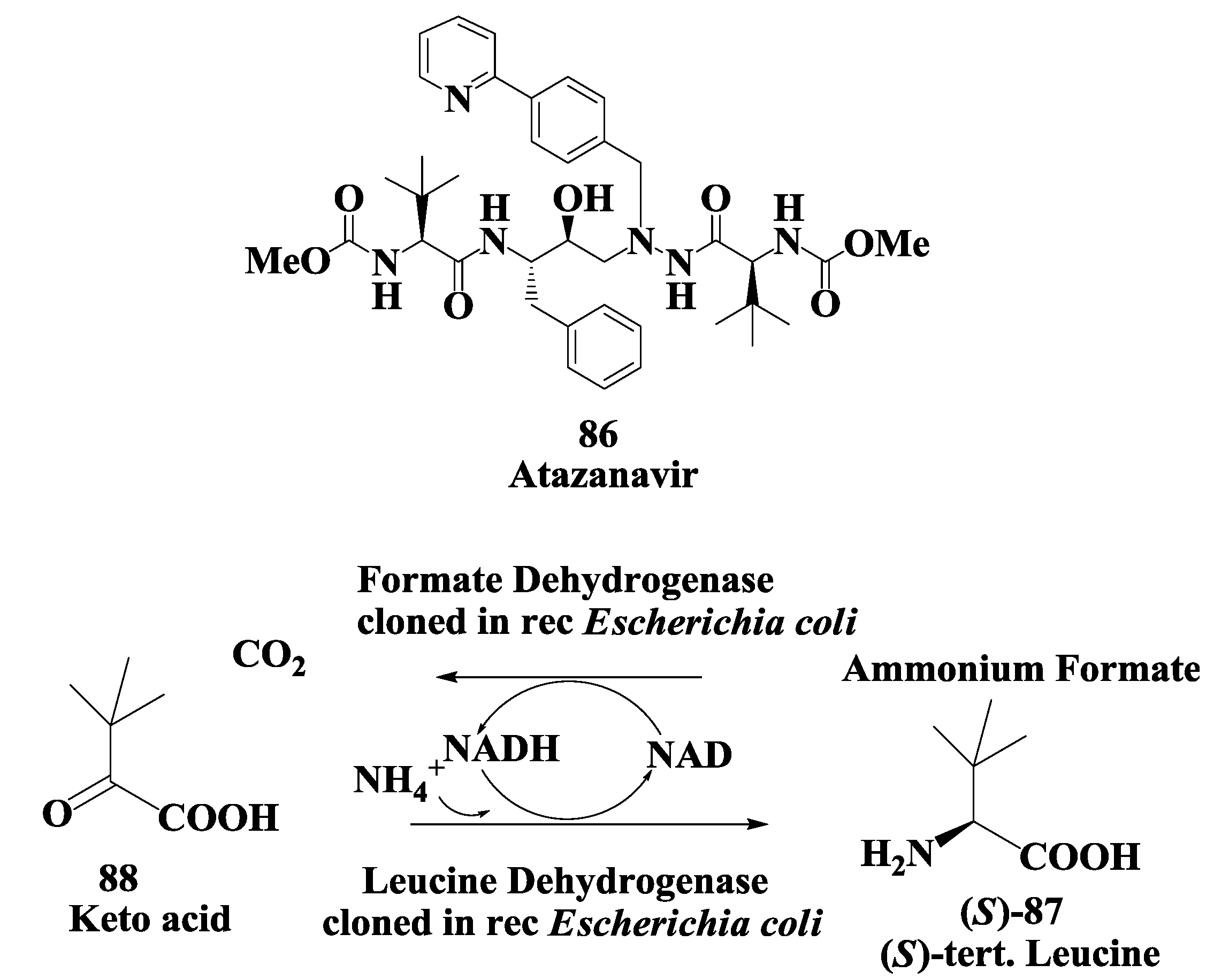
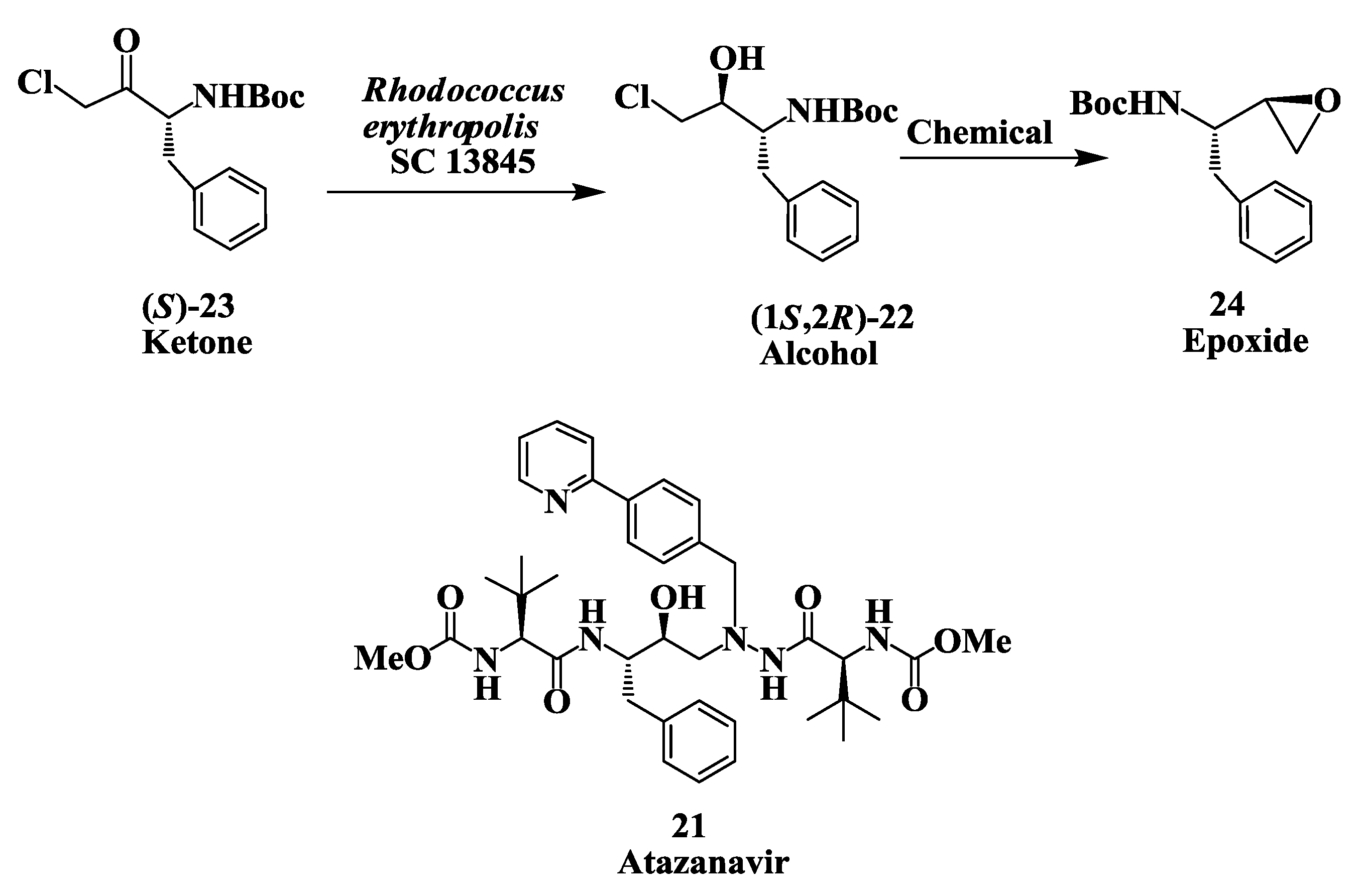
2.6. Enzymatic Preparation of Chiral Intermediates for Atazanavir
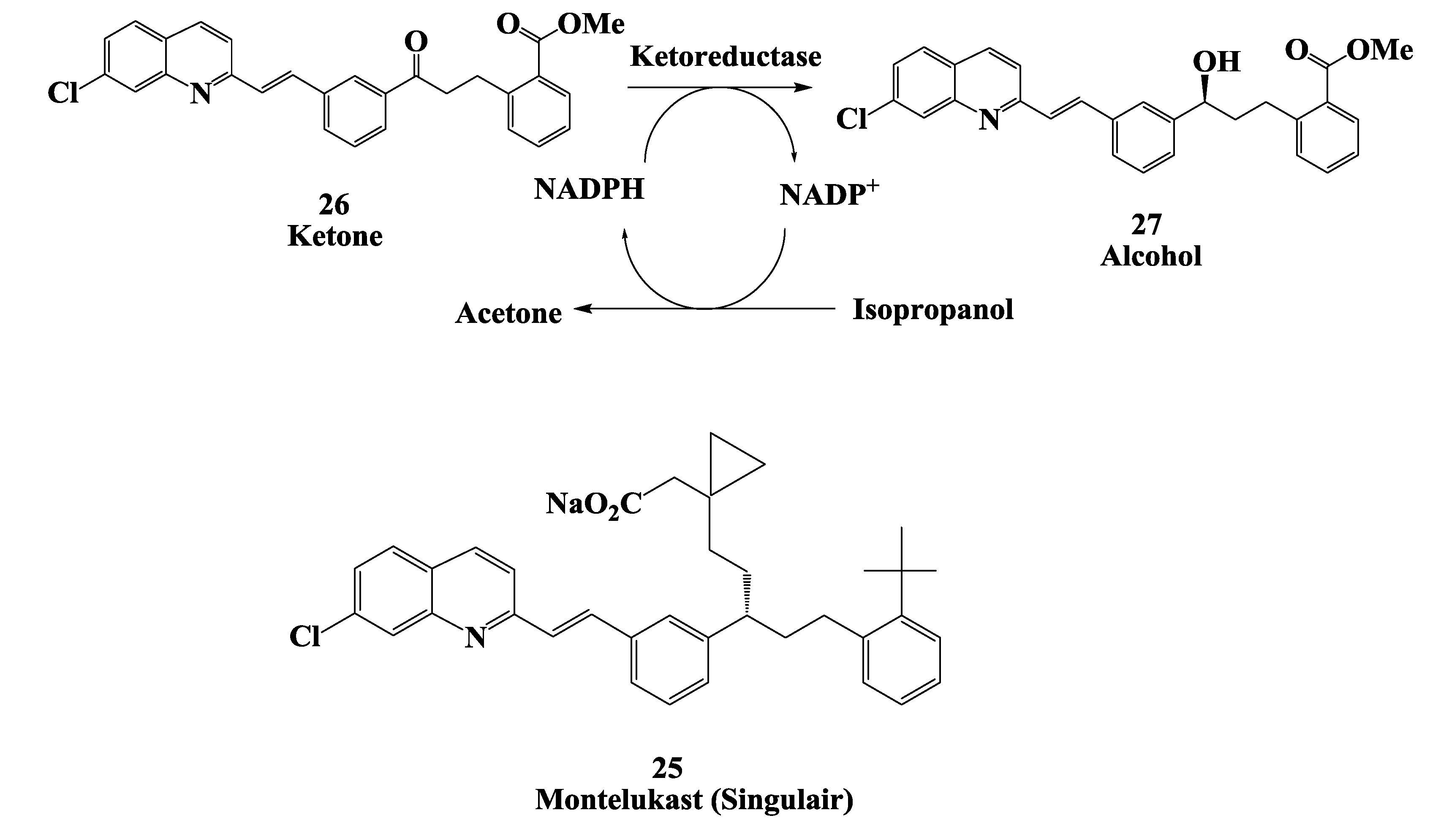
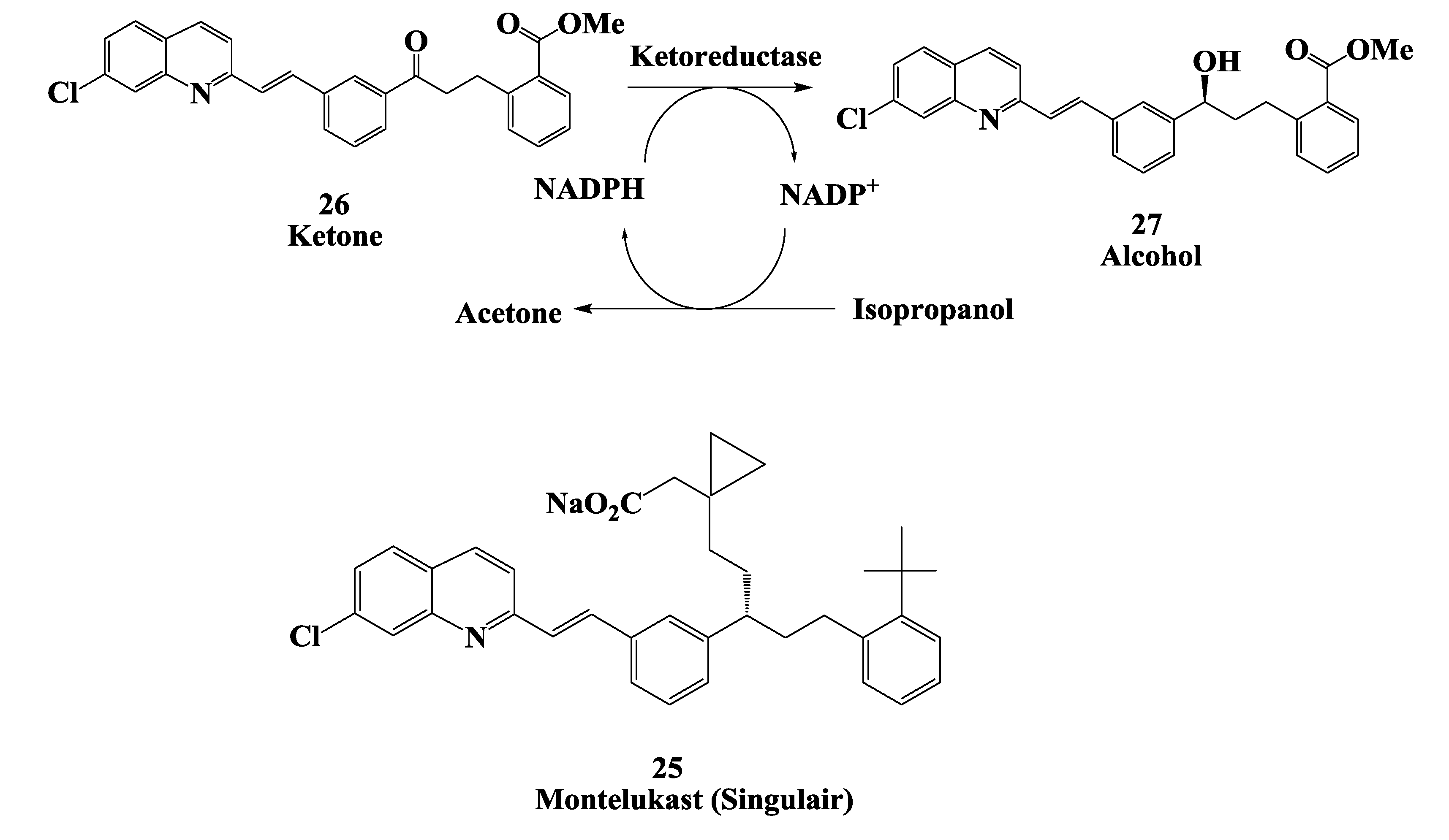
2.7. Enzymatic Reduction Process for Synthesis of Montelukast Intermediate
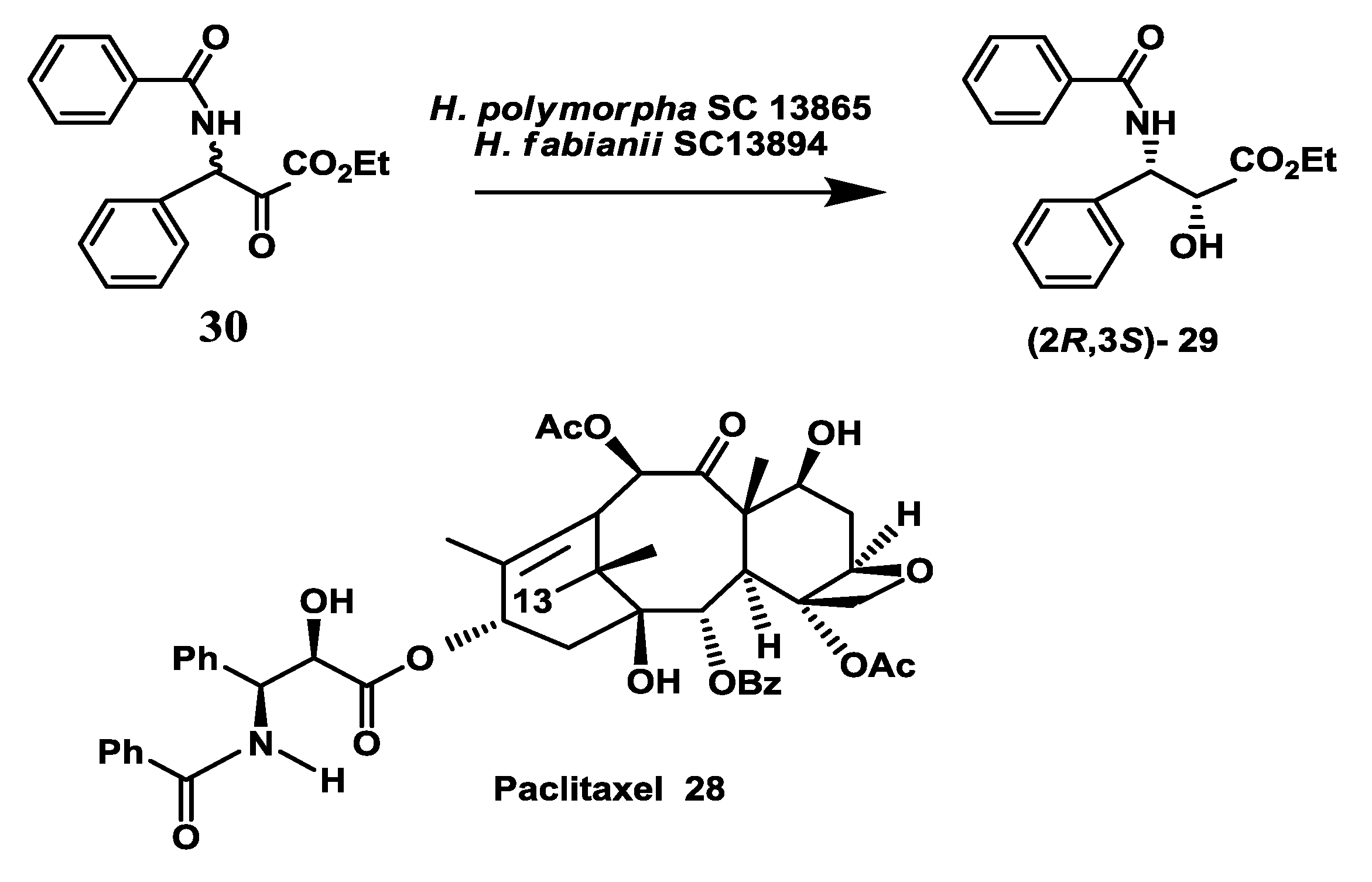
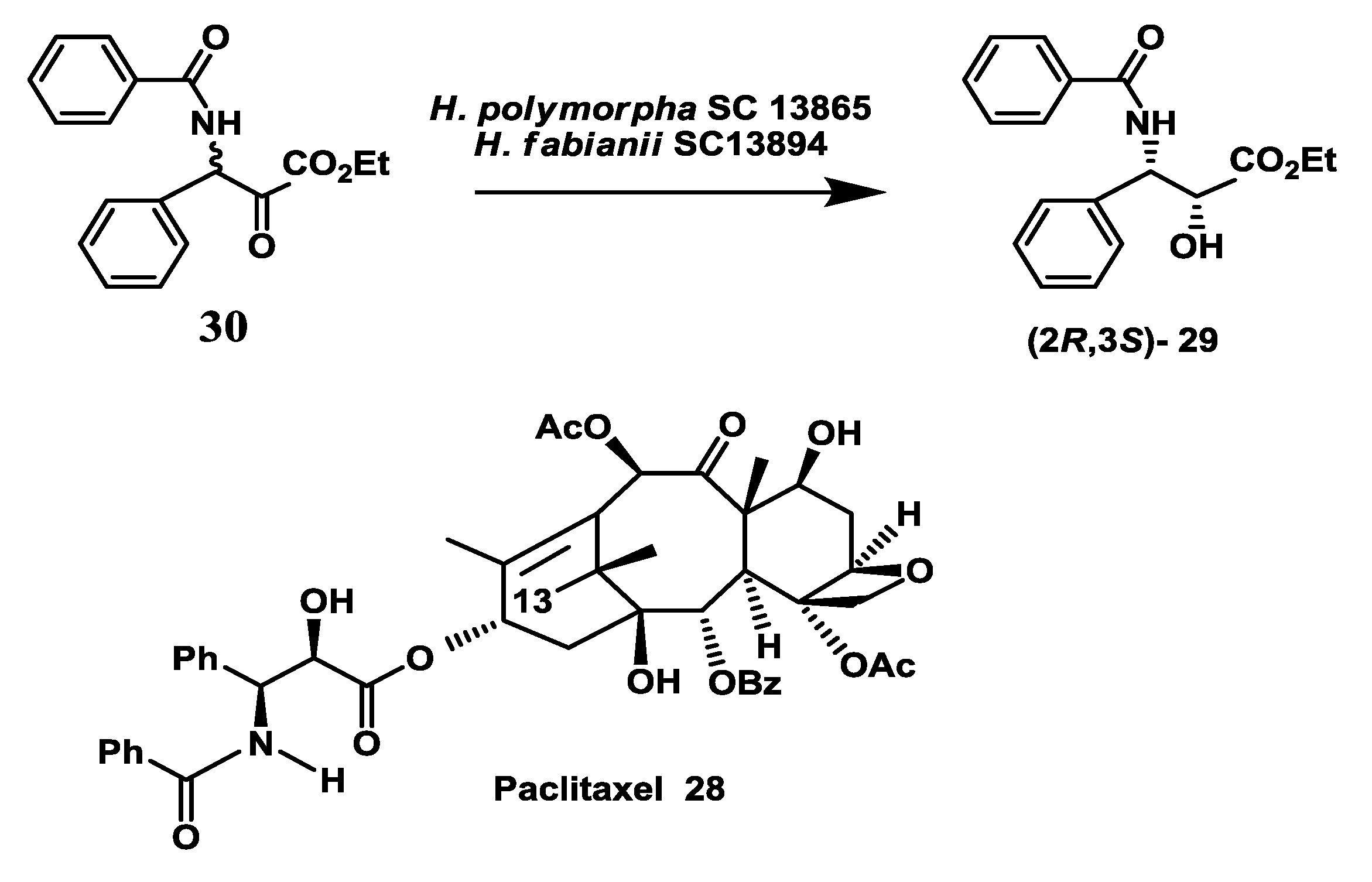
2.8. Anticancer Drug: Enzymatic Preparation of C-13 Paclitaxel Side-Chain Synthon
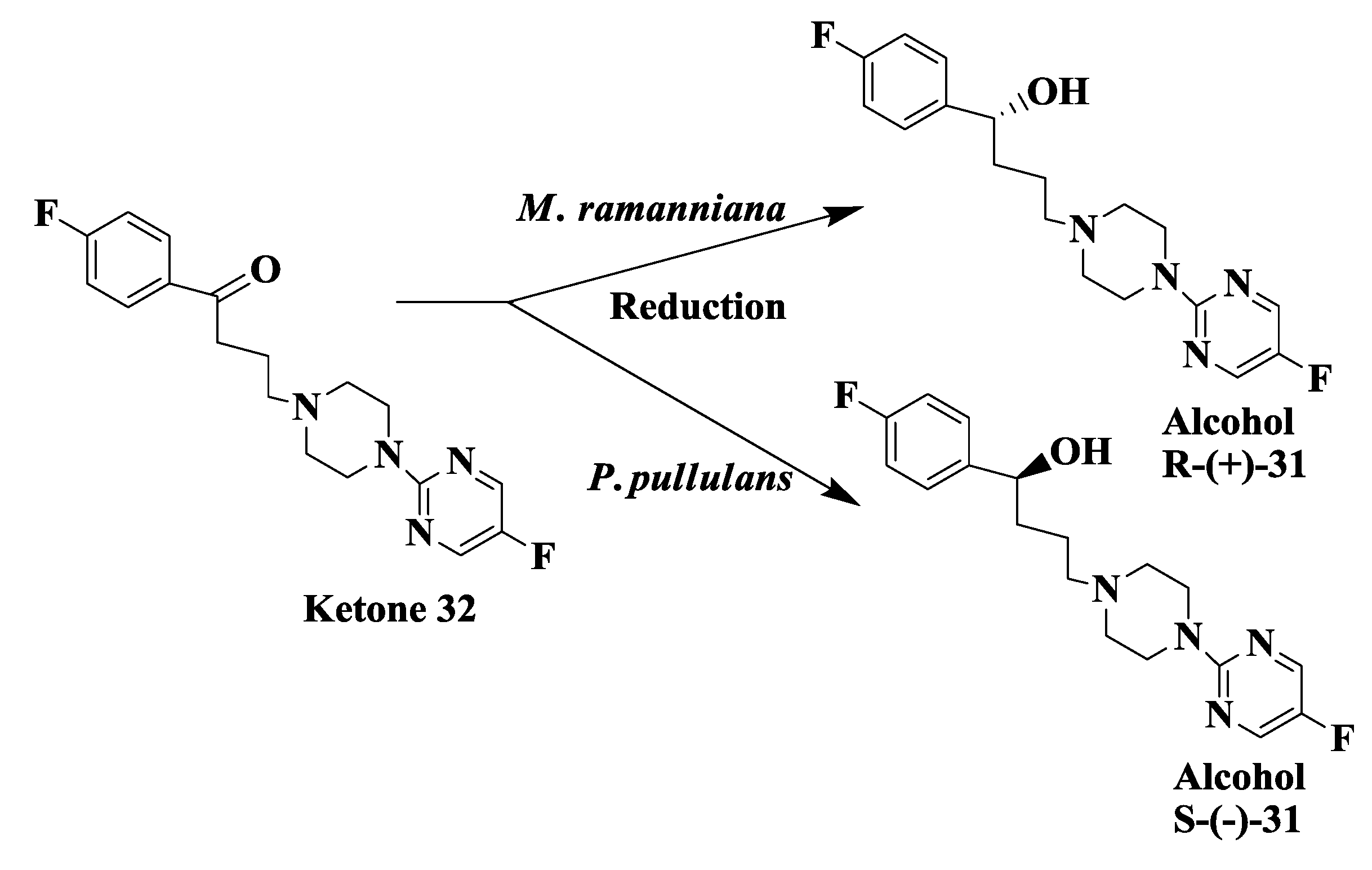
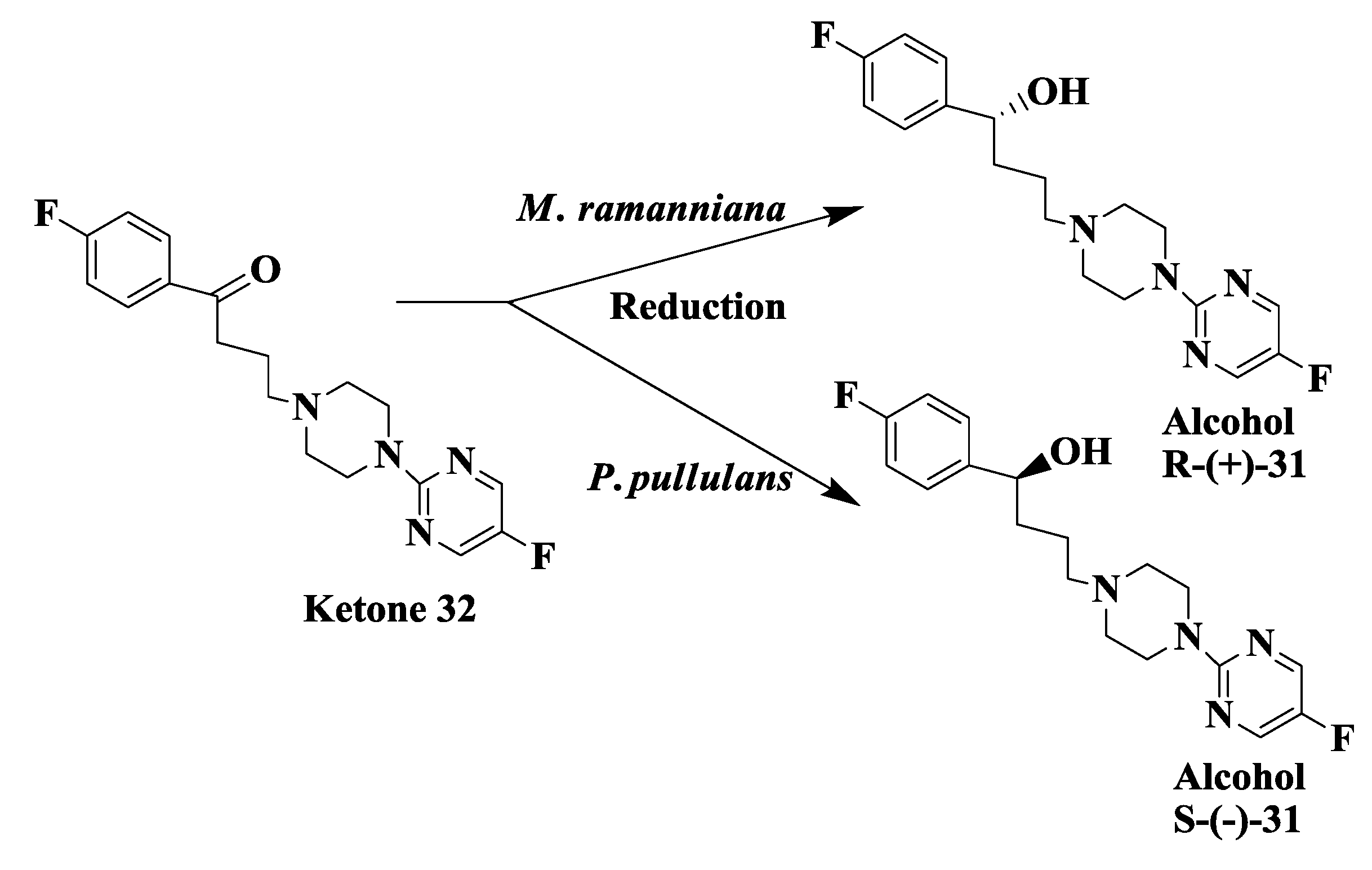
2.9. Antipsychotic Drug: Enzymatic Reduction of 1-(4-Fluorophenyl)4-[4-(5-Fluoro-2-Pyrimidinyl)1-Piperazinyl]-1-Butanone
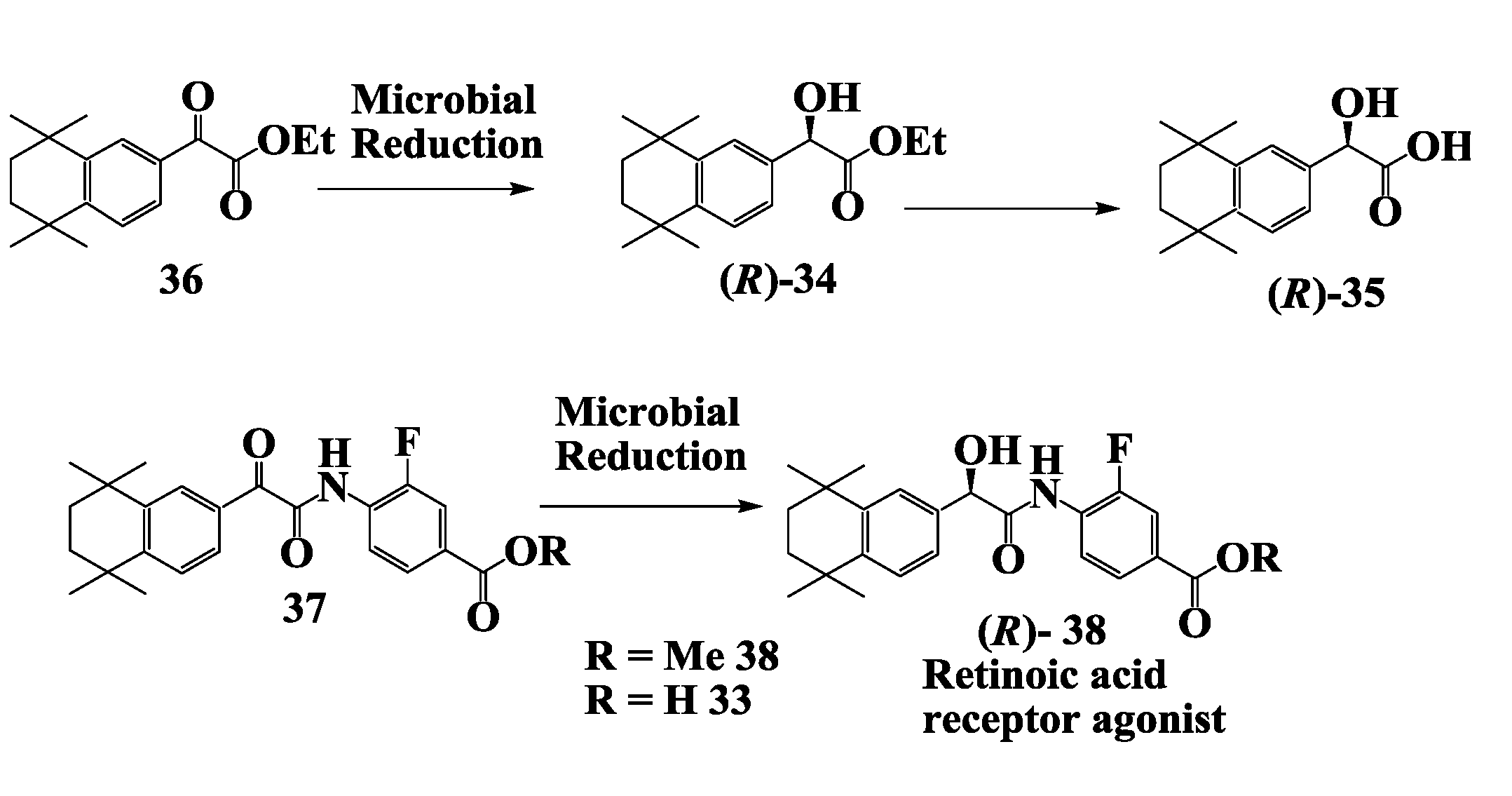
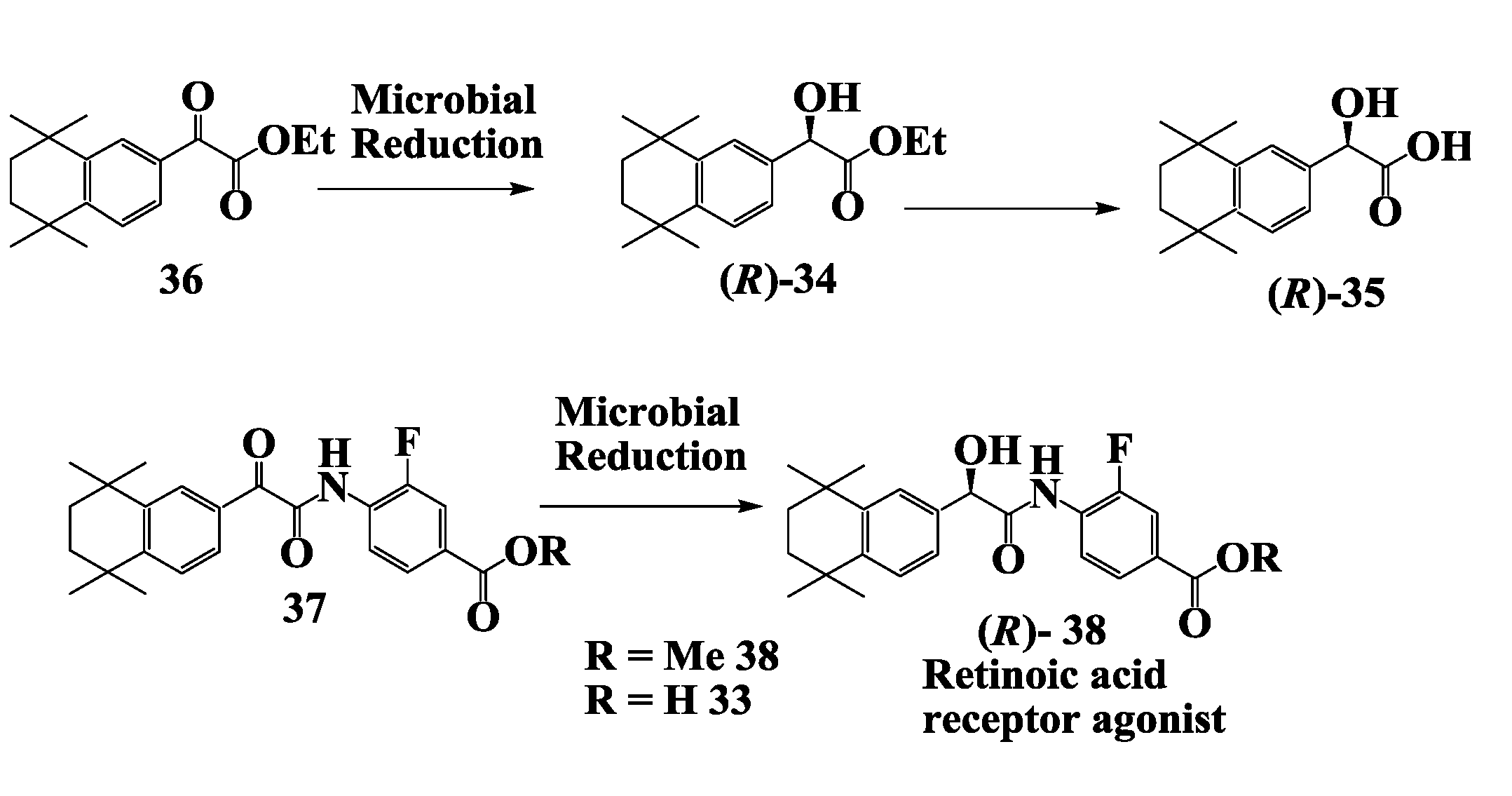
2.10. Retinoic Acid Receptpor Agonist: Enzymatic Preparation of 2-(R)-Hydroxy-2-(1',2',3',4'-Tetrahydro-1',1',4',4'-Tetramethyl-6'-Naphthalenyl)Acetate
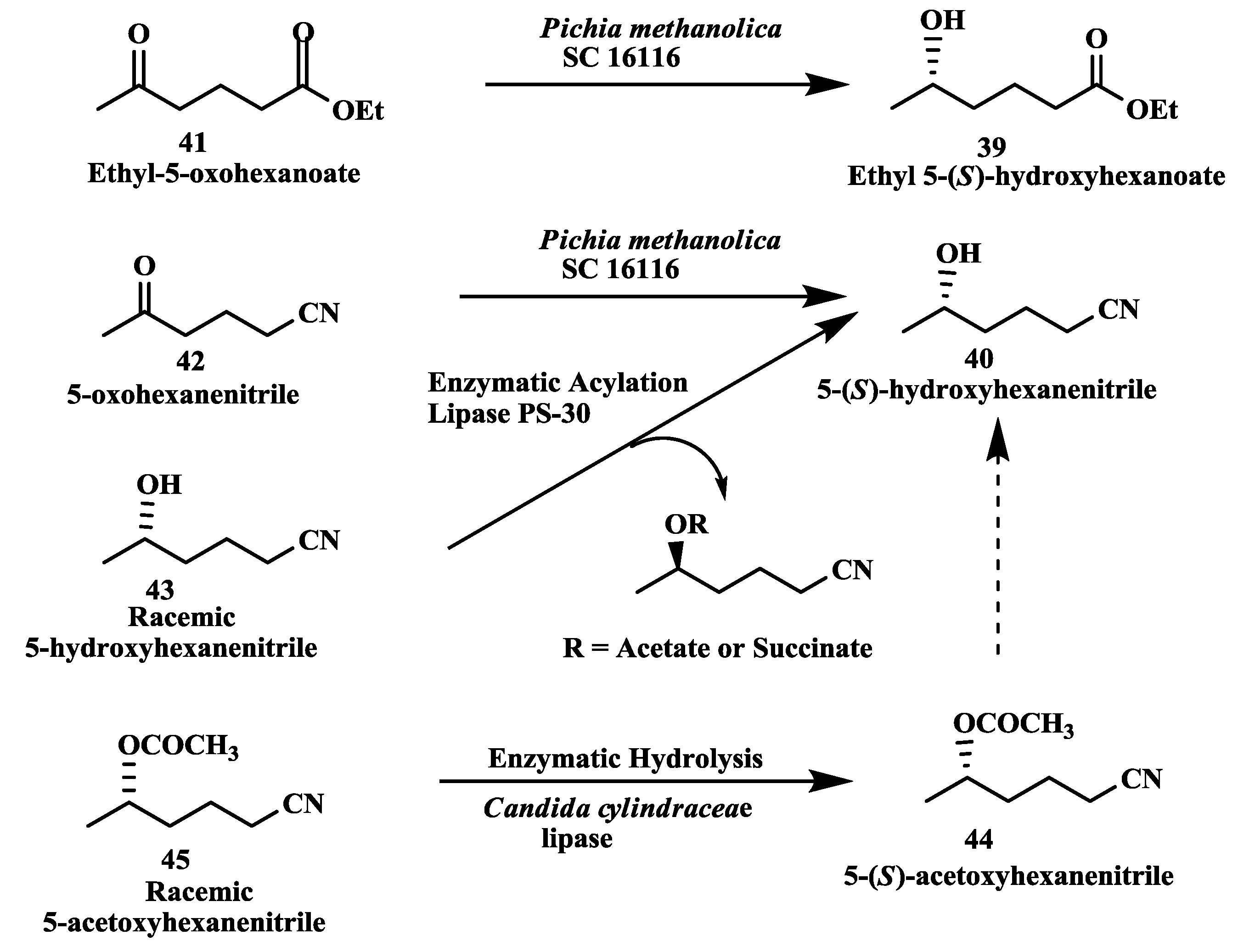
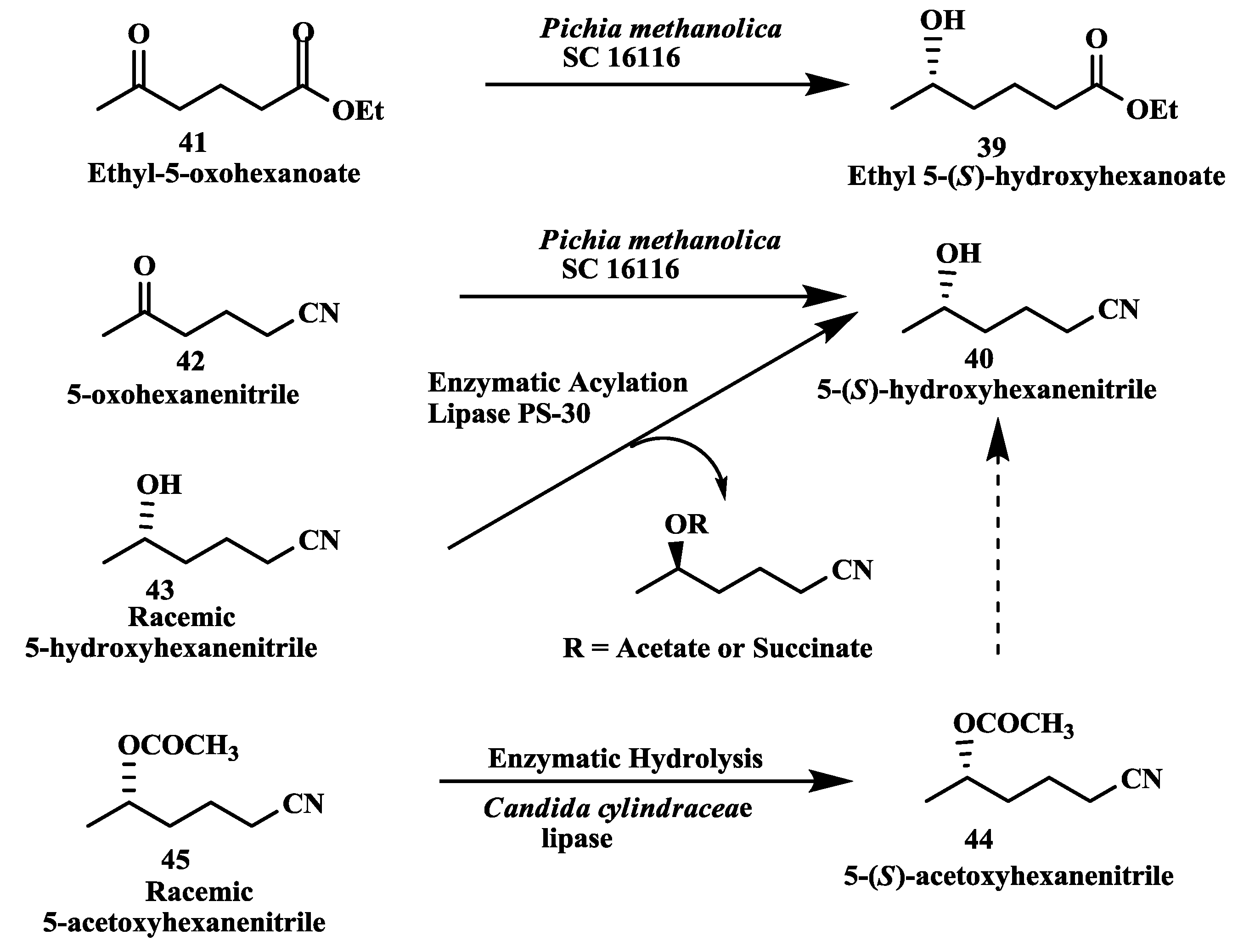
2.11. Anti-Alzheimer’s Drugs: Enzymatic Reduction of 5-Oxohexanoate and 5-Oxohexanenitrile
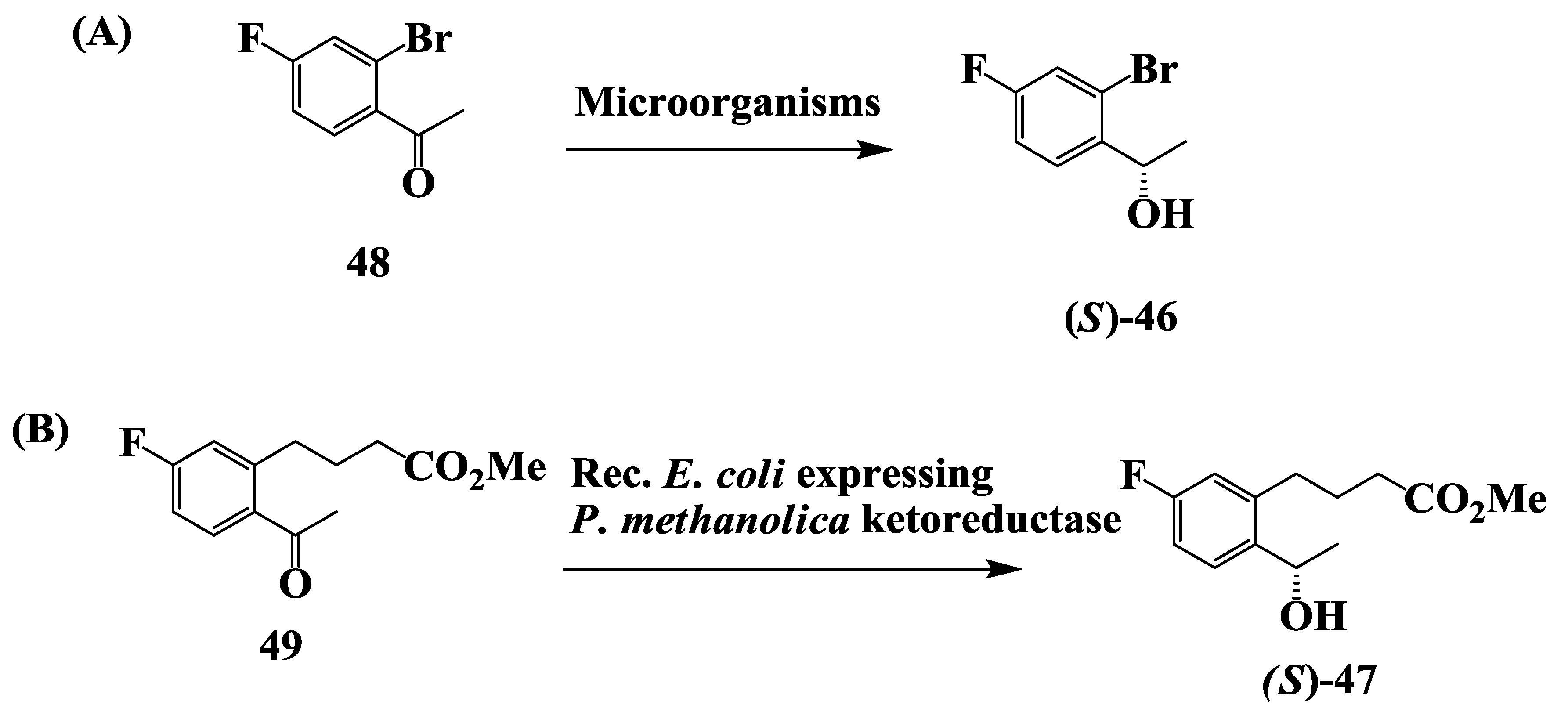
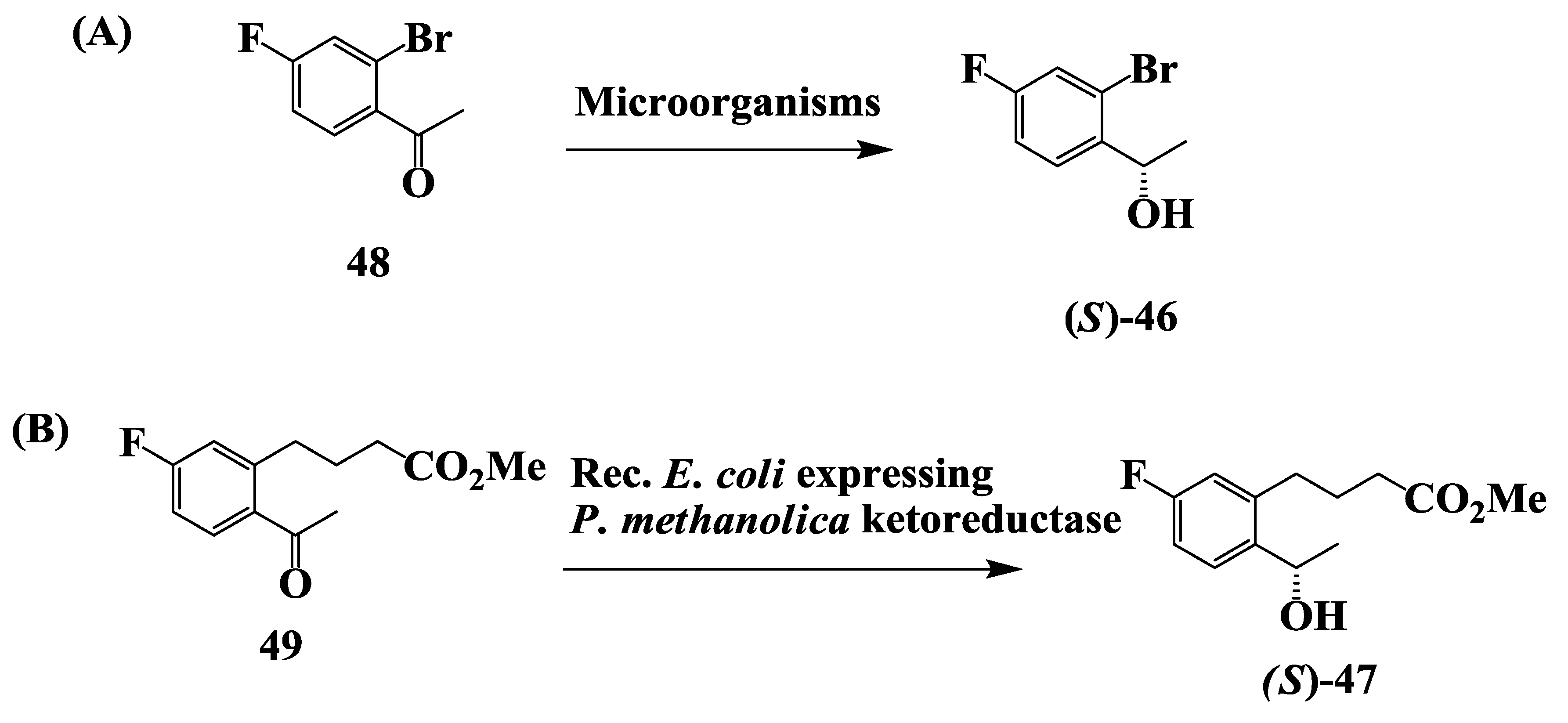
2.12. Enantioselective Microbial Reduction of Substituted Acetophenone
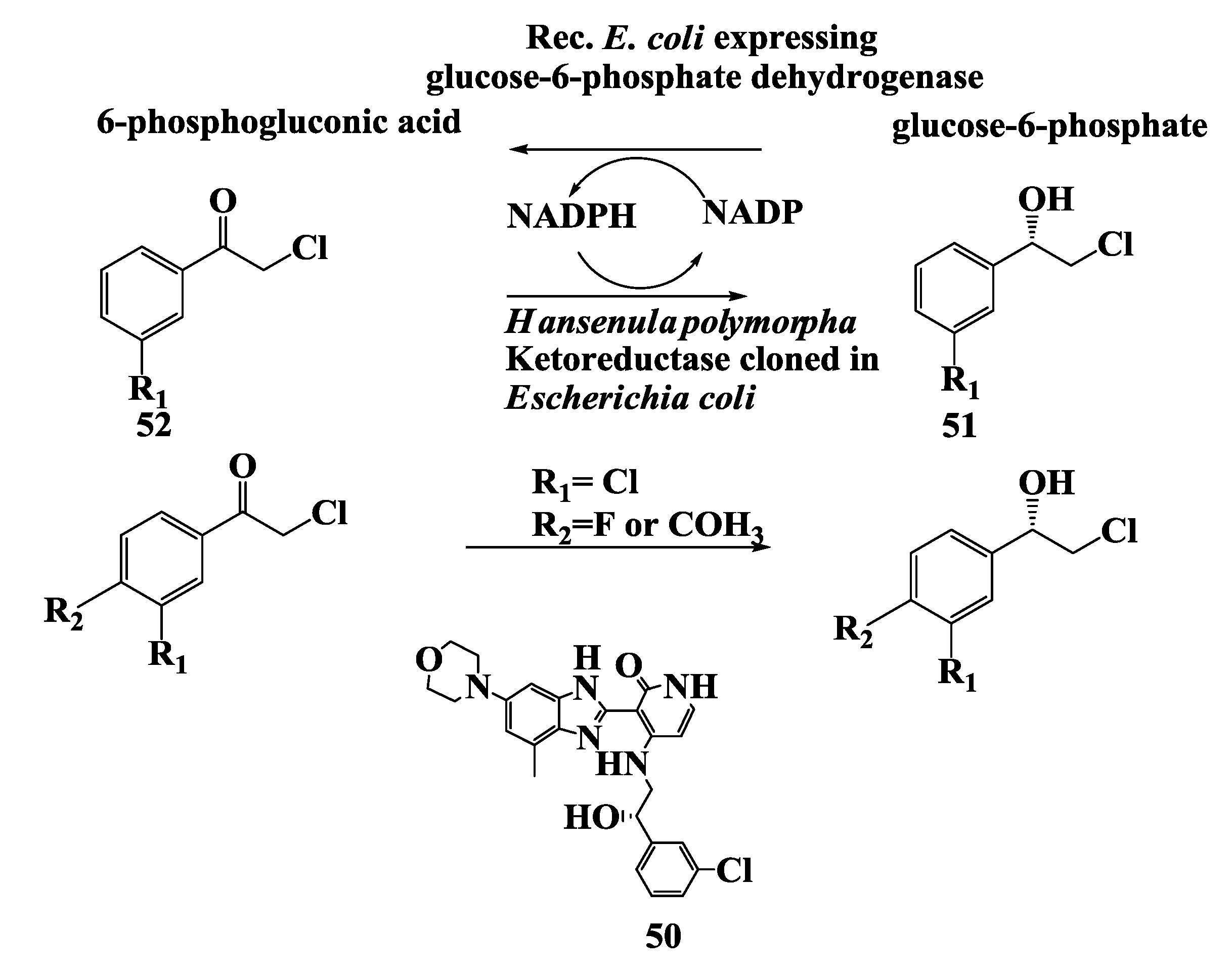
2.13. Anticancer Drug: Enzymatic Preparation of (S)-2-Chloro-1-(3-Chlorophenyl)Ethanol
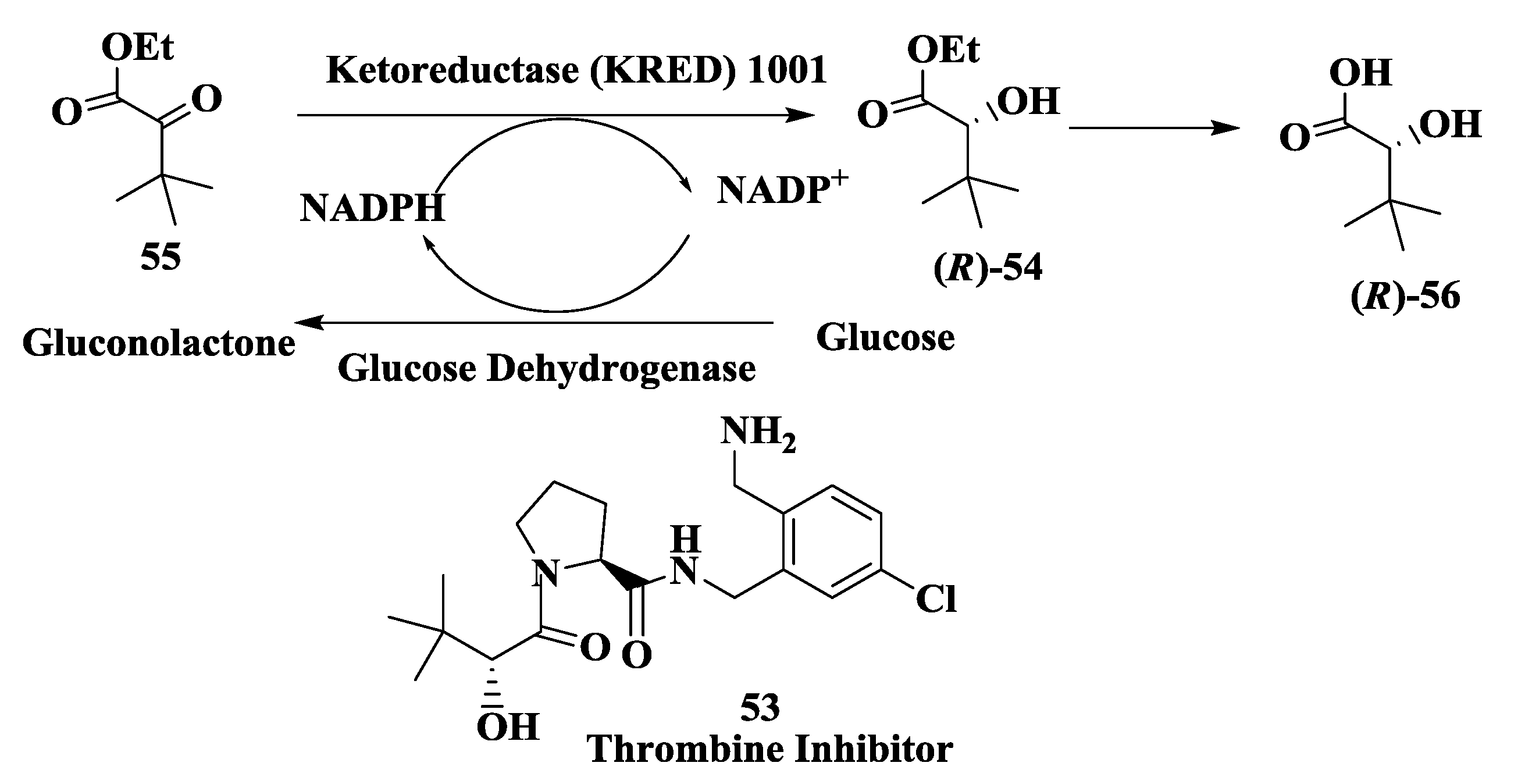
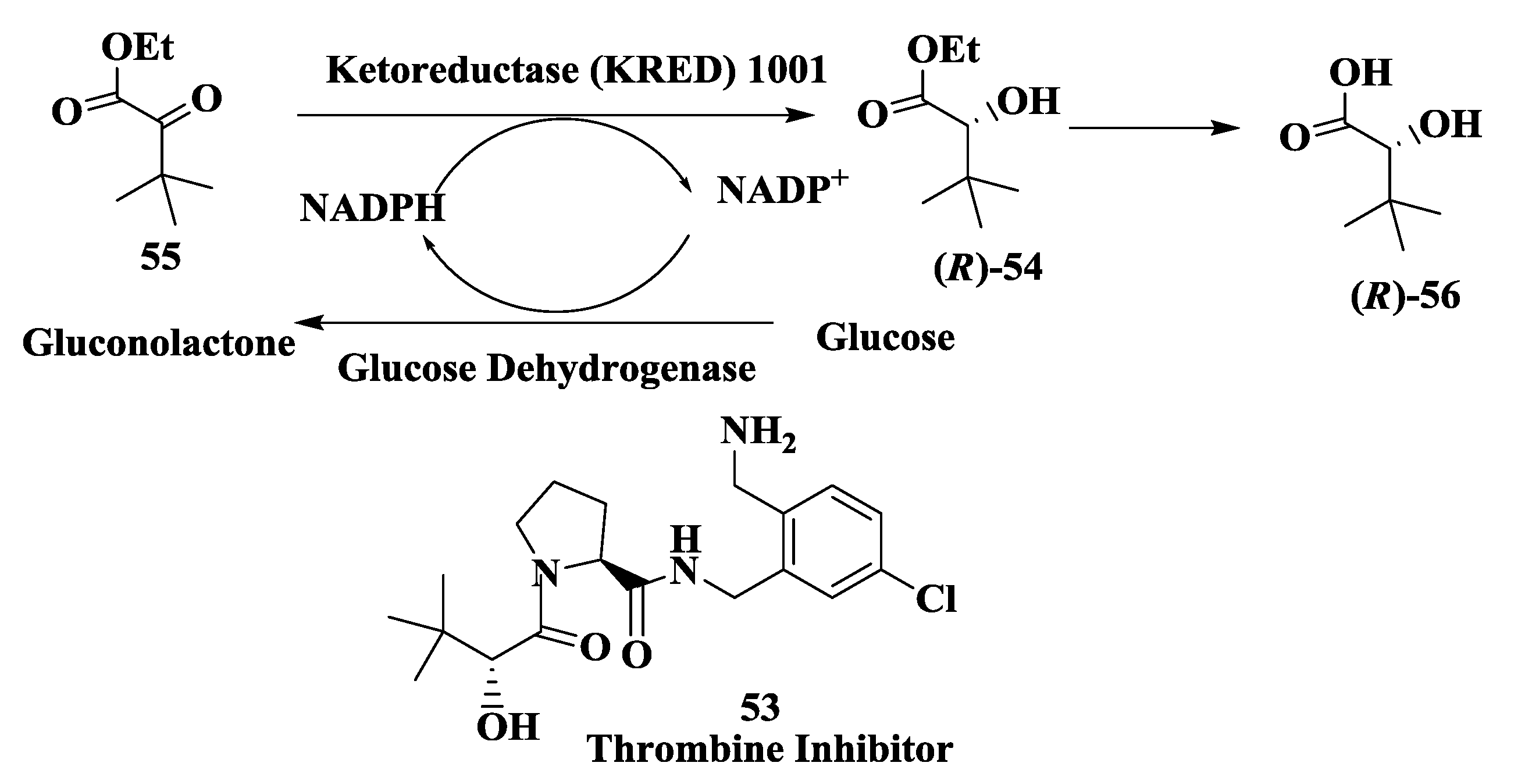
2.14. Thrombin Inhibitor: Enzymatic Preparation of (R)-2-Hydroxy-3,3-Dimethylbutanoic Acid
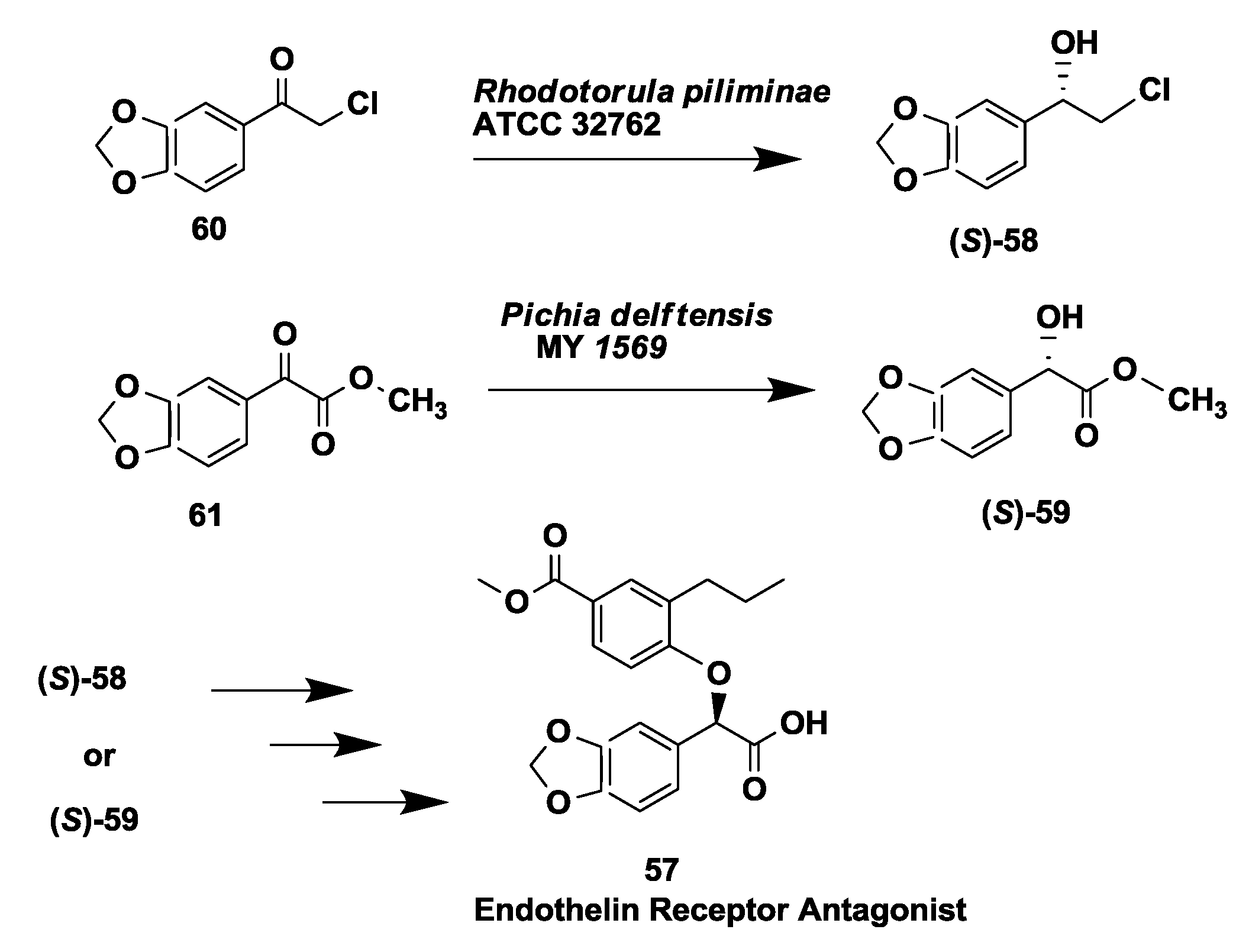
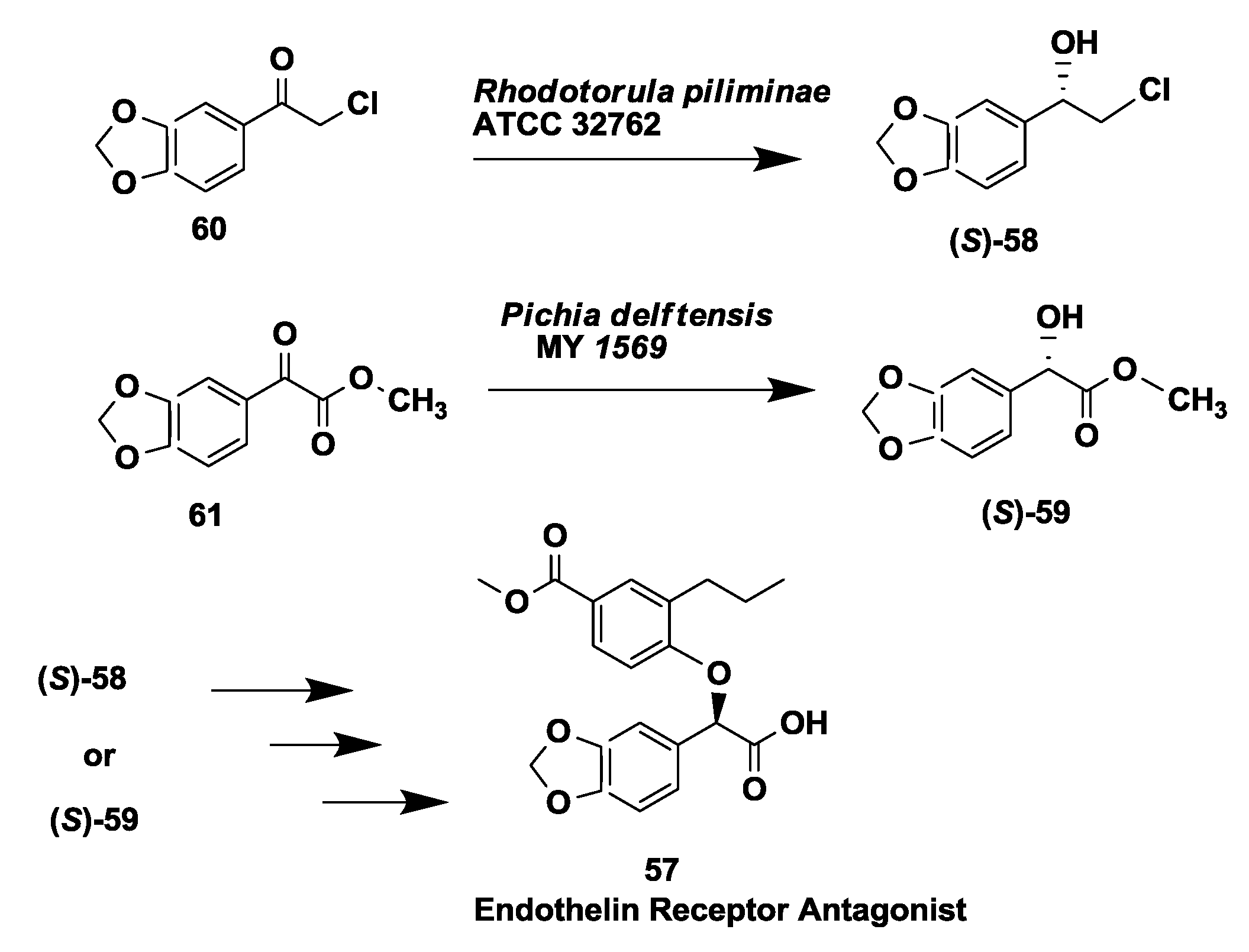
2.15. Endothelin Receptor Antagonist: Enantioselective Microbial Reduction of Keto Ester and Chloroketone
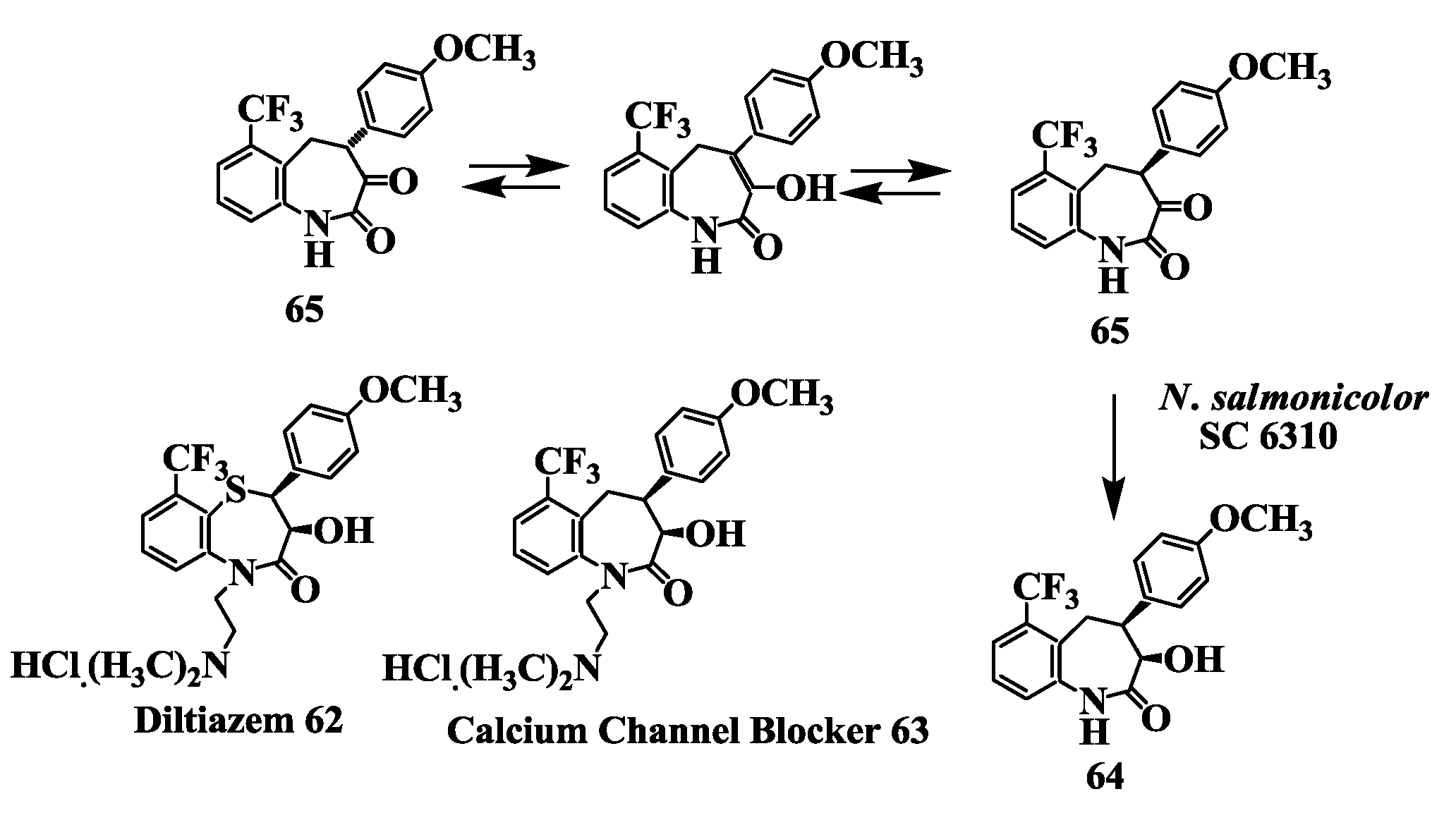
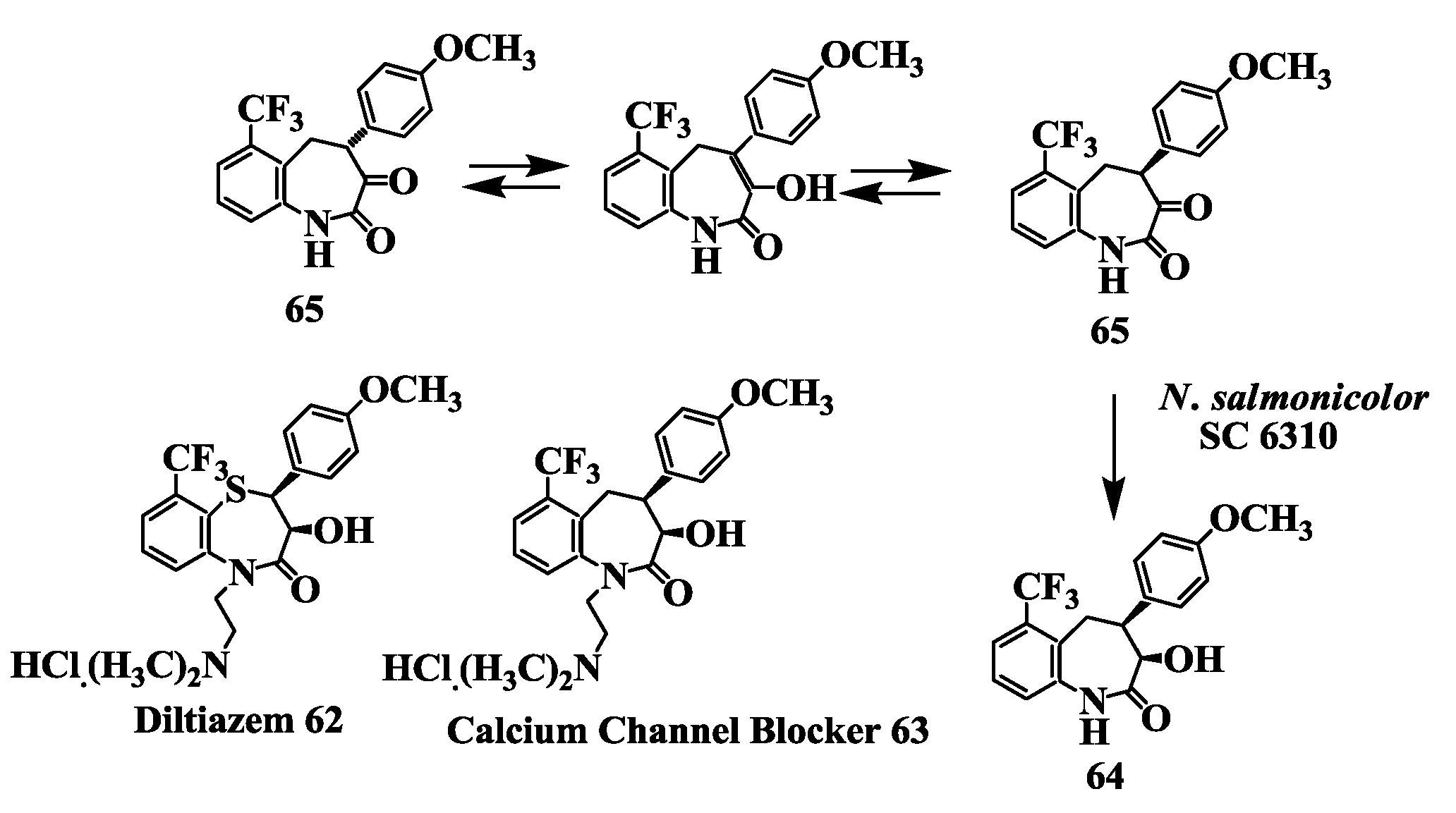
2.16. Calcium Channel Blocker: Preparation of [(3R-cis)-1,3,4,5-Tetrahydro-3-Hydroxy-4-(4-Methoxyphenyl)-6-(Trifluromethyl)-2H-1-Benzazepin-2-One]
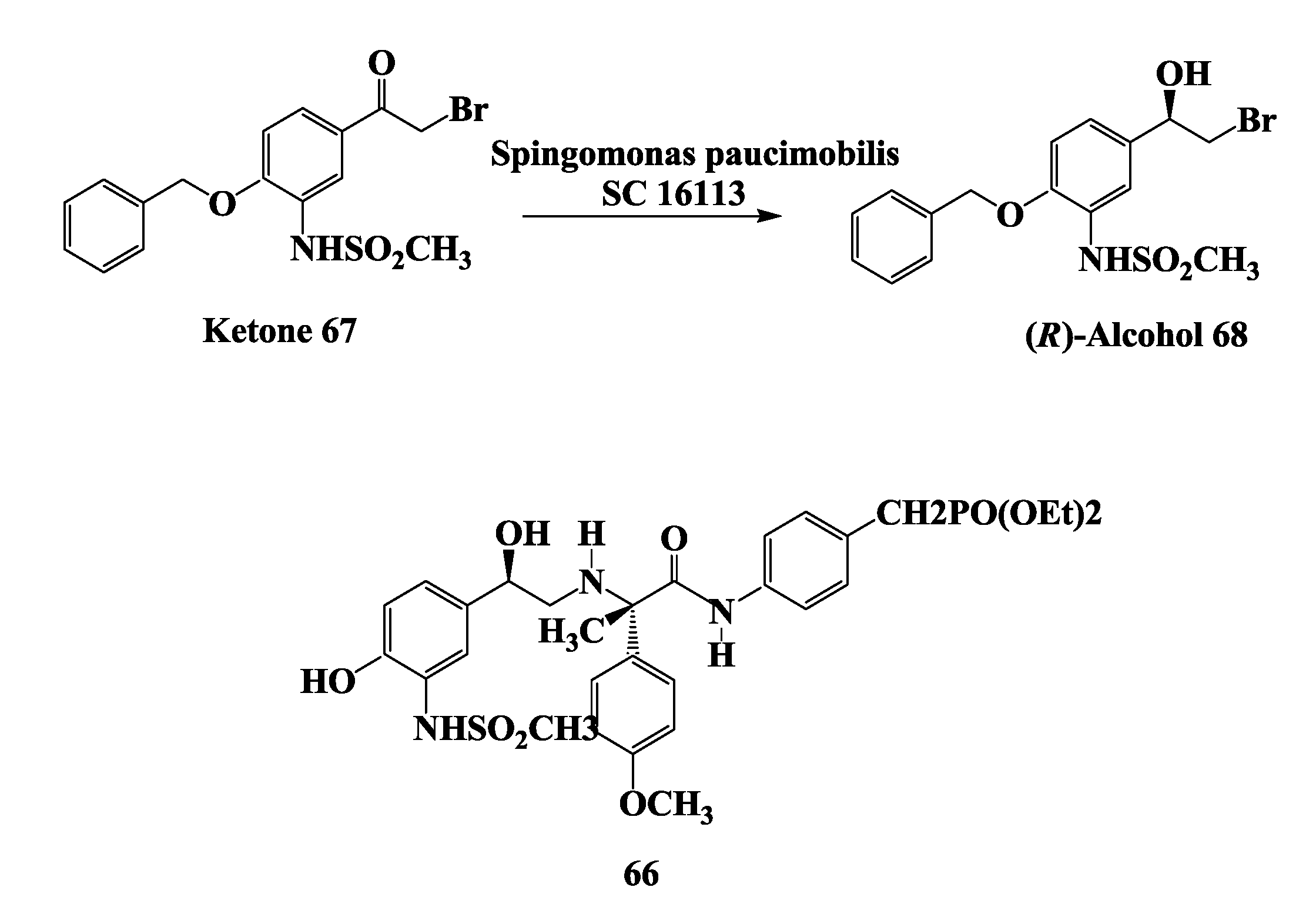
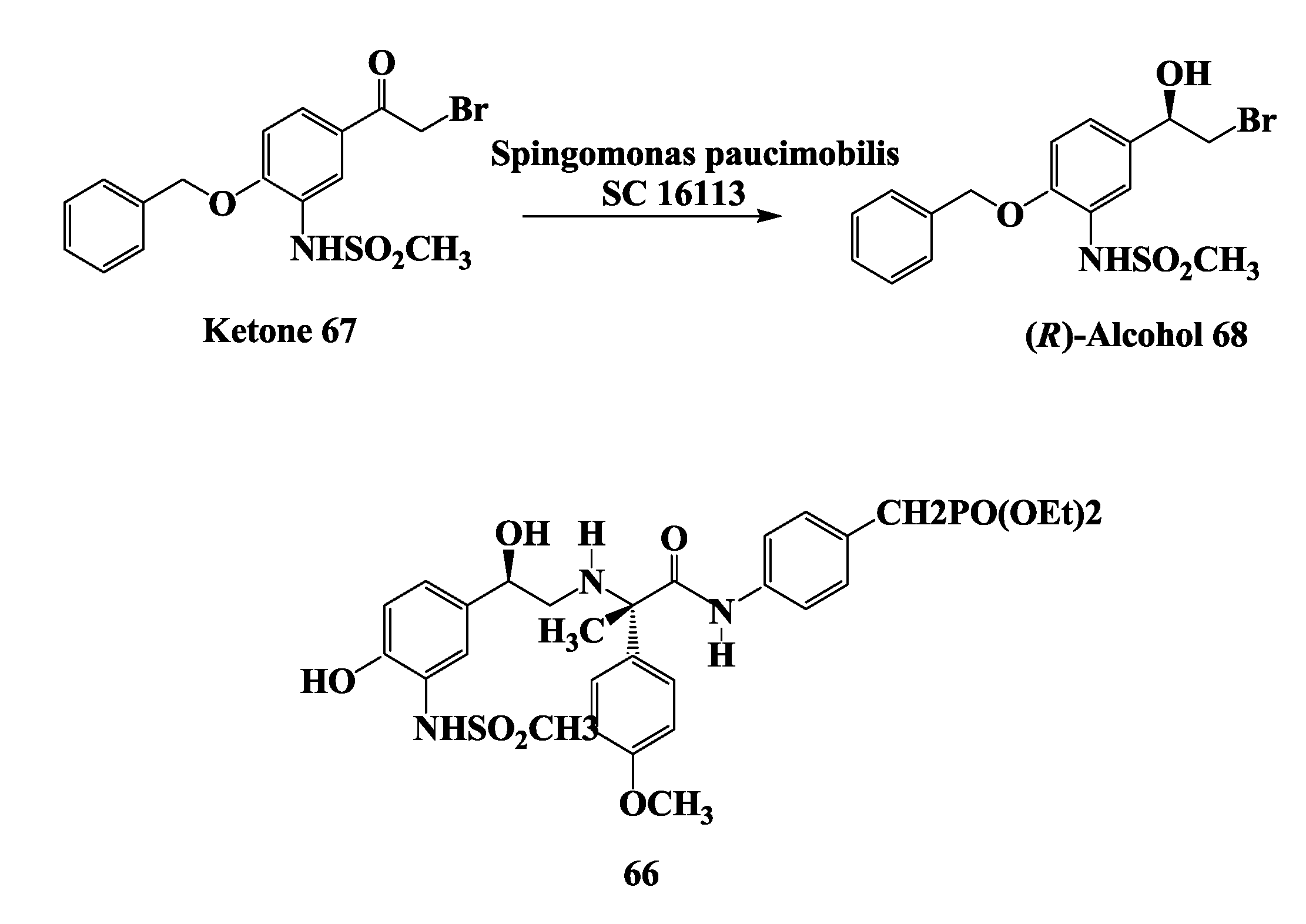
2.17. β3-Receptor Agonist: Reduction of 4-Benzyloxy-3-Methanesulfonylamino-2'-Bromo-Acetophenone
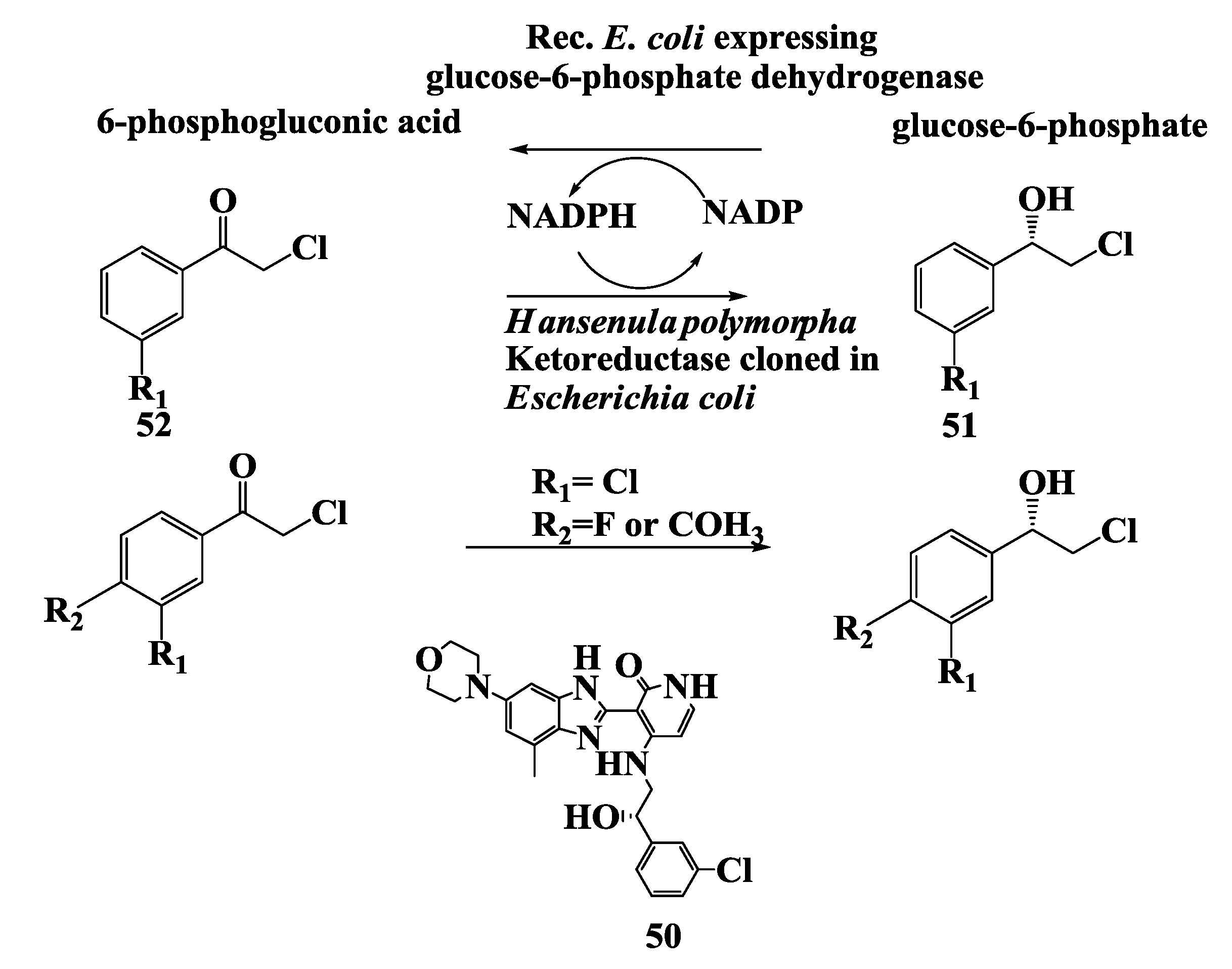
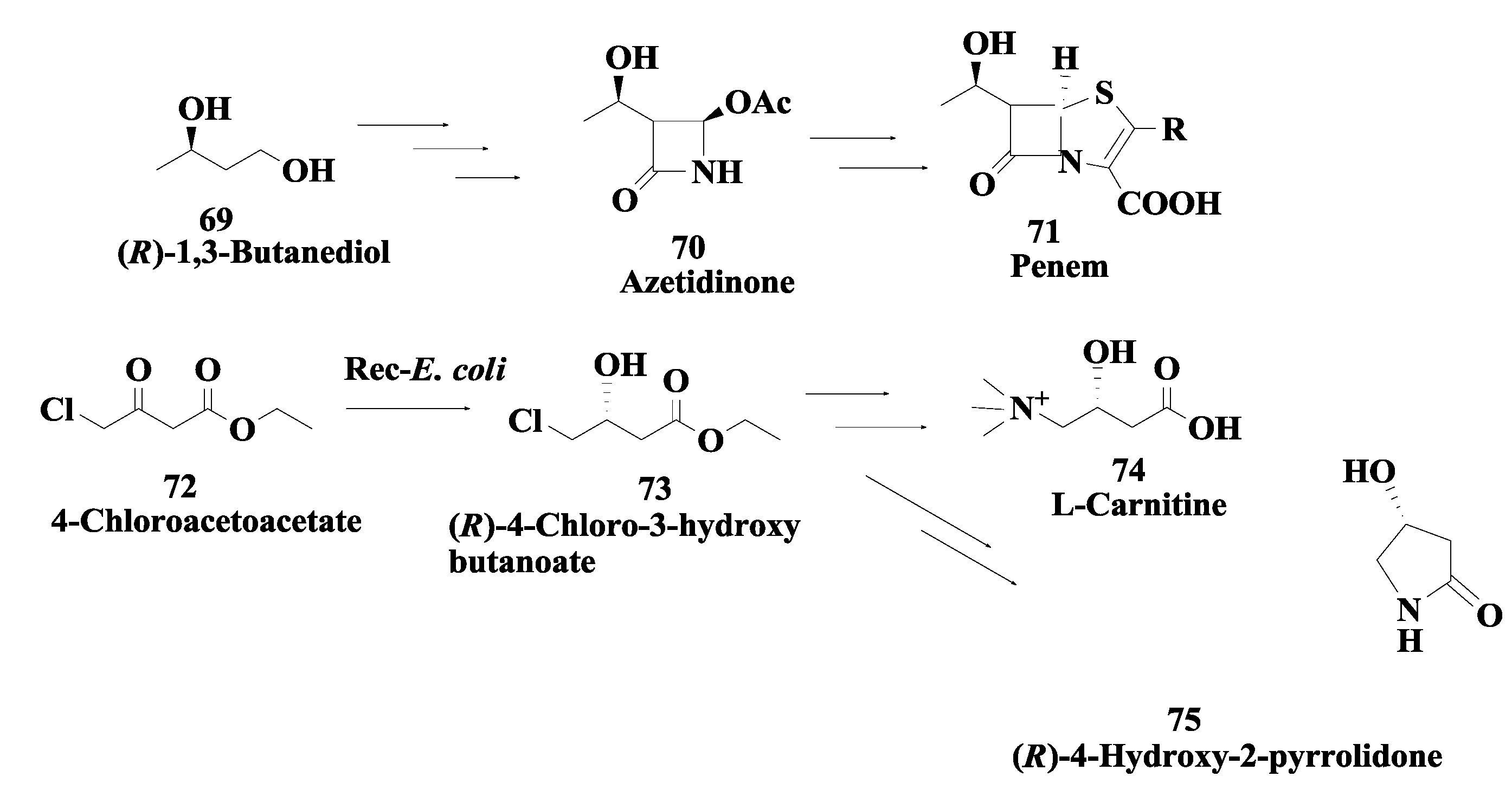
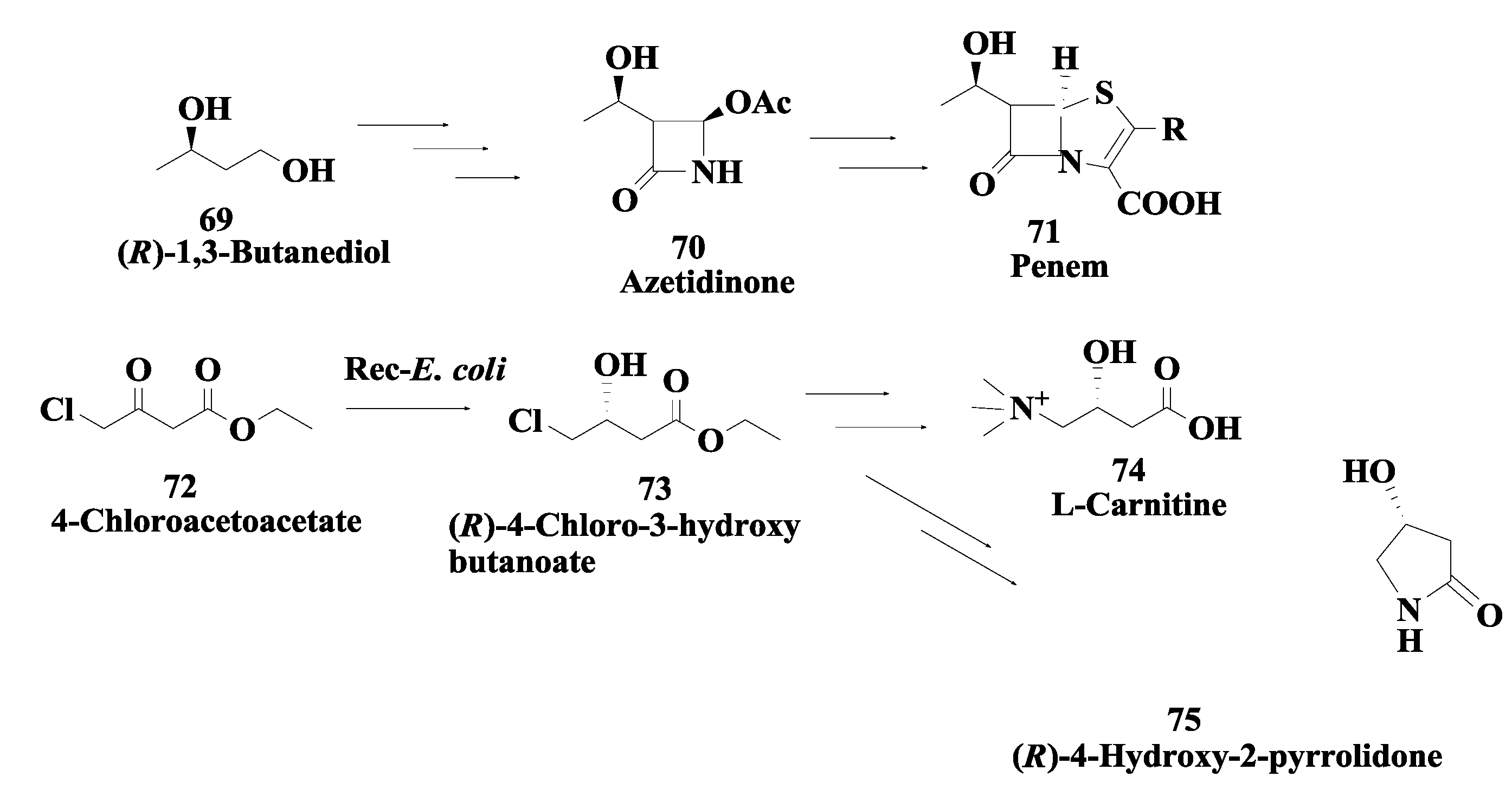
2.18. Penem and Carbapenem: Enzymatic Preparation of (R)-1,3-Butanediol and (R)-4-Chloro-3-Hydroxybutonoate
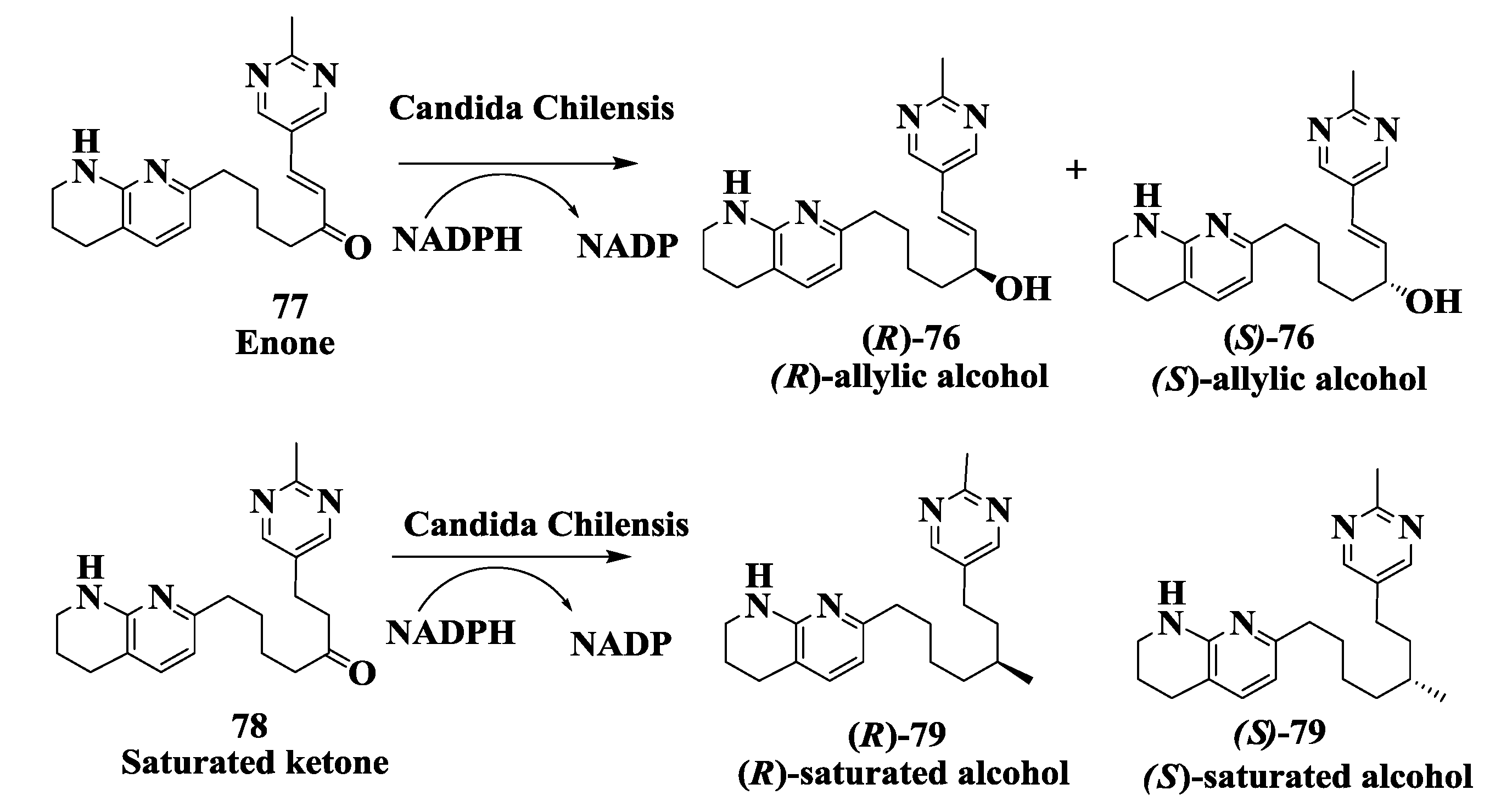
2.19. Integrin Receptor Agonist: Enzymatic Preparation of (R)-Allylic Alcohol
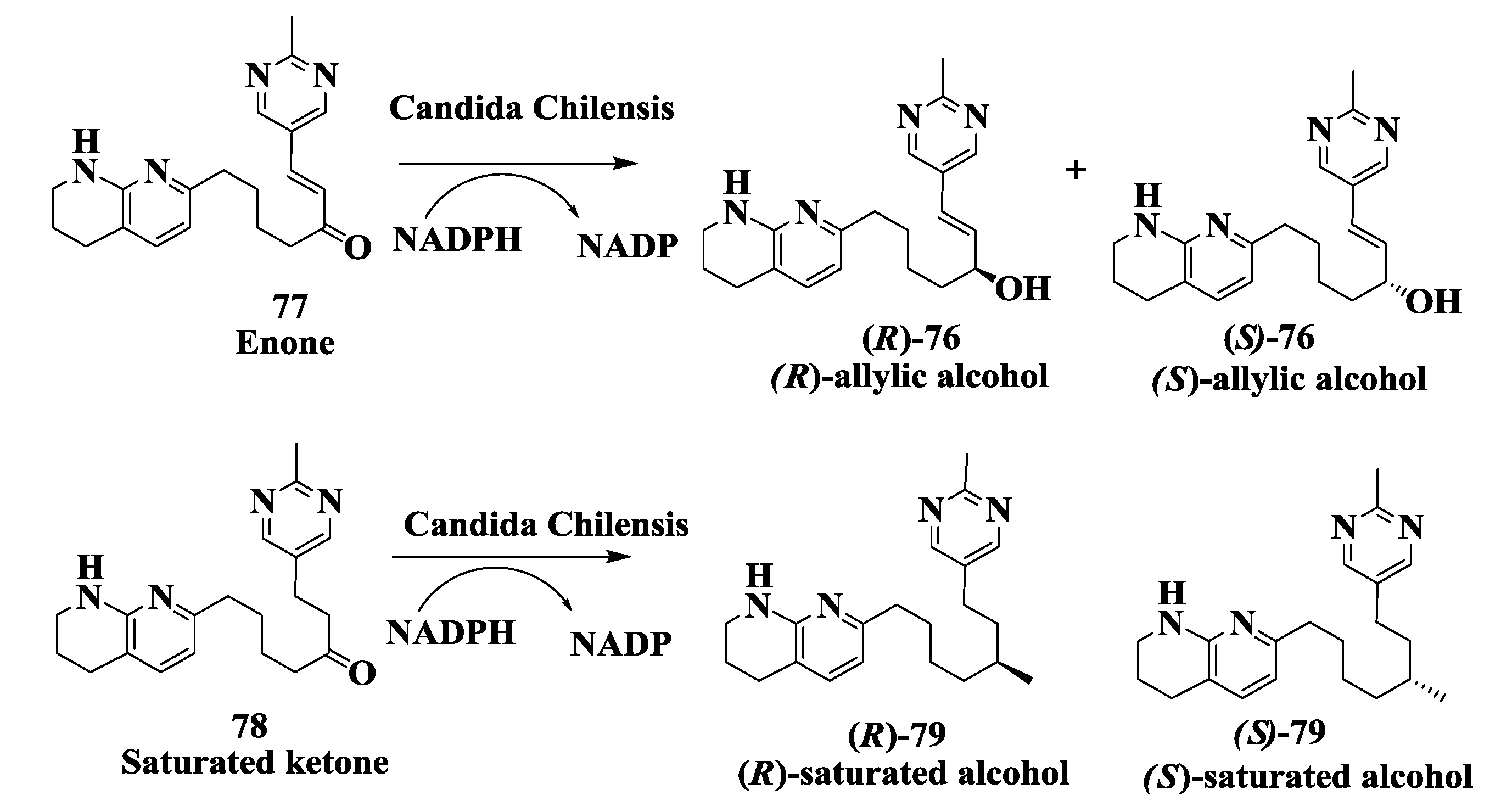
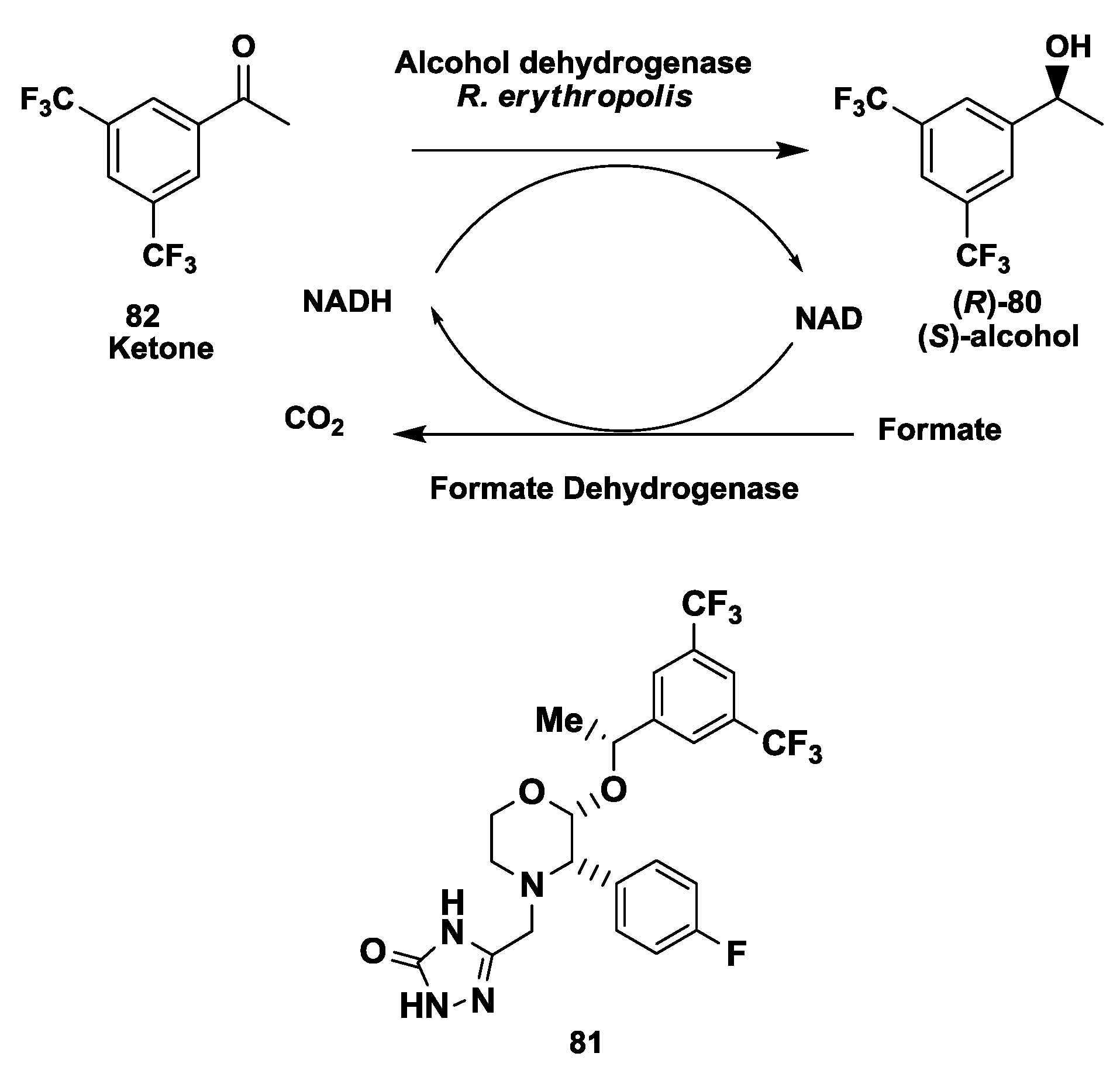
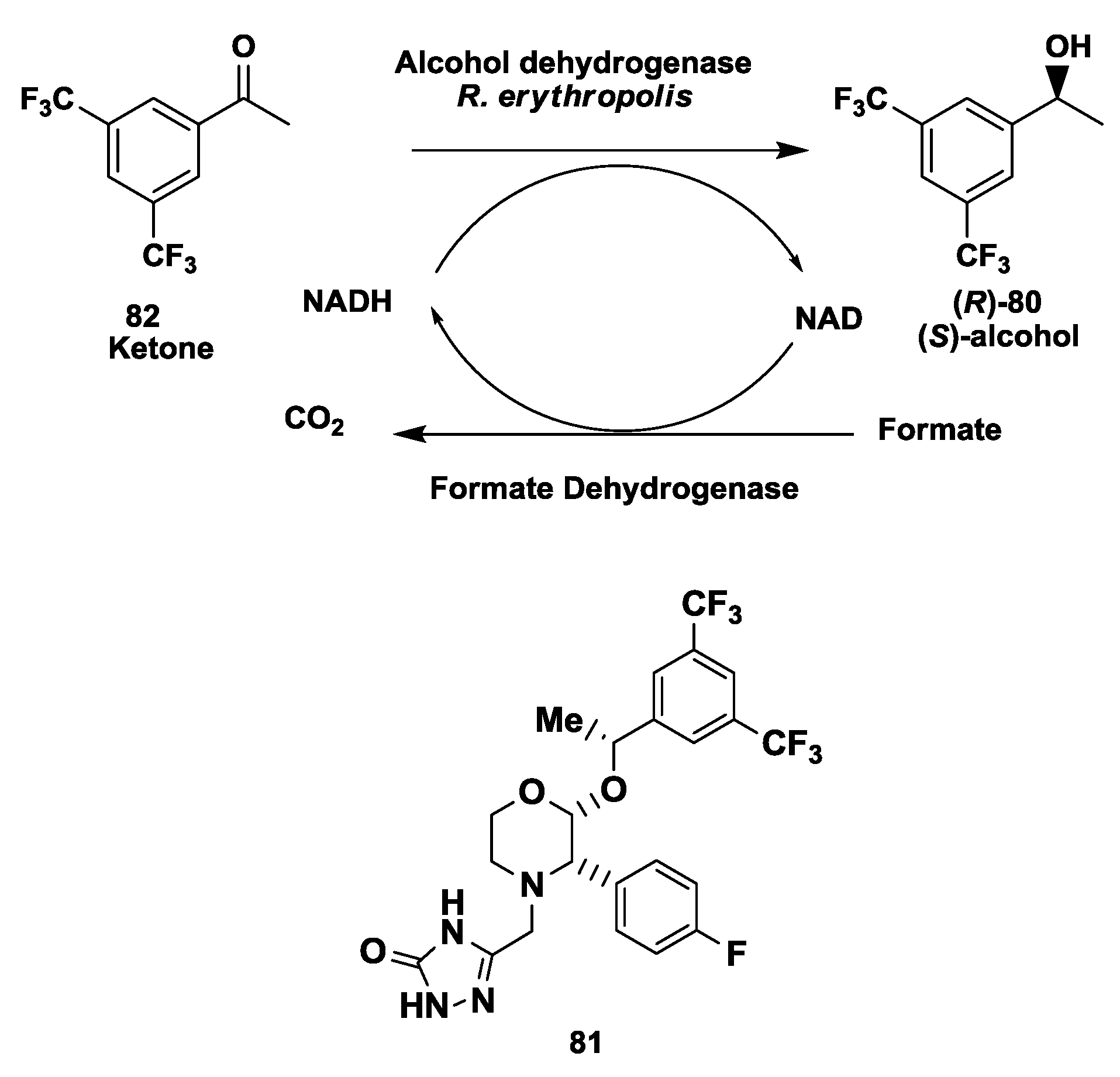
2.20. NK1 Receptor Antagonists: Enzymatic Synthesis of (S)-3,5-Bistrifluoromethylphenyl Ethanol
3. Enzymatic Preparation of Chiral Amino Acids
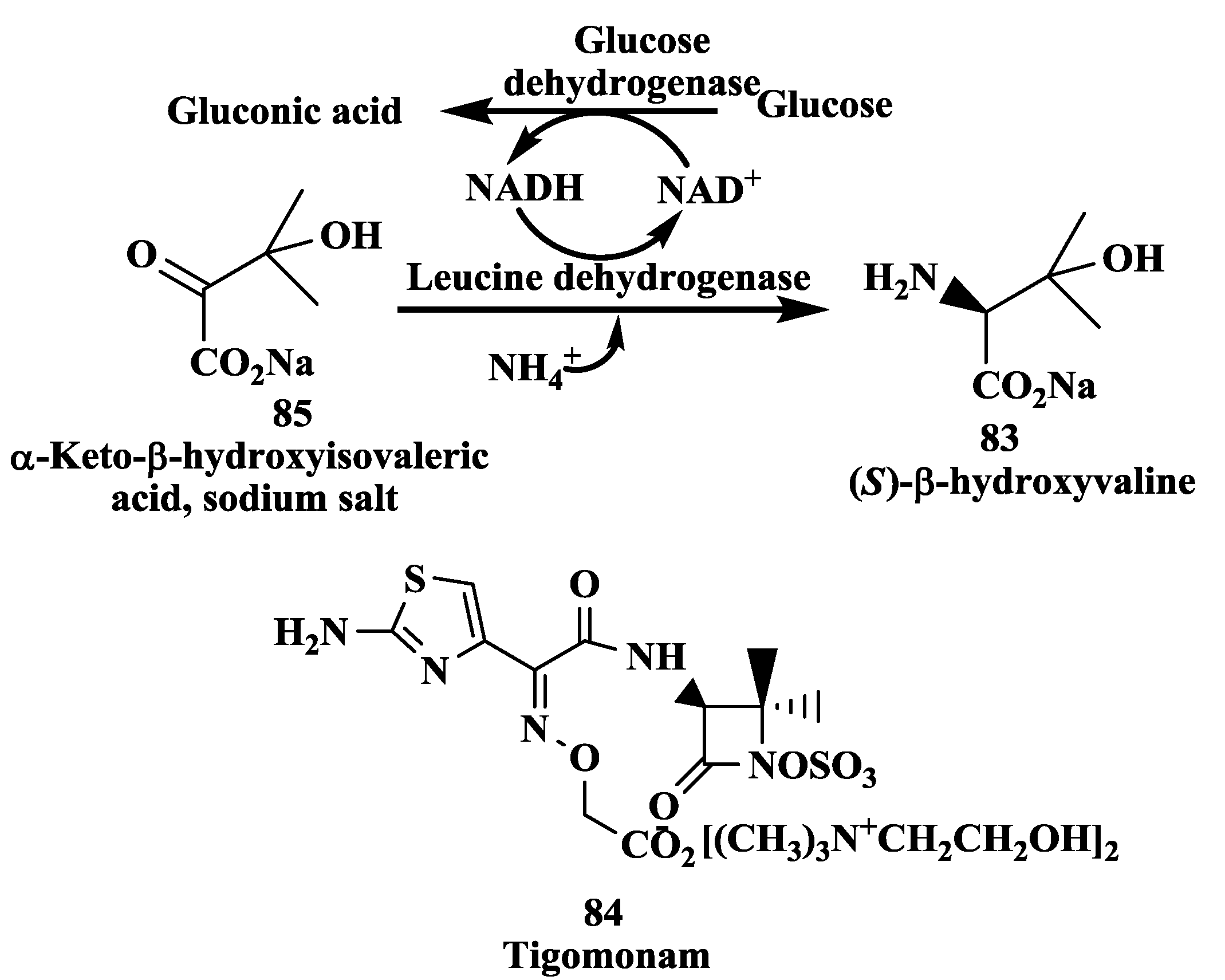
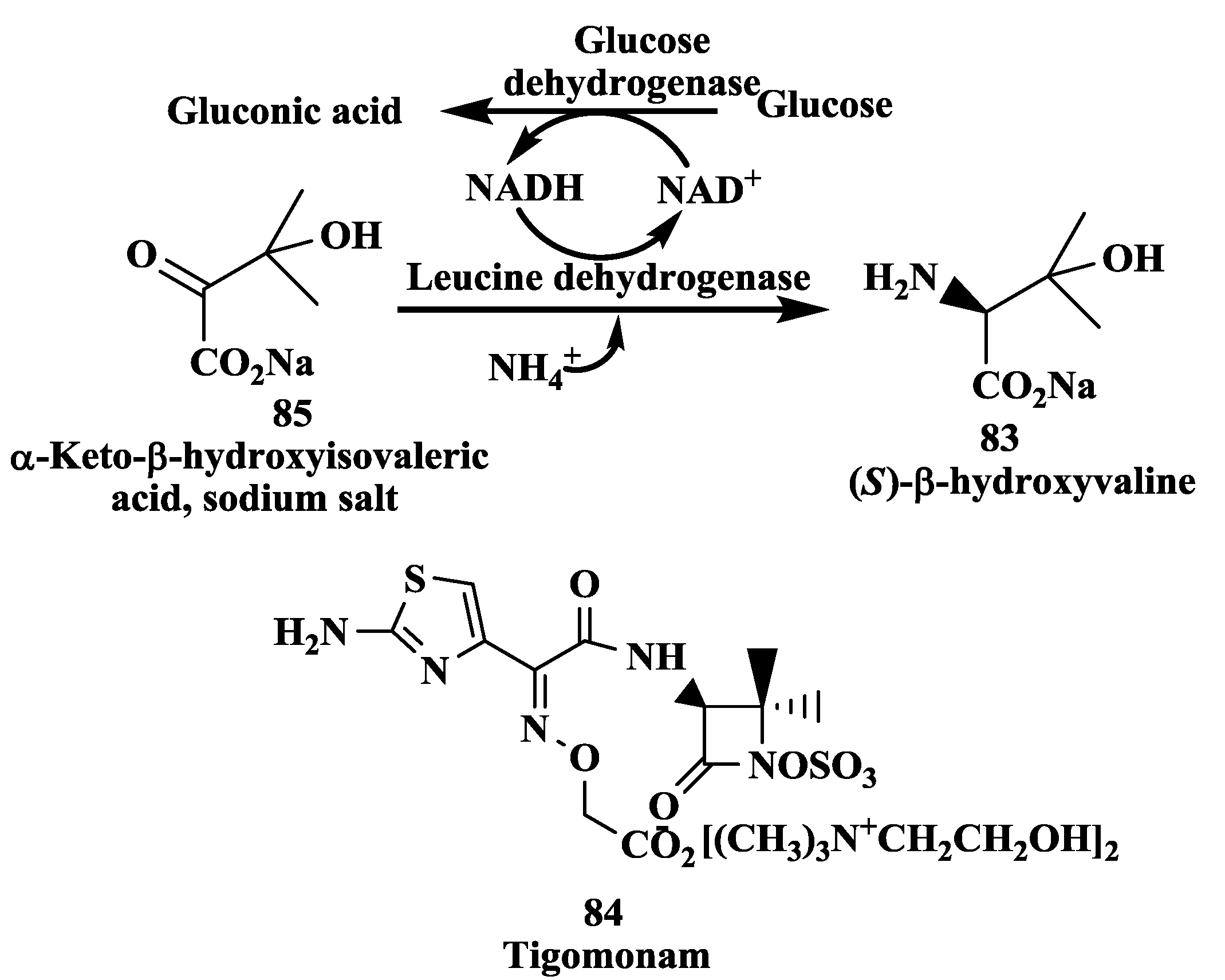
3.1. Tigemonam: Enzymatic Synthesis of (S)-β-Hydroxyvaline
3.2. Atazanavir: Enzymatic Synthesis of (S)-Tertiary-Leucine

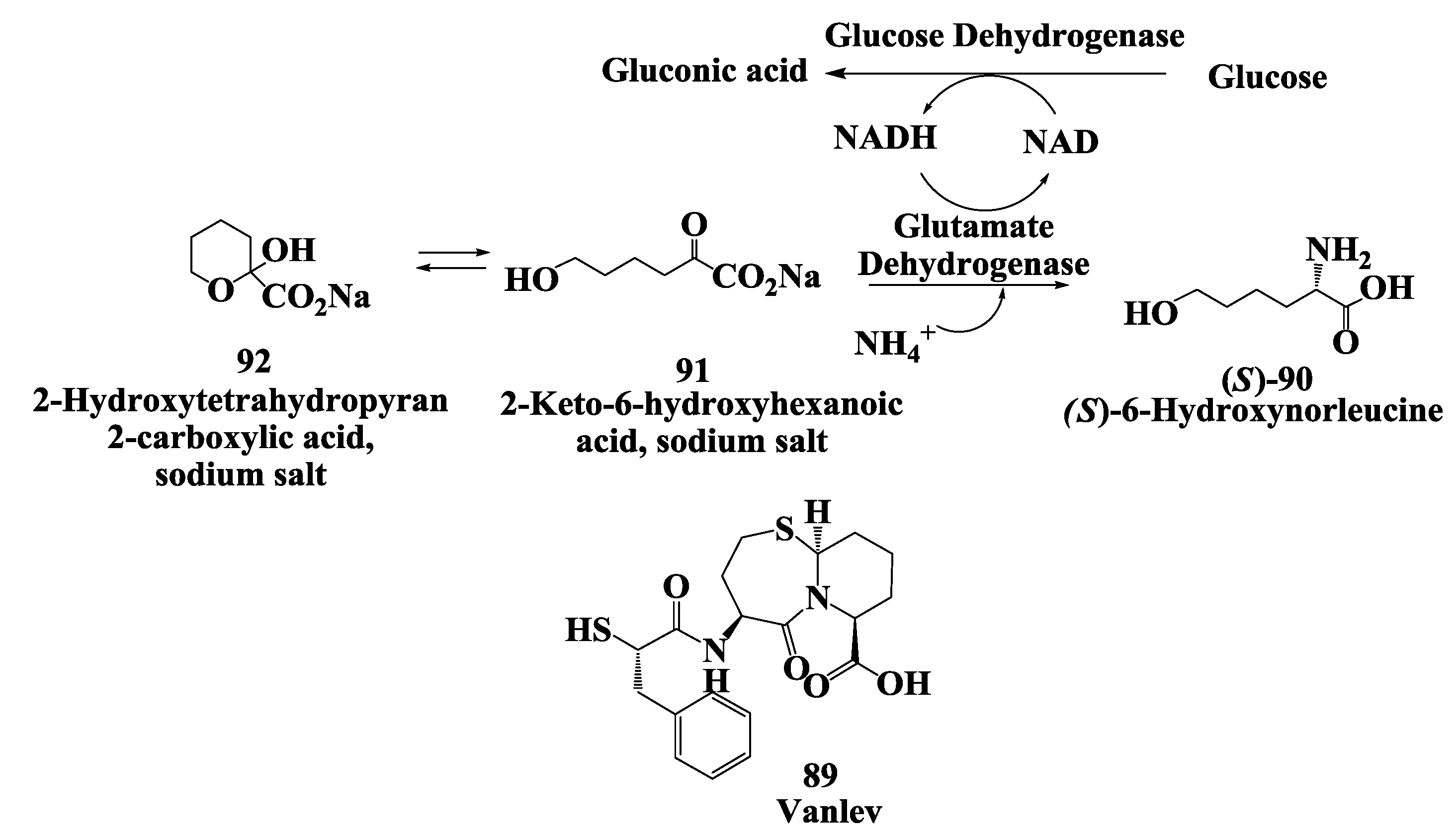
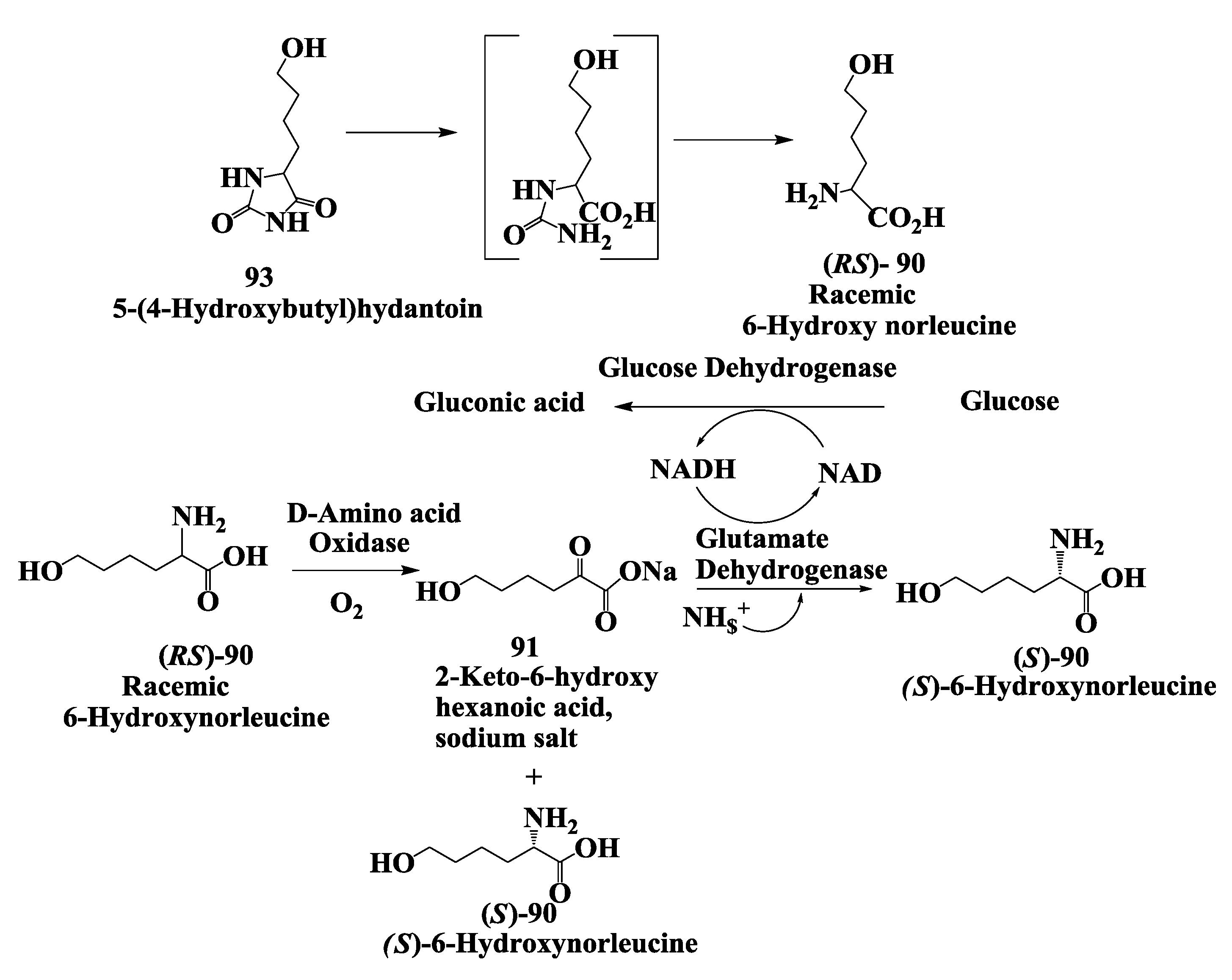
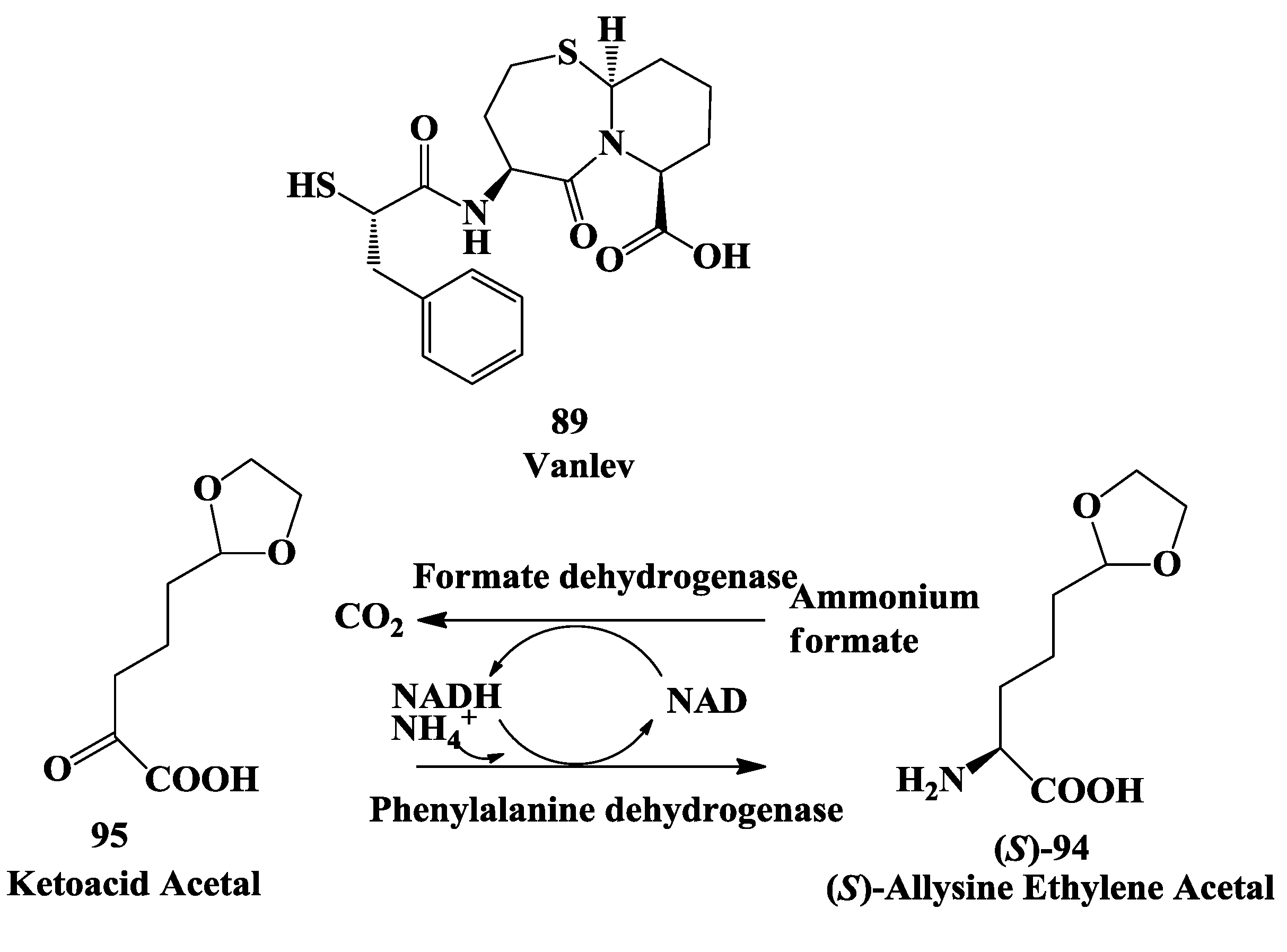
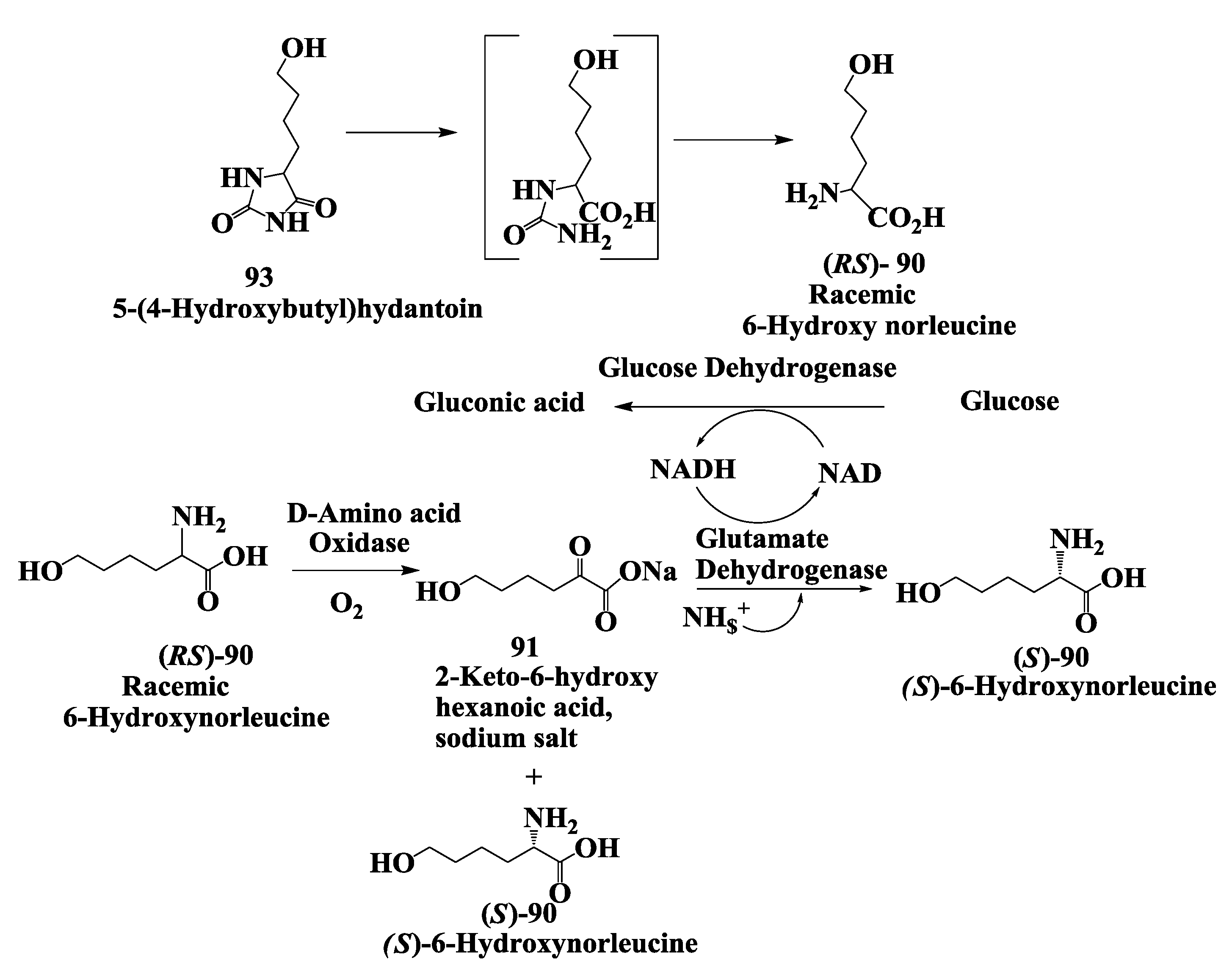
3.3. Vanlev: Enzymatic Synthesis of (S)-6-Hydroxynorleucine
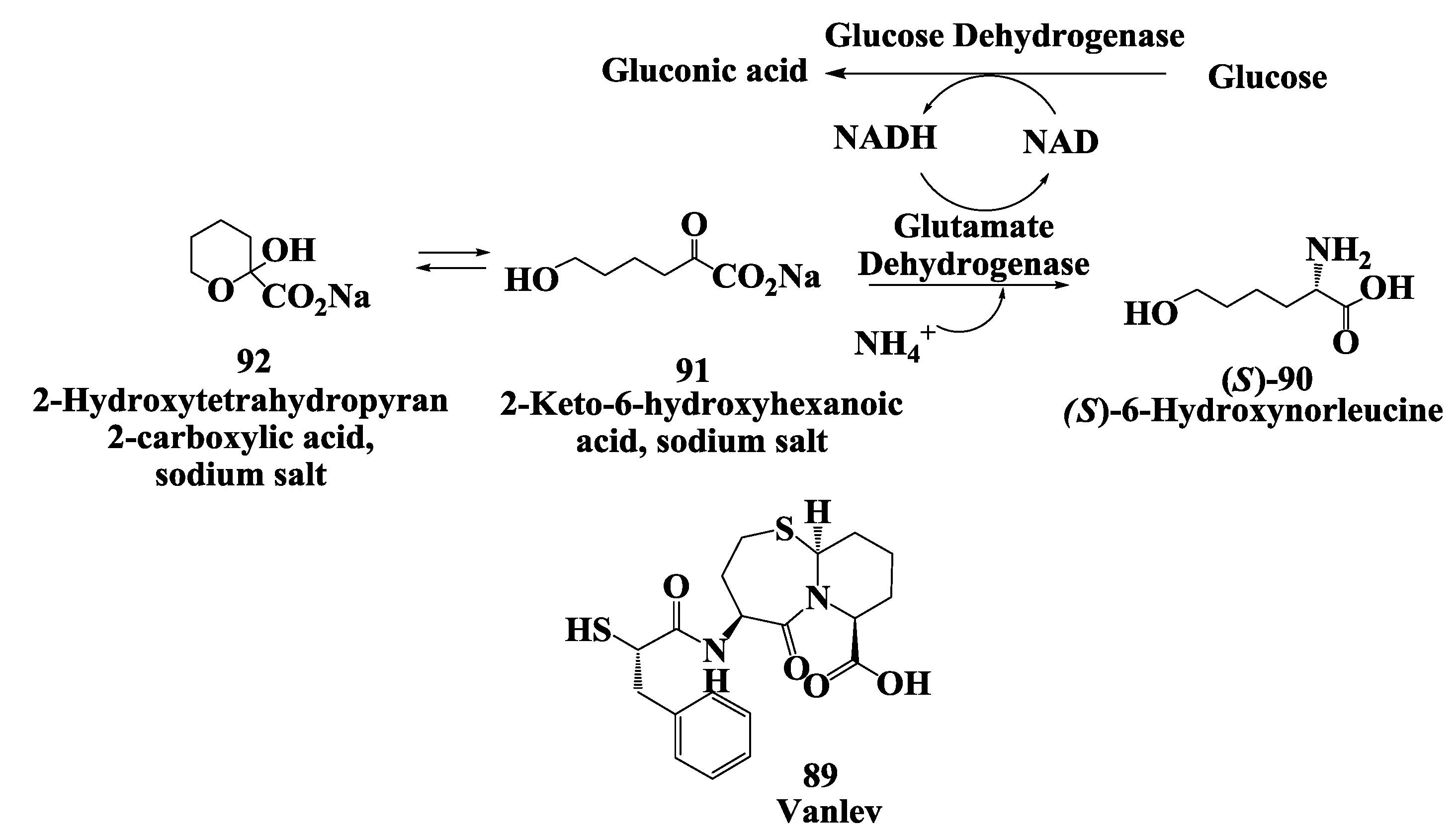
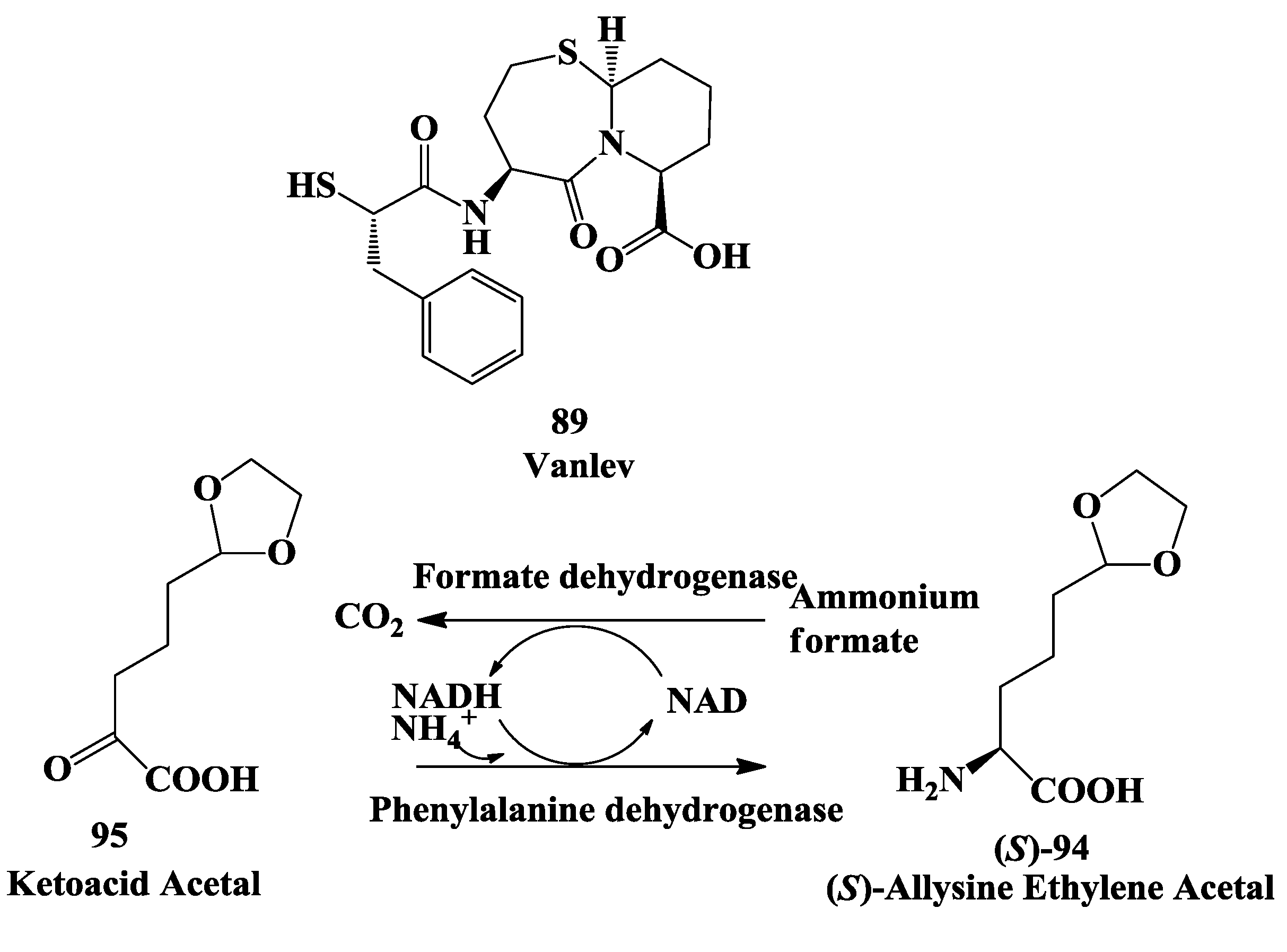
3.4. Vanlev: Enzymatic Synthesis of Allysine Ethylene Acetal
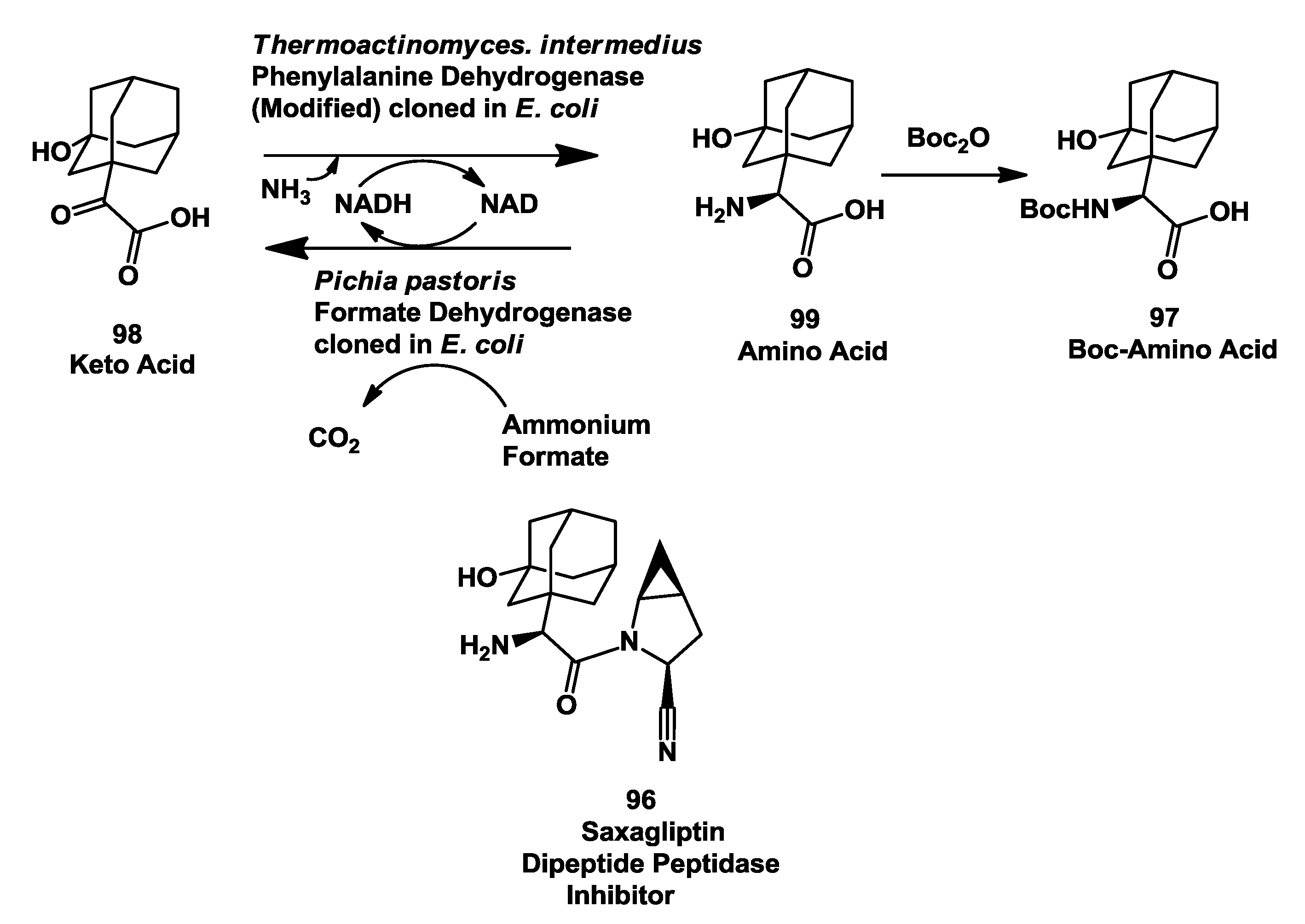
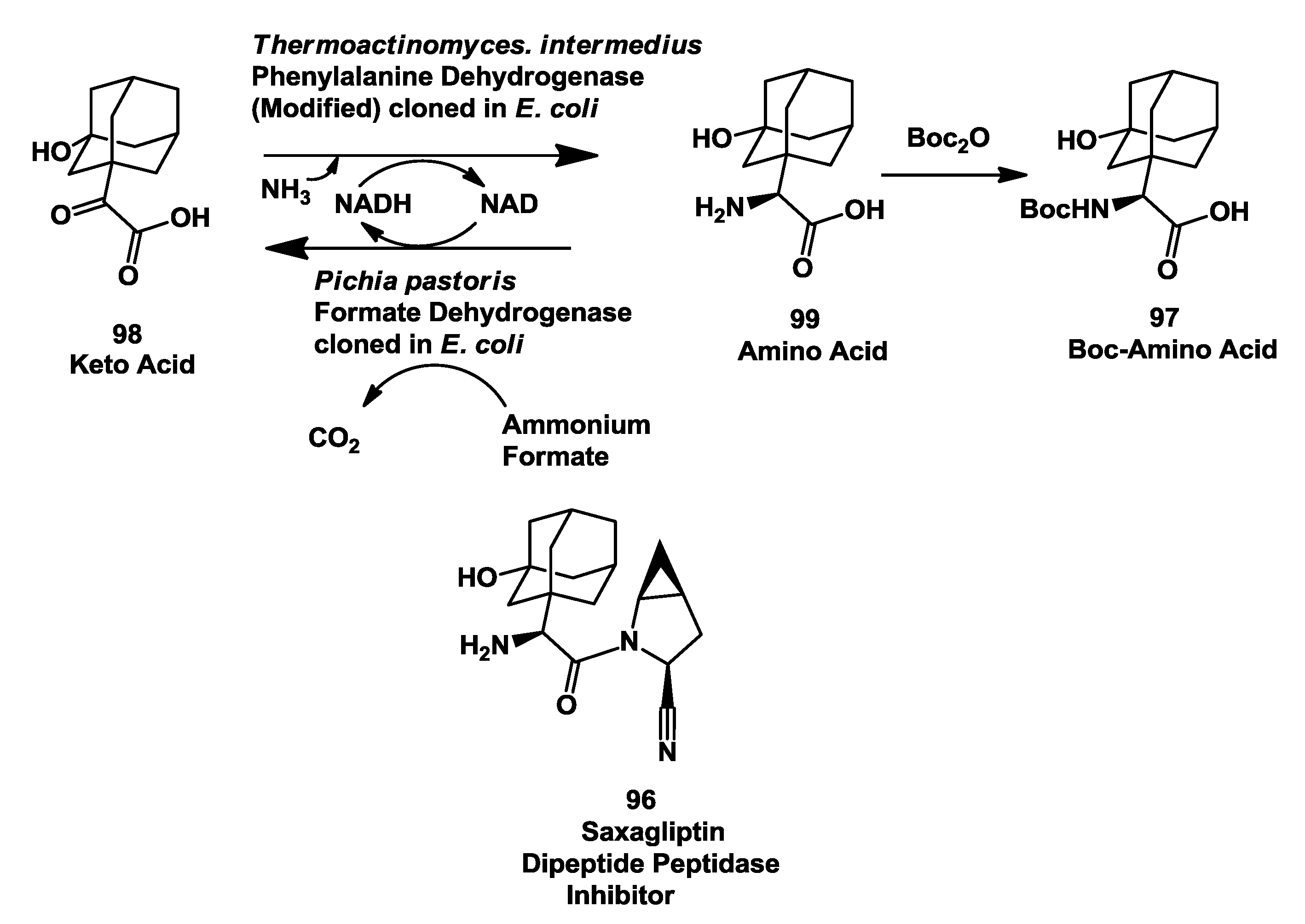
3.5. Saxagliptin: Enzymatic Reductive Amination of 2-(3-Hydroxy-1-Adamantyl)-2-Oxoethanoic Acid
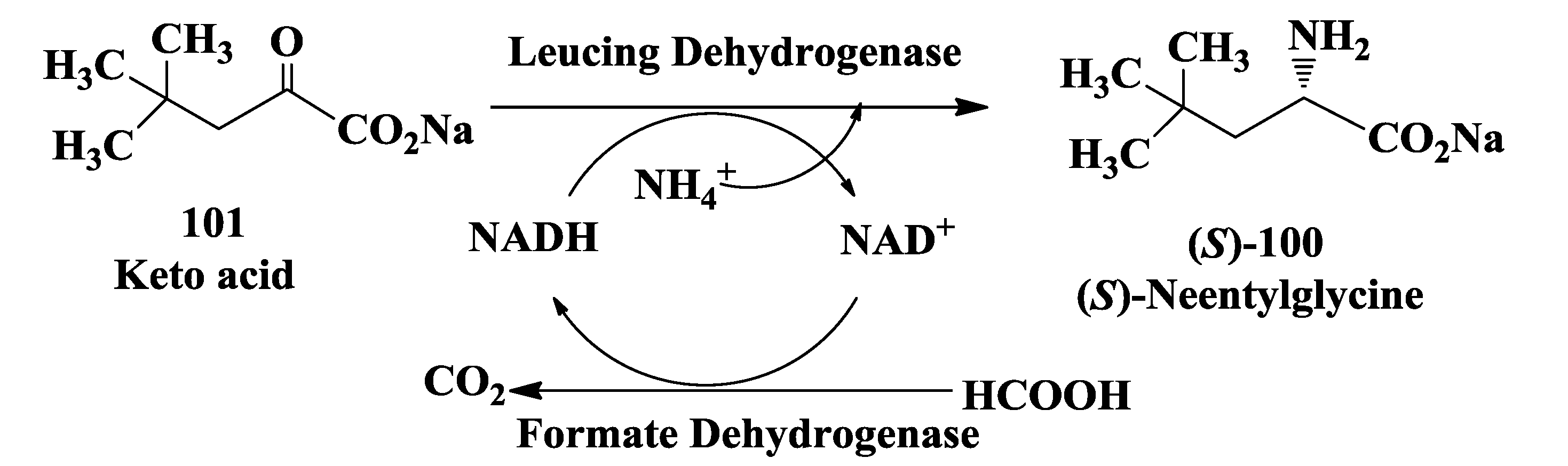
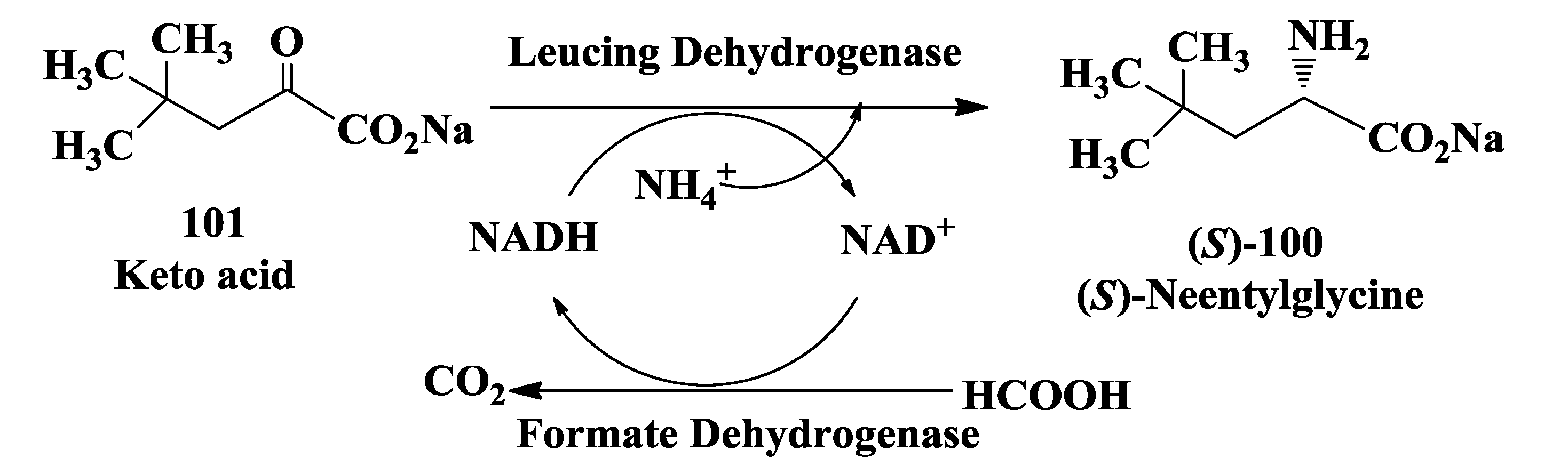
3.6. Enzymatic Synthesis of (S)-Neopentylglycine
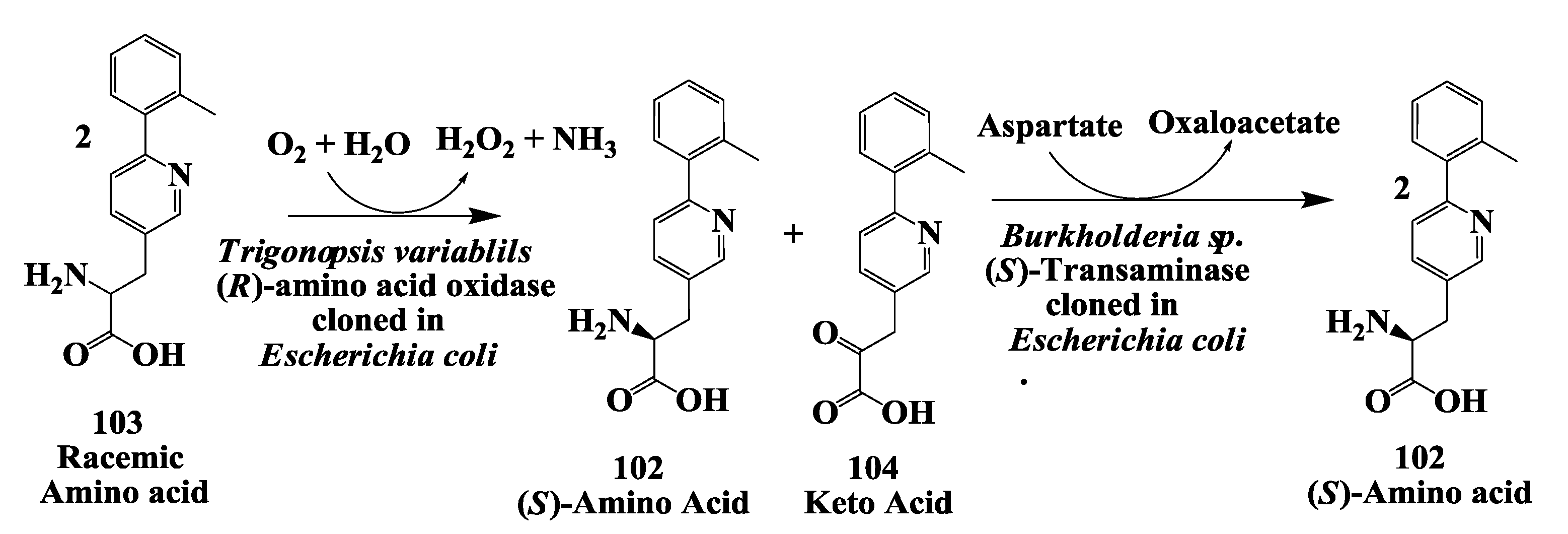
3.7. Glucogen like Peptide: Enzymatic Deracemization Racemic Amino Acid to (S)-Amino Acid
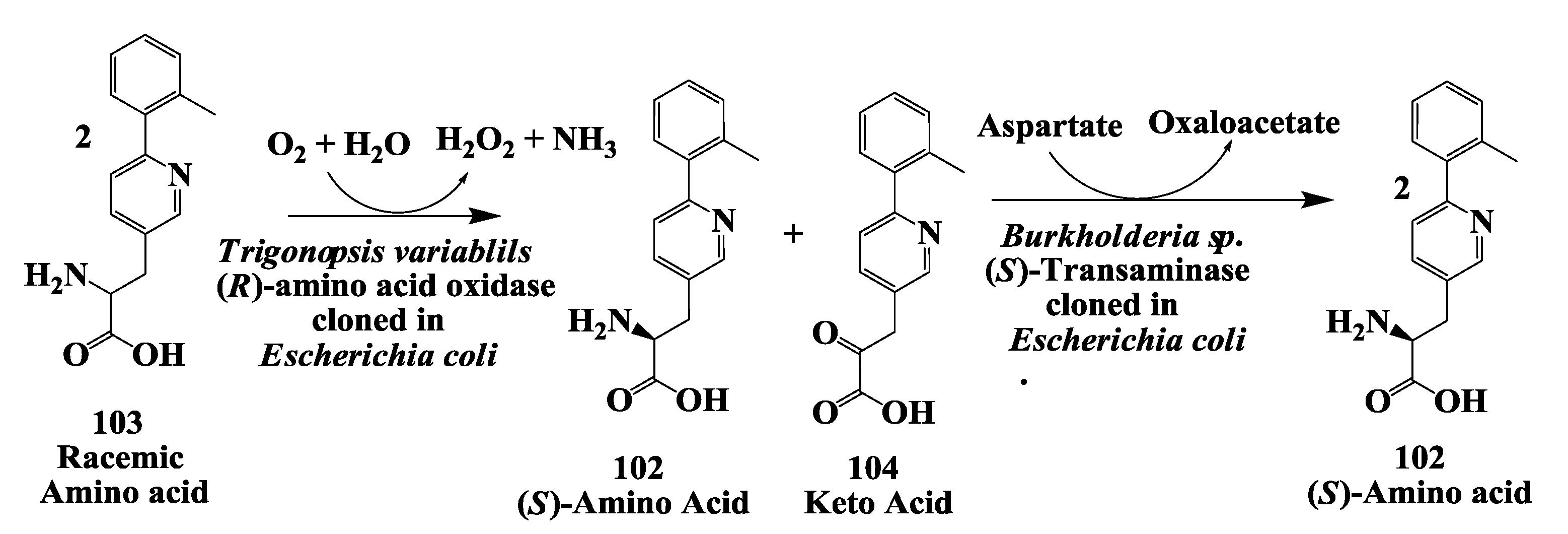
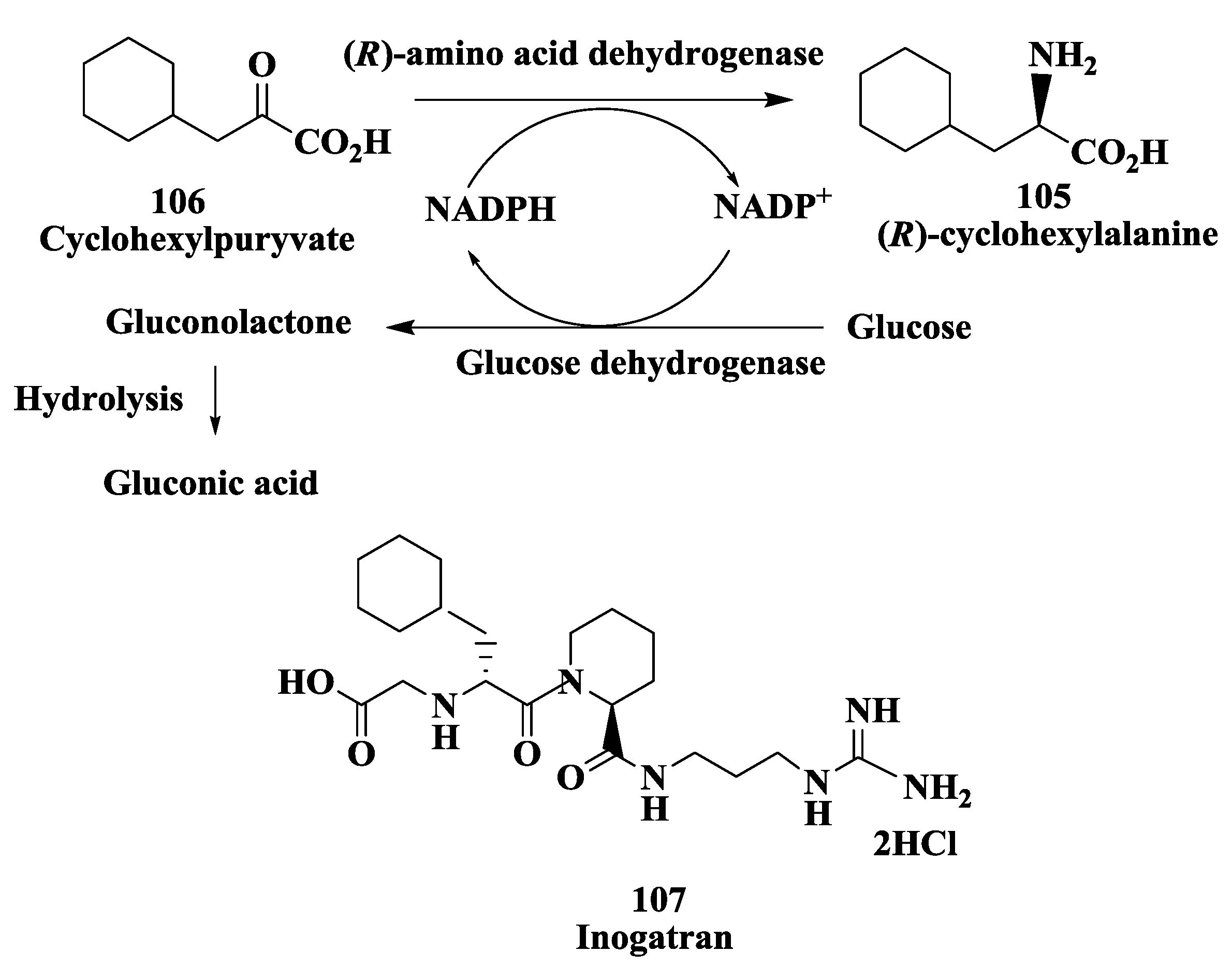
3.8. Preparation of (R)-Amino Acid
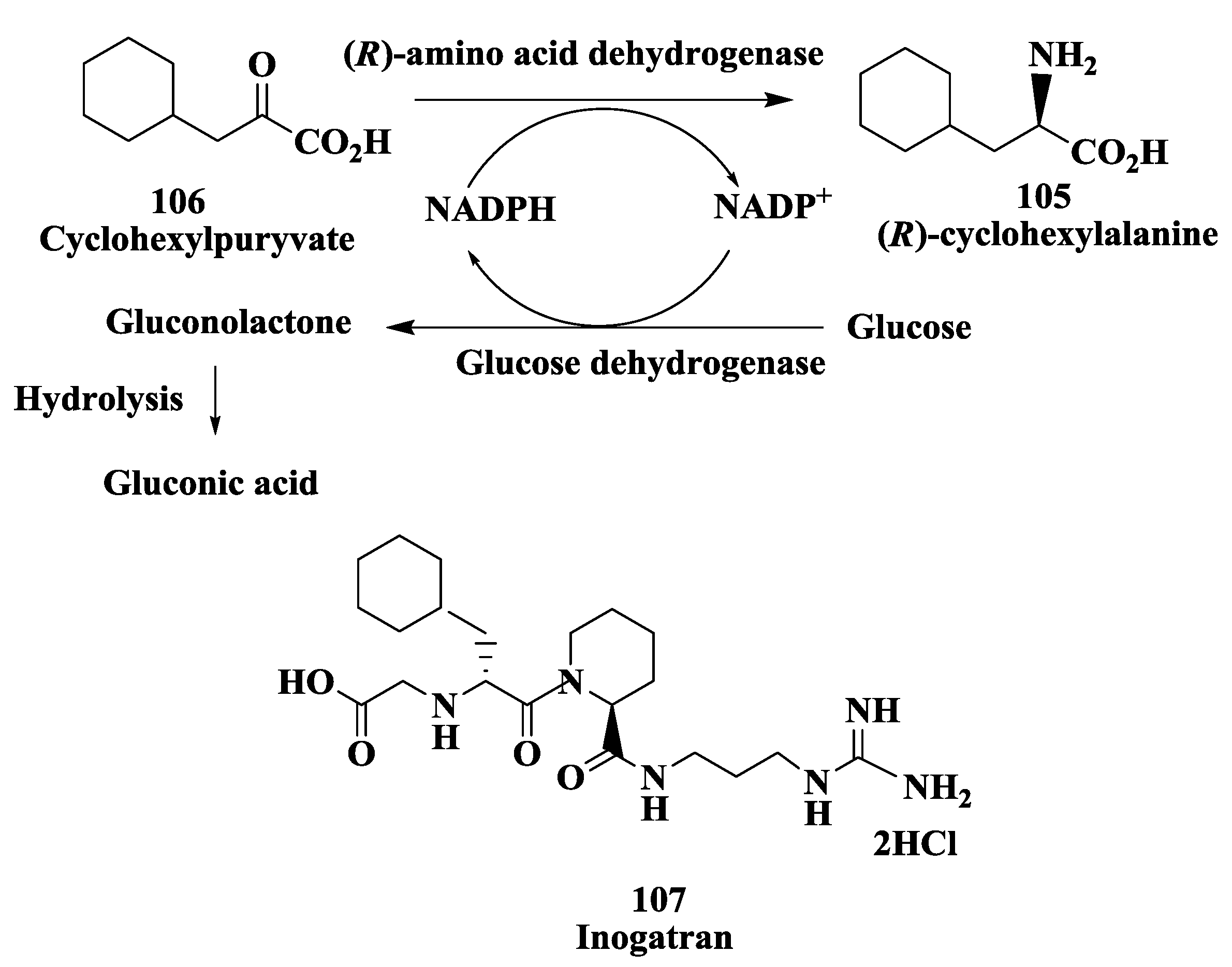
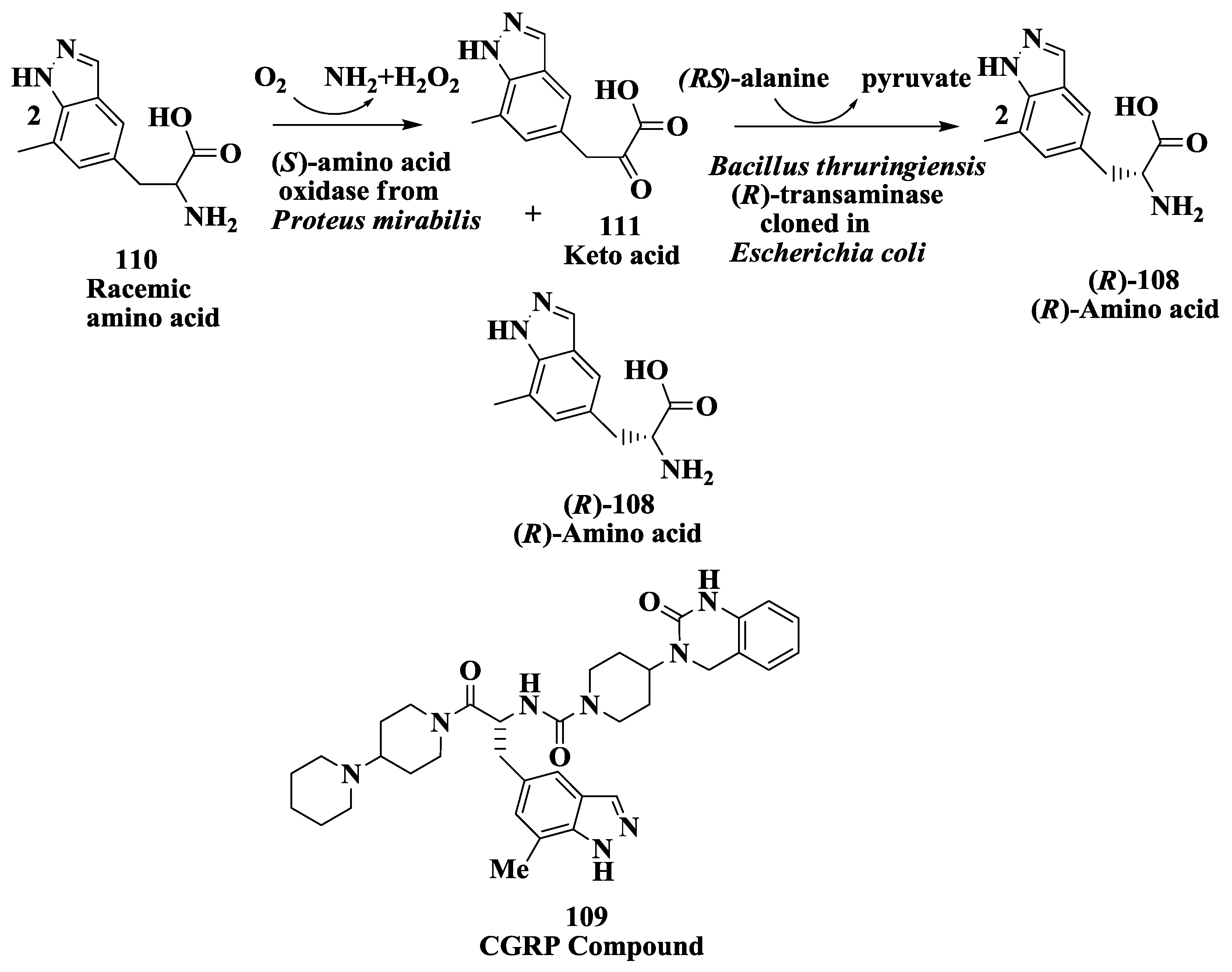
3.9. Calcitonin Gene-Related Peptide Receptors (Antimigraine Drugs): Enzymatic Deracemization Process
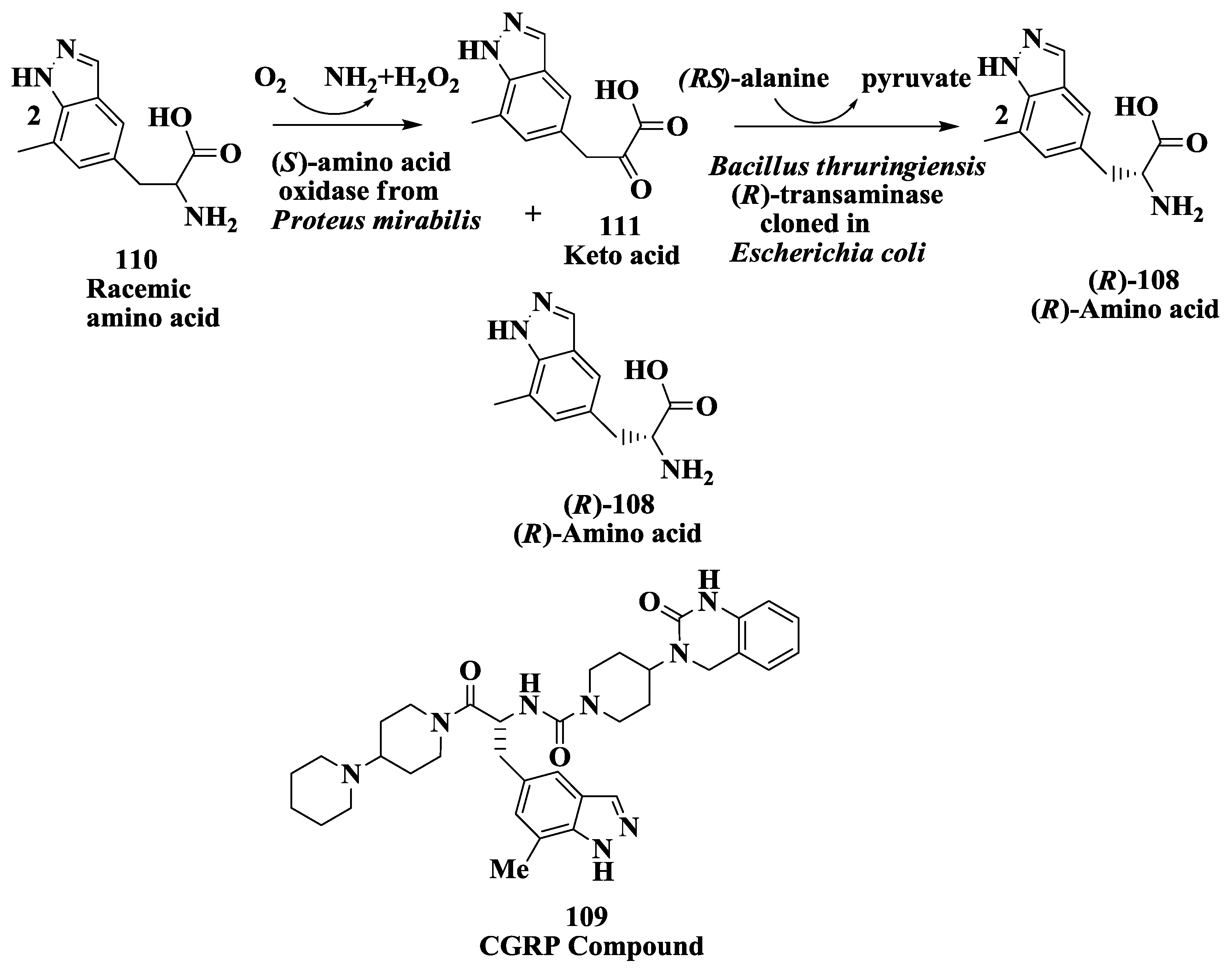
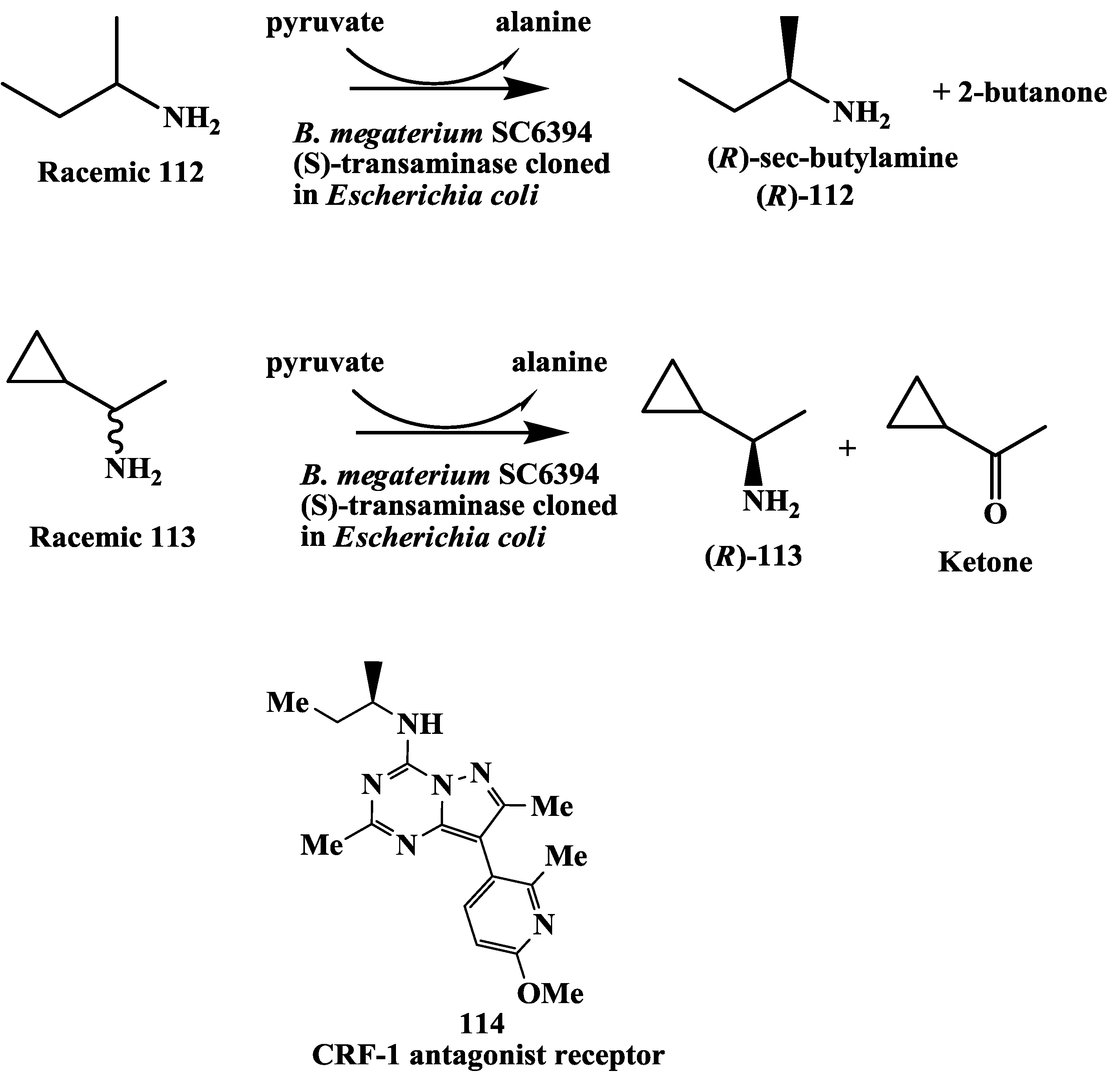
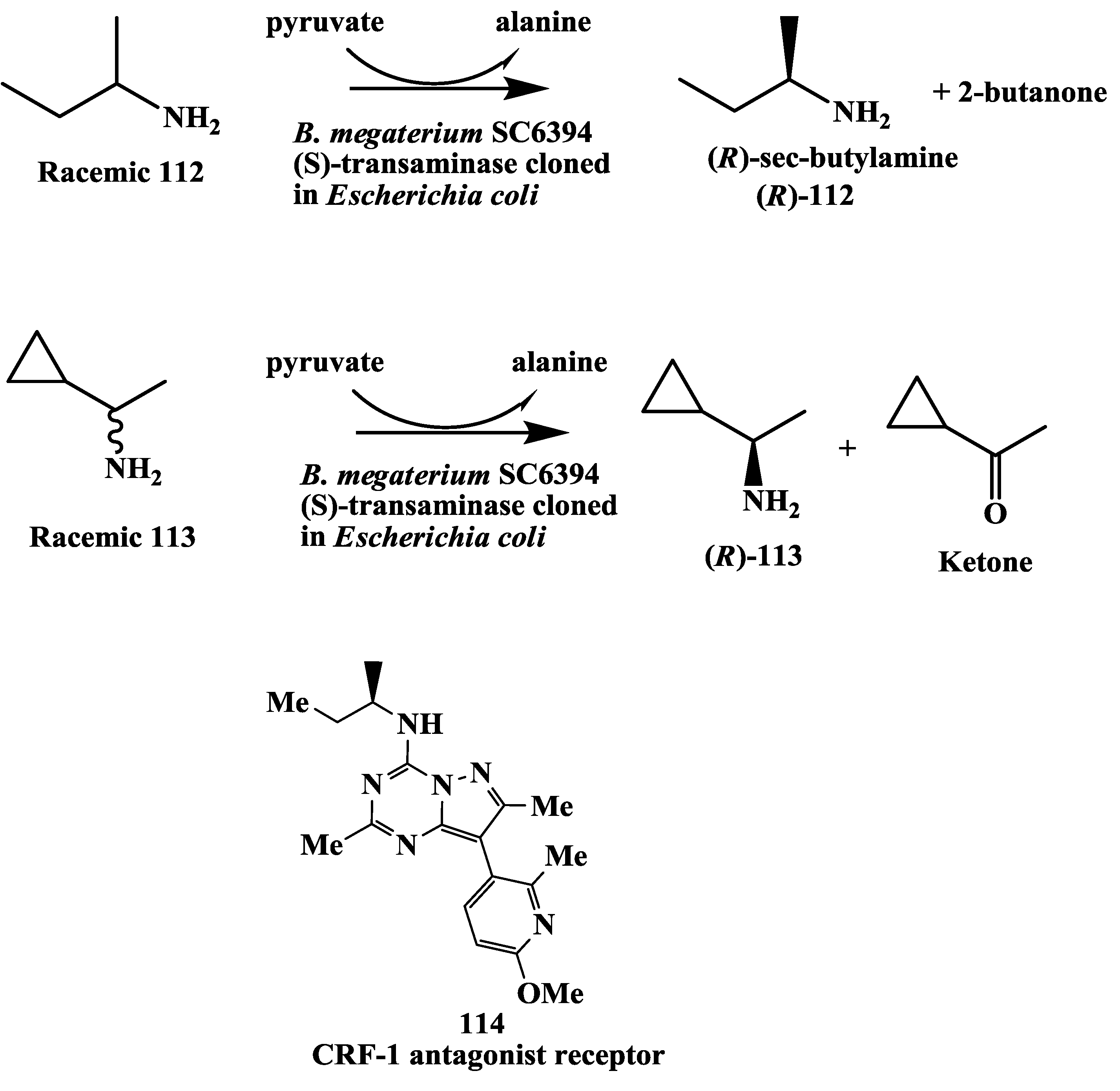
3.10. Corticotropin Releasing Factor (CRF)-1 Receptor Antagonist: Enzymatic Resolution by Transaminase
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Food and Drug Administration. FDA’s statement for the development of new stereoisomeric drugs. Chirality 1992, 4, 338–340. [CrossRef]
- Oliver, M.; Voigt, C.A.; Arnold, F.H. Enzyme engineering by directed evolution. In Enzyme Catalysis in Organic Synthesis, 2nd ed.; VCH: New York, USA, 2002; Volume 1, pp. 95–138. [Google Scholar]
- Kazlauskas, R.J. Enhancing catalytic promiscuity for biocatalysis. Curr. Opin. Chem. Biol. 2005, 9, 195–201. [Google Scholar] [CrossRef]
- Schmidt, M.; Bauman, M.; Henke, E.; Konarzycka-Bessler, M.; Bornscheuer, U.T. Directed evolution of lipases and esterases. Meth. Enzymol. 2004, 388, 199–207. [Google Scholar] [CrossRef]
- Reetz, M.T.; Torre, C.; Eipper, A.; Lohmer, R.; Hermes, M.; Brunner, B.; Maichele, A.; Brunner, B.; Arand, M.; Cronin, A.; et al. Enhancing the enantioselectivity of an epoxide hydrolase by directed evolution. Org. Lett. 2004, 6, 177–180. [Google Scholar] [CrossRef]
- Rubin-Pitel, S.B.; Zhao, H. Recent advances in biocatalysis by directed enzyme evolution. Comb. Chem. High T. Scr. 2006, 9, 247–257. [Google Scholar]
- Pollard, D.J.; Woodley, J.M. Biocatalysis for pharmaceutical intermediates: The future is now. Trends Biotechnol. 2007, 25, 66–73. [Google Scholar] [CrossRef]
- Otey, C.R.; Bandara, G.; Lalonde, J.; Takahashi, K.; Arnold, F.H. Preparation of human metabolites of propranolol using laboratory-evolved bacterial cytochromes P450. Biotechnol. Bioeng. 2006, 93, 494–499. [Google Scholar] [CrossRef]
- Huisman, G.W.; Lalonde, J.J. Enzyme evolution for chemical process applications. In Biocatalysis in the Pharmaceutical and Biotechnology Industries; Patel, R.N., Ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 717–742. [Google Scholar]
- Arnold, F.; Volkov, A. Directed evolution of biocatalysts. Curr. Opin. Chem. Biol. 1999, 3, 54–59. [Google Scholar] [CrossRef]
- Patten, P.A.; Howard, R.J.; Stemmer, W.P. Applications of DNA shuffling to pharmaceuticals and vaccines. Curr. Opin. Biotechnol. 1997, 8, 724–733. [Google Scholar] [CrossRef]
- Hibbert, E.G.; Baganz, F.; Hailes, H.C.; Ward, J.M.; Lye, G.J.; Woodley, J.M.; Dalby, P.A. Directed evolution of biocatalytic processes. Biomol. Eng. 2005, 22, 11–19. [Google Scholar] [CrossRef]
- Sylvestre, J.; Chautard, H.; Cedrone, F.M. Directed evolution of biocatalysts. Org. Process Res. Dev. 2006, 10, 562–571. [Google Scholar] [CrossRef]
- Turner, N.J. Directed evolution drives the next generation of biocatalysts. Nat. Chem. Biol. 2009, 5, 567–573. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef]
- Wells, A.S.; Finch, G.L.; Michels, P.C.; Wong, J.W. Use of enzymes in the manufacture of active pharmaceutical ingredients—A science and safety-based approach to ensure patient safety and drug quality. Org. Process Res. Dev. 2012, 16, 1986–1993. [Google Scholar] [CrossRef]
- Reetz, M.T. Biocatalysis in organic chemistry and biotechnology: Past, present, and future. J. Am. Chem. Soc. 2013, 135, 12480–12496. [Google Scholar] [CrossRef]
- Huisman, G.W.; Collier, S.J. On the development of new biocatalytic processes for practical pharmaceutical synthesis. Curr. Opin. Chem. Biol. 2013, 17, 284–292. [Google Scholar] [CrossRef]
- Bryan, M.C.; Dillon, B.; Hamann, L.G.; Hughes, G.J.; Kopach, M.E.; Peterson, E.A.; Pourashraf, M.; Raheem, I.; Richardson, P.; Richter, D.; et al. Sustainable practices in medicinal chemistry: Current state and future directions. J. Med. Chem. 2013, 56, 6007–6021. [Google Scholar] [CrossRef]
- DiCosimo, R. Nitrilases and nitrile hydratases. In Biocatalysis in the Pharmaceutical and Biotechnology Industries; Patel, R.N., Ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 1–26. [Google Scholar]
- Patel, R.N. Biocatalysis: Synthesis of chiral intermediates for pharmaceuticals. Curr. Org. Chem. 2006, 10, 1289–1321. [Google Scholar] [CrossRef]
- Simeo, Y.; Kroutil, W.; Faber, K. Biocatalytic deracemization: Dynamic resolution, stereoinversion, enantioconvergent processes, and cyclic deracemization. In Biocatalysis in the Pharmaceutical and Biotechnology Industries; Patel, R.N., Ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 27–51. [Google Scholar]
- Patel, R.N. Biocatalysis: Synthesis of key intermediates for development of pharmaceuticals. ACS Catal. 2011, 1, 1056–1074. [Google Scholar] [CrossRef]
- Turner, N.J. Enzyme catalyzed deracemization and dynamic kinetic resolution reactions. Curr. Opin. Chem. Biol. 2004, 8, 114–119. [Google Scholar] [CrossRef]
- Ishige, T.; Honda, K.; Shimizu, S. Whole organism biocatalysis. Curr. Opin. Chem. Biol. 2005, 9, 174–180. [Google Scholar] [CrossRef]
- Zhao, H.; Chockalingom, K.; Chen, Z. Directed evolution of enzymes and pathways for industrial biocatalysis. Curr. Opin. Biotechnol. 2002, 13, 104–110. [Google Scholar] [CrossRef]
- Schulze, B.; Wubbolts, M. Biocatalysis for industrial production of fine chemicals. Curr. Opin. Biotechnol. 1999, 10, 609–611. [Google Scholar] [CrossRef]
- Steinreiber, A.; Faber, K. Microbial epoxide hydrolases for preparative biotransformations. Curr. Opin. Biotechnol. 2001, 12, 552–558. [Google Scholar] [CrossRef]
- Tao, J.; Xu, J.-H. Biocatalysis in development of green pharmaceutical processes. Curr. Opin. Chem. Biol. 2009, 13, 43–50. [Google Scholar] [CrossRef]
- Patel, R.N. Biocatalytic hydrolysis (esters, amides, epoxides, nitriles) and biocatalytic dynamic kinetic resolution. In Comprehensive Chirality; 2012; Volume 10, pp. 288–317. [Google Scholar]
- Patel, R.N. Biocatalytic routes to chiral intermediates for development of drugs. In Biocatalysis for Green Chemistry and Chemical Process Development; Tao, J., Kazlauskas, R., Eds.; John Wiley & Sons, Inc.: Hoboken, USA city, 2010. [Google Scholar]
- Jajoo, H.; Mayol, R.; LaBudde, J.; Blair, I. Metabolism of the antianxiety drug buspirone in human subjects. Drug Metab. Dispos. 1989, 17, 634–640. [Google Scholar]
- Mayol, R. Buspirone Metabolite for the Alleviation of Anxiety. US6150365A, 6 June 2000. [Google Scholar]
- Yevich, J.; New, J.; Lobeck, W.; Dextraze, P.; Bernstein, E.; Taylor, D.; Yocca, F.; Eison, M.; Temple, D., Jr. Synthesis and biological characterization of α-(4-fluorophenyl)-4-(5-fluoro-2-pyrimidinyl)-1-piperazinebutanol and analogs as potential atypical antipsychotic agents. J. Med. Chem. 1992, 35, 4516–4525. [Google Scholar] [CrossRef]
- Yevich, J.; Mayol, R.; Li, J.; Yocca, F. (S)-6-Hydroxy-Buspirone for Treatment of Anxiety, Depression and Related Disorders. US2003022899, 30 January 2003. [Google Scholar]
- Patel, R.; Chu, L.; Nanduri, V.; Jianqing, L.; Kotnis, A.; Parker, W.; Liu, M.; Mueller, R. Enantioselective microbial reduction of 6-oxo-8-[4-[4-(2-pyrimidinyl)-1-piperazinyl]butyl]-8-azaspiro[4.5]decane-7,9-dione. Tetrahedron Asymmetry 2005, 16, 2778–2783. [Google Scholar] [CrossRef]
- Goldberg, S.; Nanduri, V.; Chu, L.; Johnston, R.; Patel, R. Enantioselective microbial reduction of 6-oxo-8-[4-[4-(2-pyrimidinyl)-1-piperazinyl]butyl]-8-azaspiro[4.5]decane-7,9-dione: Cloning and expression of reductases. Enzyme Microb. Technol. 2006, 39, 1441–1450. [Google Scholar] [CrossRef]
- Patel, R.N.; Banerjee, A.; McNamee, C.; Brzozowski, D.; Hanson, R.; Szarka, L. Enantioselective microbial reduction of 3,5-dioxo-6-(benzyloxy) hexanoic acid, ethyl ester. Enzyme Microb. Technol. 1993, 15, 1014–1021. [Google Scholar] [CrossRef]
- Sit, S.; Parker, R.; Motoe, I.; Balsubramanian, H.; Cott, C.; Brown, P.; Harte, W.; Thompson, M.; Wright, J. Synthesis, biological profile, and quantitative structure activity relationship of a series of novel 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. J. Med. Chem. 1990, 33, 2982–2999. [Google Scholar] [CrossRef]
- Roth, B.D. The discovery and development of atorvastatin, a potent novel hypolipidemic agent. Prog. Med. Chem. 2002, 40, 1–22. [Google Scholar] [CrossRef]
- Law, M.; Rudinka, A.R. Statin safety: A systematic review. Am. J. Cardiol. 2006, 97(8A), 52C–60C. [Google Scholar]
- McTaggart, F.; Buckett, L.; Davidson, R.; Holdgate, G.; McCormick, A.; Schneck, D.; Smith, G.; Warwick, M. Preclinical and clinical pharmacology of Rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Am. J. Cardiol. 2001, 87, 28B–32B. [Google Scholar]
- Guo, Z.; Chen, Y.; Goswami, A.; Hanson, R.L.; Patel, R.N. Synthesis of ethyl and t-butyl (3R,5S)-dihydroxy-6-benzyloxyhexanoates via diastereo- and enantioselective microbial reduction. Tetrahedron Asymmetry 2006, 17, 1589–1602. [Google Scholar] [CrossRef]
- Goldberg, S.; Guo, Z.; Chen, S.; Goswami, A.; Patel, R.N. Synthesis of ethyl-(3R,5S)-dihydroxy-6-benzyloxyhexanoates via diastereo- and enantioselective microbial reduction: Cloning and expression of ketoreductase III from Acinetobacter sp. SC 13874. Enzyme Microb. Technol. 2008, 43, 544–549. [Google Scholar] [CrossRef]
- Davis, S.; Christopher, G.; John, H.; Gray, D.; Gruber, J.; Huisman, G.; Ma, S.; Newman, L.; Sheldon, R. Enzymatic Processes for the Production of 4-Substituted 3-Hydroxybutyric Acid Derivatives. US 7807423B2, 5 October 2010. [Google Scholar]
- Jagoda, E.; Stouffer, B.; Ogan, M.; Tsay, H.M.; Turabi, N.; Mantha, S.; Yost, F.; Tu, J.I. Radioimmunoassay for hydroxyphosphinyl-3-hydroxybutanoic acid (SQ 33,600), a hypocholesterolemia agent. Ther. Drug Monit. 1993, 15, 213–219. [Google Scholar] [CrossRef]
- Wang, D.I.; Arnold, M.E.; Jemal, M.; Cohen, A.I. Determination of SQ 33,600, a phosphinic acid containing HMG CoA reductase inhibitor, in human serum by high-performance liquid chromatography combined with ionspray mass spectrometry. Biol. Mass Spectrom. 1992, 21, 189–194. [Google Scholar] [CrossRef]
- Patel, R.; McNamee, C.; Banerjee, A.; Howell, J.; Robison, R.; Szarka, L. Stereoselective reduction of β-keto ester by Geotrich candidum. Enzyme Microb. Technol. 1992, 14, 731–738. [Google Scholar] [CrossRef]
- Matsuyama, A.; Yamamoto, H.; Kobayashi, Y. Practical application of recombinant whole-cell biocatalysts for the manufacturing of pharmaceutical intermediates such as chiral alcohols. Org. Process Res. Dev. 2002, 6, 558–561. [Google Scholar] [CrossRef]
- Zalman, L.S.; Brothers, M.A.; Dragovich, P.S.; Zhou, R.; Prins, T.J.; Worland, S.T.; Patick, A.K. Inhibition of human rhinovirus-induced cytokine production by AG7088, a human rhinovirus 3C protease inhibitor. Antimicrob. Agents Chemother. 2000, 44, 1236–1241. [Google Scholar] [CrossRef]
- Dragovich, P.S.; Prins, T.J.; Zhou, R.; Webber, S.E.; Marakovits, J.T.; Fuhrman, S.A.; Patick, A.K.; Matthews, D.A.; Lee, C.A.; Ford, C.E.; et al. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 4. incorporation of P1 lactam moieties as l-glutamine replacements. J. Med. Chem. 1999, 42, 1213–1224. [Google Scholar] [CrossRef]
- Tao, J.; McGee, K. Development of a continuous enzymatic process for the preparation of (R)-3-(4-fluorophenyl)-2-hydroxy propionic acid. Org. Process Res. Dev. 2002, 6, 520–524. [Google Scholar] [CrossRef]
- Bold, G.; Faessler, A.; Capraro, H.-G.; Cozens, R.; Klimkait, T.; Lazdins, J.; JMestan, J.; Poncioni, B.; Roesel, J.; Stover, D.; et al. New aza-dipeptide analogs as potent and orally absorbed HIV-1 protease inhibitors: Candidates for clinical development. J. Med. Chem. 1998, 41, 3387–3401. [Google Scholar] [CrossRef]
- Robinson, B.S.; Riccardi, K.A.; Gong, Y.F.; Guo, Q.; Stock, D.A.; Blair, W.S.; Terry, B.J.; Deminie, C.A.; Djang, F.; Colonno, R.J.; et al. BMS-232632, a highly potent human immunodeficiency virus protease inhibitor that can be used in combination with other available antiretroviral agents. Antimicrob. Agents Chemother. 2000, 44, 2093–2099. [Google Scholar] [CrossRef]
- Patel, R.N.; Chu, L.; Mueller, R.H. Diastereoselective microbial reduction of (S)-[3-chloro-2-oxo-1-(phenylmethyl)propyl]carbamic acid, 1,1-dimethylethyl ester. Tetrahedron Asymmetry 2003, 14, 3105–3109. [Google Scholar] [CrossRef]
- Bowers, N.I.; Skonezny, P.M.; Stein, G.L.; Franceschini, T.; Chiang, S.-J.; Anderson, W.L.; You, L.; Xing, Z. Pocess for preparing (2R,3S)-1,2-epoxy-3-(protected)amino-4-substituted butane and intermediates thereof. WO 2006127180 A1, 30 November 2006. [Google Scholar]
- Xu, Z.; Singh, J.; Schwinden, M.D.; Zheng, B.; Kissick, T.P.; Patel, B.; Humora, M.J.; Quiroz, F.; Dong, L.; Hsieh, D.-M.; et al. Process research and development for an efficient synthesis of the HIV protease inhibitor BMS-232632. Org. Process Res. Dev. 2002, 6, 323–328. [Google Scholar] [CrossRef]
- King, A.O.; Corley, E.G.; Anderson, R.K.; Larsen, R.D.; Verhoeven, T.R.; Reider, P.J.; Xiang, Y.B.; Belley, M.; Leblanc, Y.; Labelle, M.; et al. An efficient synthesis of LTD4 antagonist L-699,392. J. Org. Chem. 1993, 58, 3731–3735. [Google Scholar] [CrossRef]
- Shinkai, I.; King, A.O.; Larsen, R.D. Practical asymmetric synthesis of LTD4 antagonist. Pure Appl. Chem. 1994, 66, 1551–1556. [Google Scholar] [CrossRef]
- Zhao, M.; King, A.O.; Larsen, R.D.; Verhoeven, T.R.; Reider, P.J. A convenient and economical method for the preparation of DIP-Chloride™ and its application in the asymmetric reduction of aralkyl ketones. Tetrahedron Lett. 1997, 36, 2641–2644. [Google Scholar]
- Shafiee, A.; Motamedi, H.; King, A. Purification, characterization and immobilization of an NADPH-dependent enzyme involved in the chiral specific reduction of the keto ester M, an intermediate in the synthesis of an anti-asthma drug, Montelukast, from Microbacterium campoquemadoensis (MB5614). Appl. Microbiol. Biotechnol. 1998, 49, 709–717. [Google Scholar] [CrossRef]
- Liang, J.; Lalonde, J.; Borup, B.; Mitchell, V.; Mundorff, E.; Trinh, N.; Kochrekar, D.A.; Cherat, R.N.; Pai, G.G. Development of a biocatalytic process as an alternative to the (−)-DIP-Cl-mediated asymmetric reduction of a key intermediate of Montelukast. Org. Proc. Res. Dev. 2010, 14, 193–198. [Google Scholar] [CrossRef]
- Holton, R.; Biediger, R.; Joatman, P. Semisynthesis of taxol and taxotere. In Taxol: Science and Application; Suffness, M., Ed.; CRC press: NewYork, NY, USA, 1995; pp. 97–123. [Google Scholar]
- Kingston, D. Natural taxoids: Structure and chemistry. In Taxol: Science and Application; Suffness, M., Ed.; CRC press: NewYork, NY, USA, 1995; pp. 287–317. [Google Scholar]
- Patel, R. Tour de paclitaxel: Biocatalysis for semisynthesis. Annu. Rev. Microbiol. 1995, 98, 361–395. [Google Scholar]
- Patel, R.; Banerjee, A.; Howell, J.; McNamee, C.; Brozozowski, D.; Mirfakhrae, D.; Nanduri, V.; Thottathil, J.; Szarka, L. Microbial synthesis of (2R,3S)-N-benzoyl-3-phenyl isoserine ethyl ester-a taxol side-chain synthon. Tetrahedron Asymmetry 1993, 4, 2069–2084. [Google Scholar] [CrossRef]
- Junie, J.L.; Leonard, B.E. Drugs acting on sigma and phencyclidine receptors: A review of their nature, function, and possible therapeutic importance. Clin. Neuropharmacol. 1989, 12, 353–374. [Google Scholar] [CrossRef]
- Ferris, C.D.; Hirsch, D.J.; Brooks, B.P.; Snyder, S.H. σ Receptors: From molecule to man. J. Neurochem. 1991, 57, 729–737. [Google Scholar] [CrossRef]
- Patel, R.N.; Banerjee, A.; Liu, M.; Hanson, R.; Ko, R.; Howell, J.; Szarka, L. Microbial reduction of 1-(4-fluorophenyl)-4-[4-(5-fluoro-2-pyrimidinyl)-1-piperazinyl]butan-1-one. Biotechnol. Appl. Biochem. 1993, 17, 139–153. [Google Scholar]
- Kagechika, H.; Kawachi, E.; Hashimoto, Y.; Shudo, K.; Himi, T. Retinobenzoic acids. 1. Structure-activity relationships of aromatic amides with retinoidal activity. J. Med. Chem. 1989, 32, 2583–2588. [Google Scholar]
- Kagechika, H.; Shudo, K. Retinoids. Vitamin A for clinical applications. Farumashia 1990, 26, 35–40. [Google Scholar]
- Moon, R.C.; Mehta, R.G. Anticarcinogenic effects of retinoids in animals. Adv. Exp. Med. Biol. 1986, 206, 399–411. [Google Scholar]
- Patel, R.N.; Chu, L.; Chidambaram, R.; Zhu, J.; Kant, J. Enantioselective microbial reduction of 2-oxo-2-(1',2',3',4'-tetrahydro-1',1',4',4'-tetramethyl-6'-naphthalenyl)acetic acid and its ethyl ester. Tetrahedron Asymmetry 2002, 13, 349–355. [Google Scholar] [CrossRef]
- Prasad, C.V.C.; Wallace, O.B.; Noonan, J.W.; Sloan, C.P.; Lau, W.; Vig, S.; Parker, M.F.; Smith, D.W.; Hansel, S.B.; Polson, C.T.; et al. Hydroxytriamides as potent γ-secretase inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 3361–3371. [Google Scholar]
- Nanduri, V.B.; Hanson, R.L.; Goswami, A.; Wasylyk, J.M.; LaPorte, T.L.; Katipally, K.; Chung, H.-J.; Patel, R.N. Biochemical approaches to the synthesis of ethyl 5-(S)-hydroxyhexanoate and 5-(S)-hydroxyhexanenitrile. Enzyme Microb. Technol. 2001, 28, 632–636. [Google Scholar] [CrossRef]
- Schenk, D.G.; Seubert, P. Potential treatment opportunities for Alzheimer’s disease through inhibition of secretases and Aβ immunization. J. Mol. Neurosci. 2001, 17, 259–267. [Google Scholar] [CrossRef]
- Patel, R.N.; Goswami, A.; Chu, L.; Donovan, M.J.; Nanduri, V.; Goldberg, S.; Johnston, R.; Siva, P.J.; Nielsen, B.; Fan, J.; et al. Enantioselective microbial reduction of substituted acetophenones. Tetrahedron Asymmetry 2004, 15, 1247–1258. [Google Scholar] [CrossRef]
- Wittman, M.; Carboni, J.M.; Attar, R.; Balasubramanian, B.; Balimane, P.; Brassil, P.; Beaulieu, F.; Chang, C.; Clarke, W.; Dell, J.; et al. Discovery of a 1H-Benzoimidazol-2-yl)-1H-pyridin-2-one (BMS-536924) inhibitor of insulin-like growth factor I receptor kinase with in vivo antitumor activity. J. Med. Chem. 2005, 48, 5639–5643. [Google Scholar] [CrossRef]
- Carboni, J.M.; Hurlburt, W.W.; Gottardis, M.M. Synergistic methods and compositions for treating cancer. WO 2004030625 A2, 15 April 2004. [Google Scholar]
- Beaulieu, F.; Ouellet, C.; Zimmermann, K.; Velaparthi, U.; Wittman, M.D. Novel tyrosine kinase inhibitors. WO 2004063151 A3, 9 December 2004. [Google Scholar]
- Hanson, R.L.; Goldberg, S.; Goswami, A.; Tully, T.P.; Patel, R.N. Purification and cloning of a ketoreductase used for the preparation of chiral alcohols. Adv. Synth. Catal. 2005, 347, 1073–1080. [Google Scholar] [CrossRef]
- Vacca, J.P. New advances in the discovery of thrombin and factor Xa inhibitors. Curr. Opin. Chem. Biol. 2000, 4, 394–400. [Google Scholar] [CrossRef]
- Gladwell, T.D. Bivalirudin: A direct thrombin inhibitor. Clin. Ther. 2002, 24, 38–58. [Google Scholar] [CrossRef]
- Fevig, J.M.; Wexler, R.R. Anticoagulants: Thrombin and factor Xa inhibitors. Annu. Rep. Med. Chem. 1999, 34, 81–100. [Google Scholar] [CrossRef]
- Williams, P.D.; Coburn, C.; Burgey, C.; Morrissette, M.M. Preparation of Triazolopyrimidines as Thrombin Inhibitors. WO 2002064211 A1, 22 August 2002. [Google Scholar]
- Nelson, T.D.; LeBlond, C.R.; Frantz, D.E.; Matty, L.; Mitten, J.V.; Weaver, D.G.; Moore, J.C; Kim, J.M.; Boyd, R.; Kim, P.-Y.; et al. Stereoselective synthesis of a potent thrombin inhibitor by a novel P2-P3 lactone ring opening. J. Org. Chem. 2004, 69, 3620–3627. [Google Scholar] [CrossRef]
- Fukuroda, T.; Nishikibe, M. Enhancement of pulmonary artery contraction induced by endothelin-β receptor antagonism. J. Cardiovasc. Pharmacol. 1998, 31, S169–S171. [Google Scholar] [CrossRef]
- Sumner, M.J.; Cannon, T.R.; Mundin, J.W.; White, D.G.; Watts, I.S. Endothelin ETA and ETB receptors mediate vascular smooth muscle contraction. Br. J. Pharmacol. 1992, 107, 858–860. [Google Scholar] [CrossRef]
- Krulewicz, B.; Tschaen, D.; Devine, P.; Lee, S.S.; Roberge, C.; Greasham, R.; Chartrain, M. Asymmetric biosynthesis of key aromatic intermediates in the synthesis of an endothelin receptor antagonist. Biocatal. Biotransformation 2001, 19, 267–279. [Google Scholar] [CrossRef]
- Chaffman, M.; Brogden, R.N. Diltiazem. A review of its pharmacological properties and therapeutic efficacy. Drugs 1985, 29, 387–390. [Google Scholar] [CrossRef]
- Kawai, C.; Konishi, T.; Matsuyama, E.; Okazaki, H. Comparative effects of three calcium antagonists, diatiazem, verapamil and nifedipine, on the sinoatrial and atrioventricular nodes. Experimental and clinical studies. Circulation 1981, 63, 1035–1038. [Google Scholar] [CrossRef]
- Isshiki, T.; Pegram, B.; Frohlich, E. Immediate and prolonged hemodynamic effects of TA-3090 on spontaneously hypertensive (SHR) and normal Wistar-Kyoto (WKY) rats. Cardiovasc. Drug Ther. 1988, 2, 539–544. [Google Scholar] [CrossRef]
- Das, J.; Floyd, D.M.; Kimball, D.; Duff, K.J.; Lago, M.W.; Moquin, R.V.; Gougoutas, J.Z. Benzazepinone calcium channel blockers. 3. Synthesis and structure-activity studies of 3-alkylbenzazepinones. J. Med. Chem. 1992, 35, 773–780. [Google Scholar] [CrossRef]
- Patel, R.N.; Robison, R.S.; Szarka, L.J.; Kloss, J.; Thottathil, J.K.; Mueller, R.H. Stereospecific microbial reduction of 4,5-dihydro-4-(4-methoxyphenyl)-6-(trifluoromethyl-1H-1)-benzazepin-2-one. Enzyme Microb. Technol. 1991, 13, 906–912. [Google Scholar] [CrossRef]
- Arch, J.R.S. β3-adrenoceptors and other putative atypical β-adrenoceptors. Pharmacol. Rev. Commun. 1997, 9, 141–148. [Google Scholar]
- Bloom, J.D.; Datta, M.D.; Johnson, B.D.; Wissner, A.; Bruns, M.G.; Largis, E.E.; Dolan, J.A.; Claus, T.H. Disodium (R,R)5-[2-(3-chlorophenyl)-2-hydroxyethyl]amino]propyl]-1,3-benzodioxole-2,2-dicarboxylate. A potent b-adrenergic agonist virtually specific for β3 receptors. J. Med. Chem. 1989, 35, 3081–3084. [Google Scholar]
- Fisher, L.G.; Sher, P.M.; Skwish, S.; Michael, I.M.; Seiler, S.; Dickinson, K.E.J. BMS-187257, a potent, selective, and novel heterocyclic β-3 adrenergic receptor agonist. Bioorg. Med. Chem. Lett. 1994, 6, 2253–2258. [Google Scholar]
- Patel, R.N.; Banerjee, A.; Chu, L.; Brzozowski, D.; Nanduri, V.; Szarka, L.J. Microbial synthesis of chiral intermediates for β-3-receptor agonists. J. Am. Oil Chem. Soc. 1998, 75, 1473–1482. [Google Scholar] [CrossRef]
- Iwata, H.; Tanaka, R.; Ishiguro, M. Structures of the alkaline hydrolysis products of penem antibiotic, SUN5555. J. Antibiot. 1990, 43, 901–903. [Google Scholar] [CrossRef]
- Jean-Paul Vandecasteele, J.-P. Enzymatic synthesis of l-carnitine by reduction of an achiral precursor: The problem of reduced nicotinamide adenine dinucleotide recycling. Appl. Environ. Microbiol. 1980, 39, 327–334. [Google Scholar]
- Yamamoto, H; Matsuyama, A.; Kobayashi, Y. Synthesis of (R)-1,3-butanediol by enantioselective oxidation using whole recombinant Escherichia coli cells expressing (S)-specific secondary alcohol dehydrogenase. Biosci Biotechnol Biochem. 2002, 66, 925–927. [Google Scholar] [CrossRef]
- Coleman, P.J.; Brashear, K.M.; Askew, B.C. Nonpeptide α vβ 3 antagonists. Part 11: Discovery and preclinical evaluation of potent α vβ3 antagonists for the prevention and treatment of osteoporosis. J. Med. Chem. 2004, 47, 4829–4837. [Google Scholar] [CrossRef]
- Pollard, D.J.; Telari, K.; Lane, J.; Humphrey, G.; McWilliams, C.; Nidositko, S.; Salmon, P.; Moore, J. Asymmetric reduction of alpha, beta-unsaturated ketone to (R) allylic alcohol by Candida chilensis. Biotechnol. Bioeng. 2006, 93, 674–684. [Google Scholar] [CrossRef]
- Brands, K.M.J.; Payack, J.F.; Rosen, J.D.; Nelson, T.D.; Candelario, A.; Huffman, M.A.; Zhao, M.M.; Bridgette, J.L.; Zhiguo, C.J.; Song, D.M.; et al. Efficient synthesis of NK1 receptor antagonist aprepitant using a crystallization-induced diastereoselective transformation. J. Am. Chem. Soc. 2003, 125, 2129–2135. [Google Scholar] [CrossRef]
- Pollard, D.; Truppo, M.; Pollard, J.; Chen, C.-Y.; Moore, J. Effective synthesis of (S)-3,5-bistrifluoro methylphenyl ethanol by asymmetric enzymatic reduction. Tetrahedron Asymmetry 2006, 17, 554–559. [Google Scholar] [CrossRef]
- Patel, R.N. Chemo-enzymatic synthesis of pharmaceutical intermediates. Expert Opin. Drug Dis. Dev. 2008, 3, 187–245. [Google Scholar] [CrossRef]
- Ohshima, T.; Soda, K. Stereoselective Biocatalysis: Amino Acid Dehydrogenases and Their Applications in Stereoselective Biocatalysis; Patel, R.N., Ed.; Marcel and Dekker Pub: New York, New York, USA, 2000; pp. 877–903. [Google Scholar]
- Wandrey, C.; Wichman, R.; Leuchtenberger, W. Continuous Enzymic Transformation of Water-Soluble α-Keto Carboxylic Acids into the Corresponding Amino Acids. EP 0023346 B1, 13 July 1983. [Google Scholar]
- Brunhuber, N.M.; Blanchard, J.S. The biochemistry and enzymology of amino acid dehydrogenases. Crit. Rev. Biochem. Mol. Biol. 1994, 29, 415–467. [Google Scholar] [CrossRef]
- Gordon, E.M.; Ondetti, M.A.; Pluscec, J.; Cimarusti, C.M.; Bonner, D.P.; Sykes, R.B. O-Sulfated β-lactam hydroxamic acids (monosulfactams). Novel monocyclic β-lactam antibiotics of synthetic origin. J. Am. Chem. Soc. 1982, 104, 6053–6060. [Google Scholar]
- Godfrey, J.D.; Mueller, R.H.; van Langen, D.J. β-Lactam synthesis: Cyclization versus 1,2-acyl migration-cyclization. The mechanism of the 1,2-acyl migration-cyclization. Tetrahedron Lett. 1986, 27, 2793–2796. [Google Scholar] [CrossRef]
- Hanson, R.L.; Singh, J.; Kissick, T.P.; Patel, R.N.; Szarka, L.; Mueller, R. Synthesis of L-β-hydroxyvaline from α-keto-β-hydroxyisovalerate using leucine dehydrogenase from Bacillus species. Bioorg. Chem. 1990, 18, 116–130. [Google Scholar] [CrossRef]
- Hanson, R.; Goldberg, S.; Patel, R. (unpublished results).
- Stoyan, T.; Recktenwald, A.; Kula, M.R. Cloning, sequencing and overexpression of the leucine dehydrogenase gene from Bacillus cereus. J. Biotechnol. 1997, 54, 77–80. [Google Scholar] [CrossRef]
- Menzel, A.; Werner, H.; Altenbuchner, J.; Groger, H. From enzymes to “designer bugs” in reductive amination: A new process for the synthesis of L-tert-leucine using a whole cell. Eng. Life Sci. 2004, 4, 573–576. [Google Scholar] [CrossRef]
- Robl, J.; Sun, C.; Stevenson, J.; Ryono, D.; Simpkins, L.; Cimarusti, M.; Dejneka, T.; Slusarchyk, W.; Chao, S.; Stratton, L.; et al. Dual metalloprotease inhibitors: Mercaptoacetyl-based fused heterocyclic dipeptide mimetics as inhibitors of angiotensin-converting enzyme and neutral endopeptidase. J. Med. Chem. 1997, 40, 1570–1577. [Google Scholar] [CrossRef]
- Hanson, R.L.; Schwinden, M.D.; Banerjee, A.; Brzozowski, D.B.; Chen, B.-C.; Patel, B.P.; McNamee, C.G.; Kodersha, G.A.; Kronenthal, D.R.; Patel, R.N.; et al. Enzymatic synthesis of L-6-hydroxynorleucine. Bioorg. Med. Chem. 1999, 7, 2247–2252. [Google Scholar] [CrossRef]
- Patel, R. Enzymatic synthesis of chiral intermediates for Omapatrilat, an antihypertensive drug. Biomol. Eng. 2001, 17, 167–182. [Google Scholar] [CrossRef]
- Hanson, R.L.; Howell, J.; LaPorte, T.; Donovan, M.; Cazzulino, D.; Zannella, V.; Montana, M.; Nanduri, V.; Schwarz, S.; Eiring, R.; et al. Synthesis of allylsine ethylene acetal using phenylalanine dehydrogenase from Thermoactinomyces intermedius. Enzyme Microb. Technol. 2000, 26, 348–358. [Google Scholar] [CrossRef]
- Gallwitz, B. Glucagon-like peptide-1-based therapies for the treatment of type 2 diabetes mellitus. Treat. Endocrinol. 2005, 4, 361–370. [Google Scholar] [CrossRef]
- Sinclair, E.M.; Drucker, D.J. Glucagon-like peptide 1 receptor agonists and dipeptidyl peptidase IV inhibitors: New therapeutic agents for the treatment of type 2 diabetes. Curr. Opin. Endocrinol. Diabet. 2005, 12, 146–151. [Google Scholar] [CrossRef]
- Augeri, D.J.; Robl, J.A.; Betebenner, D.A.; Magnin, D.R.; Khanna, A.; Robertson, J.G.; Wang, A.; Simpkins, L.M.; Taunk, P.; Huang, Q.; et al. A highly potent, long-acting, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2005, 48, 5025–5037. [Google Scholar] [CrossRef]
- Augeri, D.J.; Betebenner, D.A.; Hamann, L.G.; Magnin, D. R.; Robl, J.A.; Sulsky, R.B. Cyclopropyl-fused pyrrolidine-based inhibitors of dipeptidyl iv, processes for their preparation and their use. WO 2001068603 A2, 20 September 2001. [Google Scholar]
- Hanson, R.L.; Goldberg, S.L.; Brzozowski, D.B.; Tully, T.P.; Cazzulino, D.; Parker, W.L.; Lyngberg, O.K.; Vu, T.C.; Wong, M.K.; Patel, R.N. Preparation of an amino acid intermediate for the dipeptidyl peptidase IV inhibitor, saxagliptin, using a modified phenylalanine dehydro genase. Adv. Synth. Catal. 2007, 349, 1369–1378. [Google Scholar] [CrossRef]
- Groeger, H.; May, O.; Werner, H.; Menzel, A.; Altenbuchner, J. A “second-generation process” for the synthesis of L-neopentylglycine: Asymmetric reductive amination using a recombinant whole cell catalyst. Org. Process Res. Dev. 2006, 10, 666–669. [Google Scholar] [CrossRef]
- Emmanuel, M.J.; Frye, L.L.; Hickey, E.R.; Liu, W.; Morwick, T.M.; Spero, D.M.; Sun, S.; Thomson, D.S.; Ward, Y.D.; Young, E.R.R. Novel Spiroheterocyclic Compounds [Morpholine-4-Carboxylic Acid Amides of Heterocyclic Cyclohexylalanine and Neopentylglycine Derivatives and Their Analogs], Useful as Reversible Inhibitors of Cysteine Proteases such as Cathepsin S. WO 2001019816 A1 22 May 2001. [Google Scholar]
- Haque, T.S.; Ewing, W.R.; Mapelli, C.; Lee, V.G.; Sulsky, R.B.; Riexinger, D.J.; Martinez, R.L.; Zhu, Y.Z. Human Glucagon-like-Peptide-1 Modulators and Their Use in the Treatment of Diabetes and Related Conditions. WO2007082264, A2, 11 January 2007. [Google Scholar]
- Qian, F.; Ewing, W.R.; Mapelli, C.; Riexinger, D.J.; Lee, V.G.; Sulsky, R.B.; Zhu, Y.; Haque, T.S.; Martinez, R.L.; Naringrekar, V.; et al. Sustained release GLP-1 receptor modulators. US 20070099835 A1, 3 May 2007. [Google Scholar]
- Chen, Y.; Goldberg, S.L.; Hanson, R.L.; Parker, W.L.; Gill, I.; Tully, T.P.; Montana, M.; Goswami, A.; Patel, R.N. Enzymatic preparation of an (S)-amino acid from a racemic amino acid. Org. Process Res. Dev. 2011, 15, 241–248. [Google Scholar] [CrossRef]
- Straathof, A.J.J.; Panke, S.A. The production of fine chemicals by biotransformations. Curr. Opin. Biotechnol. 2002, 13, 548–556. [Google Scholar] [CrossRef]
- Bommarius, A.S.; Schwarm, M.; Drauz, K. Biocatalysis to amino acid-based chiral pharmaceuticals-examples and perspectives. J. Mol. Catal. B Enzym. 1998, 5, 1–11. [Google Scholar] [CrossRef]
- Vedha-Peters, K.; Gunawardana, M.; Rozzell, J.D.; Novick, S.J. Creation of a broad-range and highly stereoselective D-amino acid dehydrogenase for the one-step synthesis of D-amino acids. J. Am. Chem. Soc. 2006, 128, 10923–10929. [Google Scholar]
- Gustafsson, D.; Elg, M.; Lenfors, S.; Boerjesson, I.; Teger-Nilsso, A.-C. Effects of inogatran, a new low-molecular-weight thrombin inhibitor, in rat models of venous and arterial thrombosis, thrombolysis and bleeding time. Blood Coagul. Fibrinolysis 1996, 7, 69–79. [Google Scholar] [CrossRef]
- Alexandre, F.-R.; Pantaleone, D.P.; Taylor, P.P.; Fotheringham, I.G.; Ager, D.J.; Turner, N.J. Amine-boranes: Effective reducing agents for the deracemisation of DL-amino acids using L-amino acid oxidase from Proteus myxofaciens. Tetrahedron Lett. 2002, 43, 707–710. [Google Scholar]
- Chaturvedula, P.V.; Chen, L.; Civiello, R.; Degnan, A.P.; Dubowchik, G.M.; Han, X.; Jiang, J.J.; Macor, J.E.; Poindexter, G.S.; Tora, G.O.; et al. Anti-Migraine Spirocycles. US 7842808 B2, 5 January 2007. [Google Scholar]
- Han, X.; Civiello, R.L.; Conway, C.M.; Cook, D.A.; Davis, C.D.; Macci, R.; Pin, S.S.; Ren, S.X.; Schartman, R.; Signor, L.J.; Thalody, G.; Widmann, K.A.; Xu, C.; Chaturvedula, P.V.; Macor, J.E.; Dubowchik, G.M. The synthesis and SAR of calcitonin gene-related peptide (CGRP) receptor antagonists derived from tyrosine surrogates. Part 1. Bioorg Med Chem Lett. 2012, 22, 4723–4727. [Google Scholar] [CrossRef]
- Hanson, R.L.; Davis, B.L.; Goldberg, S.L.; Johnston, R.M.; Parker, W.L.; Tully, T.P.; Montana, M.A.; Patel, R.N. Enzymatic preparation of a D-amino acid from a racemic amino acid or keto acid. Org. Process Res. Dev. 2008, 12, 1119–1129. [Google Scholar] [CrossRef]
- Ising, M.; Zimmermann, U.S.; Künzel, H.E.; Uhr, M.; Foster, A.C.; Learned-Coughlin, S.M.; Holsboer, F.; Grigoriadis, D.E. High-affinity CRF1 receptor antagonist NBI-34041: Preclinical and clinical data suggest safety and efficacy in attenuating elevated stress response. Neuropsychopharmacology 2007, 32, 1941–1949. [Google Scholar] [CrossRef]
- Overstreet, D.H.; Griebel, G. Antidepressant-like effects of CRF1 receptor antagonist SSR125543 in an animal model of depression. Eur. J. Pharmacol. 2004, 497, 49–53. [Google Scholar] [CrossRef]
- Tache, Y.; Martinez, V.; Wang, L.; Million, M. CRF1 receptor signaling pathways are involved in stress-related alterations of colonic function and viscerosensitivity: Implications for irritable bowel syndrome. Br. J. Phamacol. 2004, 141, 1321–1330. [Google Scholar] [CrossRef]
- Yu, W.-L.; Lawrence, F.; Wong, H.; Lelas, S.; Zhang, G.; Lindner, M.D.; Wallace, T.; McElroy, J.; Lodge, N.J.; Gilligan, P.; et al. The pharmacology of DMP696 and DMP904, non-peptidergic CRF1 receptor antagonists. CNS Drug Rev. 2006, 11, 21–52. [Google Scholar] [CrossRef]
- Gilligan, P.J.; Clarke, T.; He, L.; Lelas, S.; Li, Y.-W.; Heman, K.; Fitzgerald, L.; Miller, K.; Zhang, G.; Marshall, A.; et al. 8-(4-Methoxyphenyl)pyrazolo[1,5-a]-1,3,5-triazines: Selective and centrally active corticotropin-releasing factor receptor-1 (CRF1) antagonists. J. Med. Chem. 2009, 52, 3084–3092. [Google Scholar] [CrossRef]
- Hanson, R.L.; Davis, B.L.; Chen, Y.; Goldberg, S.L.; Parker, W.L.; Tully, T.P.; Montana, M.A.; Patel, R.N. Preparation of (R)-amines from racemic amines with an (S)-amine transaminase from Bacillus megaterium. Adv. Synth. Catal. 2008, 350, 1367–1375. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Patel, R.N. Biocatalytic Synthesis of Chiral Alcohols and Amino Acids for Development of Pharmaceuticals. Biomolecules 2013, 3, 741-777. https://doi.org/10.3390/biom3040741
Patel RN. Biocatalytic Synthesis of Chiral Alcohols and Amino Acids for Development of Pharmaceuticals. Biomolecules. 2013; 3(4):741-777. https://doi.org/10.3390/biom3040741
Chicago/Turabian StylePatel, Ramesh N. 2013. "Biocatalytic Synthesis of Chiral Alcohols and Amino Acids for Development of Pharmaceuticals" Biomolecules 3, no. 4: 741-777. https://doi.org/10.3390/biom3040741