Angling for Uniqueness in Enzymatic Preparation of Glycosides


Abstract
:
{kind=link}
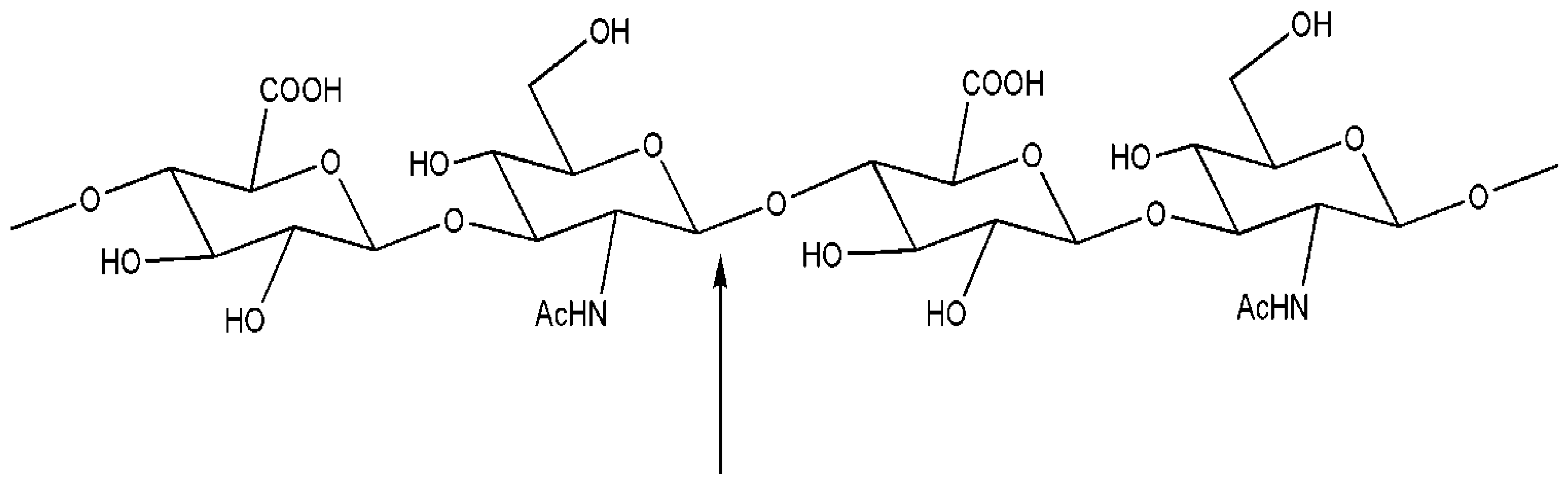{kind=link}
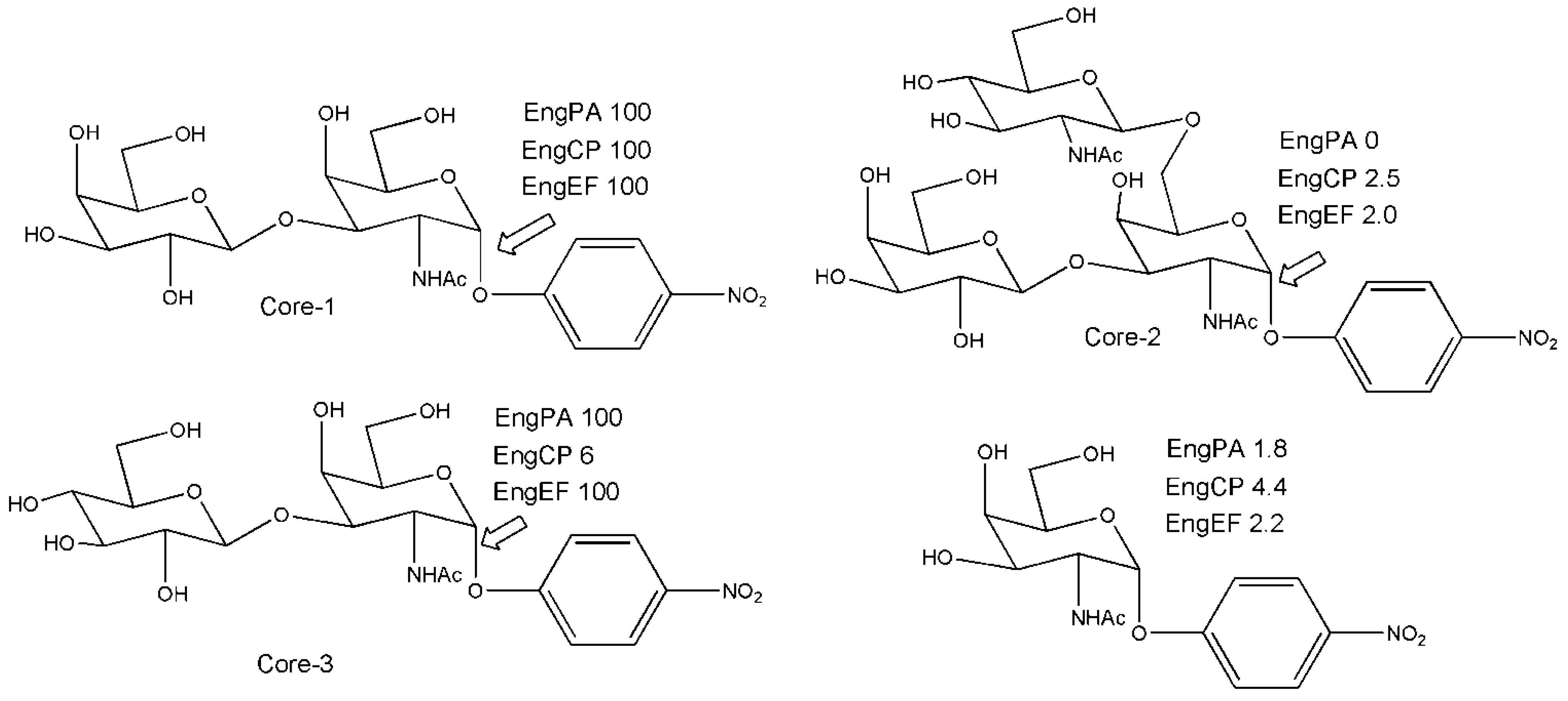{kind=link}
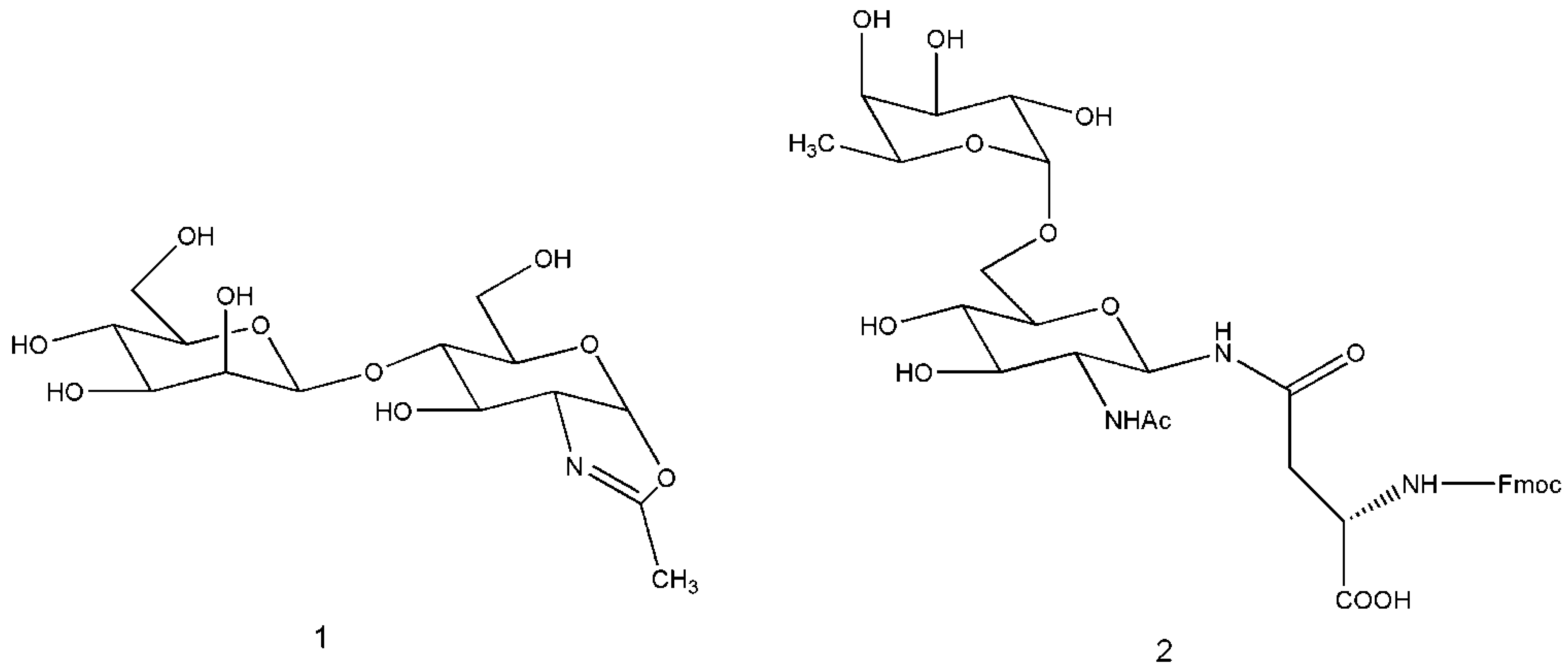{kind=link}
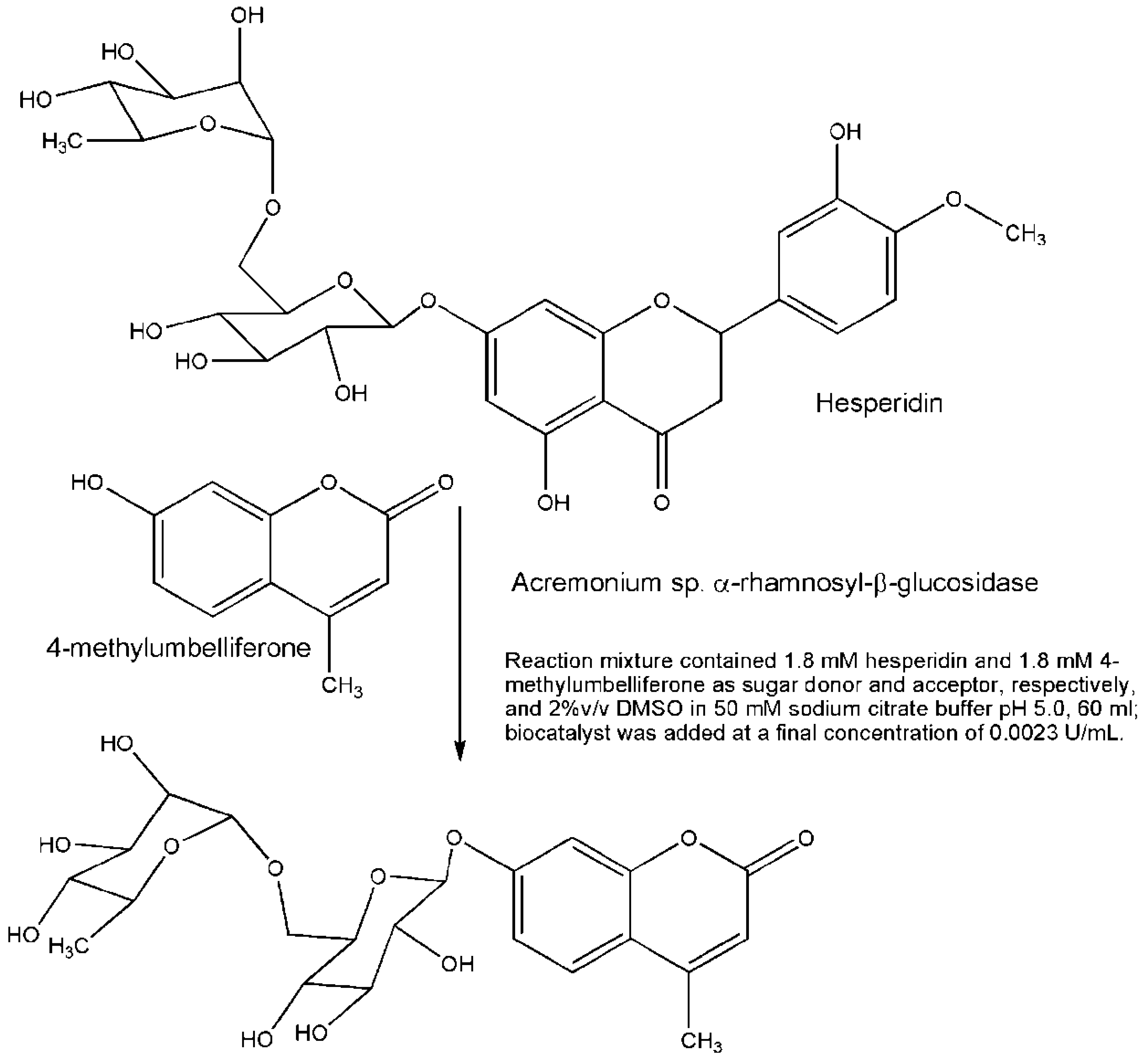{kind=link}
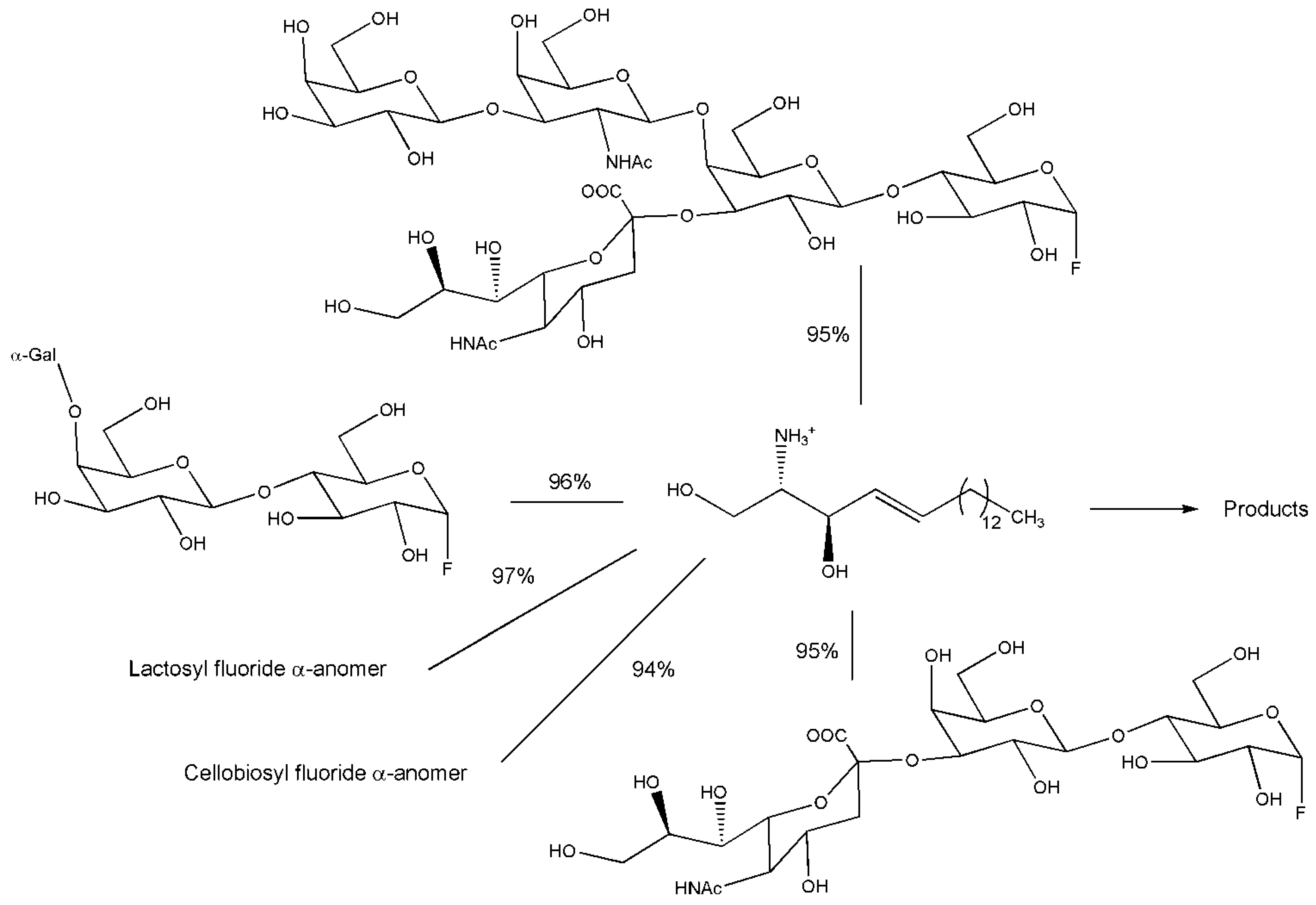{kind=link}
1. Introduction
2. Natural Enzymes for the Synthesis of Glycosidic Linkages
2.1. Interesting Transfer of Glycosyl Residues in Natural Enzymes for the Synthesis of Glycosidic Linkages
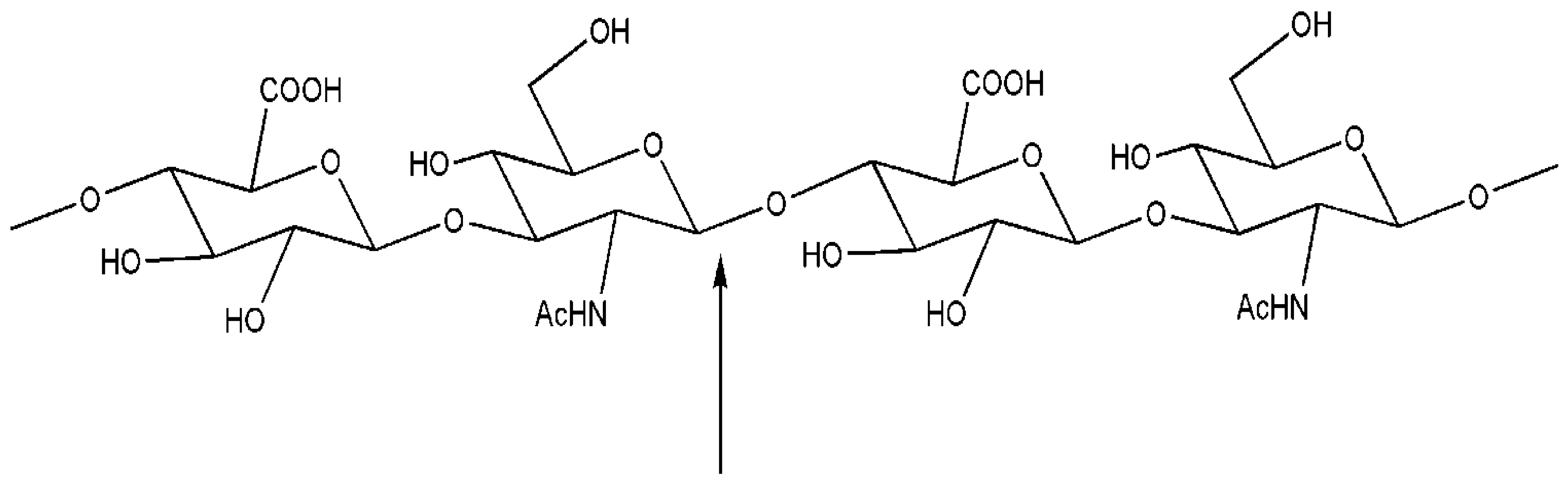
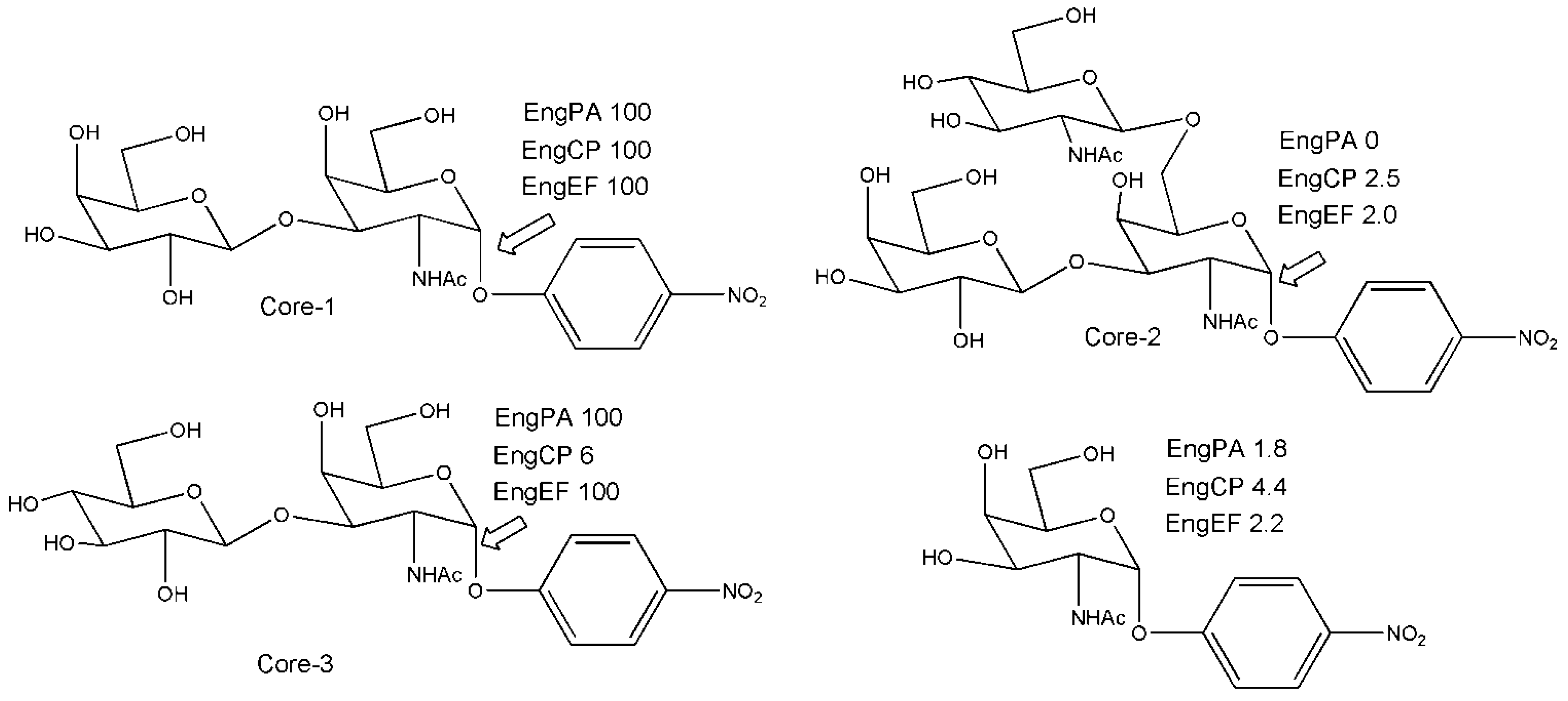
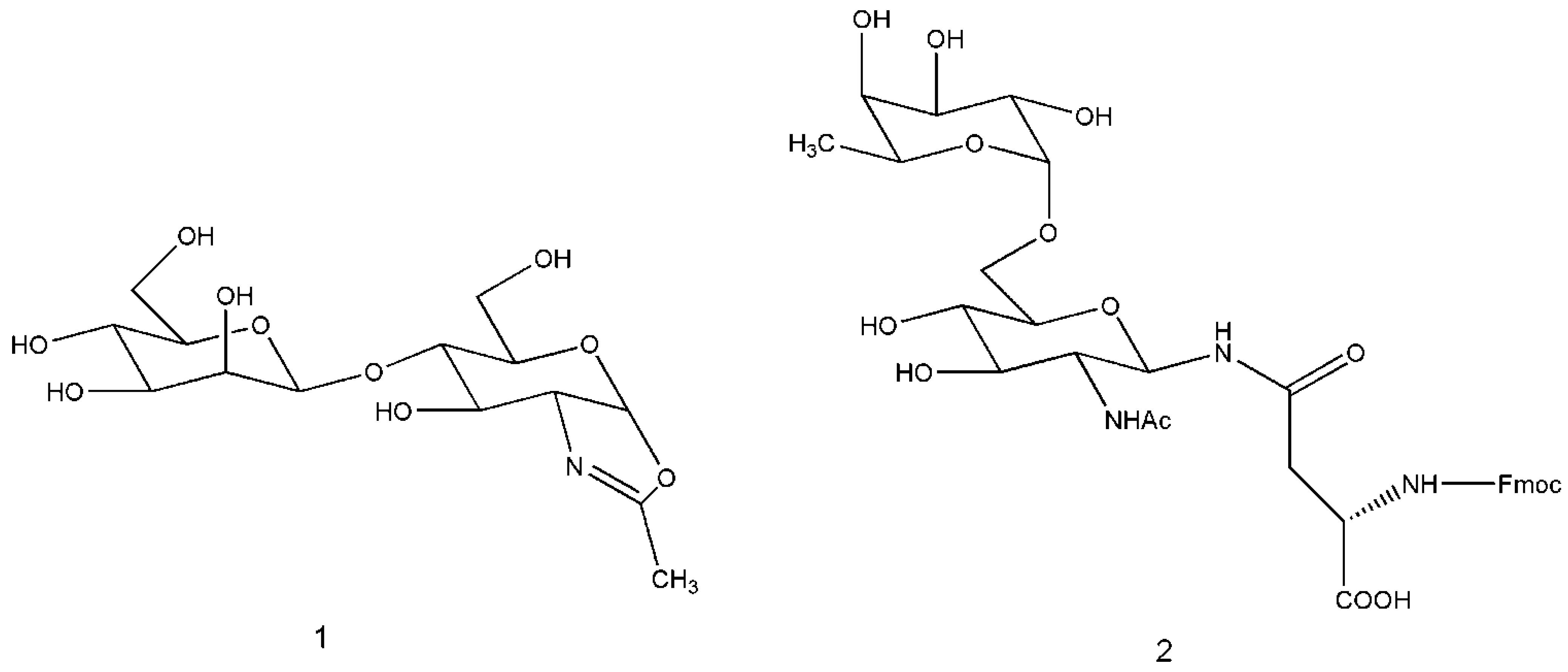
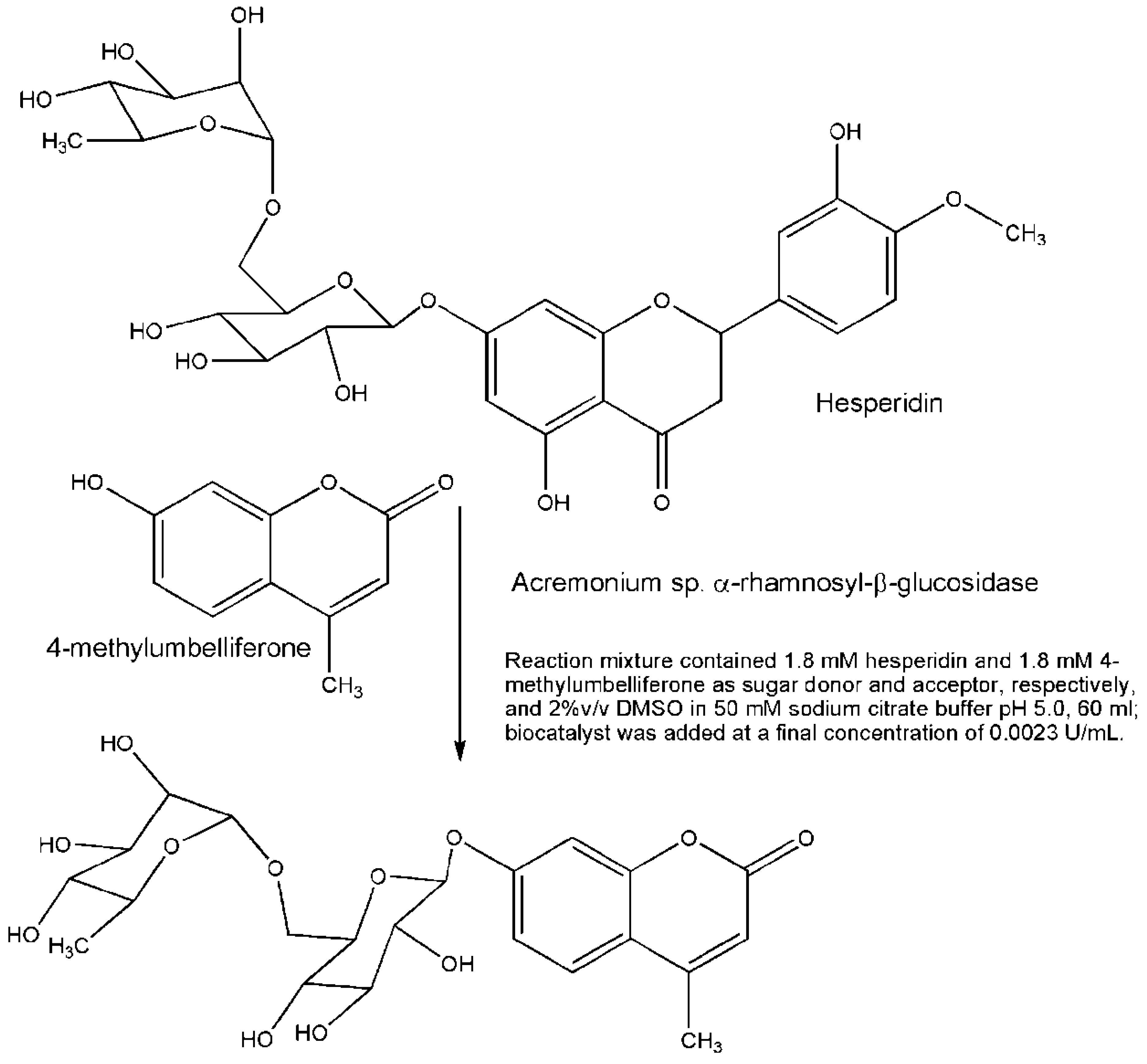
3. Engineered Enzymes
3.1. Preparative Glycosphingolipids Synthesis Operated by Glycosynthase
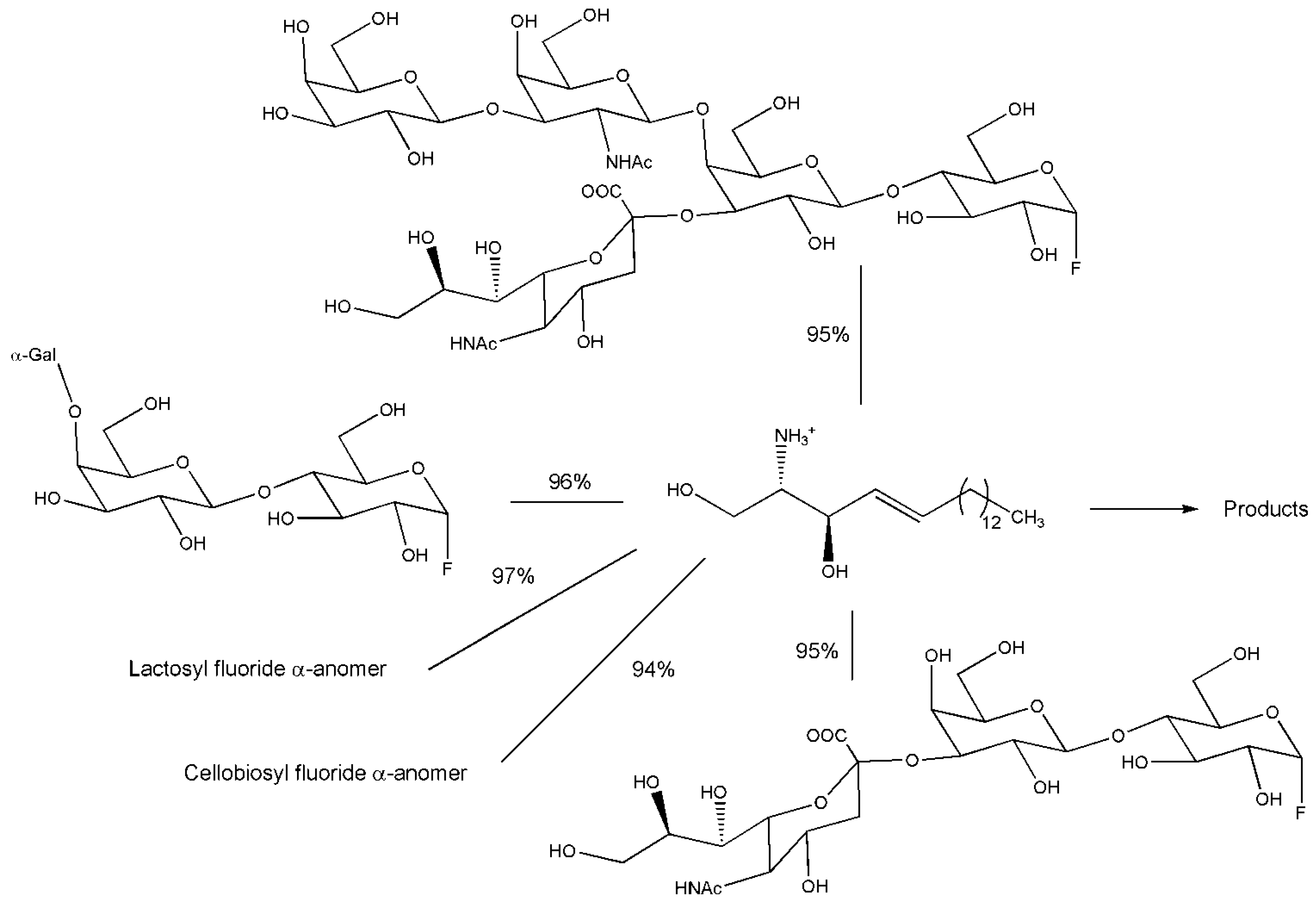
3.2. Engineering Glycoside Hydrolases not at the Active Site
4. Conclusions
Acknowledgments
References
- Bornscheuer, U.T.; Huisman, G.W.; Kazlausaks, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef]
- Riva, S. 1983–2013: The long wave of biocatalysis. Trends Biotechnol. 2013, 31, 120–121. [Google Scholar]
- Trincone, A. Potential biocatalysts originating from sea environments. J. Mol. Catal. B-Enzym. 2010, 66, 241–256. [Google Scholar] [CrossRef]
- Dumon, C.; Songa, L.; Bozonneta, S.; Fauréa, R.; O’Donohue, M.J. Progress and future prospects for pentose-specific biocatalysts in biorefining. Process Biochem. 2012, 47, 346–357. [Google Scholar] [CrossRef]
- Eklunda, E.A.; Bodeb, L.; Freeze, H.H. Diseases Associated with Carbohydrates/Glycoconjugates. In Comprehensive Glycoscience Volume 4: Cell Glycobiology and Development; Health and Disease in Glycomedicine; Kamerling, J.P., Ed.; Elsevier: N.Y., USA, 2007; Volume 4, pp. 339–371. [Google Scholar]
- Kren, V. Glycoside vs. Aglycon: The Role of Glycosidic Residue in Biological Activity. In Glycoscience, Chemistry and Chemical Biology; Fraser-Reid, B.O., Tatsuta, K., Thiem, J., Eds.; Springer-Verlag: Berlin, Heidelberg, 2008; pp. 2589–2644. [Google Scholar]
- Sears, P.; Wong, C.-H. Intervention of carbohydrate recognition by proteins and nucleic acids. Proc. Natl. Acad. Sci. USA 1996, 93, 12086–12093. [Google Scholar] [CrossRef]
- Wang, Q.; Dordick, J.S.; Linhardt, R.J. Synthesis and application of carbohydrate-containing polymers. Chem. Mater. 2002, 14, 3232–3244. [Google Scholar] [CrossRef]
- Wildman, E.C.R. Classifying Nutraceuticals. In Handbook of Nutraceuticals and Functional Foods; Wildman, E.C.R., Ed.; CRC Press: Boca Raton, London, New York Washington, D.C., USA, 2000; pp. 13–30. [Google Scholar]
- Bojarova, P.; Rosencrantz, R.R.; Elling, L.; Kren, V. Enzymatic glycosylation of multivalent scaffolds. Chem. Soc. Rev. 2013, 7, 4774–4797. [Google Scholar]
- De Roode, B.M.; Franseen, M.C.R.; van der Padt, A.; Boom, R.M. Perspectives for the industrial enzymatic production of glycosides. Biotechnol. Prog. 2003, 19, 1391–1402. [Google Scholar]
- Trincone, A.; Giordano, A. Glycosyl hydrolases and glycosyltransferases in the synthesis of oligosaccharides. Curr. Org. Chem. 2006, 10, 1163–1193. [Google Scholar] [CrossRef]
- Desmet, T.; Soetaert, W.; Bojarova, P.; Kren, V.; Dijkhuizen, L.; Eastwick-Field, V.; Schiller, A. Enzymatic glycosylation of small molecules: Challenging substrates require tailored catalysts. Chem. Eur. J. 2012, 18, 10786–10801. [Google Scholar]
- Hehre, E.J. Glycosyl transfer: A history of the concept’s development and view of its major contributions to biochemistry. Carbohydr. Res. 2001, 331, 347–368. [Google Scholar] [CrossRef]
- Kobata, A. The history of glycobiology in Japan. Glycobiology 2001, 11, 99R–105R. [Google Scholar] [CrossRef]
- Koshland, D.E. Stereochemistry and the mechanism of enzymatic reactions. Biol. Rev. 1953, 28, 416–436. [Google Scholar] [CrossRef]
- Vivian, L.Y.; Withers, S.G. Breakdown of oligosaccharides by the process of elimination. Curr. Opin. Chem. Biol. 2006, 10, 147–155. [Google Scholar] [CrossRef]
- Vuong, T.V.; Wilson, D.B. Glycoside hydrolases: Catalytic base/nucleophile diversity. Biotechnol. Bioeng. 2010, 107, 195–205. [Google Scholar] [CrossRef]
- Thibodeaux, C.J.; Melancon, C.E.; Liu, H.-W. Unusual sugar biosynthesis and natural product glycodiversification. Nature 2007, 446, 1008–1016. [Google Scholar] [CrossRef]
- Hofinger, E.S.A.; Bernhardt, G.; Buschauer, A. Kinetics of Hyal-1 and PH-20 hyaluronidases: Comparison of minimal substrates and analysis of the transglycosylation reaction. Glycobiology 2007, 17, 963–971. [Google Scholar] [CrossRef]
- Takagaki, K.; Ishido, K.; Kakizaki, I.; Iwafune, M.; Endo, M. Carriers for enzymatic attachment of glycosaminoglycan chains to peptide. Biochem. Biophys. Res. Comm. 2002, 293, 220–224. [Google Scholar] [CrossRef]
- Madokoro, M.; Ueda, A.; Kiriake, A.; Shiomi, K. Properties and cDNA cloning of a hyaluronidase from the stonefish Synanceia verrucosa venom. Toxicon 2011, 58, 285–292. [Google Scholar] [CrossRef]
- Koutsioulis, D.; Landry, D.; Guthire, E.P. Novel endo-α-N-acetylgalactosaminidases with broader substrate specificity. Glycobiology 2008, 18, 799–805. [Google Scholar] [CrossRef]
- Fujita, M.; Shoda, S.; Haneda, K.; Inazu, T.; Takegawa, K.; Yamamoto, K. A novel disaccharide substrate having 1,2-oxazoline moiety for detection of transglycosylating activity of endoglycosidases. Biochimi. Biophys. Acta 2001, 1528, 9–14. [Google Scholar]
- Huang, W.; Li, J.; Wang, L.-X. Unusual transglycosylation activity of Flavobacterium meningosepticum endoglycosidases enables convergent chemoenzymatic synthesis of core fucosylated complex N-glycopeptides. Chembiochem 2011, 11, 932–941. [Google Scholar]
- Slámová, K.; Gažák, R.; Bojarová, P.; Kulik, N.; Ettrich, R.; Pelantová, H.; Sedmera, P.; Křen, V. 4-Deoxy-substrates for β-N-acetylhexosaminidases: How to make use of their loose specificity. Glycobiology 2010, 20, 1002–1009. [Google Scholar] [CrossRef]
- Hu, Y.; Luan, H.; Liu, H.; Ge, G.; Zhou, K.; Liu, Y.; Yang, L. Acceptor specificity and transfer efficiency of a β-glycosidase from the China white jade snail. Biosci. Biotechnol. Biochem. 2009, 73, 671–676. [Google Scholar] [CrossRef]
- Roccatagliata, A.J.; Maier, M.S.; Seldes, A.M.; Iorizzi, M.; Minale, L. Starifhs saponins. Part II. Steroidal oligoglycosides from the starfish Cosmasterias lurida. J. Nat. Prod. 1994, 57, 747–754. [Google Scholar]
- Ye, M.; Yan, L.-Q.; Li, N.; Zong, M.-H. Facile and regioselective enzymatic 5β-galactosylation of pyrimidine 2β-deoxynucleosides catalyzed by β-glycosidase from bovine liver. J. Mol. Cat. B Enzym. 2012, 79, 35–40. [Google Scholar]
- Binder, W.H.; Kahlig, H.; Schmid, W. Galactosylation by use of β-galactosidase: Enzymatic syntheses of disaccharide nucleosides. Tetrahedron Asymm. 1995, 6, 1703–1710. [Google Scholar] [CrossRef]
- Andreotti, G.; Trincone, A.; Giordano, A. Convenient synthesis of β-galactosyl nucleosides using the marine β-galactosidase from Aplysia fasciata. J. Mol. Catal. B-Enzym. 2007, 47, 28–32. [Google Scholar]
- Zeng, Q.M.; Li, N.; Zong, M.H. Highly regioselective galactosylation of floxuridine catalyzed by β-galactosidase from bovine liver. Biotechnol. Lett. 2010, 32, 1251–1254. [Google Scholar]
- Andreotti, G.; Giordano, A.; Tramice, A.; Mollo, E.; Trincone, A. Hydrolyses and transglycosylations performed by purified α-D-glucosidase of the marine mollusc Aplysia fasciata. J. Biotechnol. 2006, 122, 274–284. [Google Scholar]
- Manzo, E.; Tramice, A.; Pagano, D.; Trincone, A.; Fontana, A. Chemo-enzymatic preparation of α-6-sulfoquinovosyl-1,2-O-diacylglycerols. Tetrahedron 2012, 68, 10169–10175. [Google Scholar] [CrossRef]
- Remond, C.; Plantier-Royon, R.; Aubry, N.; Maes, E.; Bliard, C.; O’Donohuea, M.J. Synthesis of pentose-containing disaccharides using a thermostable α-L-arabinofuranosidase. Carbohyd. Res. 2004, 339, 2019–2025. [Google Scholar] [CrossRef]
- Remond, C.; Plantier-Royon, R.; Aubry, N.; O’Donohuea, M.J. An original chemoenzymatic route for the synthesis of β-D-galactofuranosides using an α-L-arabinofuranosidase. Carbohydr. Res. 2005, 340, 637–644. [Google Scholar] [CrossRef]
- Sandoval, M.; Civera, C.; Berenguer, J.; García-Blanco, F.; Hernaiz, M.J. Optimised N-acetyl-D-lactosamine synthesis using Thermus thermophilus β-galactosidase in bio-solvents. Tetrahedron 2013, 69, 1148–1152. [Google Scholar]
- Mazzaferro, L.S.; Piñuel, L.; Erra-Balsells, R.; Giudicessi, S.L.; Breccia, J.D. Transglycosylation specificity of Acremonium sp. α-rhamnosyl-β-glucosidase and its application to the synthesis of the new fluorogenic substrate 4-methylumbelliferyl-rutinoside. Carbohydr. Res. 2012, 347, 69–75. [Google Scholar]
- Perugino, G.; Trincone, A.; Rossi, M.; Moracci, M. Oligosaccharide synthesis by glycosynthases. Trends Biotechnol. 2004, 22, 31–37. [Google Scholar] [CrossRef]
- Cobucci-Ponzano, B.; Moracci, M. Glycosynthases as tools for the production of glycan analogs of natural products. Nat. Prod. Rep. 2012, 29, 697–709. [Google Scholar] [CrossRef]
- Schmaltz, R.M.; Hanson, S.R.; Wong, C.-H. Enzymes in the synthesis of glycoconjugates. Chem. Rev. 2011, 111, 4259–4307. [Google Scholar] [CrossRef]
- Mocchetti, I. Exogenous gangliosides, neuronal plasticity and repair, and the neurotrophins. Cell. Mol. Life Sci. 2005, 62, 2283–2294. [Google Scholar] [CrossRef]
- Vaughan, M.D.; Johnson, K.; DeFrees, S.; Tang, X.; Warren, R.A.J.; Withers, S.G. Glycosynthase-mediated synthesis of glycosphingolipids. J. Am. Chem. Soc. 2006, 128, 6300–6301. [Google Scholar]
- Rich, J.R.; Cunningham, A-M.; Gilbert, M.; Withers, S.G. Glycosphingolipid synthesis employing a combination of recombinant glycosyltransferases and an endoglycoceramidase glycosynthase. Chem. Commun. 2011, 47, 10806–10808. [Google Scholar]
- Okada, M.; Nakayama, T.; Noguchi, A.; Yano, M.; Hemmi, H.; Nishino, T.; Ueda, T. Site-specific mutagenesis at positions 272 and 273 of the Bacillus sp. SAM1606 α-glucosidase to screen mutants with altered specificity for oligosaccharide production by transglucosylation. J. Mol. Catal. B-Enzym. 2002, 16, 265–274. [Google Scholar]
- Ki-Won, C.; Park, K.-M.; Jun, S.-Y.; Park, C.-S.; Park, K.-H.; Cha, J. Modulation of the regioselectivity of a Thermotoga neapolitana β-glucosidase by site-directed mutagenesis. J. Microbiol. Biotechnol. 2008, 18, 901–907. [Google Scholar]
- Feng, H-Y.; Drone, J.; Hoffmann, L.; Tran, V.; Tellier, C.; Rabiller, C.; Michel Dion, M. Converting a β-glycosidase into a β-transglycosidase by directed evolution. J. Biol. Chem. 2005, 280, 37088–37097. [Google Scholar]
- Tran, V.; Hoffmann, L.; Rabiller, C.; Tellier, C.; Dion, M. Rational design of a GH1 β-glycosidase to prevent self-condensation during the transglycosylation reaction. Protein Eng. Des. Sel. 2010, 23, 43–49. [Google Scholar] [CrossRef]
- Trincone, A.; Giordano, A.; Perugino, G.; Rossi, M.; Moracci, M. Highly productive autocondensation and transglycosylation reactions with Sulfolobus solfataricus glycosynthase. ChemBioChem 2005, 6, 1431–1437. [Google Scholar]
- Placier, G.; Watzlawick, H.; Rabiller, C.; Mattes, R. Evolved β-galactosidases from Geobacillus stearothermophilus with improved transgalactosylation yield for galacto-oligosaccharide production. App. Environ. Microbiol. 2009, 75, 6312–6321. [Google Scholar] [CrossRef]
- Priyadharshini, R.; Hemalatha, D.; Gunasekaran, P. Role of Val289 residue in the α-amylase of Bacillus amyloliquefaciens MTCC 610: An analysis by site directed mutagenesis. J. Microbiol. Biotechnol. 2010, 20, 563–568. [Google Scholar]
- Koné, F.M.T.; Le Bechec, M.; Sine, J.P.; Dion, M.; Tellier, C. Digital screening methodology for the directed evolution of transglycosidases. Prot. Engin. Des. Sel. 2009, 22, 37–44. [Google Scholar]
- Fer, M.; Préchoux, A.; Leroy, A.; Sassi, J.F.; Lahaye, M.; Boisset, C.; Nyvall-Collén, P.; Helbert, W. Medium-throughput profiling method for screening polysaccharide-degrading enzymes in complex bacterial extracts. J. Microbiol. Meth. 2012, 89, 222–229. [Google Scholar] [CrossRef]
- Ropartz, D.; Bodet, P.E.; Przybylski, C.; Gonnet, F.; Daniel, R.; Fer, M.; Helbert, W.; Bertrand, D.; Rogniaux, H. Performance evaluation on a wide set of matrix-assisted laser desorption ionization matrices for the detection of oligosaccharides in a high-throughput mass spectrometric screening of carbohydrate depolymerizing enzymes. Rapid Commun. Mass Spectrom. 2011, 25, 2059–2070. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Trincone, A. Angling for Uniqueness in Enzymatic Preparation of Glycosides. Biomolecules 2013, 3, 334-350. https://doi.org/10.3390/biom3020334
Trincone A. Angling for Uniqueness in Enzymatic Preparation of Glycosides. Biomolecules. 2013; 3(2):334-350. https://doi.org/10.3390/biom3020334
Chicago/Turabian StyleTrincone, Antonio. 2013. "Angling for Uniqueness in Enzymatic Preparation of Glycosides" Biomolecules 3, no. 2: 334-350. https://doi.org/10.3390/biom3020334
APA StyleTrincone, A. (2013). Angling for Uniqueness in Enzymatic Preparation of Glycosides. Biomolecules, 3(2), 334-350. https://doi.org/10.3390/biom3020334