Recent Advances in Cellular Glycomic Analyses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Recent Advances in Elementary Glycomic Analysis
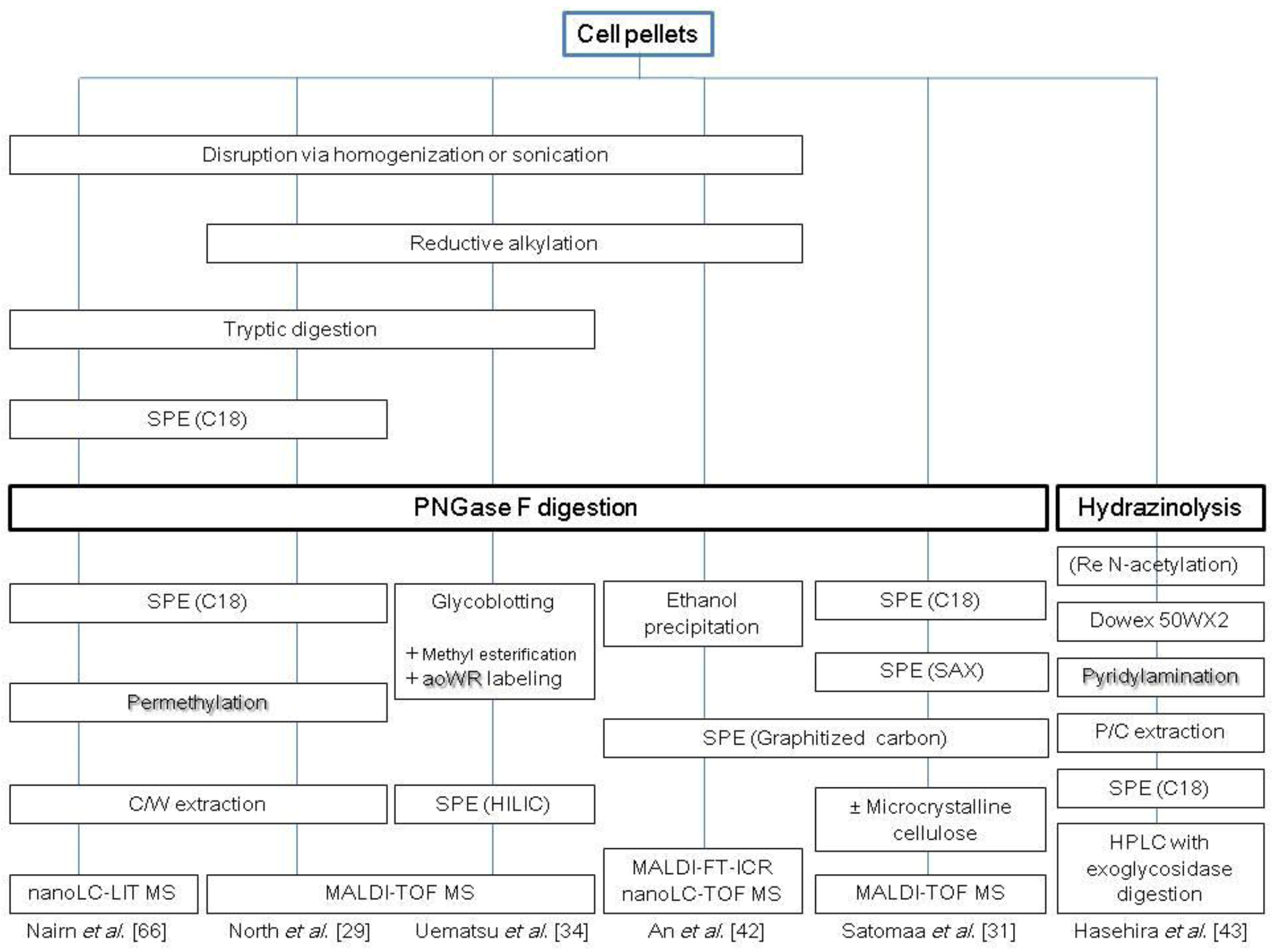
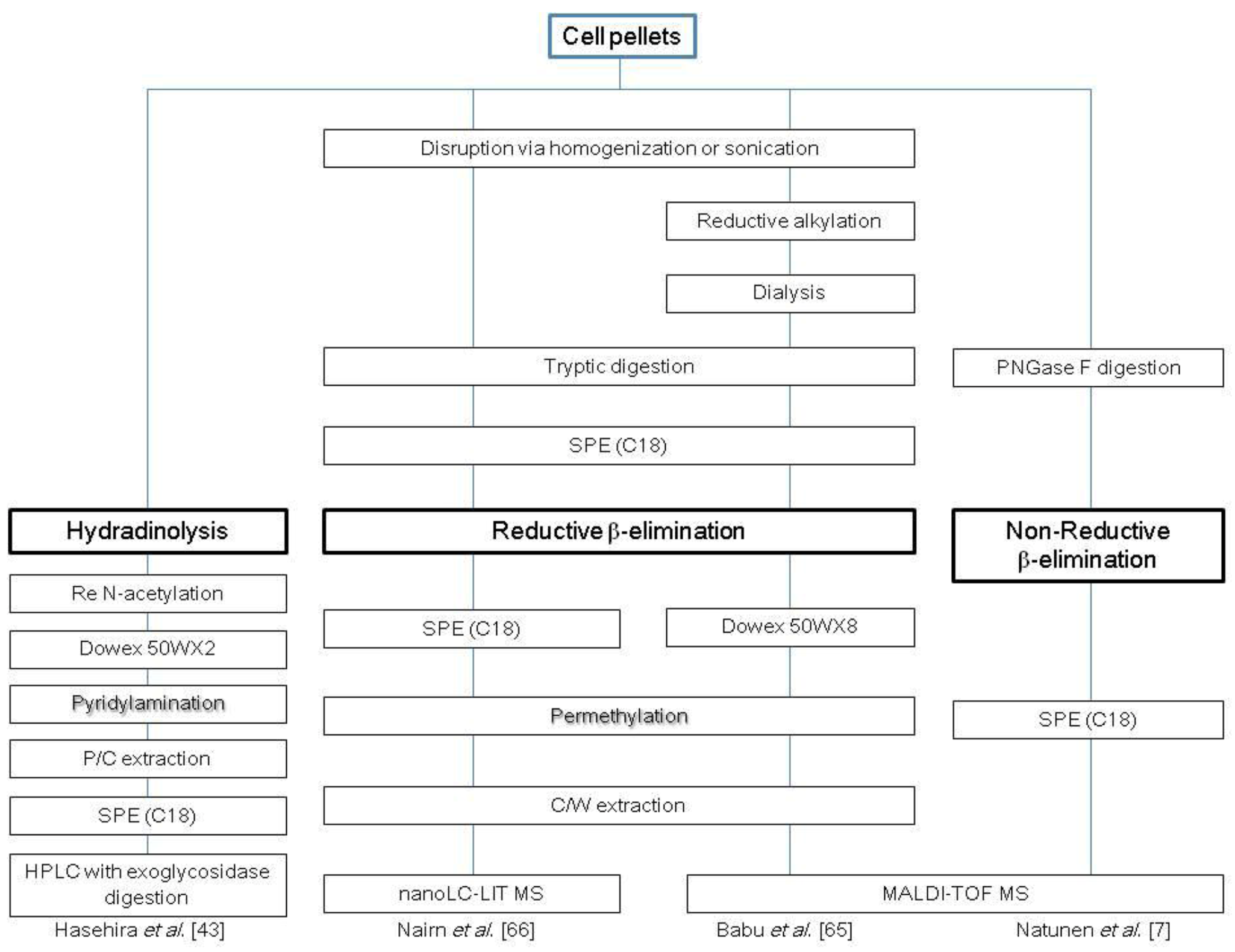
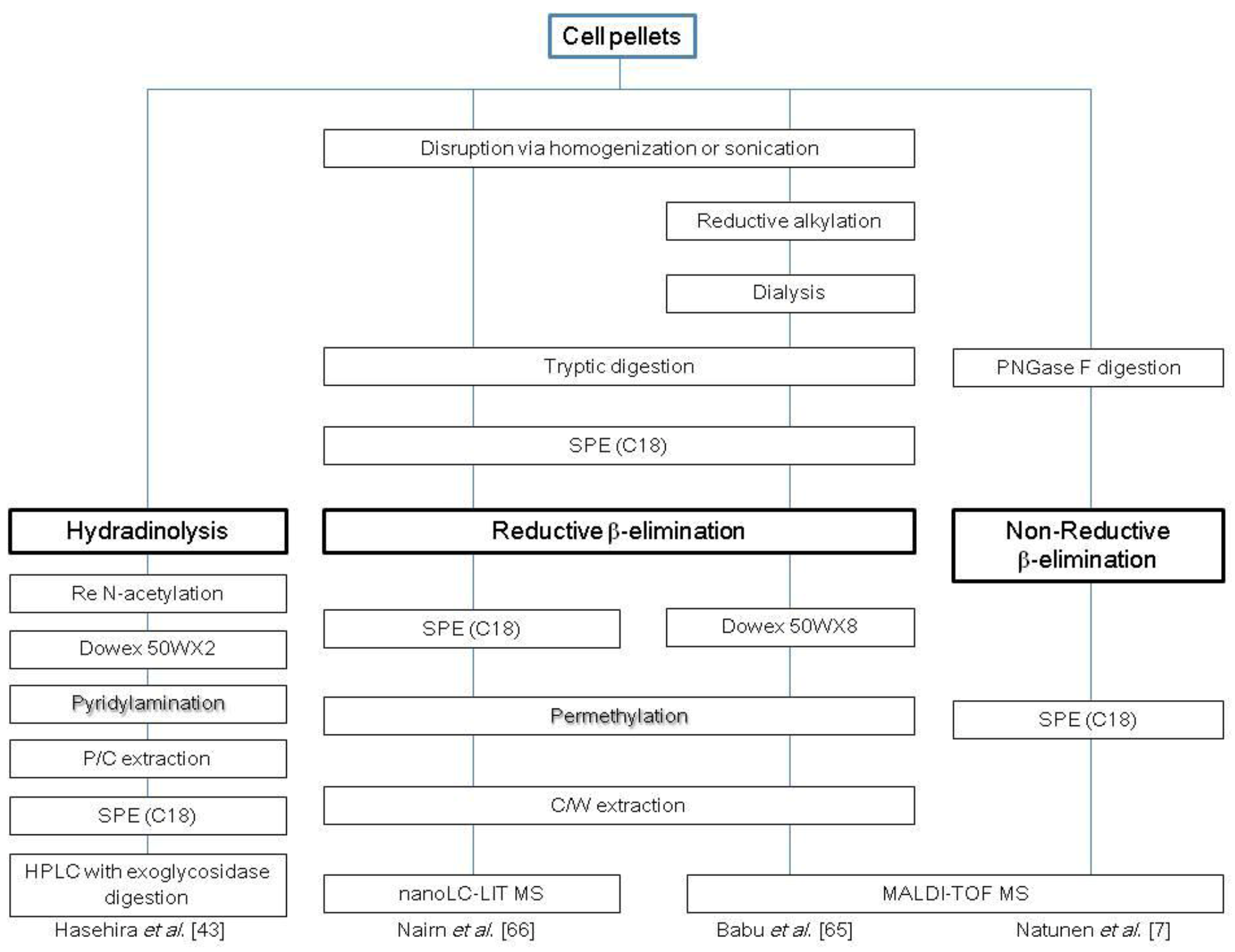
2.1. N-Glycans

2.2. O-Glycans
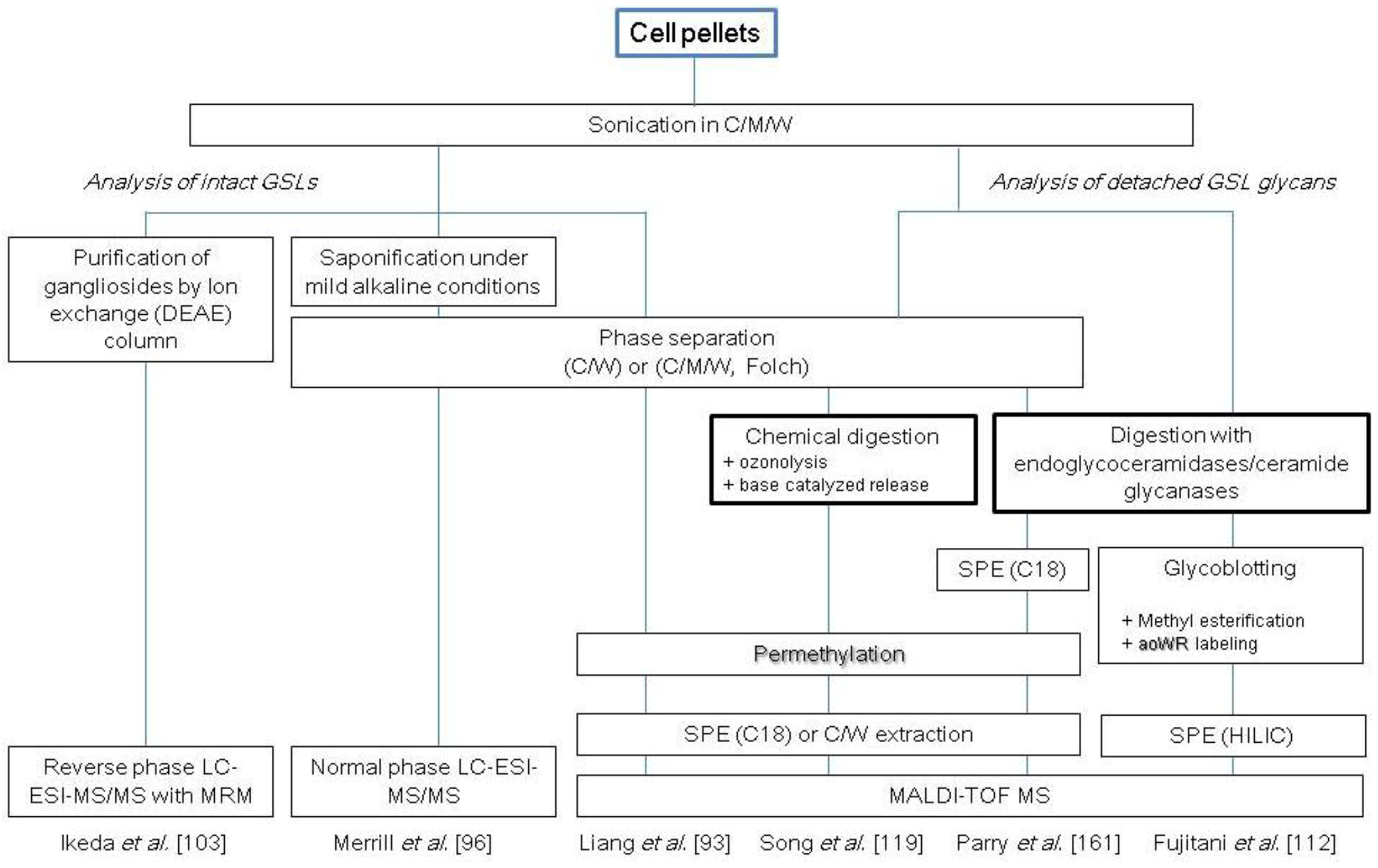
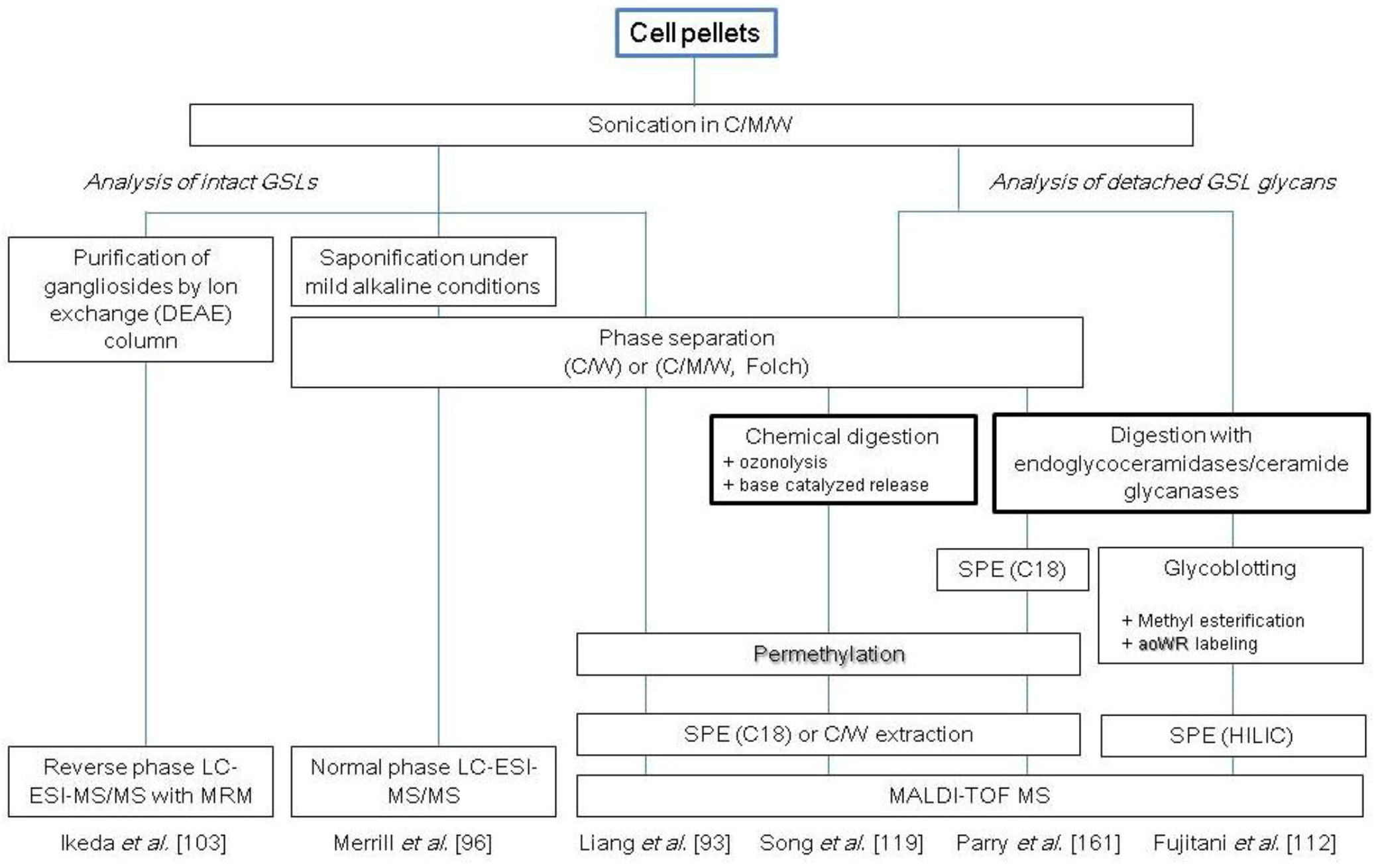
2.3. Glycosphingolipids
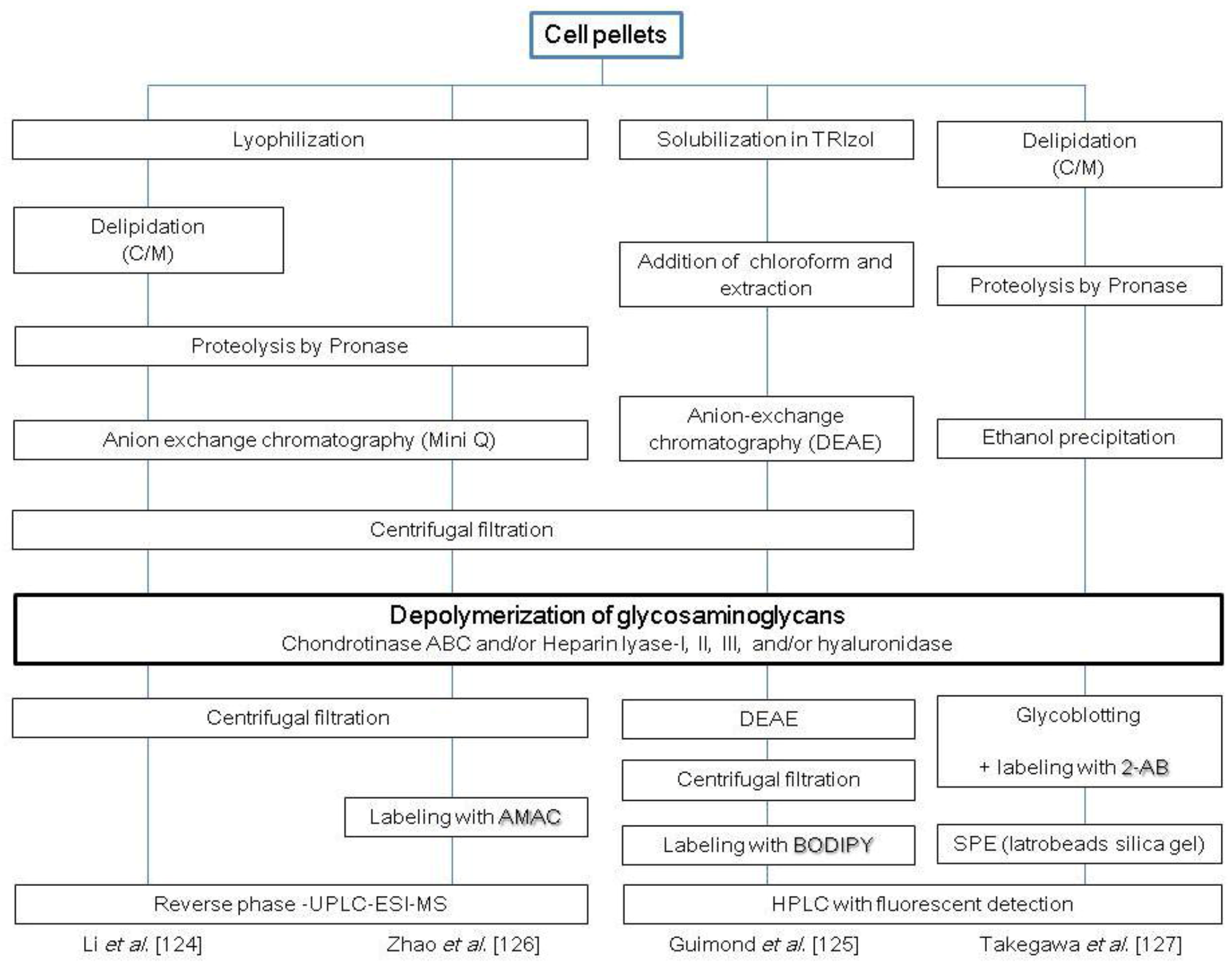
2.4. Glycosaminoglycans
3. Perspective
Acknowledgments
References
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Karsdal, M.A.; Henriksen, K.; Leeming, D.J.; Mitchell, P.; Duffin, K.; Barascuk, N.; Klickstein, L.; Aggarwal, P.; Nemirovskiy, O.; Byrjalsen, I.; et al. Biochemical markers and the FDA Critical Path: how biomarkers may contribute to the understanding of pathophysiology and provide unique and necessary tools for drug development. Biomarkers 2009, 14, 181–202. [Google Scholar] [CrossRef]
- Bertolini, F.; Mancuso, P.; Shaked, Y.; Kerbel, RS. Molecular and cellular biomarkers for angiogenesis in clinical oncology. Drug Discovery Today 2007, 12, 806–812. [Google Scholar] [CrossRef]
- Kannagi, R.; Cochran, N.A.; Ishigami, F.; Hakomori, S.; Andrews, P.W.; Knowles, B.B.; Solter, D. Stage-specific embryonic antigens (SSEA-3 and -4) are epitopes of a unique globo-series ganglioside isolated from human teratocarcinoma cells. EMBO J. 1983, 2, 2355–2361. [Google Scholar]
- Lee, J.B.; Kim, J.M.; Kim, S.J.; Park, J.H.; Hong, S.H.; Roh, S.I.; Kim, M.K.; Yoon, H.S. Comparative characteristics of three human embryonic stem cell lines. Mol. Cells 2005, 19, 31–38. [Google Scholar]
- Natunen, S.; Satomaa, T.; Pitkänen, V.; Salo, H.; Mikkola, M.; Natunen, J.; Otonkoski, T.; Valmu, L. The binding specificity of the marker antibodies Tra-1-60 and Tra-1-81 reveals a novel pluripotency-associated type 1 lactosamine epitope. Glycobiology 2011, 21, 1125–1130. [Google Scholar]
- Lanctot, P.M.; Gage, F.H.; Varki, A.P. The glycans of stem cells. Curr. Opin. Chem. Biol. 2007, 11, 373–380. [Google Scholar] [CrossRef]
- Angata, T.; Fujinawa, R.; Kurimoto, A.; Nakajima, K.; Kato, M.; Takamatsu, S.; Korekane, H.; Gao, C.X.; Ohtsubo, K.; Kitazume, S.; Taniguchi, N. Integrated approach toward the discovery of glyco-biomarkers of inflammation-related diseases. Ann. N. Y. Acad. Sci. 2012, 1253, 159–169. [Google Scholar]
- Ludwig, J.A.; Weinstein, J.N. Biomarkers in cancer staging; prognosis and treatment selection. Nat. Rev. Cancer 2005, 5, 845–856. [Google Scholar] [CrossRef]
- Rillahan, C.D.; Paulson, J.C. Glycan microarrays for decoding the glycome. Annu. Rev. Biochem. 2011, 80, 797–823. [Google Scholar] [CrossRef]
- Freeze, H.H. Genetic defects in the human glycome. Nat. Rev. Genet. 2006, 7, 537–551. [Google Scholar] [CrossRef]
- Gabius, H.J.; André, S.; Jiménez-Barbero, J.; Romero, A.; Solís, D. From lectin structure to functional glycomics: principles of the sugar code. Trends Biochem. Sci. 2011, 36, 298–313. [Google Scholar] [CrossRef]
- Stanley, P.; Cummings, R.D. Structures Common to Different Glycans. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2009; pp. 175–198. [Google Scholar]
- Furukawa, K.; Takamiya, K.; Okada, M.; Inoue, M.; Fukumoto, S.; Furukawa, K. Novel function of complex carbohydrates elucidated by the mutant mice of glycosyltransferase genes. Biochim. Biophys. Acta 2001, 1525, 1–12. [Google Scholar] [CrossRef]
- Helenius, A.; Aebi, M. Intracellular functions of N-linked glycans. Science 2001, 291, 2364–2369. [Google Scholar] [CrossRef]
- Hebert, D.N.; Molinari, M. Flagging and docking: dual roles for N-glycans in protein quality control and cellular proteostasis. Trends Biochem Sci. 2012, 37, 404–410. [Google Scholar] [CrossRef]
- Wada, Y.; Azadi, P.; Costello, C.E.; Dell, A.; Dwek, R.A.; Geyer, R.; Kakehi, K.; Karisson, N.G.; Kato, K.; Kawasaki, N.; et al. Comparison of the methods for profiling glycoprotein glycans--HUPO Human Disease Glycomics/Proteome Initiative multi-institutional study. Glycobiology 2007, 17, 411–422. [Google Scholar] [CrossRef]
- Naven, T.J.; Harvey, D.J. Effect of structure on the signal strength of oligosaccharides in matrix-assisted laser desorption/ionization mass spectrometry on time-of-flight and magnetic sector instruments. Rapid Commun. Mass Spectrom. 1996, 10, 1361–1366. [Google Scholar] [CrossRef]
- Powell, A.K.; Harvey, D.J. Stabilization of sialic acids in N-linked oligosaccharides and gangliosides for analysis by positive ion matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun. Mass Spectrom. 1996, 10, 1027–1032. [Google Scholar] [CrossRef]
- Miura, Y.; Shinohara, Y.; Furukawa, J.; Nagahori, N.; Nishimura, S. Rapid and simple solid-phase esterification of sialic acid residues for quantitative glycomics by mass spectrometry. Chem. Eur. J. 2007, 13, 4797–4804. [Google Scholar] [CrossRef]
- Dell, A.; Morris, H.R. Glycoprotein structure determination by mass spectrometry. Science 2001, 291, 2351–2356. [Google Scholar] [CrossRef]
- Kita, Y.; Miura, Y.; Furukawa, J.; Nakano, M.; Shinohara, Y.; Ohno, M.; Takimoto, A.; Nishimura, S. Quantitative glycomics of human whole serum glycoproteins based on the standardized protocol for liberating N-glycans. Mol. Cell Proteomics 2007, 6, 1437–1445. [Google Scholar] [CrossRef]
- Krishnamoorthy, L.; Mahal, L.K. Glycomic analysis: an array of technologies. ACS Chem. Biol. 2009, 4, 715–732. [Google Scholar] [CrossRef]
- Ruhaak, L.R.; Zauner, G.; Huhn, C.; Bruggink, C.; Deelder, A.M.; Wuhrer, M. Glycan labeling strategies and their use in identification and quantification. Anal. Bioanal. Chem. 2010, 397, 3457–3481. [Google Scholar] [CrossRef]
- Mechref, Y.; Kang, P.; Novotny, M.V. Solid-phase permethylation for glycomic analysis. Methods Mol. Biol. 2009, 534, 53–64. [Google Scholar] [CrossRef]
- Jeong, H.J.; Kim, Y.G.; Yang, Y.H.; Kim, B.G. High-throughput quantitative analysis of total N-glycans by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal. Chem. 2012, 84, 3453–3460. [Google Scholar] [CrossRef]
- Lemoine, J.; Chirat, F.; Domon, B. Structural analysis of derivatized oligosaccharides using post-source decay matrix-assisted laser desorption/ionization mass spectrometry. J. Mass Spectrom. 1996, 31, 908–912. [Google Scholar] [CrossRef]
- North, S.J.; Huang, H.H.; Sundaram, S.; Jang-Lee, J.; Etienne, A.T.; Trollope, A.; Chalabi, S.; Dell, A.; Stanley, P.; Haslam, S.M. Glycomics profiling of Chinese hamster ovary cell glycosylation mutants reveals N-glycans of a novel size and complexity. J. Biol Chem. 2010, 285, 5759–5775. [Google Scholar]
- Antonopoulos, A.; North, S.J.; Haslam, S.M.; Dell, A. Glycosylation of mouse and human immune cells: insights emerging from N-glycomics analyses. Biochem. Soc. Trans. 2011, 39, 1334–1340. [Google Scholar] [CrossRef]
- Satomaa, T.; Heiskanen, A.; Mikkola, M.; Olsson, C.; Blomqvist, M.; Tiittanen, M.; Jaatinen, T.; Aitio, O.; Olonen, A.; Helin, J.; et al. The N-glycome of human embryonic stem cells. BMC Cell Biol. 2009, 10, 42. [Google Scholar] [CrossRef]
- Furukawa, J.; Shinohara, Y.; Kuramoto, H.; Miura, Y.; Shimaoka, H.; Kurogochi, M.; Nakano, M.; Nishimura, S. Comprehensive approach to structural and functional glycomics based on chemoselective glycoblotting and sequential tag conversion. Anal. Chem. 2008, 80, 1094–1101. [Google Scholar]
- Uematsu, R.; Furukawa, J.; Nakagawa, H.; Shinohara, Y.; Deguchi, K.; Monde, K.; Nishimura, S. High throughput quantitative glycomics and glycoform-focused proteomics of murine dermis and epidermis. Mol. Cell. Proteomics 2005, 4, 1977–1989. [Google Scholar]
- Uematsu, R.; Shinohara, Y.; Nakagawa, H.; Kurogochi, M.; Furukawa, J.; Miura, Y.; Akiyama, M.; Shimizu, H.; Nishimura, S. Glycosylation specific for adhesion molecules in epidermis and its receptor revealed by glycoform-focused reverse genomics. Mol. Cell. Proteomics 2009, 8, 232–244. [Google Scholar]
- Amano, M.; Yamaguchi, M.; Takegawa, Y.; Yamashita, T.; Terashima, M.; Furukawa, J.; Miura, Y.; Shinohara, Y.; Iwasaki, N.; Minami, A.; Nishimura, S. Threshold in stage-specific embryonic glycotypes uncovered by a full portrait of dynamic N-glycan expression during cell differentiation. Mol. Cell Proteomics 2010, 9, 523–537. [Google Scholar] [CrossRef]
- Hase, S.; Hara, S.; Matsushima, Y. Tagging of sugars with a fluorescent compound, 2-aminopyridine. J. Biochem. 1979, 85, 217–220. [Google Scholar]
- Bigge, J.C.; Patel, T.P.; Bruce, J.A.; Goulding, P.N.; Charles, S.M.; Parekh, R.B. Nonselective and efficient fluorescent labeling of glycans using 2-amino benzamide and anthranilic acid. Anal. Biochem. 1995, 230, 229–238. [Google Scholar]
- Wuhrer, M.; de Boer, A.R.; Deelder, A.M. Structural glycomics using hydrophilic interaction chromatography (HILIC) with mass spectrometry. Mass Spectrom Rev. 2009, 28, 192–206. [Google Scholar] [CrossRef]
- Takegawa, Y.; Deguchi, K.; Keira, T.; Ito, H.; Nakagawa, H.; Nishimura, S. Separation of isomeric 2-aminopyridine derivatized N-glycans and N-glycopeptides of human serum immunoglobulin G by using a zwitterionic type of hydrophilic-interaction chromatography. J. Chromatogr. A 2006, 1113, 177–181. [Google Scholar] [CrossRef]
- Packer, N.H.; Lawson, M.A.; Jardine, D.R.; Redmond, J.W. A general approach to desalting oligosaccharides released from glycoproteins. Glycoconj. J. 1998, 15, 737–747. [Google Scholar] [CrossRef]
- Zaia, J. Mass spectrometry and glycomics. OMICS 2010, 14, 401–418. [Google Scholar] [CrossRef]
- An, H.J.; Gip, P.; Kim, J.; Wu, S.; Park, K.W.; McVaugh, C.T.; Schaffer, D.V.; Bertozzi, C.R.; Lebrilla, C.B. Extensive determination of glycan heterogeneity reveals an unusual abundance of high mannose glycans in enriched plasma membranes of human embryonic stem cells. Mol. Cell Proteomics 2012, 11. M111.010660. [Google Scholar] [CrossRef]
- Hasehira, K.; Tateno, H.; Onuma, Y.; Ito, Y.; Asashima, M.; Hirabayashi, J. Structural and quantitative evidence for dynamic glycome shift upon production of human induced pluripotent stem cells. Mol. Cell Proteomics 2012, 11, 1913–1923. [Google Scholar] [CrossRef]
- Zhang, H.; Li, X.J.; Martin, D.B.; Aebersold, R. Entification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotech. 2003, 21, 660–666. [Google Scholar]
- Baycin-Hizal, D.; Tabb, D.L.; Chaerkady, R.; Chen, L.; Lewis, N.E.; Nagarajan, H.; Sarkaria, V.; Kumar, A.; Wolozny, D.; Colao, J.; et al. Proteomic analysis of chinese hamster ovary cells. J. Proteome. Res. 2012, 11, 5265–5276. [Google Scholar]
- Sudhir, P.R.; Chen, C.H.; Pavana Kumari, M.; Wang, M.J.; Tsou, C.C.; Sung, T.Y.; Chen, J.Y.; Chen, C.H. Label-free quantitative proteomics and N-glycoproteomics analysis of KRAS-activated human bronchial epithelial cells. Mol. Cell Proteomics 2012, 11, 901–915. [Google Scholar] [CrossRef]
- Almaraz, R.T.; Tian, Y.; Bhattarcharya, R.; Tan, E.; Chen, S.H.; Dallas, M.R.; Chen, L.; Zhang, Z.; Zhang, H.; Konstantopoulos, K.; et al. Metabolic flux increases glycoprotein sialylation: implications for cell adhesion and cancer metastasis. Mol. Cell Proteomics 2012, 11. M112.017558. [Google Scholar]
- Kurogochi, M.; Matsushista, T.; Amano, M.; Furukawa, J.; Shinohara, Y.; Aoshima, M.; Nishimura, S. Sialic acid-focused quantitative mouse serum glycoproteomics by multiple reaction monitoring assay. Mol. Cell. Proteomics 2010, 9, 2354–2368. [Google Scholar] [CrossRef]
- Zhang, Y.; Yin, H.; Lu, H. Recent progress in quantitative glycoproteomics. Glycoconj. J. 2012, 29, 249–258. [Google Scholar] [CrossRef]
- Li, Y.; Tian, Y.; Rezai, T.; Prakash, A.; Lopez, M.F.; Chan, D.W.; Zhang, H. Simultaneous analysis of glycosylated and sialylated prostate-specific antigen revealing differential distribution of glycosylated prostate-specific antigen isoforms in prostate cancer tissues. Anal. Chem. 2011, 83, 240–245. [Google Scholar]
- Jensen, O.N. Interpreting the protein language using proteomics. Nat. Rev. Mol. Cell. Biol. 2006, 7, 391–403. [Google Scholar] [CrossRef]
- Gram Schjoldager, K.T.; Vester-Christensen, M.B.; Goth, C.K.; Petersen, T.N.; Brunak, S.; Bennett, E.P.; Levery, S.B.; Clausen, H. O-glycosylation modulates proprotein convertase activation of angiopoietin-like protein 3 - possible role of polypeptide GalNAc-transferase-2 in regulation of concentrations of plasma lipids. J. Biol. Chem. 2010, 285, 36293–36303. [Google Scholar]
- Kato, K.; Jeanneau, C.; Tarp, M.A.; Benet-Pages, A.; Lorenz-Depiereux, B.; Bennett, E. P.; Mandel, U.; Strom, T. M.; Clausen, H. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J. Biol. Chem. 2006, 281, 18370–18377. [Google Scholar]
- Gill, D.; Clausen, H.; Bard, F. Location, location, location: new insights into O-GalNAc protein glycosylation. Trends Cell Biol. 2010, 23, 149–158. [Google Scholar]
- Schwientek, T.; Bennett, E.P.; Flores, C.; Thacker, J.; Hollmann, M.; Reis, C.A.; Behrens, J.; Mandel, U.; Keck, B.; Schafer, M.A.; et al. Functional conservation of subfamilies of putative UDP-N-acetylgalactosamine:polypeptide N-acetylgalactosaminyltransferases in Drosophila, Caenorhabditis elegans, and mammals. One subfamily composed of l(2)35Aa is essential in Drosophila. J. Biol. Chem. 2002, 277, 22623–22638. [Google Scholar]
- Storous, G.J.; Dekker, J. Mucin-type glycoproteins. CRC Crit. Rev. Biochem. Mol. Biol. 1992, 27, 57–92. [Google Scholar] [CrossRef]
- Carlstedt, I.; Sheehan, J.K.; Corfield, A.P.; Gallagher, J.T. Mucous glycoproteins: A gel of a problem. Essays Biochem. 1985, 20, 40–76. [Google Scholar]
- Taylor-Papadimitriou, J.; Burchell, J.; Miles, D.W.; Dalziel, M. MUC1 and cancer. Biochim. Biophys. Acta 1999, 1455, 301–313. [Google Scholar]
- Park, J.H.; Nishidate, T.; Kijima, K.; Ohashi, T.; Takegawa, K.; Fujikane, T.; Hirata, K.; Nakamura, Y.; Katagiri, T. Critical roles of mucin 1 glycosylation by transactivated polypeptide N-acetylgalactosaminyltransferase 6 in mammary carcinogenesis. Cancer Res. 2010, 70, 2759–2769. [Google Scholar]
- Pedersen, J.W.; Blixt, O.; Bennett, E.P.; Tarp, M.A.; Dar, I.; Mandel, U.; Poulsen, S.S.; Pedersen, A.E.; Rasmussen, S.; Jess, P.; et al. Seromic profi ling of colorectal cancer patients with novel glycopeptide microarray. Int. J. Cancer 2011, 128, 1860–1871. [Google Scholar]
- Plummer, T.H. Jr.; Elder, J. H.; Alexander, S.; Phelan, A.W.; Tarentino, A.L. Demonstration of peptide: N-glycosidase F activity in endo-beta-N-acetylglucosaminidase F preparations. J. Biol. Chem. 1984, 259, 10700–10704. [Google Scholar]
- Dwek, R.A.; Edge, C.J.; Harvey, D.J.; Wormald, M.R.; Parekh, R.B. Analysis of glycoprotein-associated oligosaccharides. Annu. Rev. Biochem. 1993, 62, 65–100. [Google Scholar] [CrossRef]
- Wada, Y.; Dell, A.; Haslam, S.M.; Tissot, B.; Canis, K.; Azadi, P.; Backstrom, M.; Costello, C.E.; Hansson, G.C.; Hiki, Y.; et al. Comparison of methods for profi ling O-glycosylation: Human Proteome Organisation Human Disease Glycomics/Proteome Initiative multi-institutional study of IgA1. Mol. Cell. Proteomics 2010, 9, 719–727. [Google Scholar] [CrossRef]
- Carlson, D.M. Structures and immunochemical properties of oligosaccharides isolated from pig submaxillary mucins. J. Biol. Chem. 1968, 243, 616–626. [Google Scholar]
- Babu, P.; North, S. J.; Jang-Lee, J.; Chalabi, S.; Mackerness, K.; Stowell, S. R.; Cummings, R. D.; Rankin, S.; Dell, A.; Haslam, S. M. Structural characterization of neutrophil glycans by ultra sensitive mass spectrometric glycomics methodology. Glycoconj. J. 2009, 26, 975–986. [Google Scholar] [CrossRef]
- Nairn, A.V.; Aoki, K.; Dela Rosa, M.; Porterfield, M.; Lim, J.M.; Kulik, M.; Pierce, J.M.; Wells, L.; Dalton, S.; Tiemeyer, M.; et al. Regulation of Glycan Structures in Murine Embryonic Stem Cells: Combined Transcript Profiling of Glycan-Related Genes and Glycan Structural Analysis. J. Biol. Chem. 2012, 287, 37835–37856. [Google Scholar]
- Maniatis, S.; Zhou, H.; Reinhold, V. Rapid de-O-glycosylation concomitant with peptide labeling using microwave radiation and an alkyl amine base. Anal. Chem. 2010, 82, 2421–2425. [Google Scholar] [CrossRef]
- Huang, Y.; Mechref, Y.; Novotny, M.V. Microscale nonreductive release of O-linked glycans for subsequent analysis throught MALDI mass spectrometry and capillary electrophoresis. Anal. Chem. 2001, 73, 6063–6069. [Google Scholar] [CrossRef]
- Yu, G.; Zhang, Y.; Zhang, Z.; Song, L.; Wang, P.; Chai, W. Effect and limitation of excess ammonium on the release of O-glycans in reducing forms from glycoproteins under mild alkaline conditions for glycomic and functional analysis. Anal. Chem. 2010, 82, 9534–9542. [Google Scholar]
- Patel, T.; Bruce, J.; Merry, A.; Bigge, C.; Wormald, M.; Jaques, A.; Parekh, R. Use of hydrazine to release in intact and unreduced form both N- and. O-Linked oligosaccharides from glycoproteins. Biochemistry 1993, 32, 679–693. [Google Scholar]
- Merry, A.H.; Neville, D.C.; Royle, L.; Matthews, B.; Harvey, D.J.; Dwek, R.A.; Rudd, P.M. Recovery of intact 2-aminobenzamide-labeled O-glycans released from glycoproteins by hydrazinolysis. Anal. Biochem. 2002, 304, 91–99. [Google Scholar] [CrossRef]
- Tateno, H.; Toyota, M.; Saito, S.; Onuma, Y.; Ito, Y.; Hiemori, K.; Fukumura, M.; Nakasu, A.; Nakanishi, M.; Ohnuma, K.; et al. Glycome diagnosis of human induced pluripotent stem cells using lectin microarray. J. Biol. Chem. 2011, 286, 20345–20353. [Google Scholar]
- Takasaki, S.; Mizuochi, T.; Kobata, A. Hydrazinolysis of asparagine-linked sugar chains to produce free oligosaccharides. Methods Enzymol. 1982, 83, 263–268. [Google Scholar] [CrossRef]
- Kawashima, H.; Murata, T.; Yamamoto, K.; Tateishi, A.; Irimura, T.; Osawa, T. A simple method for the release of asparagine-linked oligosaccharides from a glycoprotein purified by SDS-polyacrylamide gel electrophoresis. J. Biochem. 1992, 111, 620–622. [Google Scholar]
- Zauner, G.; Koeleman, C.A.; Deelder, A.M.; Wuhrer, M. Mass spectrometric O-glycan analysis after combined O-glycan release by β-elimination and 1-phenyl-3-methyl-5-pyrazolone labeling. Biochim. Biophys. Acta 2011, 1820, 1420–1428. [Google Scholar]
- Wang, C.; Fan, W.; Zhang, P.; Wang, Z.; Huang, L. One-pot nonreductive O-glycan release and labeling with 1-phenyl-3-methyl-5-pyrazolone followed by ESI-MS analysis. Proteomics 2011, 11, 4229–4242. [Google Scholar] [CrossRef]
- Furukawa, J.-i.; Fujitani, N.; Araki, K.; Takegawa, Y.; Kodama, K.; Shinohara, Y. A versatile method for analysis of serine/threonine posttranslational modifications by β-elimination in the presence of pyrazolone analogues. Anal. Chem. 2011, 83, 9060–9067. [Google Scholar]
- Hédou, J.; Bastide, B.; Page, A.; Michalski, J.C.; Morelle, W. Mapping of O-linked beta-N-acetylglucosamine modification sites in key contractile proteins of rat skeletal muscle. Proteomics 2009, 9, 2139–2148. [Google Scholar] [CrossRef]
- Jang, H.; Kim, T.W.; Yoon, S.; Choi, S.Y.; Kang, T.W.; Kim, S.Y.; Kwon, Y.W.; Cho, E.J.; Youn, H.D. O-GlcNAc regulates pluripotency and reprogramming by directly acting on core components of the pluripotency network. Cell Stem Cell 2012, 11, 62–74. [Google Scholar] [CrossRef]
- Trinidad, J.C.; Barkan, D.T.; Gulledge, B.F.; Thalhammer, A.; Sali, A.; Schoepfer, R.; Burlingame, A.L. Global identification and characterization of both O-glcNAcylation and phosphorylation at the murine synapse. Mol. Cell Proteomics 2012, 11, 215–229. [Google Scholar] [CrossRef]
- Steentoft, C.; Vakhrushev, S.Y.; Vester-Christensen, M.B.; Schjoldager, K.T.; Kong, Y.; Bennett, E.P.; Mandel, U.; Wandall, H.; Levery, S.B.; Clausen, H. Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat. Methods 2011, 8, 977–982. [Google Scholar]
- Zauner, G.; Kozak, R.P.; Gardner, R.A.; Fernandes, D.L.; Deelder, A.M.; Wuhrer, M. Protein O-glycosylation analysis. Biol. Chem. 2012, 393, 687–708. [Google Scholar]
- Robinson, L.N.; Artpradit, C.; Raman, R.; Shriver, Z.H.; Ruchirawat, M.; Sasisekharan, R. Harnessing glycomics technologies: integrating structure with function for glycan characterization. Electrophoresis 2012, 33, 797–814. [Google Scholar]
- Hakomori, S. Structure and function of glycosphingolipids and sphingolipids: Recollections and future trends. Biochim. Biophy. Acta 2008, 1780, 325–346. [Google Scholar] [CrossRef]
- Lahiri, S.; Futerman, A.H. The metabolism and function of sphingolipids and glycosphingolipids. Cell Mol. Life Sci. 2007, 64, 2270–2284. [Google Scholar] [CrossRef]
- Hoetzl, S.; Sprong, H.; van Meer, G. van Meer, G. The way we view cellular (glyco)sphingolipids. J. Neurochem. 2007, 103, 3–13. [Google Scholar] [CrossRef]
- Hakomori, S.; Handa, K.; Iwabuchi, K.; Yamamura, S.; Prinetti, A. New insights in glycosphingolipid function: ‘‘glycosignaling domain,’’ a cell surface assembly of glycosphingolipids with signal transducer molecules, involved in cell adhesion coupled with signaling. Glycobiology 1998, 8. xi–xix. [Google Scholar] [CrossRef]
- Sorice, M.; Longo, A.; Garofalo, T.; Mattei, V.; Misasi, R.; Pavan, A. Role of GM3-enriched microdomains in signal transduction regulation in T lymphocytes. Glycoconj. J. 2004, 20, 63–70. [Google Scholar]
- Yu, S.; Withers, D.A.; Hakomori, S. Globoside-dependent adhesion of human embryonal carcinoma cells, based on carbohydrate-carbohydrate interaction, initiates signal transduction and induces enhanced activity of transcription factors AP1 and CREB. Biol. Chem. 1998, 273, 2517–2525. [Google Scholar] [CrossRef]
- Todeschini, A.R.; Hakomori, S.I. Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth through glycosynaptic microdomains. Biochim. Biophys. Acta 2008, 1780, 421–433. [Google Scholar] [CrossRef]
- Merrill, A.H. Jr. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem Rev. 2011, 111, 6387–6422. [Google Scholar] [CrossRef]
- Chang, W.W.; Lee, C.H.; Lee, P.; Lin, J.; Hsu, C.W.; Hung, J.T.; Lin, J.J.; Yu, J.C.; Shao, L.E.; Yu, J.; et al. Expression of Globo H and SSEA3 in breast cancer stem cells and the involvement of fucosyl transferases 1 and 2 in Globo H synthesis. Proc. Natl. Acad. Sci. USA 2008, 105, 11667–11672. [Google Scholar]
- Liang, Y.J.; Kuo, H.H.; Lin, C.H.; Chen, Y.Y.; Yang, B.C.; Cheng, Y.Y.; Yu, A.L.; Khoo, K.H.; Yu, J. Switching of the core structures of glycosphingolipids from globo- and lacto- to ganglio-series upon human embryonic stem cell differentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 22564–22569. [Google Scholar]
- Heimburg-Molinaro, J.; Lum, M.; Vijay, G.; Jain, M.; Almogren, A.; Rittenhouse-Olson, K. Cancer vaccines and carbohydrate epitopes. Vaccine 2011, 29, 8802–8826. [Google Scholar]
- Wuhrer, M. Glycomics using mass spectrometry. Glycoconj. J. 2013, 30, 11–22. [Google Scholar] [CrossRef]
- Merrill, A.H. Jr.; Sullards, M.C.; Allegood, J.C.; Kelly, S.; Wang, E. Sphingolipidomics: High-throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods 2005, 36, 207–224. [Google Scholar] [CrossRef]
- Ciucanu, I.; Kerek, F. A simple and rapid method for the permethylation of carbohydrates. Carbohydr. Res. 1984, 131, 209–217. [Google Scholar] [CrossRef]
- Levery, S.B. Glycosphingolipid structural analysis and glycosphingolipidomics. Methods Enzymol. 2005, 405, 300–369. [Google Scholar] [CrossRef]
- Muthing, J.; Distler, U. Advances on the compositional analysis of glycosphingolipids combining thin-layer chromatography with mass spectrometry. Mass. Spectrom. Rev. 2010, 29, 425–479. [Google Scholar]
- Flanqea, C.; Serb, A.; Sisu, E.; Zamfir, A.D. Reprint of: Chip-based nanoelectrospray mass spectrometry of brain gangliosides. Biochim. Biophy. Acta 2011, 1811, 897–917. [Google Scholar] [CrossRef]
- Gupta, V.; Bhinge, K.N.; Hosain, S.B.; Xiong, K.; Gu, X.; Shi, R.; Ho, M.Y.; Khoo, K.H.; Li, S.C.; et al. Ceramide glycosylation by glucosylceramide synthase selectively maintains the properties of breast cancer stem cells. J. Biol. Chem. 2012, 287, 37195–37205. [Google Scholar]
- Sisu, E.; Flangea, C.; Serb, A.; Rizzi, A.; Zamfir, A.D. High-performance separation techniques hyphenated to mass spectrometry for ganglioside analysis. Electrophoresis 2011, 32, 1591–1609. [Google Scholar]
- Ikeda, K.; Shimizu, T.; Taguchi, R. Targeted analysis of ganglioside and sulfatide molecular species by LC/ESI-MS/MS with theoretically expanded multiple reaction monitoring. J. Lipid Res. 2008, 49, 2678–2689. [Google Scholar] [CrossRef]
- Ikeda, K.; Taguchi, R. Highly sensitive localization analysis of gangliosides and sulfatides including structural isomers in mouse cerebellum sections by combination of laser microdissection and hydrophilic interaction liquid chromatography/electrospray ionization mass spectrometry with theoretically expanded multiple reaction monitoring. Rapid Commun. Mass Spectrom. 2010, 24, 2957–2965. [Google Scholar] [CrossRef]
- Li, S.C.; DeGasperi, R.; Muldrey, J.E.; Li, Y.T. A unique glycosphingolipid-splitting enzyme (ceramide-glycanase from leech) cleaves the linkage between the oligosaccharide and the ceramide. Biochem. Biophys. Res. Commun. 1986, 141, 346–352. [Google Scholar] [CrossRef]
- Zhou, B.; Li, S.C.; Laine, R.A.; Huang, R.T.; Li, Y.T. Isolation and characterization of ceramide glycanase from the leech, Macrobdella decora. J. Biol. Chem. 1989, 264, 12272–12277. [Google Scholar]
- Li, Y.T.; Ishikawa, Y.; Li, S.C. Occurrence of ceramide-glycanase in the earthworm; Lumbricus terrestris. Biochem. Biophys. Res. Commun. 1987, 149, 167–172. [Google Scholar] [CrossRef]
- Ito, M.; Yamagata, T. A novel glycosphingolipid-degrading enzyme cleaves the linkage between the oligosaccharide and ceramide of neutral and acidic glycosphingolipids. J. Biol. Chem. 1986, 261, 14278–14282. [Google Scholar]
- Ito, M.; Yamagata, T. Purification; characterization of glycosphingolipid-specific endoglycosidases (endoglycoceramidases) from a mutant strain of Rhodococcus sp. Evidence for three molecular species of endoglycoceramidase with different specificities. J. Biol. Chem. 1989, 264, 9510–9519. [Google Scholar]
- Ashida, H.; Yamamoto, K.; Kumagai, H.; Tochikura, T. Purification; characterization of membrane-bound endoglycoceramidase from Corynebacterium sp. Eur. J. Biochem. 1992, 205, 729–735. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Kobayashi, U.; Hijikata, A.; Sakaguchi, K.; Goda, H.M.; Tamura, T.; Okino, N.; Ito, M. Preparation and characterization of EGCase I, applicable to the comprehensive analysis of GSLs, using a rhodococcal expression system. J. Lipid. Res. 2012, 53, 2242–2251. [Google Scholar] [CrossRef]
- Fujitani, N.; Takegawa, Y.; Ishibashi, Y.; Araki, K.; Furukawa, J.; Mitsutake, S.; Igarashi, Y.; Ito, M.; Shinohara, Y. Qualitative and quantitative cellular glycomics of glycosphingolipids based on rhodococcal endoglycosylceramidase-assisted glycan cleavage; glycoblotting-assisted sample preparation; and matrix-assisted laser desorption ionization tandem time-of-flight mass spectrometry analysis. J. Biol. Chem. 2011, 286, 41669–41679. [Google Scholar]
- Heiskanen, A.; Hirvonen, T.; Salo, H.; Impola, U.; Olonen, A.; Laitinen, A.; Tiitinen, S.; Natunen, S.; Aitio, O.; Miller-Podraza, H.; et al. Glycomics of bone marrow-derived mesenchymal stem cells can be used to evaluate their cellular differentiation stage. Glycoconj. J. 2009, 26, 367–384. [Google Scholar] [CrossRef]
- Hakomori, S. Release of carbohydrates from sphingoglycolipid by osmium-catalyzed periodate oxidation, followed by treatment with mild alkali. J. Lipid Res. 1966, 7, 789–792. [Google Scholar]
- MacDonald, D.L.; Patt, L.M.; Hakomori, S. Notes on improved procedures for the chemical modification and degradation of glycosphingolipids. J. Lipid Res. 1980, 21, 642–645. [Google Scholar]
- Mylvaganam, M.; Meng, L.; Lingwood, C.A. Oxidation of glycosphingolipids under basic conditions: synthesis of glycosyl “serine acids” as opposed to “ceramide acids”. Precursors for neoglycoconjugates with increased ligand binding affinity. Biochemistry 1999, 38, 10885–10897. [Google Scholar] [CrossRef]
- Yowler, B.C.; Stoehr, S.A.; Schengrund, C.L. Oxidation and base-catalyzed elimination of the saccharide portion of GSLs having very different polarities. J. Lipid Res. 2001, 42, 659–662. [Google Scholar]
- Song, X.; Lasanajak, Y.; Xia, B.; Molinaro, J.; Rhea, J.M.; Ju, H.; Zhao, C.; Molinaro, R.J.; Cummings, R.D.; Smith, D.F. Shotgun glycomics: A microarray strategy for functional glycomics. Nat. Methods 2011, 8, 85–90. [Google Scholar]
- Song, X.; Smith, D.F.; Cummings, R.D. Nonenzymatic release of free reducing glycans from glycosphingolipids. Anal. Biochem. 2012, 429, 82–87. [Google Scholar] [CrossRef]
- Smith, R.A.; Meade, K.; Pickford, C.E.; Holley, R.J.; Merry, C.L. Glycosaminoglycans as regulators of stem cell differentiation. Biochem. Soc. Trans. 2011, 39, 383–387. [Google Scholar] [CrossRef]
- Purushothaman, A.; Sugahara, K.; Faissner, A. Chondroitin sulfate "wobble motifs" modulate maintenance and differentiation of neural stem cells and their progeny. J. Biol. Chem. 2012, 287, 2935–2942. [Google Scholar] [CrossRef]
- Quantock, A.J.; Young, R.D.; Akama, T.O. Structural and biochemical aspects of keratan sulphate in the cornea. Cell Mol. Life Sci. 2010, 67, 891–906. [Google Scholar] [CrossRef]
- Ly, M.; Leach, F.E., 3rd.; Laremore, T.N.; Toida, T.; Amster, I.J.; Linhardt, R.J. The proteoglycan bikunin has a defined sequence. Nat. Chem. Biol. 2011, 7, 827–833. [Google Scholar] [CrossRef]
- Li, B.; Liu, H.; Zhang, Z.; Stansfield, H.E.; Dordick, J.S.; Linhardt, R.J. Analysis of glycosaminoglycans in stem cell glycomics. Methods Mol. Biol. 2011, 690, 285–300. [Google Scholar] [CrossRef]
- Guimond, S.E.; Puvirajesinghe, T,M.; Skidmore, M.A.; Kalus, I.; Dierks, T.; Yates, E.A.; Turnbull, J.E. Rapid purification and high sensitivity analysis of heparan sulfate from cells and tissues: toward glycomics profiling. J. Biol. Chem. 2009, 284, 25714–25722. [Google Scholar]
- Zhao, X.; Yang, B.; Datta, P.; Gasmili, L.; Zhang, F.; Linhard, R.J. Cell-based microscale isolation of glycoaminoglycans for glycomics study. J. Carbohydr. Chem. 2012, 31, 420–435. [Google Scholar] [CrossRef]
- Takegawa, Y.; Araki, K.; Fujitani, N.; Furukawa, J.; Sugiyama, H.; Sakai, H.; Shinohara, Y. Simultaneous analysis of heparan sulfate; chondroitin/dermatan sulfates; and hyaluronan disaccharides by glycoblotting-assisted sample preparation followed by single-step zwitter-ionic-hydrophilic interaction chromatography. Anal. Chem. 2011, 83, 9443–9449. [Google Scholar]
- Izumikawa, T.; Uyama, T.; Okuura, Y.; Sugahara, K.; Kitagawa, H. Involvement of chondroitin sulfate synthase-3 (chondroitin synthase-2) in chondroitin polymerization through its interaction with chondroitin synthase-1 or chondroitin-polymerizing factor. Biochem. J. 2007, 403, 545–552. [Google Scholar] [CrossRef]
- Ernst, S.; Langer, R.; Cooney, C.L.; Sasisekharan, R. Enzymatic degradation of glycosaminoglycans. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 387–444. [Google Scholar] [CrossRef]
- Linhardt, R.J.; Rice, K.G.; Kim, Y.S.; Lohse, D.L.; Wang, H.M.; Loganathan, D. Mapping and quantification of the major oligosaccharide components of heparin. Biochem. J. 1988, 254, 781–787. [Google Scholar]
- Kitagawa, H.; Kinoshita, A.; Sugahara, K. Microanalysis of glycosaminoglycan-derived disaccharides labeled with the fluorophore 2-aminoacridone by capillary electrophoresis and high-performance liquid chromatography. Anal. Biochem. 1995, 232, 114–121. [Google Scholar] [CrossRef]
- Militsopoulou, M.; Lamari, F.N.; Hjerpe, A.; Karamanos, N.K. Determination of twelve heparin- and heparan sulfate-derived disaccharides as 2-aminoacridone derivatives by capillary zone electrophoresis using ultraviolet and laser-induced fluorescence detection. Electrophoresis 2002, 23, 1104–1109. [Google Scholar] [CrossRef]
- Kinoshita, A.; Sugahara, K. Microanalysis of glycosaminoglycan-derived oligosaccharides labeled with a fluorophore 2-aminobenzamide by high-performance liquid chromatography: application to disaccharide composition analysis and exosequencing of oligosaccharides. Anal. Biochem. 1999, 269, 367–378. [Google Scholar] [CrossRef]
- Turnbull, J.E.; Hopwood, J.J.; Gallagher, J.T. A strategy for rapid sequencing of heparan sulfate and heparin saccharides. Proc. Natl. Acad. Sci. USA 1999, 96, 2698–2703. [Google Scholar]
- Skidmore, M.A.; Guimond, S.E.; Dumax-Vorzet, A.F.; Yates, E.A.; Turnbull, J.E. Disaccharide compositional analysis of heparan sulfate and heparin polysaccharides using UV or high-sensitivity fluorescence (BODIPY) detection. Nat. Protoc. 2010, 5, 1983–1992. [Google Scholar] [CrossRef]
- Lawrence, R.; Olson, S.K.; Steele, R.E.; Wang, L.; Warrior, R.; Cummings, R.D.; Esko, J.D. Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. J. Biol. Chem. 2008, 283, 33674–33684. [Google Scholar]
- Hitchcock, A.M.; Yates, K.E.; Costello, C.E.; Zaia, J. Comparative glycomics of connective tissue glycosaminoglycans. Proteomics 2008, 8, 1384–1397. [Google Scholar] [CrossRef]
- Bowman, M.J.; Zaia, J. Comparative glycomics using a tetraplex stable-isotope coded tag. Anal. Chem. 2010, 82, 3023–3031. [Google Scholar] [CrossRef]
- Hitchkock, A.M.; Bowman, M.J.; Staples, G.O.; Zaia, J. Improved workup for glycosaminoglycan disaccharide analysis using CE with LIF detection. Electrophoresis 2008, 29, 4538–4548. [Google Scholar] [CrossRef]
- Eldridge, S.L.; Higgins, L.A.; Dickey, B.J.; Larive, C.K. Insights into the capillary electrophoresis separation of heparin disaccharides from nuclear magnetic resonance, pKa, and electrophoretic mobility measurement. Anal. Chem. 2009, 81, 7406–7415. [Google Scholar]
- Plaas, A.H.; West, L.A.; Midura, R.J. Keratan sulfate disaccharide composition determined by FACE analysis of keratanase II and endo-beta-galactosidase digestion products. Glycobiology 2001, 10, 779–790. [Google Scholar] [CrossRef]
- Korir, A.K.; Limtiaco, J.F.; Gutierrez, S.M.; Larive, C.K. Ultraperformance ion-pair liquid chromatography coupled to electrospray time-of-flight mass spectrometry for compositional profiling and quantification of heparin and heparan sulfate. Anal. Chem. 2008, 80, 1297–1306. [Google Scholar]
- Imanari, T.; Toida, T.; Koshiishi, I.; Toyoda, H. High-performance liquid chromatographic analysis of glycosaminoglycan-derived oligosaccharides. J. Chromatogr. A 1996, 720, 275–293. [Google Scholar] [CrossRef]
- Jones, C.J.; Membreno, N.; Larive, C.K. Insights into the mechanism of separation of heparin and heparan sulfate disaccharides by reverse-phase ion-pair chromatography. J. Chromatogr. A 2010, 1217, 479–488. [Google Scholar] [CrossRef]
- Staples, G.O.; Shi, X.; Zaia, J. Extended N-sulfated domains reside at the nonreducing end of heparan sulfate chains. J. Biol. Chem. 2010, 285, 18336–18343. [Google Scholar] [CrossRef]
- Volpi, N. High-performance liquid chromatography and on-line mass spectrometry detection for the analysis of chondroitin sulfates/hyaluronan disaccharides derivatized with 2-aminoacridone. Anal. Biochem. 2010, 397, 12–23. [Google Scholar] [CrossRef]
- Yang, B.; Chang, Y.; Weyers, A.M.; Sterner, E.; Linhardt, R.J. Disaccharide analysis of glycosaminoglycan mixtures by ultra-high-performance liquid chromatography-mass spectrometry. J. Chromatogr. A 2012, 1225, 91–98. [Google Scholar] [CrossRef]
- Laremore, T.N.; Leach, F.E., 3rd.; Solakyildirim, K.; Amster, I.J.; Linhardt, R.J. Glycosaminoglycan characterization by electrospray ionization mass spectrometry including fourier transform mass spectrometry. Methods Enzymol. 2010, 478, 79–108. [Google Scholar] [CrossRef]
- Uygun, B.E.; Stojsih, S.E.; Matthew, H.W. Effects of immobilized glycosaminoglycans on the proliferation and differentiation of mesenchymal stem cells. Tissue Eng. Part A 2009, 15, 3499–3512. [Google Scholar] [CrossRef]
- Kumarasuriyar, A.; Murali, S.; Nurcombe, V.; Cool, S.M. Glycosaminoglycan composition changes with MG-63 osteosarcoma osteogenesis in vitro and induces human mesenchymal stem cell aggregation. J. Cell Physiol. 2009, 218, 501–511. [Google Scholar] [CrossRef]
- Dombrowski, C.; Song, S.J.; Chuan, P.; Lim, X.; Susanto, E.; Sawyer, A.A.; Woodruff, M.A.; Hutmacher, D.W.; Nurcombe, V.; Cool, S.M. Heparan sulfate mediates the proliferation and differentiation of rat mesenchymal stem cells. Stem Cells Dev. 2009, 18, 661–670. [Google Scholar] [CrossRef]
- Nairn, A.V.; Kinoshita-Toyoda, A.; Toyoda, H.; Xie, J.; Harris, K.; Dalton, S.; Kulik, M.; Pierce, J.M.; Toida, T.; Moremen, K.W.; et al. Glycomics of proteoglycan biosynthesis in murine embryonic stem cell differentiation. J. Proteome Res. 2007, 6, 4374–4387. [Google Scholar] [CrossRef]
- Kawabe, K.; Tateyama, D.; Toyoda, H.; Kawasaki, N.; Hashii, N.; Nakao, H.; Matsumoto, S.; Nonaka, M.; Matsumura, H.; Hirose, Y.; et al. Novel Antibody for Human Induced Pluripotent Stem (hiPS) Cells and Embryonic Stem (ES) Cells Recognizes a Type of Keratan Sulfate Lacking Oversulfated Structures. Glycobiology 2012, 23, 322–336. [Google Scholar]
- Toida, T.; Hileman, R.E.; Smith, A.E.; Vlahova, P.I.; Linhardt, R.J. Enzymatic preparation of heparin oligosaccharides containing antithrombin III binding sites. J. Biol. Chem. 1996, 271, 32040–32947. [Google Scholar]
- Ly, M.; Laremore, T.N.; Linhardt, R.J. Proteoglycomics: recent progress and future challenges. OMICS 2010, 14, 389–399. [Google Scholar] [CrossRef]
- Lawrence, R.; Brown, J.R.; Al-Mafraji, K.; Lamanna, W.C,; Beitel, J.R.; Boons, G.J.; Esko, J.D.; Crawford, B.E. Disease-specific non-reducing end carbohydrate biomarkers for mucopolysaccharidoses. Nat. Chem. Biol. 2012, 8, 197–204. [Google Scholar]
- Wearne, K.A.; Winter, H.C.; O'Shea, K.; Goldstein, I.J. Use of lectins for probing differentiated human embryonic stem cells for carbohydrates. Glycobiology 2006, 16, 981–990. [Google Scholar] [CrossRef]
- Toyoda, M.; Yamazaki-Inoue, M.; Itakura, Y.; Kuno, A.; Ogawa, T.; Yamada, M.; Akutsu, H.; Takahashi, Y.; Kanzaki, S.; Narimatsu, H.; et al. Lectin microarray analysis of pluripotent and multipotent stem cells. Genes Cells 2011, 16, 1–11. [Google Scholar]
- Wang, Y.C.; Nakagawa, M.; Garitaonandia, I.; Slavin, I.; Altun, G.; Lacharite, R.M.; Nazor, K.L.; Tran, H.T.; Lynch, C.L.; Leonardo, T.R.; et al. Specific lectin biomarkers for isolation of human pluripotent stem cells identified through array-based glycomic analysis. Cell Res. 2011, 21, 1551–1563. [Google Scholar] [CrossRef]
- Yasuda, E.; Tateno, H.; Hirabayashi, J.; Iino, I.; Sako, T. Lectin microarray reveals binding profiles of Lactobacillus casei strains in a comprehensive analysis of bacterial cell wall polysaccharides. Appl. Eviron Microbiol. 2011, 77, 4539–4546. [Google Scholar] [CrossRef]
- Parry, S.; Ledger, V.; Tissot, B.; Haslam, S.M.; Scott, J.; Morris, H.R. Integrated mass spectrometric strategy for characterizing the glycans from glycosphingolipids and glycoproteins: direct identification of sialyl Lex in mice. Glycobiology 2007, 17, 646–654. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Furukawa, J.-i.; Fujitani, N.; Shinohara, Y. Recent Advances in Cellular Glycomic Analyses. Biomolecules 2013, 3, 198-225. https://doi.org/10.3390/biom3010198
Furukawa J-i, Fujitani N, Shinohara Y. Recent Advances in Cellular Glycomic Analyses. Biomolecules. 2013; 3(1):198-225. https://doi.org/10.3390/biom3010198
Chicago/Turabian StyleFurukawa, Jun-ichi, Naoki Fujitani, and Yasuro Shinohara. 2013. "Recent Advances in Cellular Glycomic Analyses" Biomolecules 3, no. 1: 198-225. https://doi.org/10.3390/biom3010198