Strategies for the Use of Poly(adenosine diphosphate ribose) Polymerase (PARP) Inhibitors in Cancer Therapy


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
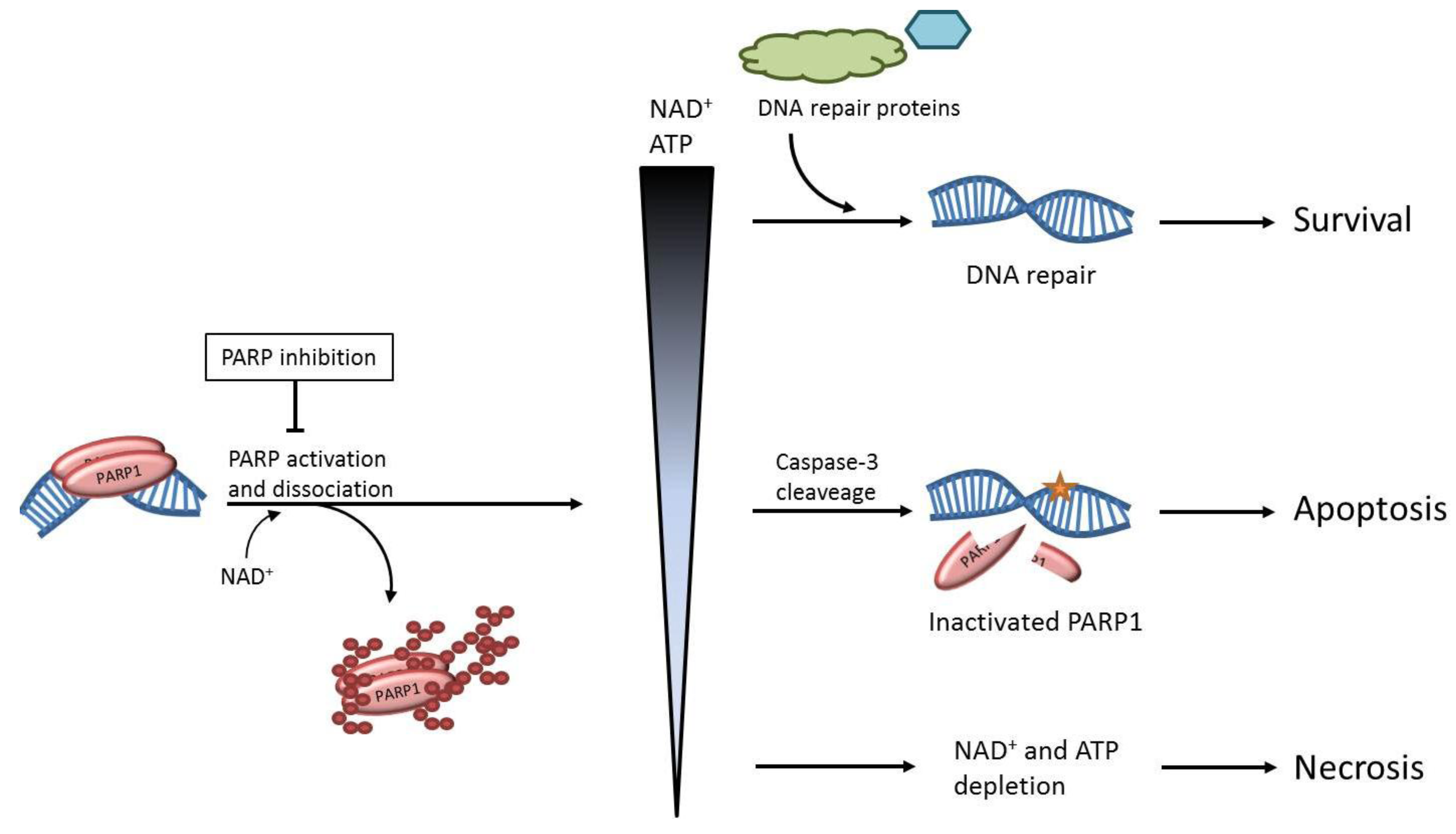
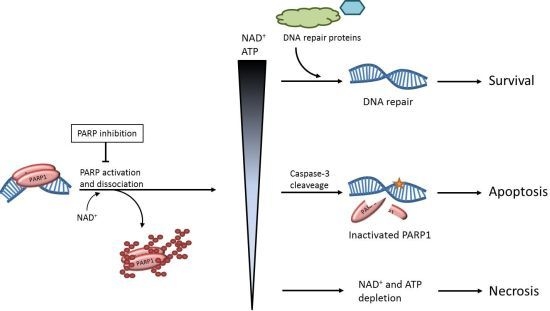
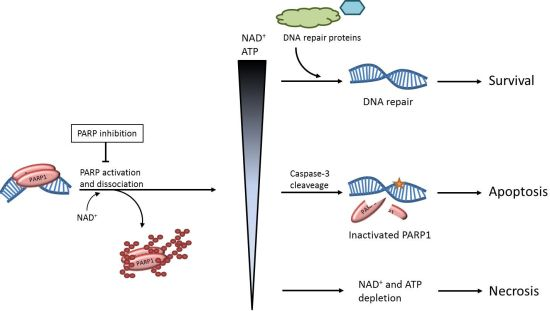
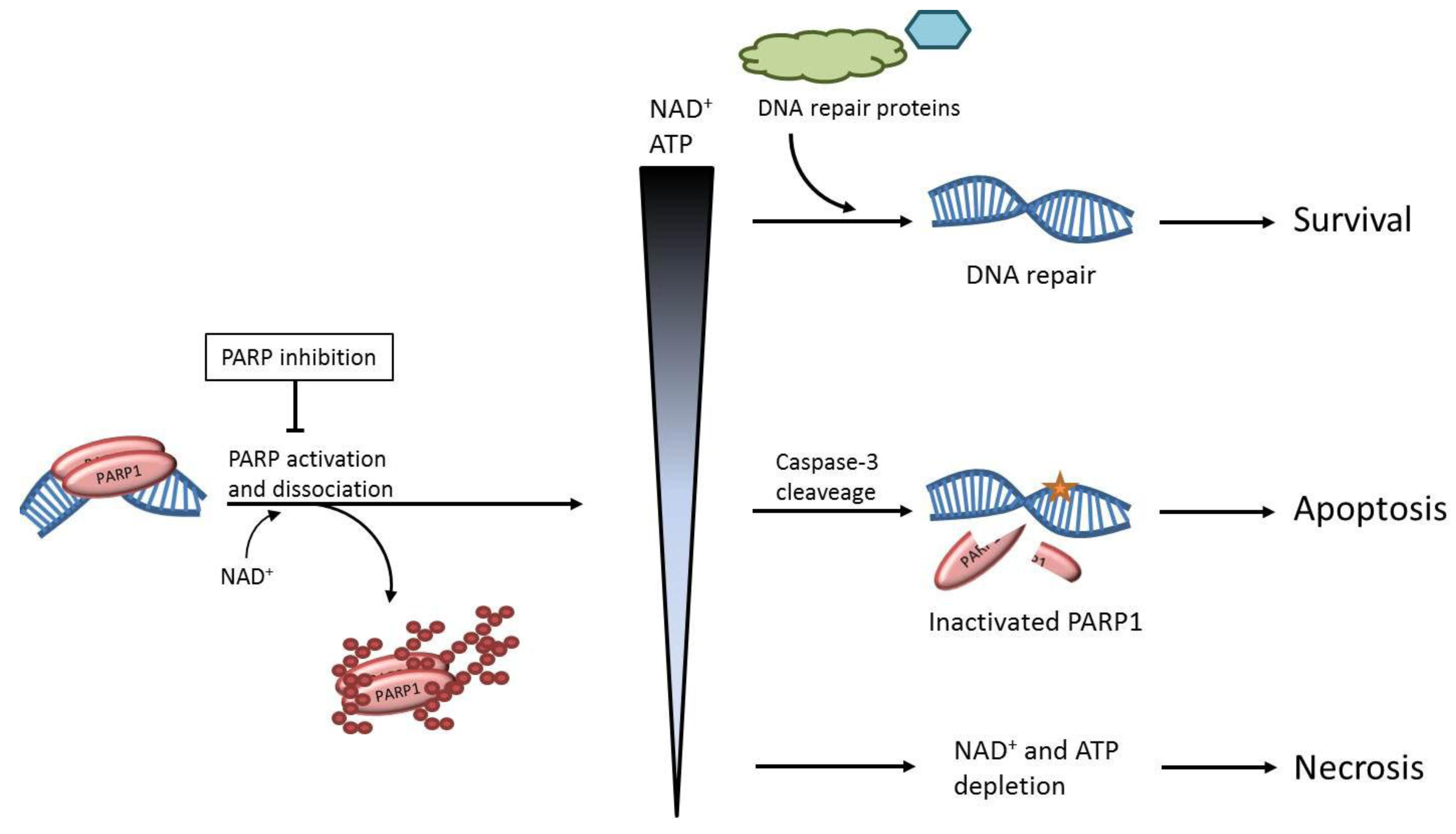
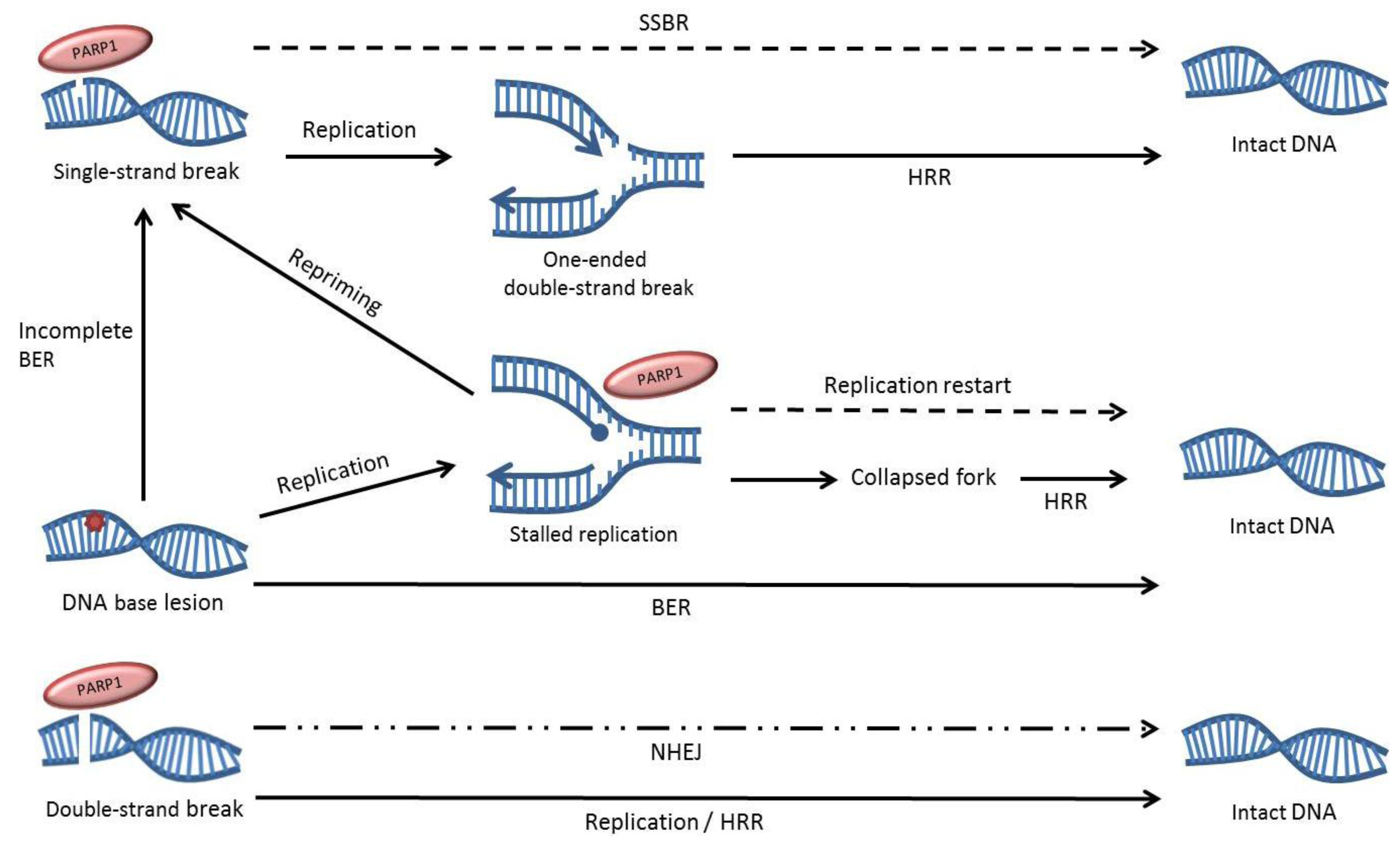
2. The role of PARP1 in DNA Repair
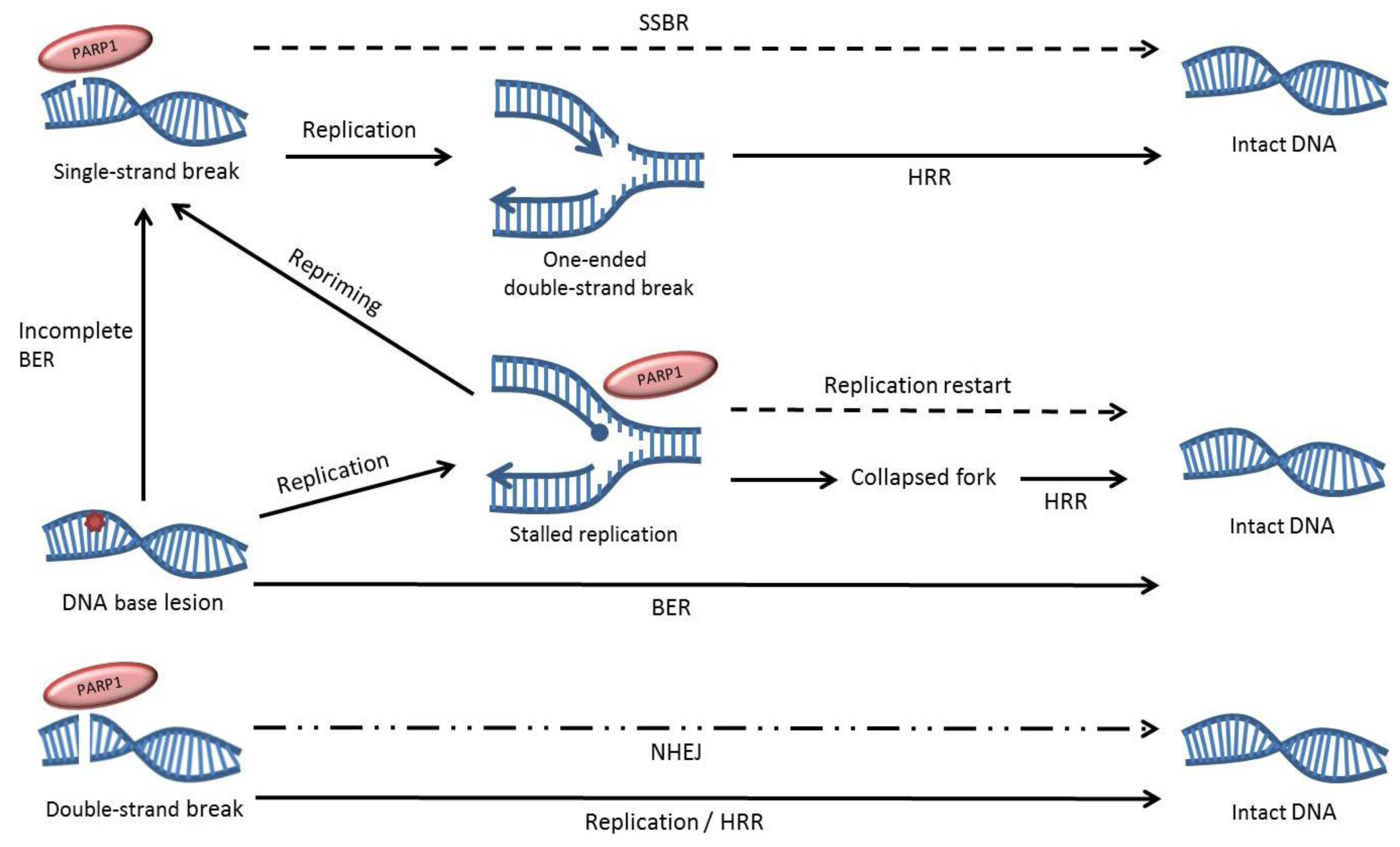
3. The Effects of PARP Inhibition on DNA Repair
4. Potential Synthetic Lethal Interactions
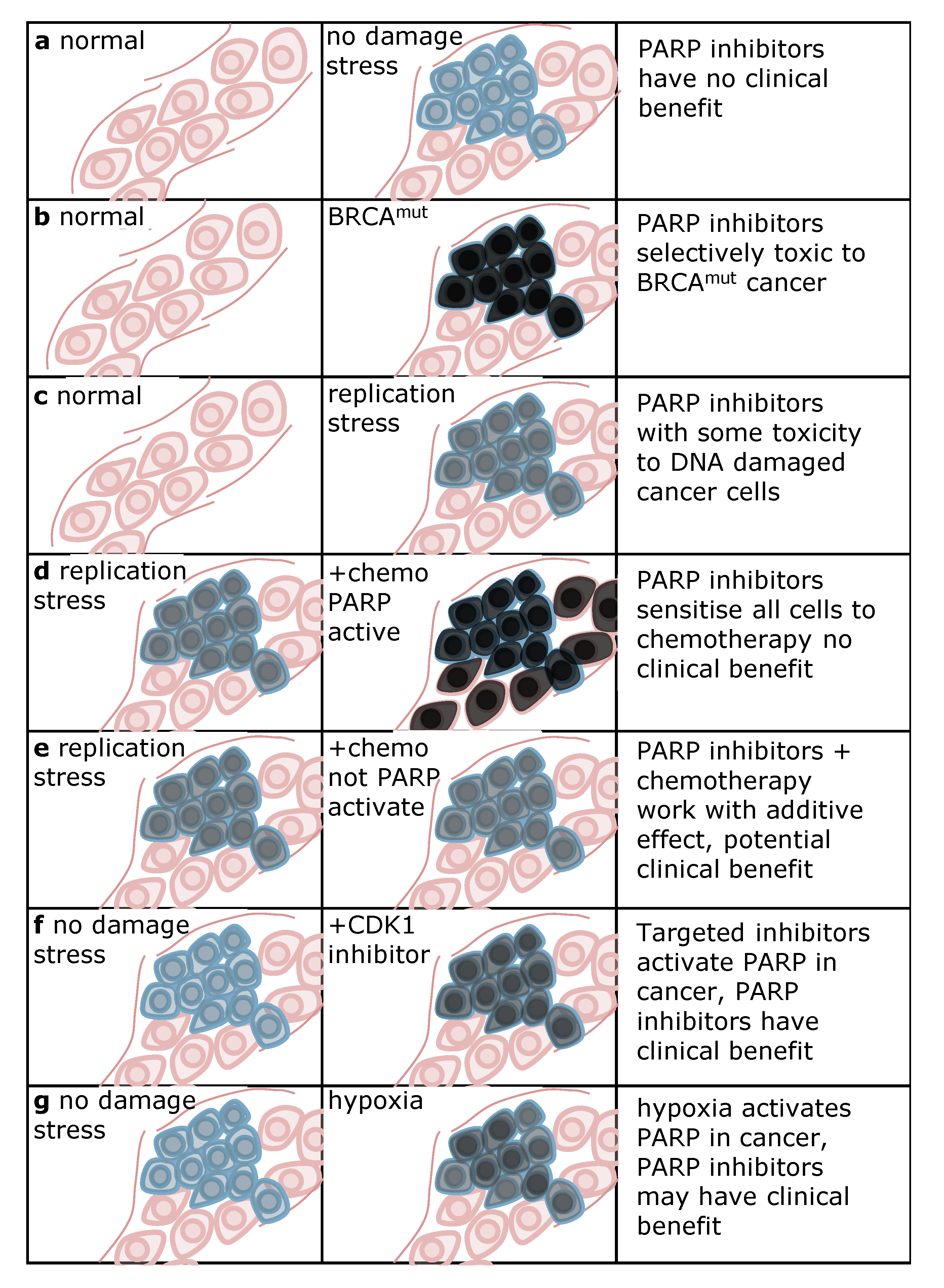
5. Strategies for Using PARP Inhibitors in the Clinic
4. Conclusions
Acknowledgments
References
- Chambon, P.; Weill, J.D.; Mandel, P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 1963, 11, 39–43. [Google Scholar] [CrossRef]
- Zahradka, P.; Ebisuzaki, K. A shuttle mechanism for DNA-protein interactions. The regulation of poly(adp-ribose) polymerase. Eur. J. Biochem. 1982, 127, 579–585. [Google Scholar] [CrossRef]
- Uchida, K.; Suzuki, H.; Maruta, H.; Abe, H.; Aoki, K.; Miwa, M.; Tanuma, S. Preferential degradation of protein-bound (adp-ribose)n by nuclear poly(adp-ribose) glycohydrolase from human placenta. J. Biol. Chem. 1993, 268, 3194–3200. [Google Scholar]
- Tanuma, S.; Yagi, T.; Johnson, G.S. Endogenous adp ribosylation of high mobility group proteins 1 and 2 and histone h1 following DNA damage in intact cells. Arch. Biochem. Biophys. 1985, 237, 38–42. [Google Scholar] [CrossRef]
- Martinez-Zamudio, R.; Ha, H.C. Histone adp-ribosylation facilitates gene transcription by directly remodeling nucleosomes. Mol. Cell. Biol. 2012, 32, 2490–2502. [Google Scholar] [CrossRef]
- Berger, N.A. Poly(adp-ribose) in the cellular response to DNA damage. Radiat. Res. 1985, 101, 4–15. [Google Scholar] [CrossRef]
- Ha, H.C.; Snyder, S.H. Poly(adp-ribose) polymerase is a mediator of necrotic cell death by atp depletion. Proc. Natl. Acad. Sci. USA 1999, 96, 13978–13982. [Google Scholar] [CrossRef]
- Liaudet, L.; Pacher, P.; Mabley, J.G.; Virag, L.; Soriano, F.G.; Hasko, G.; Szabo, C. Activation of poly(adp-ribose) polymerase-1 is a central mechanism of lipopolysaccharide-induced acute lung inflammation. Am. J. Respir. Crit. Care Med. 2002, 165, 372–377. [Google Scholar]
- Ma, Y.; Chen, H.; He, X.; Nie, H.; Sheng, C.; Wang, Q.; Xia, W.; Ying, W. Nad+ metabolism and nad+-dependent enzymes: Promising therapeutic targets for neurological diseases. Curr. Drug Targets 2012, 13, 222–229. [Google Scholar] [CrossRef]
- Moroni, F.; Cozzi, A.; Chiarugi, A.; Formentini, L.; Camaioni, E.; Pellegrini-Giampietro, D.E.; Chen, Y.; Liang, S.; Zaleska, M.M.; Gonzales, C.; et al. Long-lasting neuroprotection and neurological improvement in stroke models with new, potent and brain permeable poly(adp-ribose) polymerase inhibitors. Br. J. Pharmacol. 2012, 165, 1487–1500. [Google Scholar] [CrossRef]
- Szabo, G.; Liaudet, L.; Hagl, S.; Szabo, C. Poly(adp-ribose) polymerase activation in the reperfused myocardium. Cardiovasc. Res. 2004, 61, 471–480. [Google Scholar]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O'Connor, M.J.; et al. Inhibition of poly(adp-ribose) polymerase in tumors from brca mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of brca2-deficient tumours with inhibitors of poly(adp-ribose)polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in brca mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar]
- Gelmon, K.A.; Hirte, H.W.; Robidoux, A.; Tonkin, K.S.; Tischkowitz, M.; Swenerton, K.; Huntsman, D.; Carmichael, J.; Macpherson, E.; Oza, A.M. Can we define tumors that will respond to parp inhibitors? A phase ii correlative study of olaparib in advanced serous ovarian cancer and triple-negative breast cancer. J. Clin. Oncol. 2010, 28 suppl. 15s. abstr 3002. [Google Scholar]
- Benjamin, R.C.; Gill, D.M. Poly(adp-ribose) synthesis in vitro programmed by damaged DNA. A comparison of DNA molecules containing different types of strand breaks. J. Biol. Chem. 1980, 255, 10502–10508. [Google Scholar]
- Lindahl, T.; Satoh, M.S.; Poirier, G.G.; Klungland, A. Post-translational modification of poly(adp-ribose) polymerase induced by DNA strand breaks. Trends Biochem. Sci. 1995, 20, 405–411. [Google Scholar]
- Satoh, M.S.; Lindahl, T. Role of poly(adp-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef]
- Okano, S.; Lan, L.; Caldecott, K.W.; Mori, T.; Yasui, A. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol. Cell Biol. 2003, 23, 3974–3981. [Google Scholar] [CrossRef]
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for parp-1 for the assembly or stability of xrcc1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [Google Scholar]
- Strom, C.E.; Johansson, F.; Uhlen, M.; Al-Khalili Szigyarto, C.; Erixon, K.; Helleday, T. Poly (adp-ribose) polymerase (parp) is not involved in base excision repair but parp inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011, 39, 3166–3175. [Google Scholar]
- Allinson, S.L.; Dianova, II; Dianov, G.L. Poly(adp-ribose) polymerase in base excision repair: Always engaged, but not essential for DNA damage processing. Acta Biochim. Pol. 2003, 50, 169–179. [Google Scholar]
- Parsons, J.L.; Dianova, II; Allinson, S.L.; Dianov, G.L. Poly(adp-ribose) polymerase-1 protects excessive DNA strand breaks from deterioration during repair in human cell extracts. FEBS J. 2005, 272, 2012–2021. [Google Scholar] [CrossRef]
- D'Silva, I.; Pelletier, J.D.; Lagueux, J.; D'Amours, D.; Chaudhry, M.A.; Weinfeld, M.; Lees-Miller, S.P.; Poirier, G.G. Relative affinities of poly(adp-ribose) polymerase and DNA-dependent protein kinase for DNA strand interruptions. Biochim. Biophys. Acta 1999, 1430, 119–126. [Google Scholar] [CrossRef]
- Schultz, N.; Lopez, E.; Saleh-Gohari, N.; Helleday, T. Poly(adp-ribose) polymerase (parp-1) has a controlling role in homologous recombination. Nucleic Acids Res. 2003, 31, 4959–4964. [Google Scholar]
- Yang, Y.G.; Cortes, U.; Patnaik, S.; Jasin, M.; Wang, Z.Q. Ablation of parp-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene 2004, 23, 3872–3882. [Google Scholar] [CrossRef]
- Morrison, C.; Smith, G.C.; Stingl, L.; Jackson, S.P.; Wagner, E.F.; Wang, Z.Q. Genetic interaction between parp and DNA-pk in v(d)j recombination and tumorigenesis. Nat. Genet. 1997, 17, 479–482. [Google Scholar] [CrossRef]
- Audebert, M.; Salles, B.; Calsou, P. Involvement of poly(adp-ribose) polymerase-1 and xrcc1/DNA ligase iii in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 2004, 279, 55117–55126. [Google Scholar] [CrossRef]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. Parp-1 and ku compete for repair of DNA double strand breaks by distinct nhej pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar]
- Corneo, B.; Wendland, R.L.; Deriano, L.; Cui, X.; Klein, I.A.; Wong, S.Y.; Arnal, S.; Holub, A.J.; Weller, G.R.; Pancake, B.A.; et al. Rag mutations reveal robust alternative end joining. Nature 2007, 449, 483–486. [Google Scholar] [CrossRef]
- Wang, H.; Rosidi, B.; Perrault, R.; Wang, M.; Zhang, L.; Windhofer, F.; Iliakis, G. DNA ligase iii as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 2005, 65, 4020–4030. [Google Scholar] [CrossRef]
- Li, Z.; Otevrel, T.; Gao, Y.; Cheng, H.L.; Seed, B.; Stamato, T.D.; Taccioli, G.E.; Alt, F.W. The xrcc4 gene encodes a novel protein involved in DNA double-strand break repair and v(d)j recombination. Cell 1995, 83, 1079–1089. [Google Scholar] [CrossRef]
- Rass, E.; Grabarz, A.; Plo, I.; Gautier, J.; Bertrand, P.; Lopez, B.S. Role of mre11 in chromosomal nonhomologous end joining in mammalian cells. Nature Struct. Mol. Biol. 2009, 16, 819–824. [Google Scholar]
- Deriano, L.; Stracker, T.H.; Baker, A.; Petrini, J.H.; Roth, D.B. Roles for nbs1 in alternative nonhomologous end-joining of v(d)j recombination intermediates. Mol. Cell 2009, 34, 13–25. [Google Scholar] [CrossRef]
- Spagnolo, L.; Barbeau, J.; Curtin, N.J.; Morris, E.P.; Pearl, L.H. Visualization of a DNA-pk/parp1 complex. Nucleic Acids Res. 2012, 40, 4168–4177. [Google Scholar] [CrossRef]
- Galande, S.; Kohwi-Shigematsu, T. Poly(adp-ribose) polymerase and ku autoantigen form a complex and synergistically bind to matrix attachment sequences. J. Biol. Chem. 1999, 274, 20521–20528. [Google Scholar]
- Ariumi, Y.; Masutani, M.; Copeland, T.D.; Mimori, T.; Sugimura, T.; Shimotohno, K.; Ueda, K.; Hatanaka, M.; Noda, M. Suppression of the poly(adp-ribose) polymerase activity by DNA-dependent protein kinase in vitro. Oncogene 1999, 18, 4616–4625. [Google Scholar] [CrossRef]
- Cheng, Q.; Barboule, N.; Frit, P.; Gomez, D.; Bombarde, O.; Couderc, B.; Ren, G.S.; Salles, B.; Calsou, P. Ku counteracts mobilization of parp1 and mrn in chromatin damaged with DNA double-strand breaks. Nucleic Acids Res. 2011, 39, 9605–9619. [Google Scholar]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. Parp is activated at stalled forks to mediate mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-dependent degradation of stalled DNA replication forks is prevented by brca2 and parp1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef]
- Gottipati, P.; Vischioni, B.; Schultz, N.; Solomons, J.; Bryant, H.E.; Djureinovic, T.; Issaeva, N.; Sleeth, K.; Sharma, R.A.; Helleday, T. Poly(adp-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 2010, 70, 5389–5398. [Google Scholar] [CrossRef]
- Groth, P.; Auslander, S.; Majumder, M.M.; Schultz, N.; Johansson, F.; Petermann, E.; Helleday, T. Methylated DNA causes a physical block to replication forks independently of damage signalling, o(6)-methylguanine or DNA single-strand breaks and results in DNA damage. J. Mol. Biol. 2010, 402, 70–82. [Google Scholar] [CrossRef]
- Lundin, C.; North, M.; Erixon, K.; Walters, K.; Jenssen, D.; Goldman, A.S.; Helleday, T. Methyl methanesulfonate (mms) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 2005, 33, 3799–3811. [Google Scholar]
- Wang, Z.Q.; Auer, B.; Stingl, L.; Berghammer, H.; Haidacher, D.; Schweiger, M.; Wagner, E.F. Mice lacking adprt and poly(adp-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995, 9, 509–520. [Google Scholar] [CrossRef]
- Shall, S.; de Murcia, G. Poly(adp-ribose) polymerase-1: What have we learned from the deficient mouse model? Mutat. Res. 2000, 460, 1–15. [Google Scholar] [CrossRef]
- Conde, C.; Mark, M.; Oliver, F.J.; Huber, A.; de Murcia, G.; Menissier-de Murcia, J. Loss of poly(adp-ribose) polymerase-1 causes increased tumour latency in p53-deficient mice. EMBO J. 2001, 20, 3535–3543. [Google Scholar]
- Ame, J.C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Hoger, T.; Menissier-de Murcia, J.; de Murcia, G. Parp-2, a novel mammalian DNA damage-dependent poly(adp-ribose) polymerase. J. Biol. Chem. 1999, 274, 17860–17868. [Google Scholar]
- Menissier de Murcia, J.; Ricoul, M.; Tartier, L.; Niedergang, C.; Huber, A.; Dantzer, F.; Schreiber, V.; Ame, J.C.; Dierich, A.; LeMeur, M.; et al. Functional interaction between parp-1 and parp-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003, 22, 2255–2263. [Google Scholar] [CrossRef]
- Jagtap, P.; Szabo, C. Poly(adp-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440. [Google Scholar]
- Godon, C.; Cordelieres, F.P.; Biard, D.; Giocanti, N.; Megnin-Chanet, F.; Hall, J.; Favaudon, V. Parp inhibition versus parp-1 silencing: Different outcomes in terms of single-strand break repair and radiation susceptibility. Nucleic Acids Res. 2008, 36, 4454–4464. [Google Scholar] [CrossRef]
- Curtin, N.J. Parp inhibitors for cancer therapy. Expert Rev. Mol. Med. 2005, 7, 1–20. [Google Scholar]
- Helleday, T. The underlying mechanism for the parp and brca synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef]
- Fisher, A.E.; Hochegger, H.; Takeda, S.; Caldecott, K.W. Poly(adp-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(adp-ribose) glycohydrolase. Mol. Cell Biol. 2007, 27, 5597–5605. [Google Scholar]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(adp-ribose) polymerase (parp) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411. [Google Scholar] [CrossRef]
- Saintigny, Y.; Delacote, F.; Vares, G.; Petitot, F.; Lambert, S.; Averbeck, D.; Lopez, B.S. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001, 20, 3861–3870. [Google Scholar] [CrossRef]
- Serrano, M.A.; Li, Z.; Dangeti, M.; Musich, P.R.; Patrick, S.; Roginskaya, M.; Cartwright, B.; Zou, Y. DNA-pk, atm and atr collaboratively regulate p53-rpa interaction to facilitate homologous recombination DNA repair. Oncogene 2012. [Google Scholar] [CrossRef]
- Tobin, L.A.; Robert, C.; Nagaria, P.; Chumsri, S.; Twaddell, W.; Ioffe, O.B.; Greco, G.E.; Brodie, A.H.; Tomkinson, A.E.; Rassool, F.V. Targeting abnormal DNA repair in therapy-resistant breast cancers. Mol. Cancer Res. : MCR 2012, 10, 96–107. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of brca2-deficient tumours with inhibitors of poly(adp-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar]
- Buisson, R.; Dion-Cote, A.M.; Coulombe, Y.; Launay, H.; Cai, H.; Stasiak, A.Z.; Stasiak, A.; Xia, B.; Masson, J.Y. Cooperation of breast cancer proteins palb2 and piccolo brca2 in stimulating homologous recombination. Nature Struct. Mol. Biol. 2010, 17, 1247–1254. [Google Scholar] [CrossRef]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-Perry, M.; Snape, K.; et al. Germline mutations in rad51d confer susceptibility to ovarian cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef]
- Vilar, E.; Bartnik, C.M.; Stenzel, S.L.; Raskin, L.; Ahn, J.; Moreno, V.; Mukherjee, B.; Iniesta, M.D.; Morgan, M.A.; Rennert, G.; et al. Mre11 deficiency increases sensitivity to poly(adp-ribose) polymerase inhibition in microsatellite unstable colorectal cancers. Cancer Res. 2011, 71, 2632–2642. [Google Scholar]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O'Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(adp-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of 'brcaness' in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar]
- Dedes, K.J.; Wetterskog, D.; Mendes-Pereira, A.M.; Natrajan, R.; Lambros, M.B.; Geyer, F.C.; Vatcheva, R.; Savage, K.; Mackay, A.; Lord, C.J.; et al. Pten deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to parp inhibitors. Science Trans. Med. 2010, 2, 53ra75. [Google Scholar] [CrossRef]
- Bryant, H.E.; Helleday, T. Inhibition of poly (adp-ribose) polymerase activates atm which is required for subsequent homologous recombination repair. Nucleic Acids Res. 2006, 34, 1685–1691. [Google Scholar]
- Hoglund, A.; Stromvall, K.; Li, Y.; Forshell, L.P.; Nilsson, J.A. Chk2 deficiency in myc overexpressing lymphoma cells elicits a synergistic lethal response in combination with parp inhibition. Cell Cycle 2011, 10, 3598–3607. [Google Scholar] [CrossRef]
- Mitchell, C.; Park, M.; Eulitt, P.; Yang, C.; Yacoub, A.; Dent, P. Poly(adp-ribose) polymerase 1 modulates the lethality of chk1 inhibitors in carcinoma cells. Mol. Pharmacol. 2010, 78, 909–917. [Google Scholar] [CrossRef]
- Fraser, M.; Zhao, H.; Luoto, K.R.; Lundin, C.; Coackley, C.; Chan, N.; Joshua, A.M.; Bismar, T.A.; Evans, A.; Helleday, T.; et al. Pten deletion in prostate cancer cells does not associate with loss of rad51 function: Implications for radiotherapy and chemotherapy. Clin. Cancer Res. 2012, 18, 1015–1027. [Google Scholar] [CrossRef]
- Johnson, N.; Li, Y.C.; Walton, Z.E.; Cheng, K.A.; Li, D.; Rodig, S.J.; Moreau, L.A.; Unitt, C.; Bronson, R.T.; Thomas, H.D.; et al. Compromised cdk1 activity sensitizes brca-proficient cancers to parp inhibition. Nat. Med. 2011, 17, 875–882. [Google Scholar] [CrossRef]
- McLellan, J.L.; O'Neil, N.J.; Barrett, I.; Ferree, E.; van Pel, D.M.; Ushey, K.; Sipahimalani, P.; Bryan, J.; Rose, A.M.; Hieter, P. Synthetic lethality of cohesins with parps and replication fork mediators. PLoS Genetics 2012, 8, e1002574. [Google Scholar] [CrossRef]
- Taniguchi, T.; Tischkowitz, M.; Ameziane, N.; Hodgson, S.V.; Mathew, C.G.; Joenje, H.; Mok, S.C.; D'Andrea, A.D. Disruption of the fanconi anemia-brca pathway in cisplatin-sensitive ovarian tumors. Nat. Med. 2003, 9, 568–574. [Google Scholar] [CrossRef]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.S.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar]
- Wickramanyake, A.; Bernier, G.; Pennil, C.; Casadei, S.; Agnew, K.J.; Stray, S.M.; Mandell, J.; Garcia, R.L.; Walsh, T.; King, M.C.; et al. Loss of function germline mutations in rad51d in women with ovarian carcinoma. Gynecol. Oncol. 2012, 127, 552–555. [Google Scholar]
- Byers, L.A.; Wang, J.; Nilsson, M.B.; Fujimoto, J.; Saintigny, P.; Yordy, J.; Giri, U.; Peyton, M.; Fan, Y.H.; Diao, L.; et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including parp1. Cancer Discov. 2012, 2, 798–811. [Google Scholar] [CrossRef]
- Galia, A.; Calogero, A.E.; Condorelli, R.; Fraggetta, F.; La Corte, A.; Ridolfo, F.; Bosco, P.; Castiglione, R.; Salemi, M. Parp-1 protein expression in glioblastoma multiforme. Eur. J. Histochem.: EJH 2012, 56, e9. [Google Scholar]
- Rojo, F.; Garcia-Parra, J.; Zazo, S.; Tusquets, I.; Ferrer-Lozano, J.; Menendez, S.; Eroles, P.; Chamizo, C.; Servitja, S.; Ramirez-Merino, N.; et al. Nuclear parp-1 protein overexpression is associated with poor overall survival in early breast cancer. ESMO 2012, 23, 1156–1164. [Google Scholar]
- Nosho, K.; Yamamoto, H.; Mikami, M.; Taniguchi, H.; Takahashi, T.; Adachi, Y.; Imamura, A.; Imai, K.; Shinomura, Y. Overexpression of poly(adp-ribose) polymerase-1 (parp-1) in the early stage of colorectal carcinogenesis. Eur. J. Cancer 2006, 42, 2374–2381. [Google Scholar]
- Ibrahim, Y.H.; Garcia-Garcia, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzman, M.; Grueso, J.; et al. Pi3k inhibition impairs brca1/2 expression and sensitizes brca proficient triple negative breast cancer to parp inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef]
- Juvekar, A.; Burga, L.N.; Hu, H.; Lunsford, E.P.; Ibrahim, Y.H.; Balmana, J.; Rajendran, A.; Papa, A.; Spencer, K.; Lyssiotis, C.A.; et al. Combining a pi3k inhibitor with a parp inhibitor provides an effective therapy for a mouse model of brca1-related breast cancer. Cancer Discov. 2012, 2, 1048–1063. [Google Scholar] [CrossRef]
- Kimbung, S.; Biskup, E.; Johansson, I.; Aaltonen, K.; Ottosson-Wadlund, A.; Gruvberger-Saal, S.; Cunliffe, H.; Fadeel, B.; Loman, N.; Berglund, P.; et al. Co-targeting of the pi3k pathway improves the response of brca1 deficient breast cancer cells to parp1 inhibition. Cancer Lett. 2012, 319, 232–241. [Google Scholar] [CrossRef]
- Chan, N.; Pires, I.M.; Bencokova, Z.; Coackley, C.; Luoto, K.R.; Bhogal, N.; Lakshman, M.; Gottipati, P.; Oliver, F.J.; Helleday, T.; et al. Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res. 2010, 70, 8045–8054. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ström, C.E.; Helleday, T. Strategies for the Use of Poly(adenosine diphosphate ribose) Polymerase (PARP) Inhibitors in Cancer Therapy. Biomolecules 2012, 2, 635-649. https://doi.org/10.3390/biom2040635
Ström CE, Helleday T. Strategies for the Use of Poly(adenosine diphosphate ribose) Polymerase (PARP) Inhibitors in Cancer Therapy. Biomolecules. 2012; 2(4):635-649. https://doi.org/10.3390/biom2040635
Chicago/Turabian StyleStröm, Cecilia E., and Thomas Helleday. 2012. "Strategies for the Use of Poly(adenosine diphosphate ribose) Polymerase (PARP) Inhibitors in Cancer Therapy" Biomolecules 2, no. 4: 635-649. https://doi.org/10.3390/biom2040635