
Exploiting Differences in Heme Biosynthesis between Bacterial Species to Screen for Novel Antimicrobials
Abstract
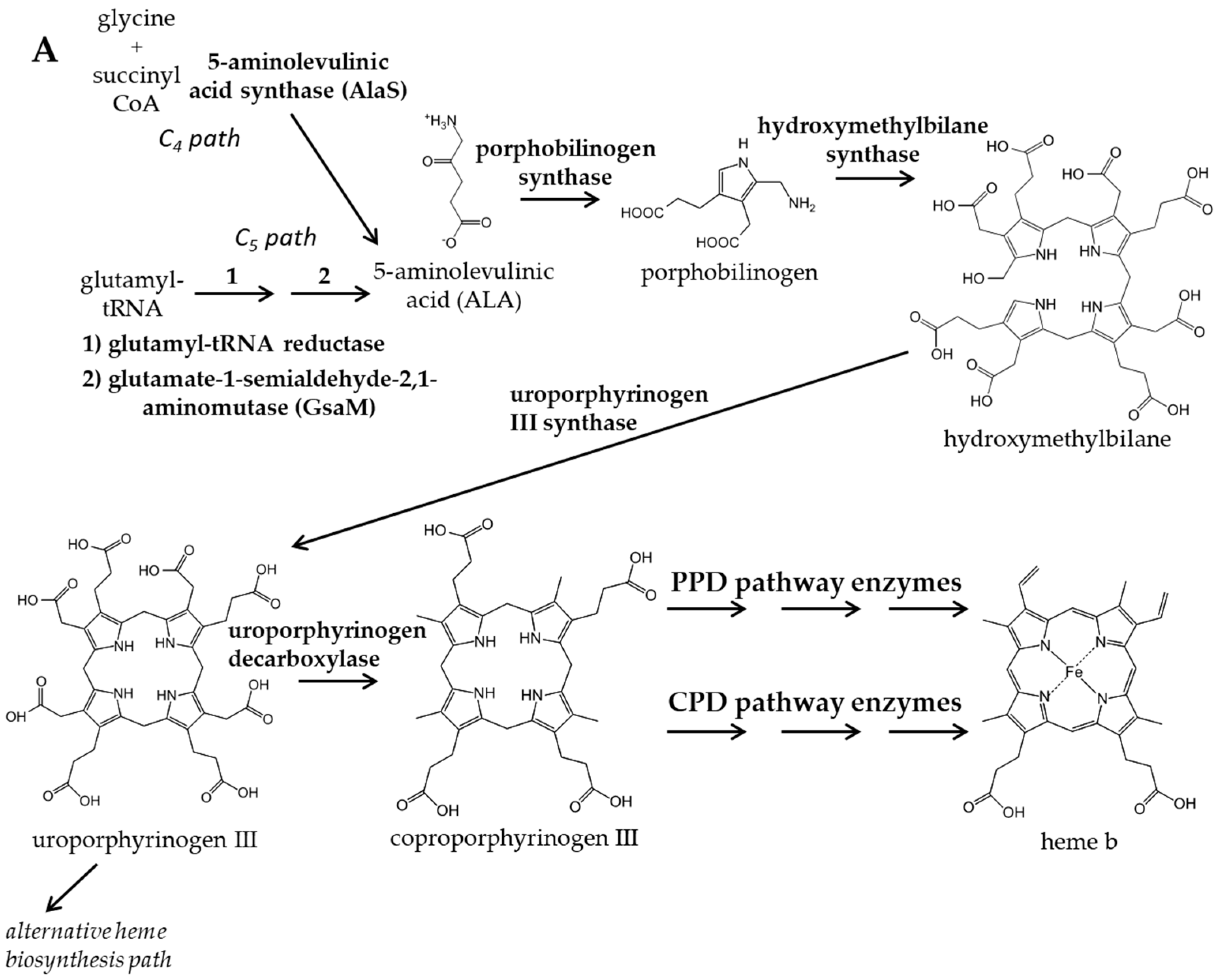
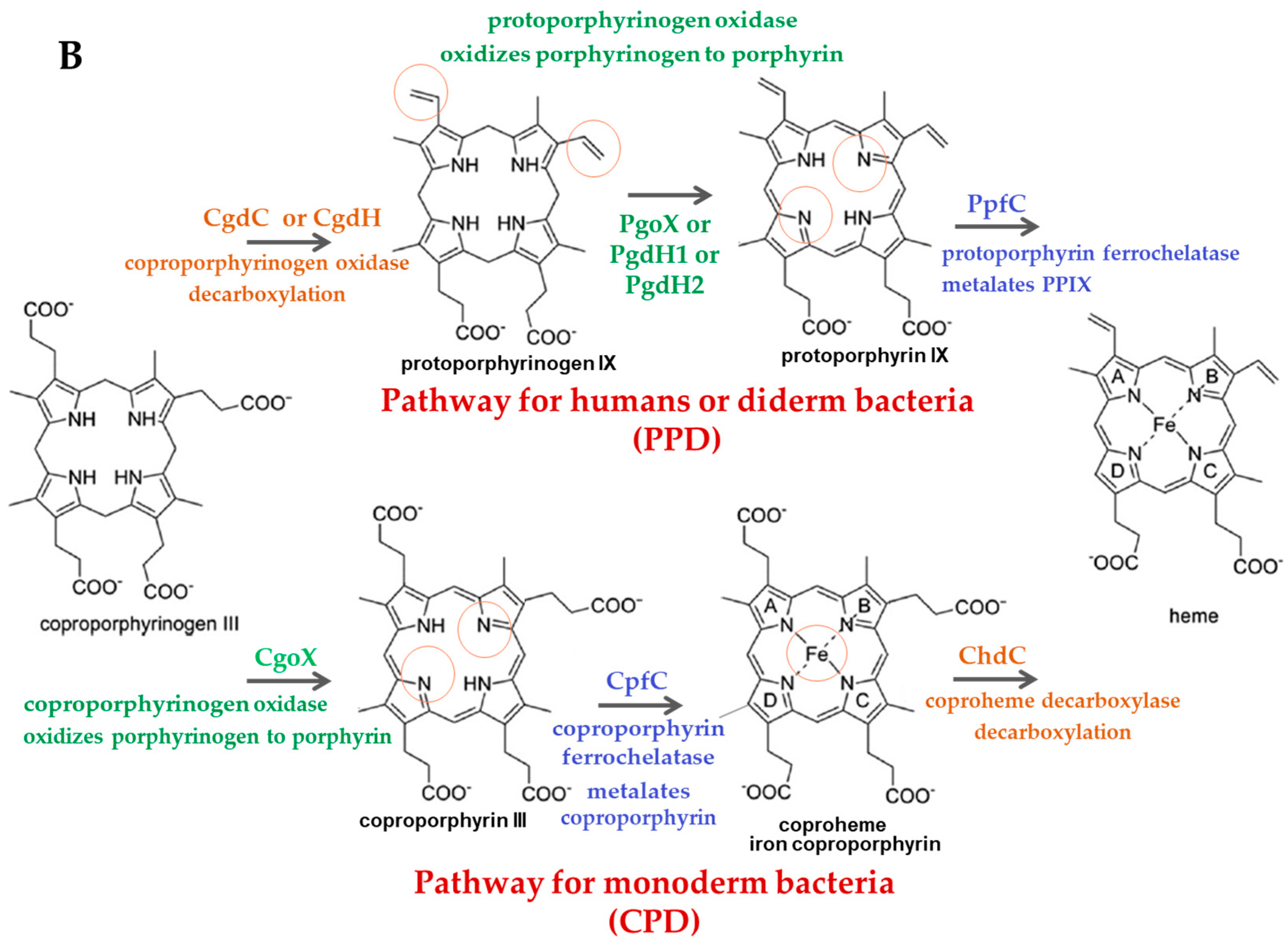
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Recombineering of Sa-CPD-YFP
2.2.1. Preparation of Linear DNA Fragments for Homologous Recombination
2.2.2. Making MC4100-YFP (AmpR) Efficient for Homologous Recombination
2.2.3. Preparation of Electrocompetent Cells and Section Media for Recombination
2.2.4. Selection of tetA-sacB Integrants
2.2.5. Counter-Selection (Replacement of tetA-sacB)
2.3. qPCR
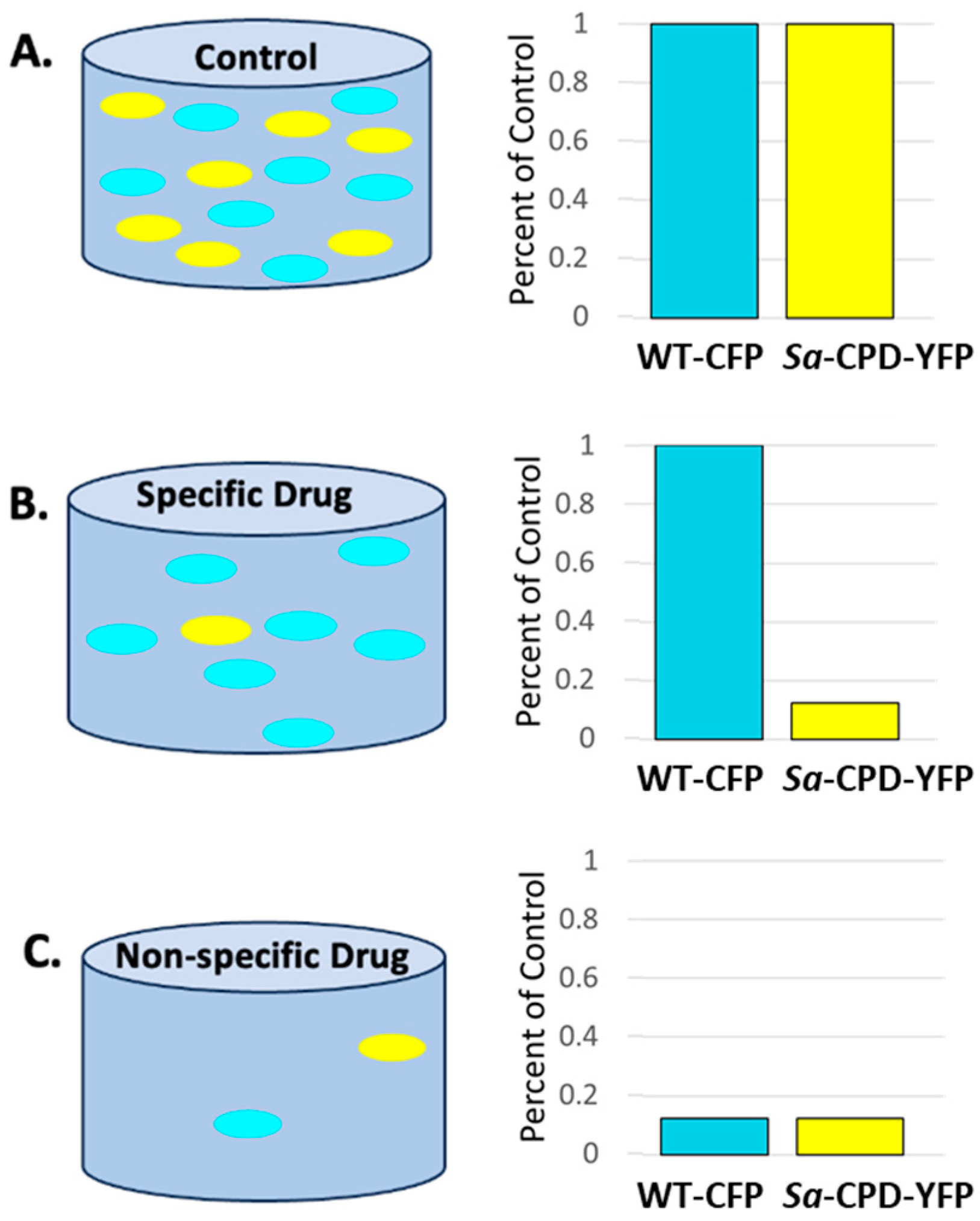
2.4. The Screen
2.5. Calculations of Z’ Factors for the screen
2.6. Titration of Screen Hits
2.7. Determination of Background Signal for Prodiginine Family Compounds
2.8. UPLC Assays for Heme and Porphyrins
2.9. Spectral Analysis of Hemes A, B, and C
2.10. Assay to Determine the Impact of Inhibitors on Heme Biosynthesis
2.11. In Silico Ligand–Protein Binding Analysis
3. Results and Discussion
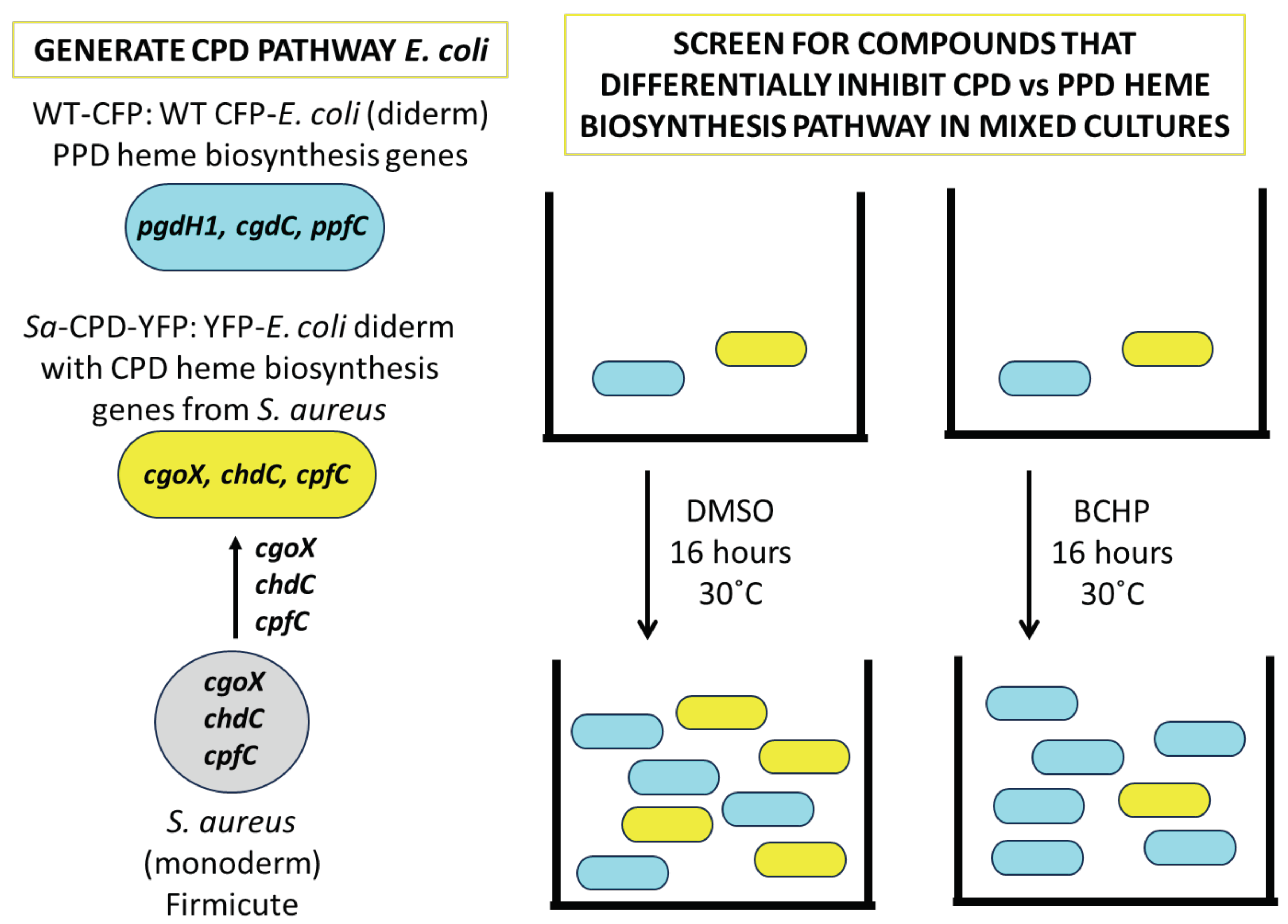
3.1. Creation and Validation of Bacterial Strains Used for Competitive Bacterial Viability Assay
3.2. The Screen-Competitive Bacterial Viability Assay Successfully Identified Specific Hits
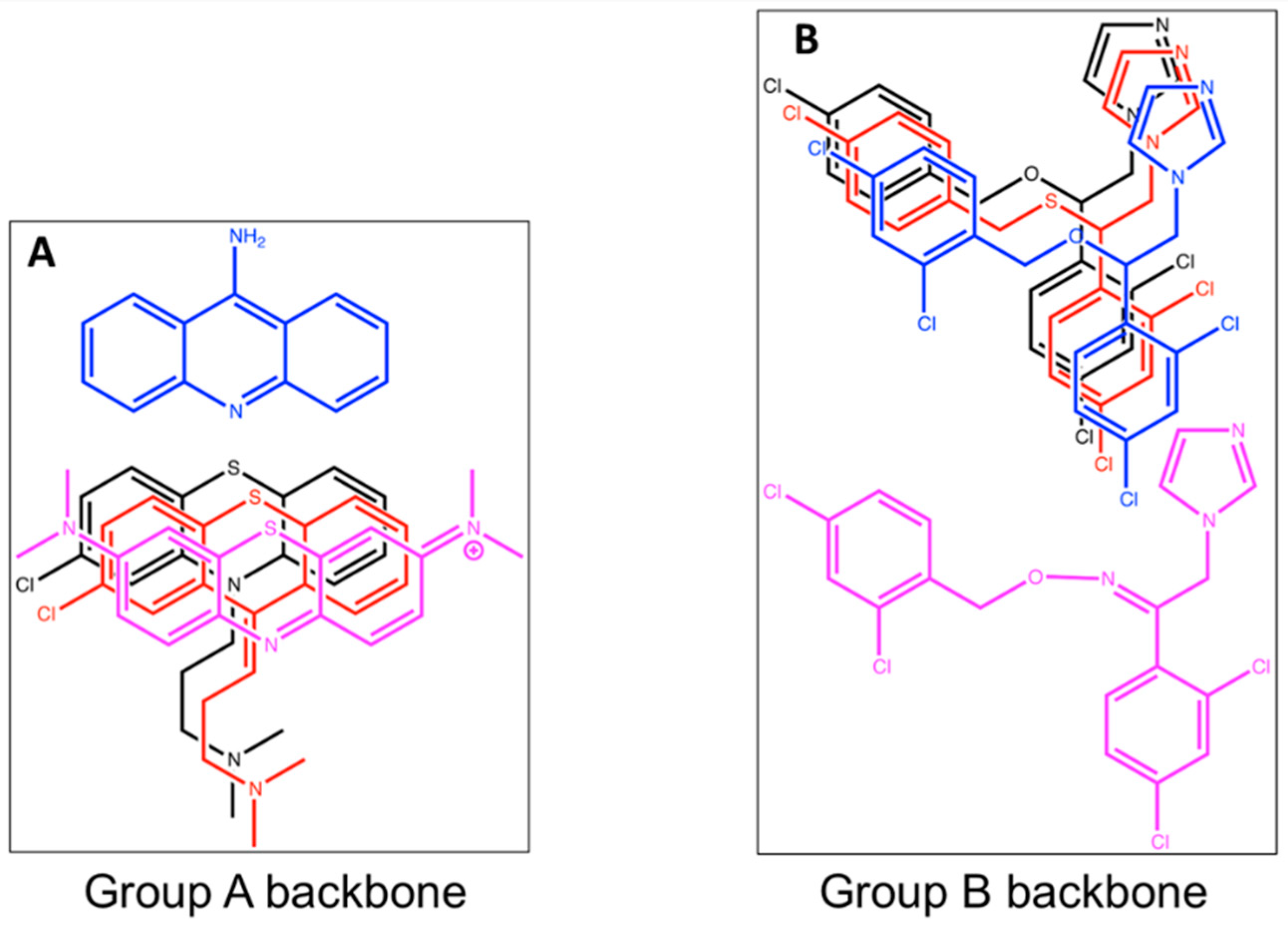
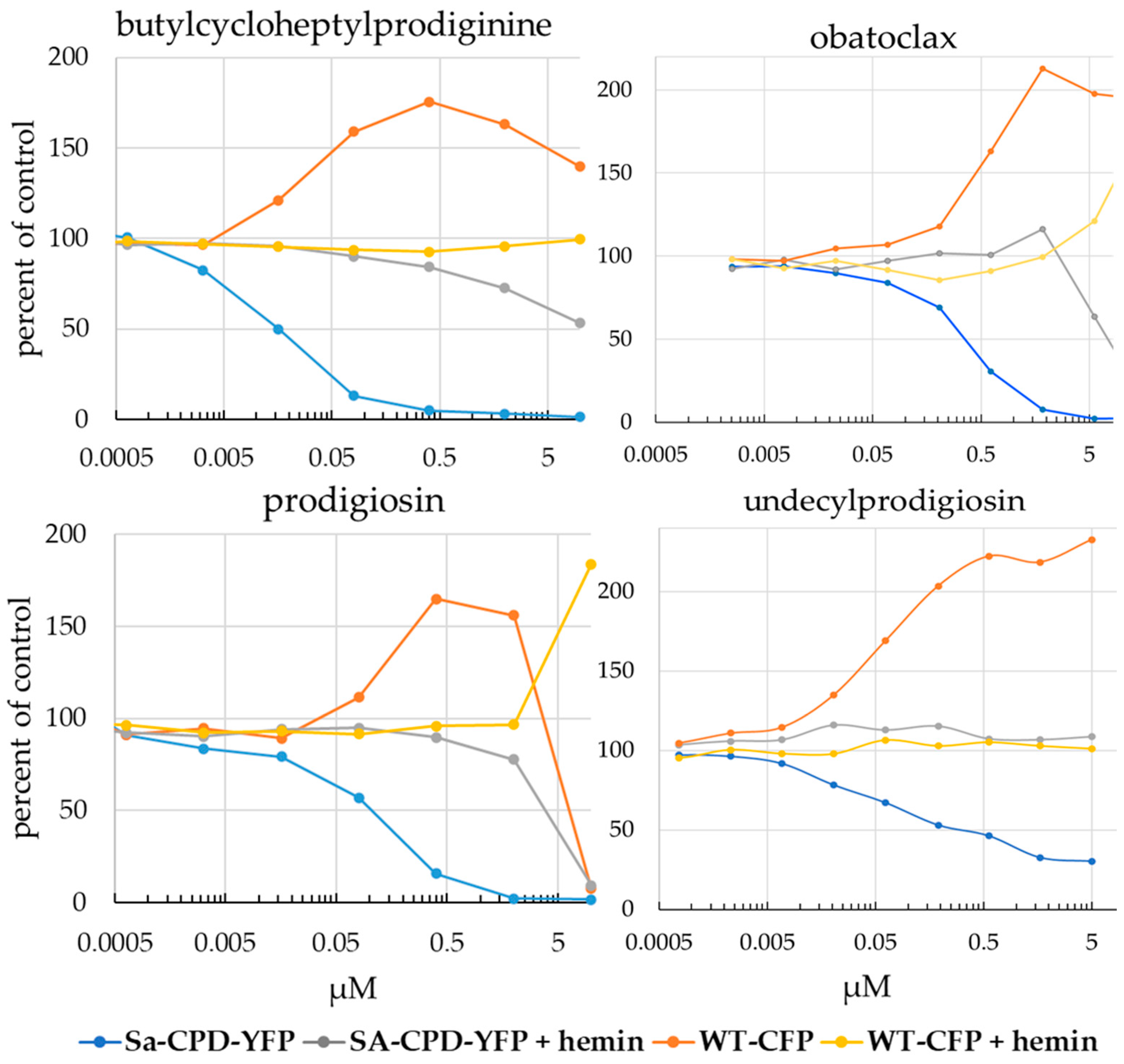
3.3. Analysis of Screen Hits by Titration in a Competitive Viability Assay Identified Two Strong Inhibitors
3.4. Prodigiosin and BCHP Inhibition Is Not an Artifact of Fluorescence Interference
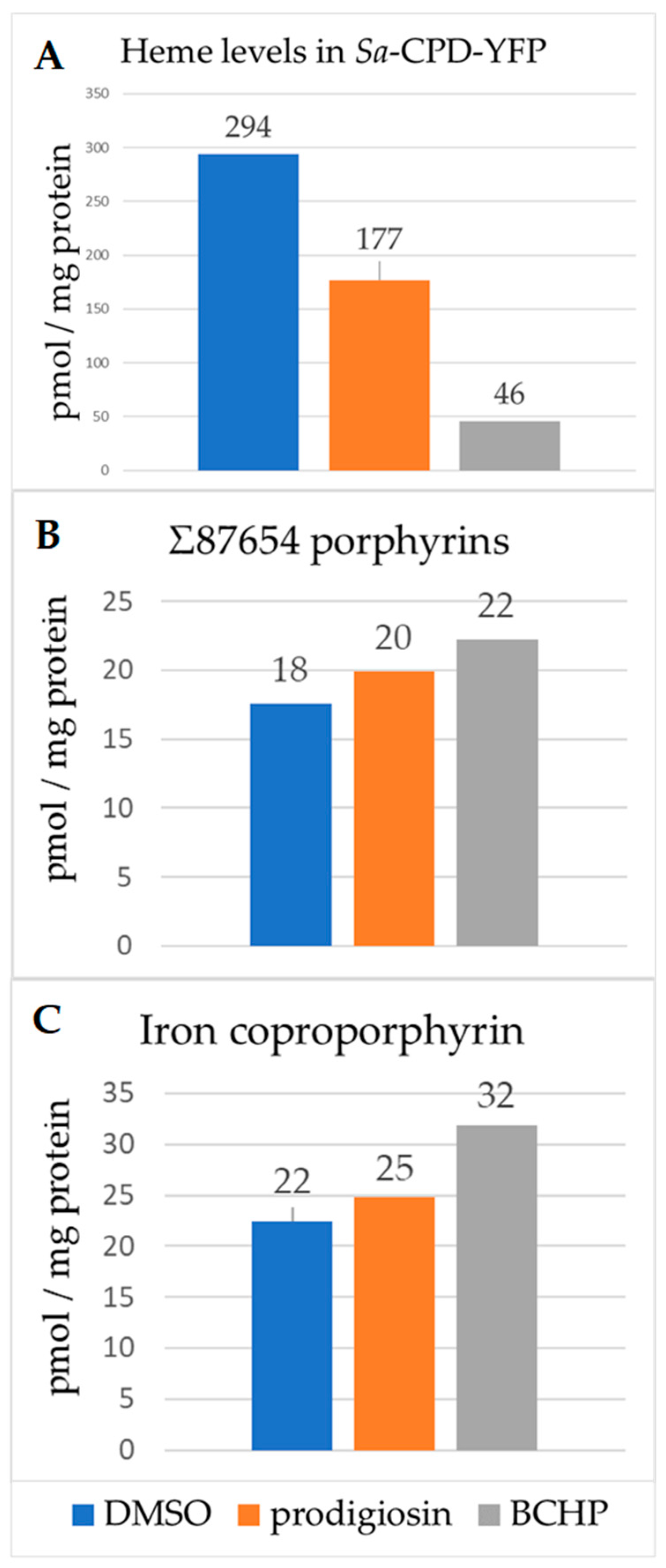
3.5. Heme and Porphyrin Analysis
3.6. Measured Antimicrobial Activity against Cultures of S. aureus Newman
3.7. In Silico Docking of Butylcycloheptylprodiginine and Prodigiosin with a ChdC Protein Structure Is Consistent with the Hypothesis That These Molecules Inhibit by Binding to the ChdC Active Site
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Shimizu, T.; Huang, D.; Yan, F.; Stranava, M.; Bartosova, M.; Fojtikova, V.; Martinkova, M. Gaseous O2, NO, and CO in signal transduction: Structure and function relationships of heme-based gas sensors and heme-redox sensors. Chem. Rev. 2015, 115, 6491–6533. [Google Scholar] [CrossRef]
- Shimizu, T.; Lengalova, A.; Martinek, V.; Martinkova, M. Heme: Emergent roles of heme in signal transduction, functional regulation and as catalytic centres. Chem. Soc. Rev. 2019, 48, 5624–5657. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Visca, P. Scavenging of reactive nitrogen species by mycobacterial truncated hemoglobins. Methods Enzymol. 2008, 436, 317–337. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Bolognesi, M.; Milani, M.; Guertin, M.; Visca, P. Mycobacterial truncated hemoglobins: From genes to functions. Gene 2007, 398, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Stauff, D.L.; Skaar, E.P. The heme sensor system of Staphylococcus aureus. Contrib. Microbiol. 2009, 16, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.J.; Stauff, D.L.; Pishchany, G.; Bezbradica, J.S.; Gordy, L.E.; Iturregui, J.; Anderson, K.L.; Dunman, P.M.; Joyce, S.; Skaar, E.P. A Staphylococcus aureus regulatory system that responds to host heme and modulates virulence. Cell Host Microbe 2007, 1, 109–119. [Google Scholar] [CrossRef]
- Parish, T.; Schaeffer, M.; Roberts, G.; Duncan, K. HemZ is essential for heme biosynthesis in Mycobacterium tuberculosis. Tuberculosis 2005, 85, 197–204. [Google Scholar] [CrossRef]
- Mayfield, J.A.; Hammer, N.D.; Kurker, R.C.; Chen, T.K.; Ojha, S.; Skaar, E.P.; DuBois, J.L. The chlorite dismutase (HemQ) from Staphylococcus aureus has a redox-sensitive heme and is associated with the small colony variant phenotype. J. Biol. Chem. 2013, 288, 23488–23504. [Google Scholar] [CrossRef]
- Melter, O.; Radojevic, B. Small colony variants of Staphylococcus aureus—Review. Folia Microbiol. 2010, 55, 548–558. [Google Scholar] [CrossRef]
- Hammer, N.D.; Reniere, M.L.; Cassat, J.E.; Zhang, Y.; Hirsch, A.O.; Indriati Hood, M.; Skaar, E.P. Two heme-dependent terminal oxidases power Staphylococcus aureus organ-specific colonization of the vertebrate host. mBio 2013, 4, e00241-13. [Google Scholar] [CrossRef]
- von Eiff, C.; Heilmann, C.; Proctor, R.A.; Woltz, C.; Peters, G.; Gotz, F. A site-directed Staphylococcus aureus hemB mutant is a small-colony variant which persists intracellularly. J. Bacteriol. 1997, 179, 4706–4712. [Google Scholar] [CrossRef] [PubMed]
- Hao, G.F.; Zuo, Y.; Yang, S.G.; Yang, G.F. Protoporphyrinogen oxidase inhibitor: An ideal target for herbicide discovery. Chimia 2011, 65, 961–969. [Google Scholar] [CrossRef]
- Coomber, S.A.; Jones, R.M.; Jordan, P.M.; Hunter, C.N. A putative anaerobic coproporphyrinogen III oxidase in Rhodobacter sphaeroides. I. Molecular cloning, transposon mutagenesis and sequence analysis of the gene. Mol. Microbiol. 1992, 6, 3159–3169. [Google Scholar] [CrossRef]
- Layer, G.; Moser, J.; Heinz, D.W.; Jahn, D.; Schubert, W.D. Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of Radical SAM enzymes. EMBO J. 2003, 22, 6214–6224. [Google Scholar] [CrossRef] [PubMed]
- Layer, G.; Reichelt, J.; Jahn, D.; Heinz, D.W. Structure and function of enzymes in heme biosynthesis. Protein Sci. 2010, 19, 1137–1161. [Google Scholar] [CrossRef] [PubMed]
- Boynton, T.O.; Daugherty, L.E.; Dailey, T.A.; Dailey, H.A. Identification of Escherichia coli HemG as a novel, menadione-dependent flavodoxin with protoporphyrinogen oxidase activity. Biochemistry 2009, 48, 6705–6711. [Google Scholar] [CrossRef]
- Boynton, T.O.; Gerdes, S.; Craven, S.H.; Neidle, E.L.; Phillips, J.D.; Dailey, H.A. Discovery of a gene involved in a third bacterial protoporphyrinogen oxidase activity through comparative genomic analysis and functional complementation. Appl. Environ. Microbiol. 2011, 77, 4795–4801. [Google Scholar] [CrossRef]
- Hamza, I.; Dailey, H.A. One ring to rule them all: Trafficking of heme and heme synthesis intermediates in the metazoans. Biochim. Biophys. Acta 2012, 1823, 1617–1632. [Google Scholar] [CrossRef]
- Kannangara, C.G.; Gough, S.P.; Bruyant, P.; Hoober, J.K.; Kahn, A.; von Wettstein, D. tRNA(Glu) as a cofactor in delta-aminolevulinate biosynthesis: Steps that regulate chlorophyll synthesis. Trends Biochem. Sci. 1988, 13, 139–143. [Google Scholar] [CrossRef]
- Ishida, T.; Yu, L.; Akutsu, H.; Ozawa, K.; Kawanishi, S.; Seto, A.; Inubushi, T.; Sano, S. A primitive pathway of porphyrin biosynthesis and enzymology in Desulfovibrio vulgaris. Proc. Natl. Acad. Sci. USA 1998, 95, 4853–4858. [Google Scholar] [CrossRef]
- Bali, S.; Lawrence, A.D.; Lobo, S.A.; Saraiva, L.M.; Golding, B.T.; Palmer, D.J.; Howard, M.J.; Ferguson, S.J.; Warren, M.J. Molecular hijacking of siroheme for the synthesis of heme and d1 heme. Proc. Natl. Acad. Sci. USA 2011, 108, 18260–18265. [Google Scholar] [CrossRef] [PubMed]
- Bali, S.; Palmer, D.J.; Schroeder, S.; Ferguson, S.J.; Warren, M.J. Recent advances in the biosynthesis of modified tetrapyrroles: The discovery of an alternative pathway for the formation of heme and heme d 1. Cell Mol. Life Sci. 2014, 71, 2837–2863. [Google Scholar] [CrossRef] [PubMed]
- Kuhner, M.; Haufschildt, K.; Neumann, A.; Storbeck, S.; Streif, J.; Layer, G. The alternative route to heme in the methanogenic archaeon Methanosarcina barkeri. Archaea 2014, 2014, 327637. [Google Scholar] [CrossRef]
- Dailey, H.A.; Gerdes, S.; Dailey, T.A.; Burch, J.S.; Phillips, J.D. Noncanonical coproporphyrin-dependent bacterial heme biosynthesis pathway that does not use protoporphyrin. Proc. Natl. Acad. Sci. USA 2015, 112, 2210–2215. [Google Scholar] [CrossRef] [PubMed]
- Streit, B.R.; Celis, A.I.; Shisler, K.; Rodgers, K.R.; Lukat-Rodgers, G.S.; DuBois, J.L. Reactions of Ferrous Coproheme Decarboxylase (HemQ) with O(2) and H(2)O(2) Yield Ferric Heme b. Biochemistry 2017, 56, 189–201. [Google Scholar] [CrossRef]
- Sebastiani, F.; Michlits, H.; Lier, B.; Becucci, M.; Furtmuller, P.G.; Oostenbrink, C.; Obinger, C.; Hofbauer, S.; Smulevich, G. Reaction intermediate rotation during the decarboxylation of coproheme to heme b in C. diphtheriae. Biophys. J. 2021, 120, 3600–3614. [Google Scholar] [CrossRef]
- Hegreness, M.; Shoresh, N.; Hartl, D.; Kishony, R. An equivalence principle for the incorporation of favorable mutations in asexual populations. Science 2006, 311, 1615–1617. [Google Scholar] [CrossRef]
- Torella, J.P.; Chait, R.; Kishony, R. Optimal drug synergy in antimicrobial treatments. PLoS Comput. Biol. 2010, 6, e1000796. [Google Scholar] [CrossRef]
- Sabri, S.; Steen, J.A.; Bongers, M.; Nielsen, L.K.; Vickers, C.E. Knock-in/Knock-out (KIKO) vectors for rapid integration of large DNA sequences, including whole metabolic pathways, onto the Escherichia coli chromosome at well-characterised loci. Microb. Cell Fact. 2013, 12, 60. [Google Scholar] [CrossRef]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef]
- Li, X.T.; Thomason, L.C.; Sawitzke, J.A.; Costantino, N.; Court, D.L. Positive and negative selection using the tetA-sacB cassette: Recombineering and P1 transduction in Escherichia coli. Nucleic Acids Res. 2013, 41, e204. [Google Scholar] [CrossRef] [PubMed]
- Clifford, R.J.; Milillo, M.; Prestwood, J.; Quintero, R.; Zurawski, D.V.; Kwak, Y.I.; Waterman, P.E.; Lesho, E.P.; Mc Gann, P. Detection of bacterial 16S rRNA and identification of four clinically important bacteria by real-time PCR. PLoS ONE 2012, 7, e48558. [Google Scholar] [CrossRef]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Peter, F.; Growcock, G.; Strunc, G. Fluorometric determination of erythrocyte protoporphyrin in blood, a comparison between direct (hematofluorometric) and indirect (extraction) methods. Clin. Chem. 1978, 24, 1515–1517. [Google Scholar] [CrossRef]
- Yuan, X.; Rietzschel, N.; Kwon, H.; Walter Nuno, A.B.; Hanna, D.A.; Phillips, J.D.; Raven, E.L.; Reddi, A.R.; Hamza, I. Regulation of intracellular heme trafficking revealed by subcellular reporters. Proc. Natl. Acad. Sci. USA 2016, 113, E5144–E5152. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, S.; Mlynek, G.; Milazzo, L.; Puhringer, D.; Maresch, D.; Schaffner, I.; Furtmuller, P.G.; Smulevich, G.; Djinovic-Carugo, K.; Obinger, C. Hydrogen peroxide-mediated conversion of coproheme to heme b by HemQ-lessons from the first crystal structure and kinetic studies. FEBS J. 2016, 283, 4386–4401. [Google Scholar] [CrossRef] [PubMed]
- Chait, R.; Shrestha, S.; Shah, A.K.; Michel, J.B.; Kishony, R. A differential drug screen for compounds that select against antibiotic resistance. PLoS ONE 2010, 5, e15179. [Google Scholar] [CrossRef]
- Celis, A.I.; Choby, J.E.; Kentro, J.; Skaar, E.P.; DuBois, J.L. Control of Metabolite Flux during the Final Steps of Heme b Biosynthesis in Gram-Positive Bacteria. Biochemistry 2019, 58, 5259–5270. [Google Scholar] [CrossRef]
- Darshan, N.; Manonmani, H.K. Prodigiosin and its potential applications. J. Food Sci. Technol. 2015, 52, 5393–5407. [Google Scholar] [CrossRef]
- Ryoyama, C. Failure of protective action of sodium and potassium ions against heat inactivation of Escherichia coli L-asparaginase. Biochim. Biophys. Acta 1972, 268, 539–541. [Google Scholar] [CrossRef]
- Ouellet, H.; Johnston, J.B.; Ortiz de Montellano, P.R. The Mycobacterium tuberculosis cytochrome P450 system. Arch. Biochem. Biophys. 2010, 493, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Milano, A.; Pasca, M.R.; Provvedi, R.; Lucarelli, A.P.; Manina, G.; Ribeiro, A.L.; Manganelli, R.; Riccardi, G. Azole resistance in Mycobacterium tuberculosis is mediated by the MmpS5-MmpL5 efflux system. Tuberculosis 2009, 89, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Mirnejad, R.; Asadi, A.; Khoshnood, S.; Mirzaei, H.; Heidary, M.; Fattorini, L.; Ghodousi, A.; Darban-Sarokhalil, D. Clofazimine: A useful antibiotic for drug-resistant tuberculosis. Biomed. Pharmacother. 2018, 105, 1353–1359. [Google Scholar] [CrossRef]
- Vjecha, M.J.; Tiberi, S.; Zumla, A. Accelerating the development of therapeutic strategies for drug-resistant tuberculosis. Nat. Rev. Drug Discov. 2018, 17, 607–608. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hemin | PPIX | Porphyrin Σ87654 | Uro | Hepta | Hexa | Penta | Copro | |
|---|---|---|---|---|---|---|---|---|
| E. coli Strain | pmol/mg protein | pmol/mg protein | pmol/mg protein | % mol | % mol | % mol | % mol | % mol |
| parental MC4100-YFP | 463.5 | 0.4 | 0.4 | 74.9 | 1.2 | 0 | 0 | 23.9 |
| S. aureus triple mutant * | 48.5 | 68.6 | 265.8 | 58.1 | 5.6 | 1.7 | 7.6 | 27 |
| S. aureus triple mutant plus cgdH KO ‡ | 46.6 | 3.7 | 449.3 | 59.3 | 7.0 | 1.9 | 6.8 | 25.1 |
| Treatment | Sa-CPD-YFP | WT-CFP | Sa-CPD-YFP + Hemin | WT-CFP + Hemin |
|---|---|---|---|---|
| BCHP | 4 | 149 | 70 | 99 |
| prodigiosin | 2 | 46 | 73 | 110 |
| Compound | Inhibition 0 µM | Inhibition 2 µM | Inhibition 10 µM |
|---|---|---|---|
| Chlorprothixene | 0 | 0 | 0 |
| Diphenyl urea | 0 | 0 | 0 |
| Benzethonium | 0 | 76% | 93% |
| Clotrimazole | 0 | 40% | 70% |
| Miconazole | 0 | 79% | 83% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jackson, L.K.; Dailey, T.A.; Anderle, B.; Warren, M.J.; Bergonia, H.A.; Dailey, H.A.; Phillips, J.D. Exploiting Differences in Heme Biosynthesis between Bacterial Species to Screen for Novel Antimicrobials. Biomolecules 2023, 13, 1485. https://doi.org/10.3390/biom13101485
Jackson LK, Dailey TA, Anderle B, Warren MJ, Bergonia HA, Dailey HA, Phillips JD. Exploiting Differences in Heme Biosynthesis between Bacterial Species to Screen for Novel Antimicrobials. Biomolecules. 2023; 13(10):1485. https://doi.org/10.3390/biom13101485
Chicago/Turabian StyleJackson, Laurie K., Tammy A. Dailey, Brenden Anderle, Martin J. Warren, Hector A. Bergonia, Harry A. Dailey, and John D. Phillips. 2023. "Exploiting Differences in Heme Biosynthesis between Bacterial Species to Screen for Novel Antimicrobials" Biomolecules 13, no. 10: 1485. https://doi.org/10.3390/biom13101485