
FLVCR1a Controls Cellular Cholesterol Levels through the Regulation of Heme Biosynthesis and Tricarboxylic Acid Cycle Flux in Endothelial Cells
 ,
,  , , ,
, , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Gene Silencing and Overexpression
2.3. Measurement of Heme Concentration
2.4. Mice
2.5. LSEC Isolation
2.6. EC Isolation
2.7. Crystal Violet Assay
2.8. Membrane Fluidity Assay
2.9. Membrane Cholesterol Content
2.10. Filipin Staining
2.11. Total (Intracellular) Cholesterol
2.12. De Novo Synthesis of Farnesyl Pyrophosphate (FPP) and Geranylgeranyl Pyrophosphate (GGPP)
2.13. De Novo Synthesis of Cholesterol
2.14. RNA Extraction and Real-Time PCR Analysis
2.15. Western Blot
2.16. Mitochondrial Extraction and ALAS Activity
2.17. C-Isotope Labeling Experiments
2.18. Bioinformatic Analyses
2.19. Statistical Analyses
3. Results
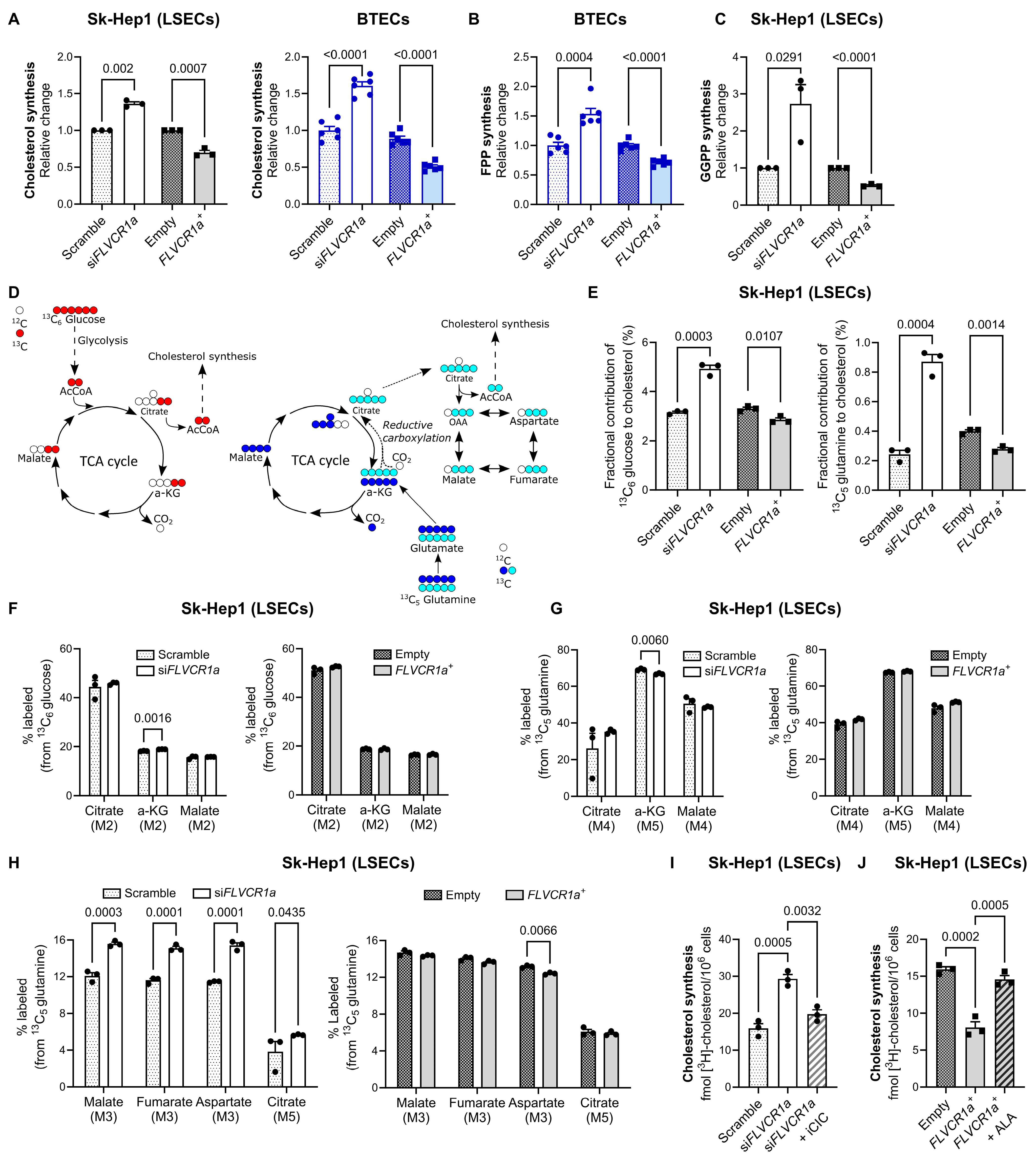
3.1. FLVCR1a Is a Positive Regulator of Heme Synthesis in Endothelial Cells
3.2. FLVCR1a Expression Regulates Cholesterol Production in Endothelial Cells
3.3. FLVCR1a Modulates Cellular Cholesterol Levels
3.4. FLVCR1a Modulates Cellular Cholesterol Levels In Vivo
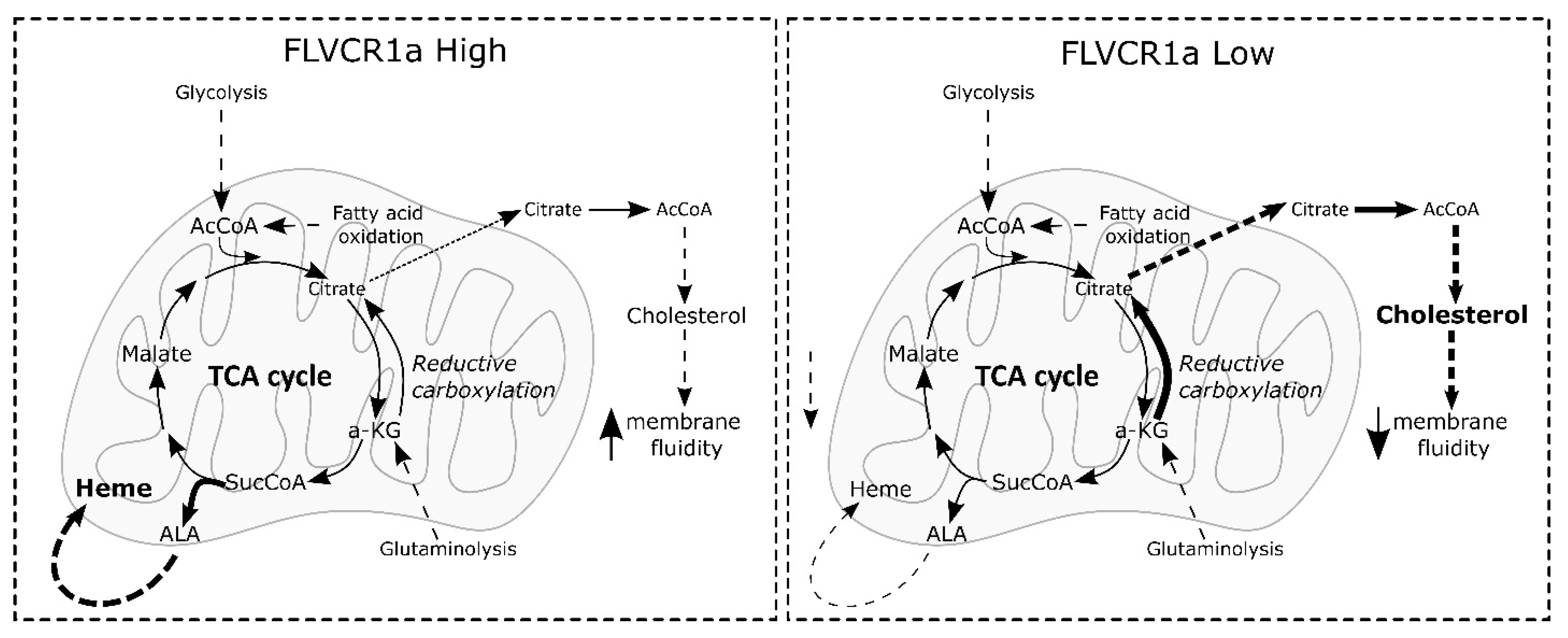
4. Discussion
5. Declaration of Generative AI and AI-Assisted Technologies in the Writing Process
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Quigley, J.G.; Burns, C.C.; Anderson, M.M.; Lynch, E.D.; Sabo, K.M.; Overbaugh, J.; Abkowitz, J.L. Cloning of the cellular receptor for feline leukemia virus subgroup C (FeLV-C), a retrovirus that induces red cell aplasia. Blood 2000, 95, 1093–1099. [Google Scholar] [CrossRef]
- Drew, D.; North, R.A.; Nagarathinam, K.; Tanabe, M. Structures and General Transport Mechanisms by the Major Facilitator Superfamily (MFS). Chem. Rev. 2021, 121, 5289–5335. [Google Scholar] [CrossRef]
- Quigley, J.G.; Yang, Z.; Worthington, M.T.; Phillips, J.D.; Sabo, K.M.; Sabath, D.E.; Berg, C.L.; Sassa, S.; Wood, B.L.; Abkowitz, J.L. Identification of a human heme exporter that is essential for erythropoiesis. Cell 2004, 118, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Fiorito, V.; Allocco, A.L.; Petrillo, S.; Gazzano, E.; Torretta, S.; Marchi, S.; Destefanis, F.; Pacelli, C.; Audrito, V.; Provero, P.; et al. The heme synthesis-export system regulates the tricarboxylic acid cycle flux and oxidative phosphorylation. Cell Rep. 2021, 35, 109252. [Google Scholar] [CrossRef]
- Arnold, P.K.; Finley, L.W.S. Regulation and function of the mammalian tricarboxylic acid cycle. J. Biol. Chem. 2023, 299, 102838. [Google Scholar] [CrossRef]
- Bartz, F.; Kern, L.; Erz, D.; Zhu, M.; Gilbert, D.; Meinhof, T.; Wirkner, U.; Erfle, H.; Muckenthaler, M.; Pepperkok, R.; et al. Identification of cholesterol-regulating genes by targeted RNAi screening. Cell Metab. 2009, 10, 63–75. [Google Scholar] [CrossRef]
- Vandekeere, S.; Dubois, C.; Kalucka, J.; Sullivan, M.R.; García-Caballero, M.; Goveia, J.; Chen, R.; Diehl, F.F.; Bar-Lev, L.; Souffreau, J.; et al. Serine Synthesis via PHGDH Is Essential for Heme Production in Endothelial Cells. Cell Metab. 2018, 28, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Shetty, T.; Sishtla, K.; Park, B.; Repass, M.J.; Corson, T.W. Heme Synthesis Inhibition Blocks Angiogenesis via Mitochondrial Dysfunction. iScience 2020, 23, 101391. [Google Scholar] [CrossRef]
- Petrillo, S.; Chiabrando, D.; Genova, T.; Fiorito, V.; Ingoglia, G.; Vinchi, F.; Mussano, F.; Carossa, S.; Silengo, L.; Altruda, F.; et al. Heme accumulation in endothelial cells impairs angiogenesis by triggering paraptosis. Cell Death Differ. 2018, 25, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; De Giorgio, F.; Bertino, F.; Garello, F.; Bitonto, V.; Longo, D.L.; Mercurio, S.; Ammirata, G.; Allocco, A.L.; Fiorito, V.; et al. Endothelial cells require functional FLVCR1a during developmental and adult angiogenesis. Angiogenesis 2023, 26, 365–384. [Google Scholar] [CrossRef]
- Byfield, F.J.; Aranda-Espinoza, H.; Romanenko, V.G.; Rothblat, G.H.; Levitan, I. Cholesterol depletion increases membrane stiffness of aortic endothelial cells. Biophys. J. 2004, 87, 3336–3343. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Staiculescu, M.C.; Hampel, P.; Levitan, I.; Forgacs, G. How cholesterol regulates endothelial biomechanics. Front. Physiol. 2012, 3, 426. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, B.J.; Denis, M.; Genest, J. Cellular physiology of cholesterol efflux in vascular endothelial cells. Circulation 2004, 110, 2881–2888. [Google Scholar] [CrossRef] [PubMed]
- Immanuel, J.; Yun, S. Vascular Inflammatory Diseases and Endothelial Phenotypes. Cells 2023, 12, 1640. [Google Scholar] [CrossRef] [PubMed]
- Grange, C.; Bussolati, B.; Bruno, S.; Fonsato, V.; Sapino, A.; Camussi, G. Isolation and characterization of human breast tumor-derived endothelial cells. Oncol. Rep. 2006, 15, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Bussolati, B.; Deambrosis, I.; Russo, S.; Deregibus, M.C.; Camussi, G. Altered angiogenesis and survival in human tumor-derived endothelial cells. FASEB J. 2003, 17, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Bertino, F.; Firestone, K.; Bellacchio, E.; Jackson, K.E.; Asamoah, A.; Hersh, J.; Fiorito, V.; Destefanis, F.; Gonser, R.; Tucker, M.E.; et al. Heme and sensory neuropathy: Insights from novel mutations in the heme exporter feline leukemia virus subgroup C receptor 1. Pain 2019, 160, 2766–2775. [Google Scholar] [CrossRef]
- Sinclair, P.R.; Gorman, N.; Jacobs, J.M. Measurement of heme concentration. Curr. Protoc. Toxicol. 2001, Chapter 8, Unit 8.3. [Google Scholar] [CrossRef]
- Vinchi, F.; Ingoglia, G.; Chiabrando, D.; Mercurio, S.; Turco, E.; Silengo, L.; Altruda, F.; Tolosano, E. Heme exporter FLVCR1a regulates heme synthesis and degradation and controls activity of cytochromes P450. Gastroenterology 2014, 146, 1325–1338. [Google Scholar] [CrossRef]
- Rausa, M.; Pagani, A.; Nai, A.; Campanella, A.; Gilberti, M.E.; Apostoli, P.; Camaschella, C.; Silvestri, L. Bmp6 expression in murine liver non parenchymal cells: A mechanism to control their high iron exporter activity and protect hepatocytes from iron overload? PLoS ONE 2015, 10, e0122696. [Google Scholar] [CrossRef]
- Liang, L.; Wang, H.; Yao, J.; Wei, Q.; Lu, Y.; Wang, T.; Cao, X. NPC1 Deficiency Contributes to Autophagy-Dependent Ferritinophagy in HEI-OC1 Auditory Cells. Front. Mol. Biosci. 2022, 9, 952608. [Google Scholar] [CrossRef] [PubMed]
- Campia, I.; Gazzano, E.; Pescarmona, G.; Ghigo, D.; Bosia, A.; Riganti, C. Digoxin and ouabain increase the synthesis of cholesterol in human liver cells. Cell Mol. Life Sci. 2009, 66, 1580–1594. [Google Scholar] [CrossRef]
- Riganti, C.; Pinto, H.; Bolli, E.; Belisario, D.C.; Calogero, R.A.; Bosia, A.; Cavallo, F. Atorvastatin modulates anti-proliferative and pro-proliferative signals in Her2/neu-positive mammary cancer. Biochem. Pharmacol. 2011, 82, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Salaroglio, I.C.; Belisario, D.C.; Akman, M.; La Vecchia, S.; Godel, M.; Anobile, D.P.; Ortone, G.; Digiovanni, S.; Fontana, S.; Costamagna, C.; et al. Mitochondrial ROS drive resistance to chemotherapy and immune-killing in hypoxic non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2022, 41, 243. [Google Scholar] [CrossRef]
- Bergonia, H.A.; Franklin, M.R.; Kushner, J.P.; Phillips, J.D. A method for determining δ-aminolevulinic acid synthase activity in homogenized cells and tissues. Clin. Biochem. 2015, 48, 788–795. [Google Scholar] [CrossRef]
- Nolan, D.J.; Ginsberg, M.; Israely, E.; Palikuqi, B.; Poulos, M.G.; James, D.; Ding, B.S.; Schachterle, W.; Liu, Y.; Rosenwaks, Z.; et al. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev. Cell 2013, 26, 204–219. [Google Scholar] [CrossRef]
- Manicardi, N.; Fernández-Iglesias, A.; Abad-Jordà, L.; Royo, F.; Azkargorta, M.; Ortega-Ribera, M.; Sanfeliu-Redondo, D.; Martínez-Alcocer, A.; Elortza, F.; Hessheimer, A.J.; et al. Transcriptomic Profiling of the Liver Sinusoidal Endothelium during Cirrhosis Reveals Stage-Specific Secretory Signature. Cancers 2021, 13, 2688. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.; Gao, J.H.; Zhao, C.; Tong, H.; Zheng, S.P.; Huang, Z.Y.; Liu, R.; Tang, C.W.; Li, J. SK-Hep1: Not hepatocellular carcinoma cells but a cell model for liver sinusoidal endothelial cells. Int. J. Clin. Exp. Pathol. 2018, 11, 2931–2938. [Google Scholar]
- Petrillo, S.; De Giorgio, F.; Kopecka, J.; Genova, T.; Fiorito, V.; Allocco, A.L.; Bertino, F.; Chiabrando, D.; Mussano, F.; Altruda, F.; et al. Endothelial Heme Dynamics Drive Cancer Cell Metabolism by Shaping the Tumor Microenvironment. Biomedicines 2021, 9, 1557. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef]
- Anastasiou, D.; Cantley, L.C. Breathless cancer cells get fat on glutamine. Cell Res. 2012, 22, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Sachar, M.; Anderson, K.E.; Ma, X. Protoporphyrin IX: The Good, the Bad, and the Ugly. J. Pharmacol. Exp. Ther. 2016, 356, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.E.; Homann, U. Cell surface area regulation and membrane tension. J. Membr. Biol. 2001, 179, 79–102. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.L.; Wang, L.; Pan, H.T.; Zhang, T.R.; Chen, Y.H.; Xu, S.J.; Mao, X.L.; Li, S.W. Animal and Organoid Models of Liver Fibrosis. Front. Physiol. 2021, 12, 666138. [Google Scholar] [CrossRef]
- Fagoonee, S.; Arigoni, M.; Manco, M.; Olivero, M.; Bizzaro, F.; Magagnotti, C.; Andolfo, A.; Miniscalco, B.; Forni, M.; Todeschi, S.; et al. Circulating Extracellular Vesicles Contain Liver-Derived RNA Species as Indicators of Severe Cholestasis-Induced Early Liver Fibrosis in Mice. Antioxid. Redox Signal 2022, 36, 480–504. [Google Scholar] [CrossRef] [PubMed]
- Ponka, P. Cell biology of heme. Am. J. Med. Sci. 1999, 318, 241–256. [Google Scholar] [CrossRef]
- Mercurio, S.; Petrillo, S.; Chiabrando, D.; Bassi, Z.I.; Gays, D.; Camporeale, A.; Vacaru, A.; Miniscalco, B.; Valperga, G.; Silengo, L.; et al. The heme exporter Flvcr1 regulates expansion and differentiation of committed erythroid progenitors by controlling intracellular heme accumulation. Haematologica 2015, 100, 720–729. [Google Scholar] [CrossRef]
- Kenny, T.C.; Khan, A.; Son, Y.; Yue, L.; Heissel, S.; Sharma, A.; Pasolli, H.A.; Liu, Y.; Gamazon, E.R.; Alwaseem, H.; et al. Integrative genetic analysis identifies FLVCR1 as a plasma-membrane choline transporter in mammals. Cell Metab. 2023, 35, 1057–1071.e12. [Google Scholar] [CrossRef]
- Tsuchiya, M.; Tachibana, N.; Nagao, K.; Tamura, T.; Hamachi, I. Organelle-selective click labeling coupled with flow cytometry allows pooled CRISPR screening of genes involved in phosphatidylcholine metabolism. Cell Metab. 2023, 35, 1072–1083.e9. [Google Scholar] [CrossRef]
- Ha, H.T.T.; Sukumar, V.K.; Chua, J.W.B.; Nguyen, D.T.; Nguyen, T.Q.; Lim, L.H.K.; Cazenave-Gassiot, A.; Nguyen, L.N. Mfsd7b facilitates choline transport and missense mutations affect choline transport function. Cell Mol. Life Sci. 2023, 81, 3. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Zheng, L.; Folger, O.; Rajagopalan, K.N.; MacKenzie, E.D.; Jerby, L.; Micaroni, M.; Chaneton, B.; Adam, J.; Hedley, A.; et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 2011, 477, 225–228. [Google Scholar] [CrossRef]
- Biancur, D.E.; Kapner, K.S.; Yamamoto, K.; Banh, R.S.; Neggers, J.E.; Sohn, A.S.W.; Wu, W.; Manguso, R.T.; Brown, A.; Root, D.E.; et al. Functional Genomics Identifies Metabolic Vulnerabilities in Pancreatic Cancer. Cell Metab. 2021, 33, 199–210.e8. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.G.; Chudnovskiy, A.; Baudrier, L.; Prizer, B.; Liu, Y.; Ostendorf, B.N.; Yamaguchi, N.; Arab, A.; Tavora, B.; Timson, R.; et al. Functional Genomics In Vivo Reveal Metabolic Dependencies of Pancreatic Cancer Cells. Cell Metab. 2021, 33, 211–221.e6. [Google Scholar] [CrossRef]
- Mao, R.; Meng, S.; Gu, Q.; Araujo-Gutierrez, R.; Kumar, S.; Yan, Q.; Almazan, F.; Youker, K.A.; Fu, Y.; Pownall, H.J.; et al. AIBP Limits Angiogenesis Through γ-Secretase-Mediated Upregulation of Notch Signaling. Circ. Res. 2017, 120, 1727–1739. [Google Scholar] [CrossRef]
- Bogachkov, Y.Y.; Chen, L.; Le Master, E.; Fancher, I.S.; Zhao, Y.; Aguilar, V.; Oh, M.J.; Wary, K.K.; DiPietro, L.A.; Levitan, I. LDL induces cholesterol loading and inhibits endothelial proliferation and angiogenesis in Matrigels: Correlation with impaired angiogenesis during wound healing. Am. J. Physiol. Cell Physiol. 2020, 318, C762–C776. [Google Scholar] [CrossRef] [PubMed]
- Rohlenova, K.; Goveia, J.; García-Caballero, M.; Subramanian, A.; Kalucka, J.; Treps, L.; Falkenberg, K.D.; de Rooij, L.P.M.H.; Zheng, Y.; Lin, L.; et al. Single-Cell RNA Sequencing Maps Endothelial Metabolic Plasticity in Pathological Angiogenesis. Cell Metab. 2020, 31, 862–877.e14. [Google Scholar] [CrossRef] [PubMed]
- Kuo, A.; Checa, A.; Niaudet, C.; Jung, B.; Fu, Z.; Wheelock, C.E.; Singh, S.A.; Aikawa, M.; Smith, L.E.; Proia, R.L.; et al. Murine endothelial serine palmitoyltransferase 1 (SPTLC1) is required for vascular development and systemic sphingolipid homeostasis. Elife 2022, 11, e78861. [Google Scholar] [CrossRef]
- Rodriguez-Vita, J.; Morales-Ruiz, M. Down the liver sinusoidal endothelial cell (LSEC) hole. Is there a role for lipid rafts in LSEC fenestration? Hepatology 2013, 57, 1272–1274. [Google Scholar] [CrossRef]
- Svistounov, D.; Warren, A.; McNerney, G.P.; Owen, D.M.; Zencak, D.; Zykova, S.N.; Crane, H.; Huser, T.; Quinn, R.J.; Smedsrød, B.; et al. The Relationship between fenestrations, sieve plates and rafts in liver sinusoidal endothelial cells. PLoS ONE 2012, 7, e46134. [Google Scholar] [CrossRef]
- Simionescu, N.; Lupu, F.; Simionescu, M. Rings of membrane sterols surround the openings of vesicles and fenestrae, in capillary endothelium. J. Cell Biol. 1983, 97, 1592–1600. [Google Scholar] [CrossRef]
- Allocco, A.L.; Bertino, F.; Petrillo, S.; Chiabrando, D.; Riganti, C.; Bardelli, A.; Altruda, F.; Fiorito, V.; Tolosano, E. Inhibition of Heme Export and/or Heme Synthesis Potentiates Metformin Anti-Proliferative Effect on Cancer Cell Lines. Cancers 2022, 14, 1230. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manco, M.; Ammirata, G.; Petrillo, S.; De Giorgio, F.; Fontana, S.; Riganti, C.; Provero, P.; Fagoonee, S.; Altruda, F.; Tolosano, E. FLVCR1a Controls Cellular Cholesterol Levels through the Regulation of Heme Biosynthesis and Tricarboxylic Acid Cycle Flux in Endothelial Cells. Biomolecules 2024, 14, 149. https://doi.org/10.3390/biom14020149
Manco M, Ammirata G, Petrillo S, De Giorgio F, Fontana S, Riganti C, Provero P, Fagoonee S, Altruda F, Tolosano E. FLVCR1a Controls Cellular Cholesterol Levels through the Regulation of Heme Biosynthesis and Tricarboxylic Acid Cycle Flux in Endothelial Cells. Biomolecules. 2024; 14(2):149. https://doi.org/10.3390/biom14020149
Chicago/Turabian StyleManco, Marta, Giorgia Ammirata, Sara Petrillo, Francesco De Giorgio, Simona Fontana, Chiara Riganti, Paolo Provero, Sharmila Fagoonee, Fiorella Altruda, and Emanuela Tolosano. 2024. "FLVCR1a Controls Cellular Cholesterol Levels through the Regulation of Heme Biosynthesis and Tricarboxylic Acid Cycle Flux in Endothelial Cells" Biomolecules 14, no. 2: 149. https://doi.org/10.3390/biom14020149