Development and Application of Multidimensional HPLC Mapping Method for O-linked Oligosaccharides


Abstract
: Glycosylation improves the solubility and stability of proteins, contributes to the structural integrity of protein functional sites, and mediates biomolecular recognition events involved in cell-cell communications and viral infections. The first step toward understanding the molecular mechanisms underlying these carbohydrate functionalities is a detailed characterization of glycan structures. Recently developed glycomic approaches have enabled comprehensive analyses of N-glycosylation profiles in a quantitative manner. However, there are only a few reports describing detailed O-glycosylation profiles primarily because of the lack of a widespread standard method to identify O-glycan structures. Here, we developed an HPLC mapping method for detailed identification of O-glycans including neutral, sialylated, and sulfated oligosaccharides. Furthermore, using this method, we were able to quantitatively identify isomeric products from an in vitro reaction catalyzed by N-acetylglucosamine-6O-sulfotransferases and obtain O-glycosylation profiles of serum IgA as a model glycoprotein.1. Introduction
Glycosylation is one of most ubiquitous post-translational modifications. Carbohydrate moieties, which are typically found on asparagine or serine/threonine residues, are associated with an increase in solubility and stability of proteins, structural integrity of protein functional sites, and mediation of biomolecular recognition events involved in cell-cell communications and viral infections [1,2]. Since glycans affect the serum half-life of proteins and functional protein–protein interactions, glycosylation is currently considered to be a crucial factor in the design and development of biopharmaceuticals [3,4,5]. To address the detailed molecular basis of the functional roles of protein glycosylation, the first step is identifying the glycan structures expressed on the proteins [6,7,8,9,10]. Recently developed glycomic approaches using chromatographic and mass spectrometric (MS) techniques have enabled comprehensive analyses of N-glycosylation profiles [11,12]. For example, a multidimensional HPLC mapping method has been developed for quantitative N-glycosylation profiling at molecular, cellular, and tissue levels, enabling isomeric N-glycan structures [13,14]. In this method, identification of individual N-glycans is based on their elution positions on three types of HPLC columns. The accumulated HPLC data of approximately 500 different N-glycans are now available by using the web application GALAXY ( http://www.glycoanalysis.info/galaxy2/ENG/index.jsp) [15], and the applicability of this method has been extended to sialylated, glucronylated, and sulfated oligosaccharides [16,17,18].
However, few reports describe the detailed O-glycosylation profiles with linkage information due to the lack of widespread standard methods for unambiguous identification of O-glycan structures [19,20,21]. The HPLC elution conditions employed in the current GALAXY protocols are not applicable to the profiling of O-glycans, because they frequently include smaller saccharides, e.g., mono- and di-saccharides, in contrast to the generally larger N-glycans. In view of this situation, we herein attempted to develop HPLC-based profiling of O-glycans for their detailed structural identification. By chromatographic and mass spectrometric analyses in conjunction with several exoglycosidase treatments in vitro, we successfully collected HPLC data for 27 different O-glycans including neutral, sialyl, and sulfated oligosaccharides, which were isolated from natural sources and/or by in vitro enzymatic reactions. By applying this extended HPLC map, we have obtained O-glycosylation profiles of serum immunoglobulin A (IgA) as a model glycoprotein. Furthermore, we characterized branch specificity in the sulfation reaction catalyzed by human N-acetylglucosamine-6O-sulfotransferases (GlcNAc6ST)-1.
2. Experimental
2.1. Materials
Materials used for the experiments were purchased from the sources indicated below: Glycoamidase A from sweet almond, β-galactosidase and β-N-acetylhexosaminidase from jack bean were purchased from Seikagaku Kogyo Co. (Tokyo, Japan). α-Galactosidase from green coffee bean was purchased from Oxford Glycosystems Inc. (Bedford, MA, USA) (currently available in Prozyme (Hayward, CA, USA)). α-Sialidase from Arthrobacter ureafaciens was purchased from Nacalai Tesque (Kyoto, Japan). α2,3-Sialidase from Salmonella typhimurium was purchased from Takara Bio Inc. (Otsu, Japan). Recombinat α2,3-sialyltransferase and α2,6-sialyltransferase were purchased from Calbiochem (San Diego, CA, USA). Colostrum IgA, porcine stomach mucin, trypsin, and chymotrypsin were purchased from Sigma Chemical Co. (St. Louis, MO, USA).
2-Aminopyridine-derivatized (PA) isomalto-oligosaccharides were prepared from glucose oligomers (1-20) (Seikagaku Kogyo Co., Tokyo, Japan), fucose (Fuc), galactose (Gal), N-acetylgalactosamine (GalNAc) (Seikagaku Kogyo Co., Tokyo, Japan), glucose (Glc), N-acetylglucosamine (GlcNAc), mannose (Man), Galβ1-3GalNAc (Calbiochem, Schwalbach, Germany), and Galβ1-3(Fucα1-2)GalNAc. Four types of O-glycosylated peptides—Galβ1-4GlcNAcβ1-6(Neu5Acα2-3Galβ1-3)GalNAcα 1-peptide (AHGVT*SAPDTRK; asterisks indicate glycosylation sites)-FAM (5-carboxyfluorescein), Galβ1-4GlcNAcβ1-6(Galβ1-3)GalNAcα1-peptide-FAM, GlcNAcβ1 -6(GlcNAcβ1-3Galβ 1-3)GalNAcα1-peptide-FAM, and Neu5Acα2-6(Galβ1-3)GalNAc-peptide-FAM were purchased from GlycoGene (Tsukuba, Japan).
2.2. Purification of IgA from Human Serum
Human serum (1 mL) was diluted in 10 mL of 0.01 M phosphate buffer (pH 7.4) containing 0.15 M NaCl and absorbed on jacalin-agarose columns (1 mL) (Thermo Scientific, San Jose, CA, USA). After the column was thoroughly washed with 10 mL of 0.01 M phosphate buffer (pH 7.4) containing 0.15 M NaCl and 0.8 M glucose, lectin-binding proteins were eluted with 10 mL of 0.01 M phosphate buffer (pH 7.4) containing 0.1 M melibiose as described previously [22]. After the eluate was concentrated using an AMICON Ultra-15 centrifugal filter unit (Millipore, Billerica, MA, USA), serum IgA was purified with a Superose 12 gel filtration column (GE Healthcare, Uppsala, Sweden) equilibrated with 0.01 M phosphate buffer (pH 7.4) containing 0.15 M NaCl. The purified IgA was desalted with a PD-10 column (GE Healthcare) according to the manufacturer's instructions and then lyophilized for glycosylation profiling.
2.3. Sulfation Reaction by GlcNAc6ST-1
COS7 cells grown in 75 cm2 culture flasks (Corning, Corning, NY) were transfected with 10 μg of relevant plasmid, pcDNA-GlcNAc6ST-1 [23] using Lipofectamine Plus (Invitrogen, Carlsbad, CA, USA) according to manufacturer's instructions. After 24 h of culture in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum, the medium was replaced with DMEM containing 2% IgG-free fetal calf serum. The cells were further cultured for 96 h. Subsequently, the culture medium was collected and concentrated to 1 mL using Amicon Ultra-15 (Millipore). The recombinant protein A-fused GlcNAc6ST-1 expressed in the medium was adsorbed to IgG-Sepharose (20 μL resin/1 mL of culture medium) at 4 °C for 3 h. The resin was collected by centrifugation and washed three times with 50 mM Tris-HCl (pH 7.5). Finally, the resin was suspended in 20 μL of 50 mM Tris-HCl (pH 7.5) and used for enzymatic reaction. The glycopeptide GlcNAcβ1-6(GlcNAcβ1-3Galβ1-3)GalNAcα1-peptide-FAM was utilized as an acceptor substrate. The standard reaction mixture was composed of 1 μmol of Tris-HCl (pH 7.5), 0.4 μmol of MnCl2, 0.08 μmol of AMP, 24 μmol of NaF, 50 pmol of glycopeptide, 300 pmol of 3′-phosphoadenosine 5′-phosphosulfate, 0.1% Triton X-100, and 20 μL of the fusion protein suspension in a final volume of 40 μL. After incubation at 37 °C for 1, 5, 24, and 48 h, the individual reaction mixtures were applied to a TSK gel ODS-80s HPLC column (TOSOH, Tokyo, Japan) at a flow rate of 1.0 mL/min at 25 °C using two solvents: G and H. G comprised water containing 0.05% trifluoroacetic acid and H comprised acetonitrile-2-propanol (2:1, v/v) containing 0.05% trifluoroacetic acid. The column was equilibrated with 90% solvent G and 10% solvent H. The time for gradient elution was 0–40 min with a linear gradient of 10%–15% D. The glycopeptides were detected by fluorescence using excitation and emission wavelengths of 492 and 520 nm, respectively.
2.4. Liberation of O-glycans from Glycoproteins
The O-glycans were released from glycoproteins and glycopeptides by β-elimination using hydrazine for a convenient modification with 2-aminopyridine. Lyophilized glycoproteins (∼250 μg) or glycopeptides (∼5 μg) were dissolved in 1 mL of distilled anhydrous hydrazine with a water content of less than 1% (v/v) in 10 mL glass tube, incubated at 60 °C for 6 h and quenched by 9 mL of 50 mM ammonium acetate buffer (pH 7) with slight modification of the previous literature [24]. The excess hydrazine, peptides, and other reagents were removed and N-acetylated using a graphite carbon column (GL-Pak Carbograph, GL Science, Tokyo, Japan) according to the literature [25]. The hydrazine solution was mixed with 3 mL of 50 mM ammonium acetate buffer (pH 7) and loaded onto the GL-Pak Carbograph column. After the column was washed with 15 mL of 50 mM ammonium acetate buffer (pH 7.0), the released glycans were eluted with 5 mL of a mixture of 50 mM ammonium acetate buffer (pH 7.0):acetonitrile containing 2% acetic anhydride (40:60).
2.5. Pyridylamination
The released O-linked saccharides, as well as those commercially obtained, were labeled with 2-aminopyridine as described previously [26]. Ten volumes of acetonitrile were added to one volume of reaction mixture. The excess PA reagents were removed with a MonoSpin NH2 desalting column (GL Science). After the column was equilibrated with 200 μL of acetonitrile, the PA reaction mixture was loaded onto the column. The column was washed with acetonitrile three times. Then, the PA-saccharides were eluted with 100 μL of water and subsequently dried under vacuum.
2.6. HPLC and MS Analyses
Three types of HPLC columns were used for the separation of PA-saccharides. In each step, PA-saccharides were detected by fluorescence using excitation and emission wavelengths of 310 and 380 nm, respectively. The PA-saccharide mixture was firstly separated on a Mono-Q HR 5/5 anion-exchange column (GE Healthcare) at 30 °C with a flow rate of 1.0 mL/min using two solvents, A and B. Solvent A was aqueous ammonia (pH 9.0) and solvent B was a 50 mM ammonium acetate solution (pH 9.0). The column was equilibrated with solvent A. The gradient elution parameters were 0–3 min, linear gradient 0%–12% B; 3–17 min, linear gradient 12%–40% B; 17–22 min, linear gradient 40%–100% B. Each oligosaccharide was separated according to its anionic charges.
In the second step, each fraction separated from the Mono-Q column was collected, evaporated, and then applied to a Decelosil C30-HG-5 (C30) column (Nomura Chemical Co., Ltd., Seto, Japan). Elution was performed at a flow rate of 1.0 mL/min at 25 °C using two solvents, C and D. Solvent C was 0.1 M ammonium acetate buffer (pH 6.0) containing 0.01% 1-butanol and solvent D was 0.1 M ammonium acetate buffer (pH 6.0) containing 1% 1-butanol. The column was equilibrated with solvent C. The gradient elution parameters were 0–51 min, linear gradient 0%–50% D and 51–63 min, linear gradient 50%–100% D.
In the third step, individual peak fractions from the C30 column were isolated using a TSK gel amide-80 size fractionation column (TOSOH). In this system, two solvents were used at 25 °C. Solvent E was composed of 3% acetic acid in water with triethylamine (pH 7.3) and acetonitrile, 15:85 by volume. Solvent F was composed of 3% acetic acid in water with triethylamine (pH 7.3). The column was equilibrated with solvent E. The gradient elution parameters were 0–5 min, linear gradient 0%–20% F and 5–17 min, linear gradient 20%–44% F.
The HPLC elution times were represented by glucose units (GUs) on the columns calibrated with a PA-derivatized isomalto-oligosaccharides mixture. The structures of the PA-saccharides were characterized by HPLC mapping in conjunction with exoglycosidase treatments and matrix-assisted laser desorption/ionization time of flight (MALDI-TOF-MS) analysis using a MALDI-TOF-MS spectrometer (AXIMA-CFR; Shimadzu, Kyoto, Japan). Collision-induced dissociation spectra of PA-oligosaccharides were acquired using a MALDI-quadrupole ion trap (QIT)-TOF-MS spectrometer (AXIMA-QIT; Shimadzu). All analytical procedures used in this work, including sulfation, sialylation, several glycosidase treatments, and MALDI-TOF-MS analyses have been described previously [16,17,27,28,29].
3. Results and Discussion
3.1. Collection of HPLC Data of O-linked Saccharides
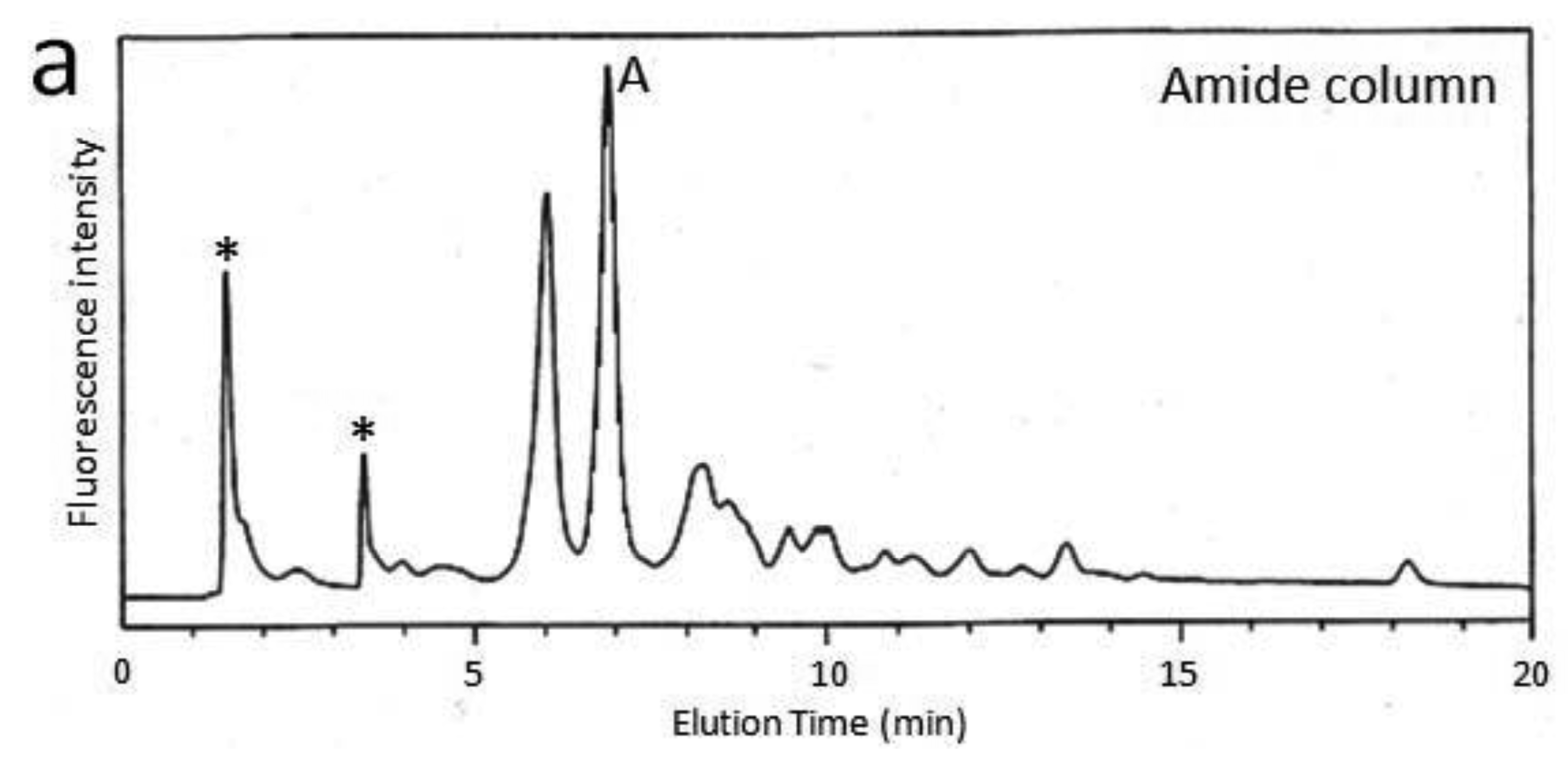
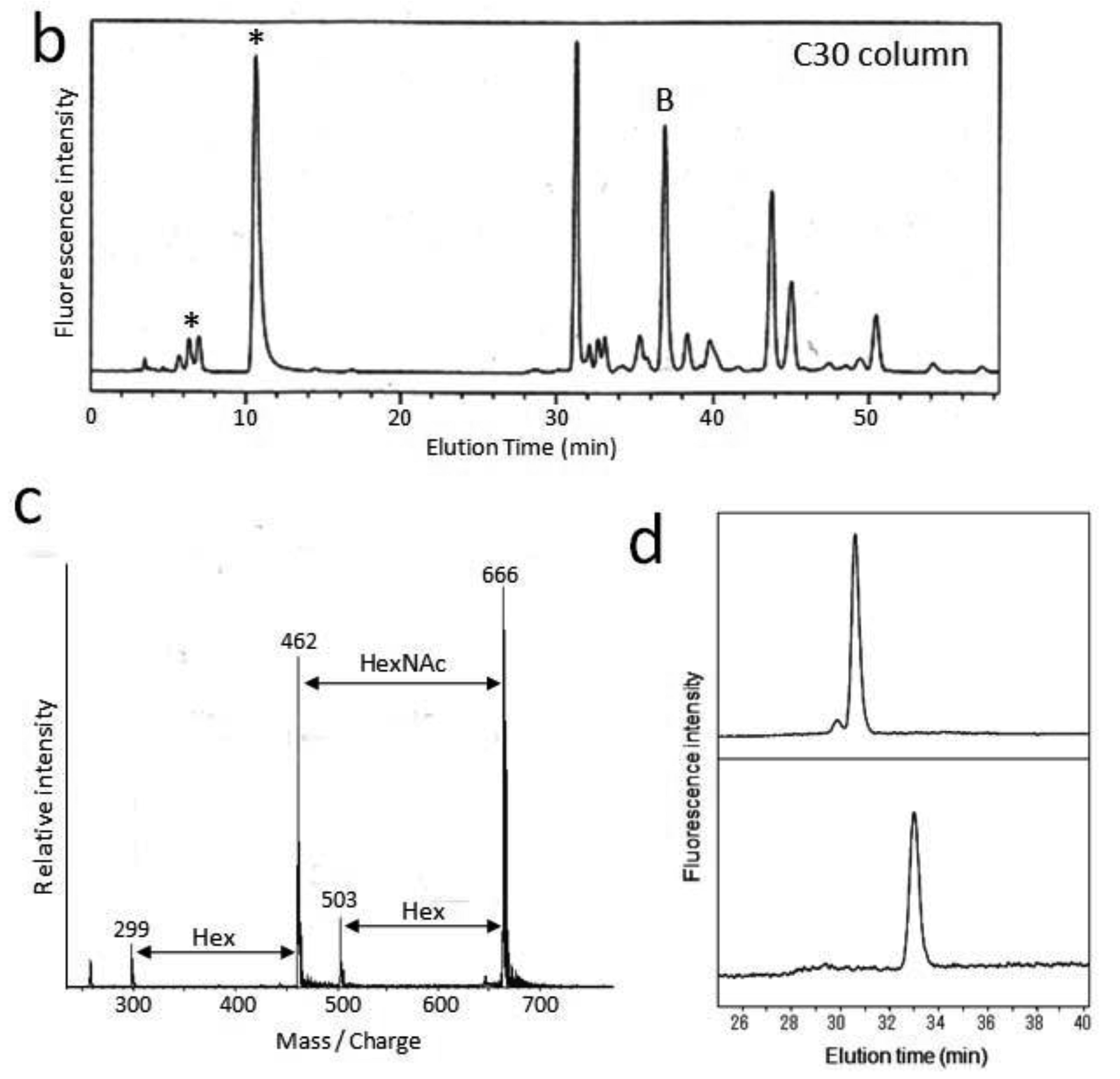
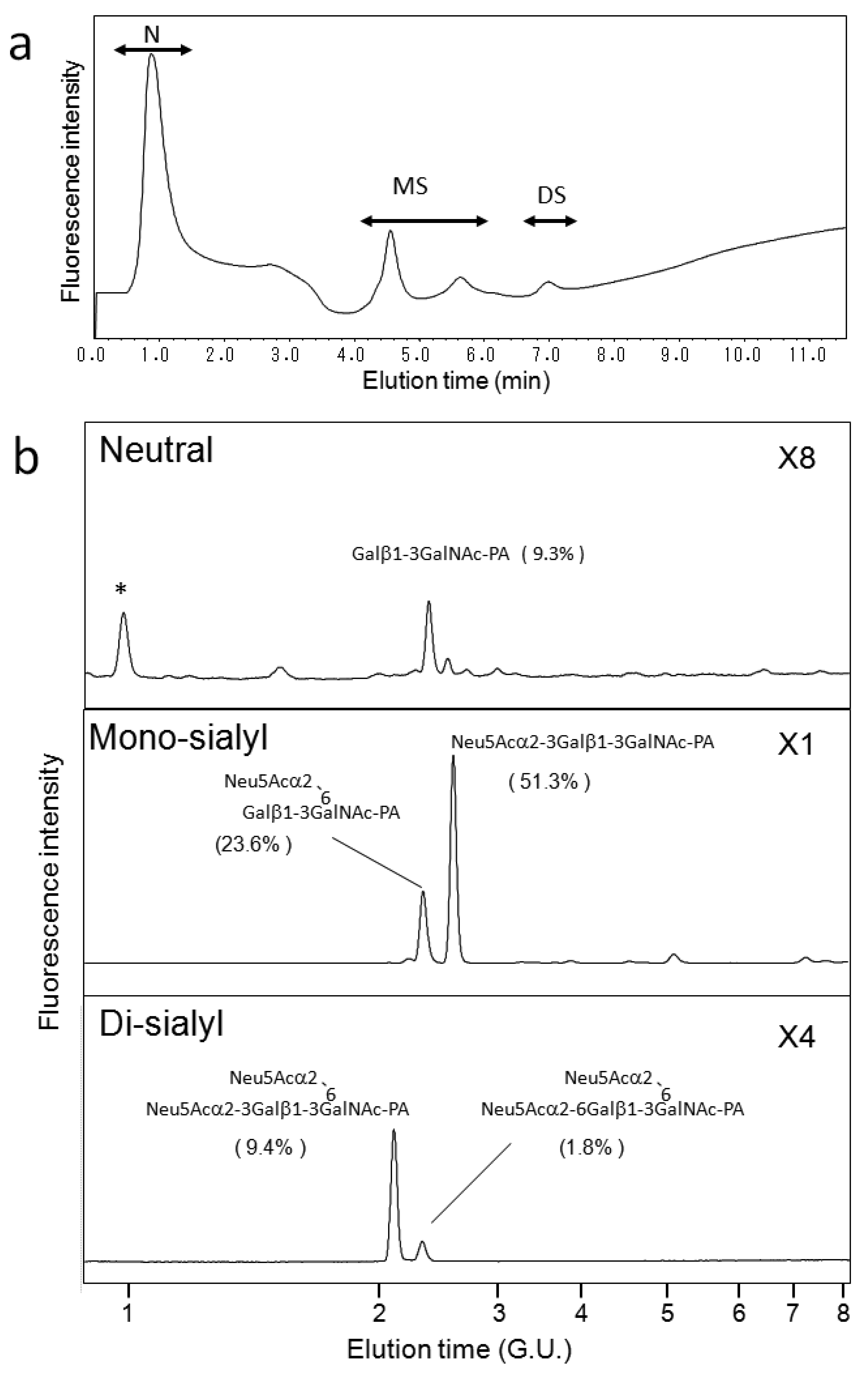
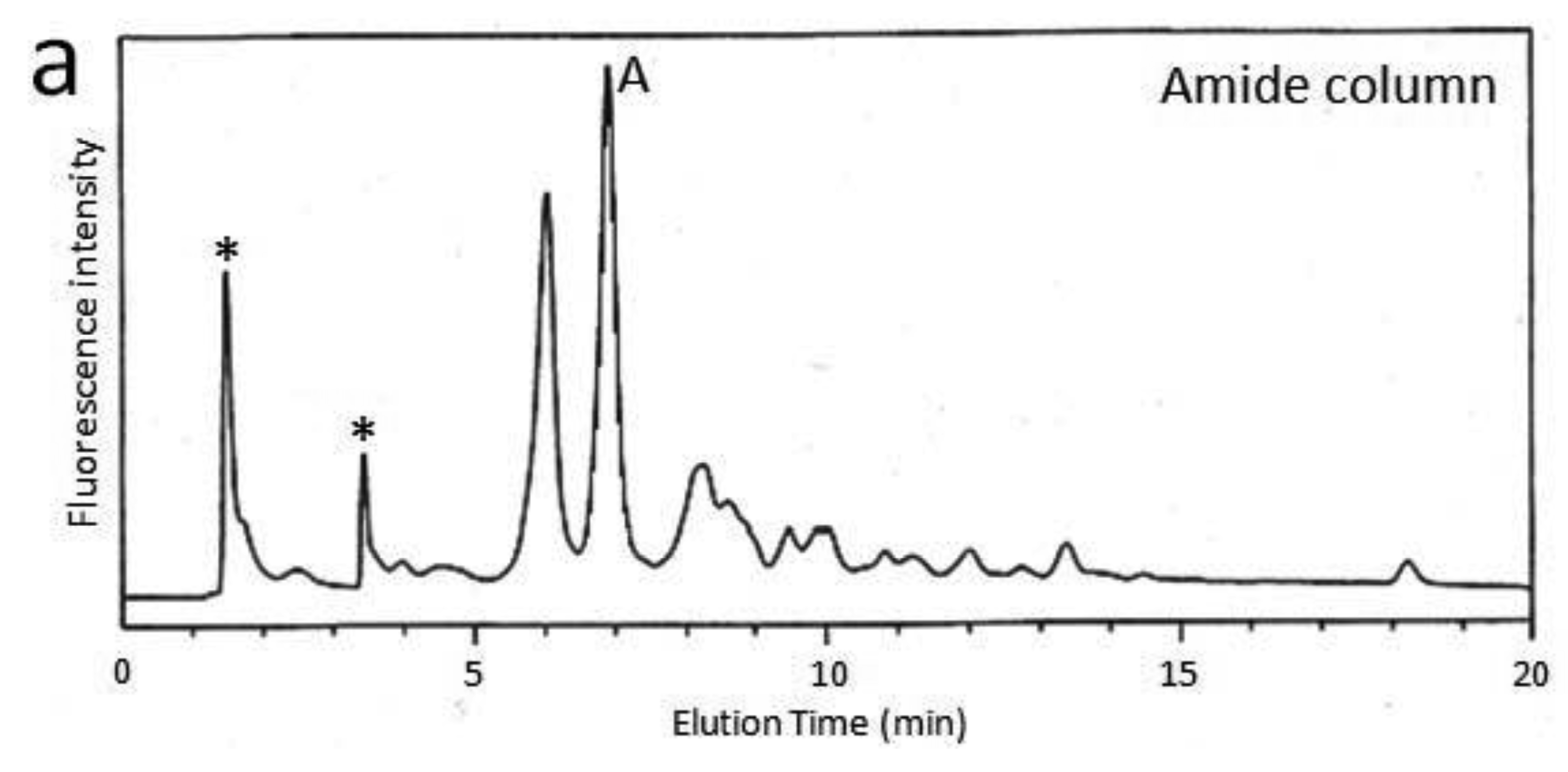
First, we attempted to make an HPLC map of the standard PA-saccharide. The PA tag was attached to the commercially obtained saccharides Fuc, Gal, GalNAc, Glc, GlcNac, Man, Galβ1-3GalNAc, and Galβ1-3(Fucα1-2)GalNAc. In addition, four types of O-glycosylated peptides were treated with hydrazine, and the released oligosaccharides were labeled with 2-aminopyridine. The PA-saccharides thus prepared were subjected to amide and C30 columns to record their elution times (Table 1). Furthermore, pyridylaminated O-glycans were prepared from colostrum IgA and porcine stomach mucin, and their structures were identified by chromatographic analyses combined with exoglycosidase treatments and MALDI-TOF-MS. The structural identification of these O-glycans would be exemplified by a glycan derived from mucin. Since no sialylated O-glycans were detected in the glycosylation profile of mucin on a Mono-Q column (data not shown), the PA-glycans were directly applied to an amide column. Figure 1a shows the O-glycosylation profile of the mucin on the amide column in which two major O-glycans were found. Then, fraction A was applied to a C30 column, giving rise to several peaks including B (Figure 1b). The elution times of the PA-O-glycan in fraction B are represented as 3.2 GU on the amide column and 4.6 GU on the C30 column. The molecular mass of this glycan was determined by MALDI-TOF-MS analysis as 665 Da, which corresponds to (Hex)1(HexNAc)2PA (Figure 1c). The fragment ions indicated that the PA-O-glycan exhibits the branching structure Hex-(HexNAc-)HexNAc-PA. Finally, the glycan corresponding to fraction B was treated with β1,3-galactosidase and then applied to a C30 column, giving rise to a new fraction. The elution time of the glycan corresponding to this fraction coincided with that of the reference PA-glycan, GlcNAcβ1-6GalNAc-PA. On the basis of all these data, we concluded that the glycan corresponding to fraction B was Galβ1-3(GlcNAcβ1-6)GalNAc-PA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PA-Saccharides | GU (amide) | GU (C30) | Molecular mass (Da) a |
|---|---|---|---|
| GalNAc-PA | 0.8 | 3.0 | 300 |
| Man-PA | 0.8 | 0.8 | 259 |
| GlcNAc-PA | 1.0 | 2.7 | 300 |
| Gal-PA | 1.2 | 0.4 | 259 |
| Fuc-PA | 0.7 | 1.5 | 243 |
| Galβ1-3GalNAc-PA | 1.7 | 2.4 | 462 |
| GlcNAcβ1-6GalNAc-PA | 2.4 | 6.3 | 503 |
| Galβ1-3(Fucα1-2)GalNAc-PA | 2.4 | 8.5 | 608 |
| GlcNAcβ1-3Galβ1-3(GlcNAcβ1-6)GalNAc-PA | 3.0 | 2.9 | 868 |
| Galβ1-3(Galβ1-4GlcNAcβ1-6)GalNAc-PA | 3.4 | 4.0 | 827 |
| Neu5Acα2-3Galβ1-3(Galβ1-4GlcNAcβ1-6)GalNAc-PA | 3.2 | 4.3 | 1118 |
| Galβ1-3(Neu5Acα2-6)GalNAc-PA | 1.6 | 2.3 | 753 |
aAverage mass calculated from the m/z values of [M+H]+, [M+Na]+, and [M-H]– ions for PA-saccharides.

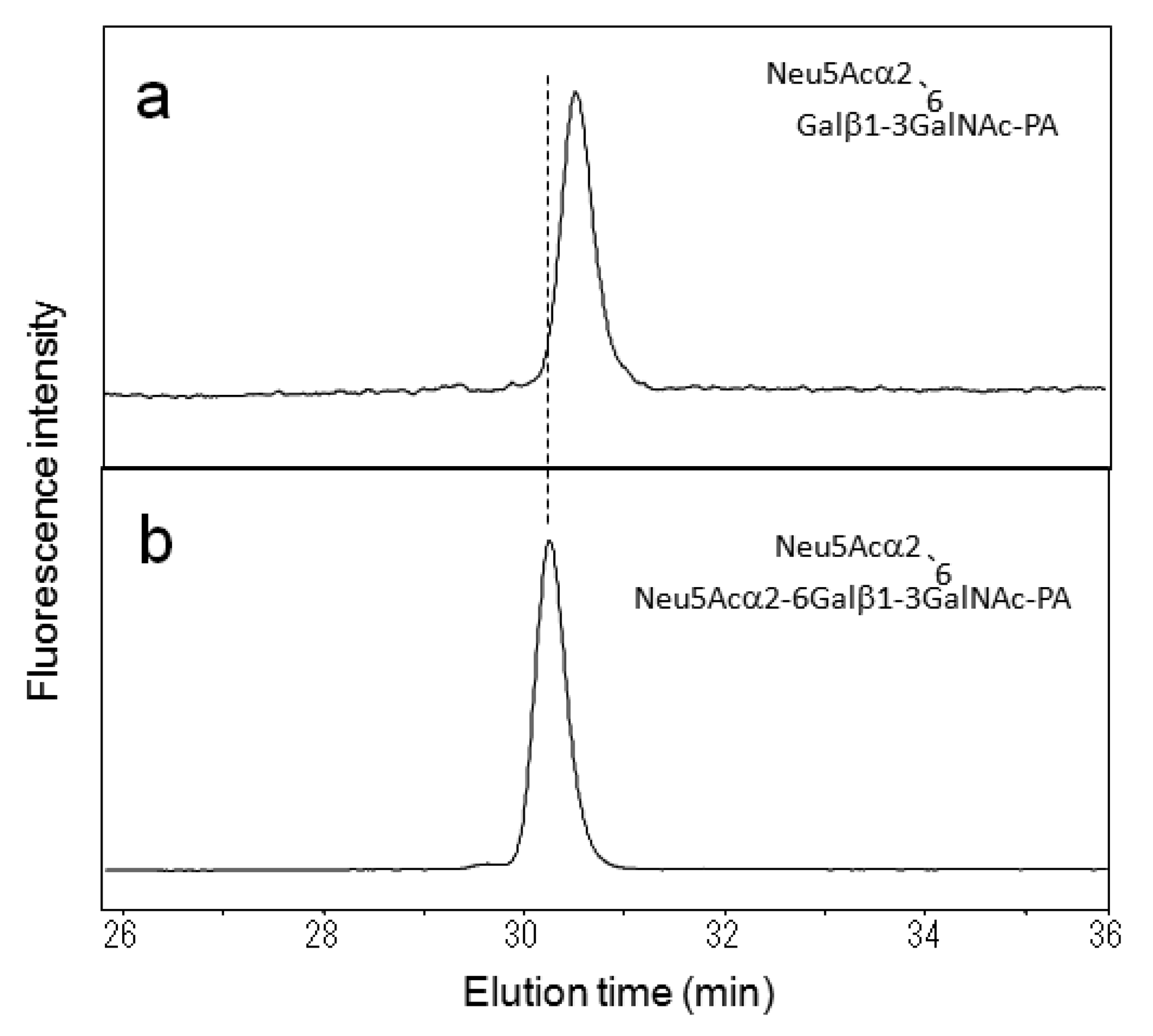
With similar methodology, we identified eight types of O-glycans derived from mucin and IgA glycoproteins and recorded their elution times (Table 2). Using the HPLC data as a guide, these O-glycans could be strategically collected from glycoproteins and further derivatized by glycosidase glycosyltransferase and sulfotransferase treatments in vitro, giving rise to a variety of standard PA-oligosaccharides. For example, the mono-sialyl PA-oligosaccharide Galβ1-3(Neu5Acα2-6)GalNAc-PA was treated with α2,6-silayltransferase, giving rise to di-sialyl PA-oligosaccharide Neu5Acα2-6Galβ1-3(Neu5Acα2-6)GalNAc-PA, which was eluted differently from the reaction precursor on the C30 column (Figure 2). Similarly, we collected the HPLC data of seven kinds of PA-O-glycans (Table 3). Finally, we made an HPLC map containing 16 neutral, seven sialylated, and four sulfated O-glycans (Figure 3).
| PA-Saccharides | GU (amide) | GU (C30) | Molecular mass (Da) a | Source |
|---|---|---|---|---|
| Fucα1-2Galβ1-3GalNAc-PA | 2.4 | 5.3 | 608 | Porcine stomach mucin |
| Galβ1-4GlcNAcβ1-4Galβ1-3(GlcNAcβ1-4Galβ1-4GlcNAcβ1-6)GalNAc-PA | 6.0 | 5.2 | 1395 | Porcine stomach mucin |
| GalNAcα1-3(Fucα1-2)Galβ1-3GalNAc-PA | 3.0 | 12.8 | 811 | Porcine stomach mucin |
| Galβ1-3GlcNAcβ1-6GalNAc-PA | 2.6 | 6.1 | 665 | Porcine stomach mucin |
| GalNAcα1-3Galβ1-3GalNAc-PA | 2.6 | 3.0 | 665 | Porcine stomach mucin |
| Galβ1-3(GlcNAcβ1-6)GalNAc-PA | 3.2 | 4.6 | 665 | Porcine stomach mucin |
| Neu5Acα2-3Galβ1-3GalNAc-PA | 2.1 | 2.6 | 753 | Colostrum IgA |
| Neu5Acα2-3Galβ1-3(Neu5Acα2-6)GalNAc-PA | 2.7 | 2.2 | 1044 | Colostrum IgA |
aAverage mass calculated from the m/z values of [M+H]+, [M+Na]+, and [M-H]– ions for PA-saccharides.

| PA-Saccharides | GU (amide) | GU (C30) | Molecular mass (Da) a |
|---|---|---|---|
| Neu5Acα2-3Galβ1-3(GlcNAcβ1-6)GalNAc-PA | 2.2 | 4.1 | 956 |
| Galβ1-3(Neu5Acα2-3Galβ1-4GlcNAcβ1-6)GalNAc-PA | 3.2 | 4.2 | 1118 |
| Neu5Acα2-6Galβ1-3(Neu5Acα2-6)GalNAc-PA | 2.7 | 2.3 | 1044 |
| HSO3-6GlcNAcβ1-6GalNAc-PA | 2.2 | 6.9 | 583 |
| Galβ1-4GlcNAcβ1-4Galβ1-3(HSO3-6GlcNAcβ1-4Galβ1-4GlcNAcβ1-6)GalNAc-PA | 5.6 | 4.9 | 1475 |
| GlcNAcβ1-3Galβ1-3(HSO3-6GlcNAcβ1-6)GalNAc-PA | 2.6 | 3.7 | 948 |
| Galβ1-3(HSO3-6GlcNAcβ1-6)GalNAc-PA | 1.5 | 5.2 | 745 |
aAverage mass calculated from the m/z values of [M+H]+, [M+Na]+, and [M−H]− ions for PA-saccharides.

The HPLC map thus established facilitates the quantitative O-glycosylation profiling with discriminating isomeric structures of O-glycans, which would be difficult to perform by MS-based approaches. The HPLC-based O-glycosylation profiling methods so far reported need much longer elution times (more than 2 h) or employ different elution conditions between neutral and acidic O-glycans [19,20]. Our developed HPLC map is able to deal with neutral and anionic O-glycans (including sulfated O-glycans whose HPLC data have not been reported previously) with the same protocol using a shorter elution time (within 1 h) and therefore would be advantageous in comparison to previously reported methods.
3.2. Branch Specificity of GlcNAc6ST-1
The HPLC map thus established will facilitate structural identification of sulfated O-glycans. To date, five types of sulfotransferases (termed GlcNAc6STs) have been reported to catalyze a sulfate group on the C6 position of GlcNAc residues [30]. Although spatio-temporal expression patterns of these enzymes have been extensively characterized, their reaction specificities are not fully understood. We herein applied the developed HPLC data to examination of the branch specificity of enzymatic sulfation catalyzed by human GlcNAc6ST-1, which was expressed by COS7 cells as a fusion protein with protein A [23].
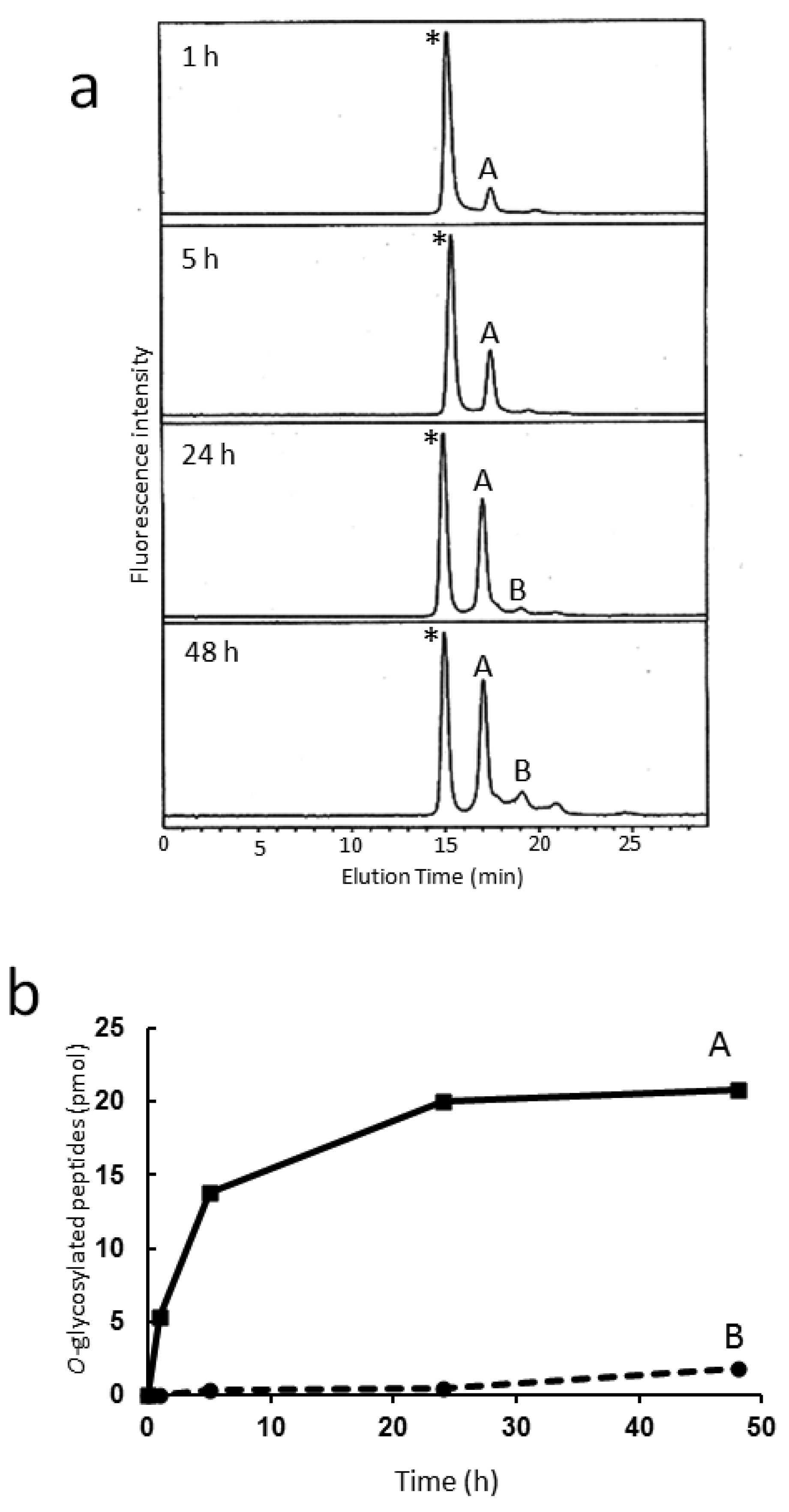
Figure 4a shows the time-dependent change of reverse-phase HPLC elution profiles for the reaction mixture of the in vitro sulfation catalyzed by this recombinant enzyme using a fluorescent glycopeptide, GlcNAcβ1-6(GlcNAcβ1-3Galβ1-3)GalNAcα1-peptide-FAM, as an acceptor. MALDI-TOF-MS indicated that two reaction products, A and B, were isomeric glycopeptides that possessed a single sulfate group. For unambiguous identification of the isomeric structures of sulfated O-glycans, fractions A and B were subjected to the developed HPLC mapping. As a result, the PA-glycans derived from fractions A and B were identified as GlcNAcβ1-6(HSO3-GlcNAcβ1-3Galβ1-3)GalNAc-PA and HSO3-GlcNAcβ1-6(GlcNAcβ1-3Galβ1-3)GalNAc-PA, respectively. This result clearly indicates that GlcNAc6ST-1 selectively catalyzed sulfation at the β1-6-linked GlcNAc residue in comparison with the remaining β1-3-linked GlcNAc residue, consistent with the previous report that preferential sulfation occurs with core 2 (GlcNAcβ1-6(Galβ1-3)GalNAc) rather than core 3 (GlcNAcβ1-3Galβ1-3GalNAc) as the acceptor [23]. Thus, our HPLC map is a useful tool for detailed characterization of substrate and reaction specificities of glycosyltransferases and sulfotransferases, leading to a better understanding of their detailed functional roles.
3.3. O-glycosylation Profiling of Serum IgA
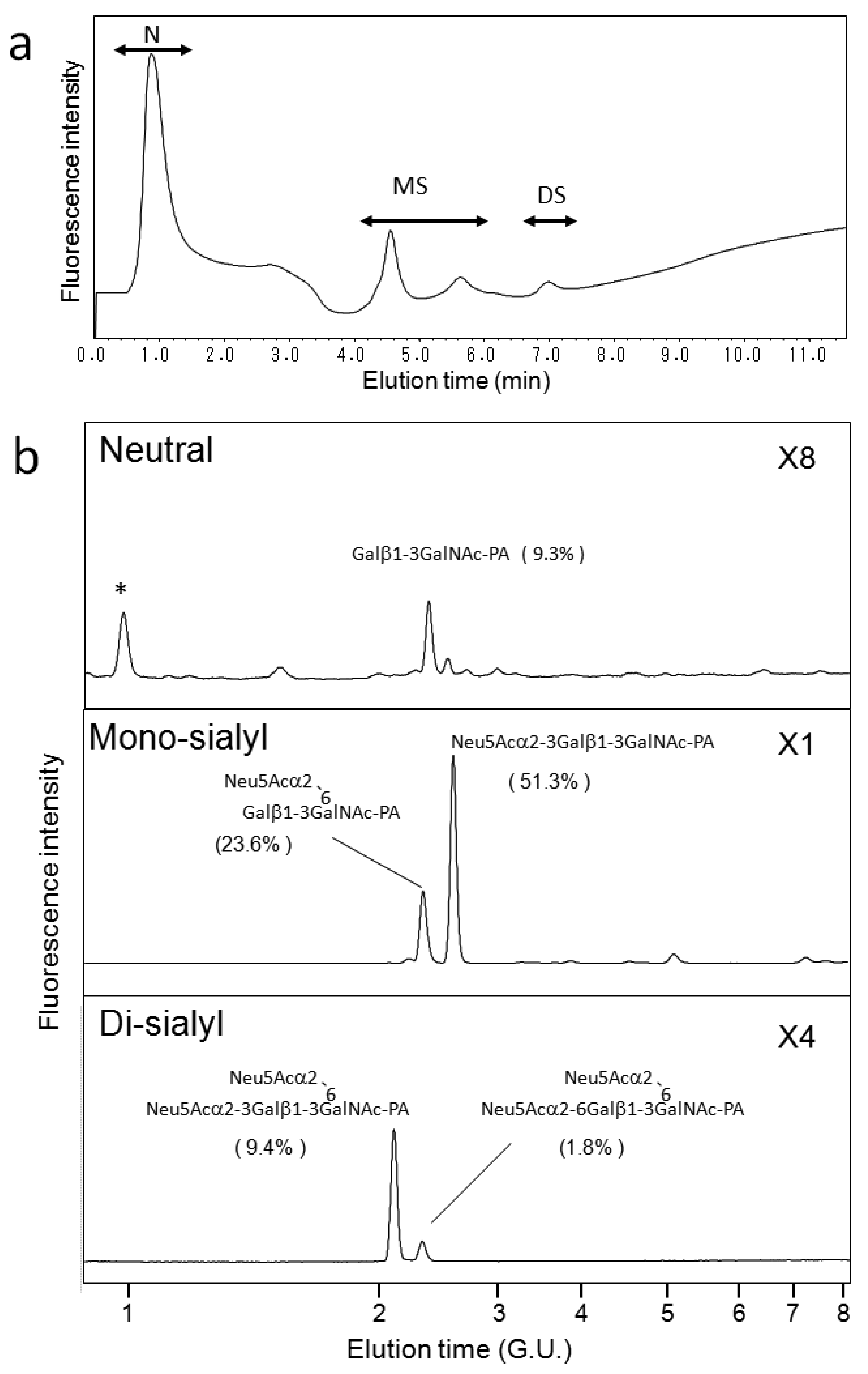
We also applied our HPLC map to O-glycosylation profiling of human serum IgA, which possesses nine O-glycosylation sites at the hinge region [31,32]. Galactose depletion of O-glycans at the IgA hinge has been observed in the serum of patients with IgA nephropathy [31,32]. Figure 5a shows a typical elution profile on a Mono-Q column of the PA-O-glycans derived from the IgA sample, which were separated according to the degrees of sialylation. Each fraction was further separated on a C30 column as shown in Figure 5b. Individual fractions separated by the C30 column were further separated on an amide-silica column. The PA-oligosaccharides were identified on the basis of coincidence of the elution data with those in the HPLC map established in the present study. The incidence of O-glycan structures derived from serum IgA is indicated in Figure 5b. To date, IgA O-glycosylation has been characterized by lectin blotting, mass spectroscopy, and chromatographic separation [21,31,32,33]. These studies have described O-glycan structures Galβ1-3GalNAc, Neu5Acα2-3Galβ1-3GalNAc, Galβ1-3(Neu5Acα2-6)GalNAc-PA, and Neu5Acα2-3Galβ1-3(Neu5Acα2-6)GalNAc. To the best of our knowledge, the present study is the first to identify the di-sialyl O-glycan Neu5Acα2-6Galβ1-3(Neu5Acα2-6)GalNAc in serum IgA. The HPLC map developed in the present study enables us to distinguish the isomeric structures of sialyl O-glycans, offering quantitative information for O-glycosylation profiling.

4. Conclusions
In the present study, we developed an HPLC mapping method for detailed structural identification of O-glycans in neutral, sialylated, and sulfated forms. Furthermore, using this method, we were able to quantitatively identify isomeric products from an in vitro reaction catalyzed by human GlcNAc6ST-1 and obtain O-glycosylation profiles of human serum IgA as a model glycoprotein. The HPLC map will provide a glycomics tool for unambiguous identification and quantitative profiling of O-glycans expressed on a variety of proteins of physiological and pathological interest.
Acknowledgments
This work was supported in part by Grants-in-Aid for Young Scientists B (H.Y.) and Scientific Research B (K.K.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), and the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NIBIO) (K.K.). We thank Kazuhiro Ikenaka (National Institute for Physiological Sciences) for his helpful remarks and suggestions about the purification of PA-O-glycans. We also thank Yoshiki Yamaguchi (RIKEN) for his support in the MS/MS analyses. We also appreciate Uchimura (Research Institute, National Center for Geriatrics and Gerontology) and Kannangi (Aichi Medical University) for the gift of the expression vector of GlcNAc6ST-1 and for the useful discussion. Glycosylation profiling of human serum IgA has been approved by the ethics committee of Nagoya City University.
References
- Varki, A. Biological roles of oligosaccharides: All of the theories are correct. Glycobiology 1993, 3, 97–130. [Google Scholar]
- Sharon, N. Lectins: Carbohydrate-specific reagents and biological recognition molecules. J. Biol. Chem. 2007, 282, 2753–2764. [Google Scholar]
- Fadda, E.; Woods, R.J. Molecular simulations of carbohydrates and protein-carbohydrate interactions: motivation, issues and prospects. Drug Discov. Today 2010, 15, 596–609. [Google Scholar]
- Kawashima, H.; Petryniak, B.; Hiraoka, N.; Mitoma, J.; Huckaby, V.; Nakayama, J.; Uchimura, K.; Kadomatsu, K.; Muramatsu, T.; Lowe, J.B.; Fukuda, M. N-acetylglucosamine-6-O-sulfotransferases 1 and 2 cooperatively control lymphocyte homing through L-selectin ligand biosynthesis in high endothelial venules. Nat. Immunol. 2005, 6, 1096–1104. [Google Scholar]
- Mizushima, T.; Yagi, H.; Takemoto, E.; Shibata-Koyama, M.; Isoda, Y.; Iida, S.; Masuda, K.; Satoh, M.; Kato, K. Structural basis for improved efficacy of therapeutic antibodies upon defucosylation of their Fc glycans. Genes Cells 2011, 16, 1071–1080. [Google Scholar]
- Aoki, K.; Perlman, M.; Lim, J.M.; Cantu, R.; Wells, L.; Tiemeyer, M. Dynamic developmental elaboration of N-linked glycan complexity in the Drosophila melanogaster embryo. J. Biol. Chem. 2007, 282, 9127–9142. [Google Scholar]
- Cipollo, J.F.; Awad, A.M.; Costello, C.E.; Hirschberg, C.B. N-Glycans of Caenorhabditis elegans are specific to developmental stages. J. Biol. Chem. 2005, 280, 26063–26072. [Google Scholar]
- Guérardel, Y.; Chang, L.Y.; Maes, E.; Huang, C.J.; Khoo, K.H. Glycomic survey mapping of zebrafish identifies unique sialylation pattern. Glycobiology 2006, 16, 244–257. [Google Scholar]
- Takemoto, T.; Natsuka, S.; Nakakita, S.; Hase, S. Expression of complex-type N-glycans in developmental periods of zebrafish embryo. Glycoconj. J. 2005, 22, 21–26. [Google Scholar]
- Yagi, H.; Nakagawa, M.; Takahashi, N.; Kondo, S.; Matsubara, M.; Kato, K. Neural complex-specific expression of xylosyl N-glycan in Ciona intestinalis. Glycobiology 2008, 18, 145–151. [Google Scholar]
- Marino, K.; Bones, J.; Kattla, J.J.; Rudd, P.M. A systematic approach to protein glycosylation analysis: a path through the maze. Nat. Chem. Biol. 2010, 6, 713–723. [Google Scholar]
- Wada, Y.; Azadi, P.; Costello, C.E.; Dell, A.; Dwek, R.A.; Geyer, H.; Geyer, R.; Kakehi, K.; Karlsson, N.G.; Kato, K.; et al. Comparison of the methods for profiling glycoprotein glycans-HUPO Human Disease Glycomics/Proteome Initiative multi-institutional study. Glycobiology 2007, 17, 411–422. [Google Scholar]
- Takahashi, N.; Nakagawa, H.; Fujikawa, K.; Kawamura, Y.; Tomiya, N. Three-dimensional elution mapping of pyridylaminated N-linked neutral and sialyl oligosaccharides. Anal. Biochem. 1995, 226, 139–146. [Google Scholar]
- Takahashi, N.; Yagi, H.; Kato, K. The Two/Three Dimensional HPLC Mapping Method for the Identification of N-glycan Structures. In Comprehensive Glycoscience; Kamerling, J.P., Ed.; Elsevier: Amsterdam, the Netherlands, 2007; Volume 2, pp. 283–300. [Google Scholar]
- Takahashi, N.; Kato, K. GALAXY(Glycoanalysis by the three axes of MS and chromatography): A web application that assists structural analyses of N-glycans. Trends Glycosci. Glycotechnol. 2003, 15, 235–251. [Google Scholar]
- Yagi, H.; Kato, K. Multidimensional HPLC mapping method for the structural analysis of anionic N-glycans. Trends Glycosci. Glycotechnol. 2009, 21, 95–104. [Google Scholar]
- Yagi, H.; Takahashi, N.; Yamaguchi, Y.; Kimura, N.; Uchimura, K.; Kannagi, R.; Kato, K. Development of structural analysis of sulfated N-glycans by multi-dimensional HPLC mapping methods. Glycobiology 2005, 15, 1051–1060. [Google Scholar]
- Yagi, H.; Yamada, K.; Ohono, E.; Utsumi, M.; Yamaguchi, Y.; Kurimoto, E.; Takahashi, N.; Oka, S.; Kawasaki, T.; Kato, K. Development and application of high performance liquid chromatography map of glucuronyl N-glycans. Open Glycosci. 2008, 1, 8–18. [Google Scholar]
- Kuraya, N.; Hase, S. Analysis of pyridylaminated O-linked sugar chains by two-dimensional sugar mapping. Anal. Biochem. 1996, 233, 205–211. [Google Scholar]
- Royle, L.; Mattu, T.S.; Hart, E.; Langridge, J.I.; Merry, A.H.; Murphy, N.; Harvey, D.J.; Dwek, R.A.; Rudd, P.M. An analytical and structural database provides a strategy for sequencing O-glycans from microgram quantities of glycoproteins. Anal. Biochem. 2002, 304, 70–90. [Google Scholar]
- Wada, Y.; Dell, A.; Haslam, S.M.; Tissot, B.; Canis, K.; Azadi, P.; Backstrom, M.; Costello, C.E.; Hansson, G.C.; Hiki, Y.; et al. Comparison of methods for profiling O-glycosylation: Human proteome organisation human disease glycomics/proteome initiative multi-institutional study of IgA1. Mol. Cell Proteomics 2010, 9, 719–727. [Google Scholar]
- Hiki, Y.; Iwase, H.; Kokubo, T.; Horii, A.; Tanaka, A.; Nishikido, J.; Hotta, K.; Kobayashi, Y. Association of asialo-galactosyl β1 -3N-acetylgalactosamine on the hinge with a conformational instability of Jacalin-reactive immunoglobulin A1 in immunoglobulin A nephropathy. J. Am. Soc. Nephrol. 1996, 7, 955–960. [Google Scholar]
- Uchimura, K.; El-Fasakhany, F.M.; Hori, M.; Hemmerich, S.; Blink, S.E.; Kansas, G.S.; Kanamori, A.; Kumamoto, K.; Kannagi, R.; Muramatsu, T. Specificities of N-acetylglucosamine-6-O-sulfotransferases in relation to L-selectin ligand synthesis and tumor-associated enzyme expression. J. Biol. Chem. 2002, 277, 3979–3984. [Google Scholar]
- Patel, T.; Bruce, J.; Merry, A.; Bigge, C.; Wormald, M.; Jaques, A.; Parekh, R. Use of hydrazine to release in intact and unreduced form both N- and O-linked oligosaccharides from glycoproteins. Biochemistry 1993, 32, 679–693. [Google Scholar]
- Tanabe, K.; Ikenaka, K. In-column removal of hydrazine and N-acetylation of oligosaccharides released by hydrazionolysis. Anal. Biochem. 2006, 348, 324–326. [Google Scholar]
- Yamamoto, S.; Hase, S.; Fukuda, S.; Sano, O.; Ikenaka, T. Structures of the sugar chains of interferon-gamma produced by human myelomonocyte cell line HBL-38. J. Biochem. 1989, 105, 547–555. [Google Scholar]
- Nakagawa, H.; Kawamura, Y.; Kato, K.; Shimada, I.; Arata, Y.; Takahashi, N. Identification of neutral and sialyl N-linked oligosaccharide structures from human serum glycoproteins using three kinds of high-performance liquid chromatography. Anal. Biochem. 1995, 226, 130–138. [Google Scholar]
- Takahashi, N.; Lee, K.B.; Nakagawa, H.; Tsukamoto, Y.; Kawamura, Y.; Li, Y.T.; Lee, Y.C. Enzymatic sialylation of N-linked oligosaccharides using an α-(2,3)-specific trans-sialidase from Trypanosoma cruzi: Structural identification using a three-dimensional elution mapping technique. Anal. Biochem. 1995, 230, 333–342. [Google Scholar]
- Yagi, H.; Yasukawa, N.; Yu, S.Y.; Guo, C.T.; Takahashi, N.; Takahashi, T.; Bukawa, W.; Suzuki, T.; Khoo, K.H.; Suzuki, Y.; et al. The expression of sialylated high-antennary N-glycans in edible bird's nest. Carbohydr. Res. 2008, 343, 1373–1377. [Google Scholar]
- Taniguchi, N.; Honke, K.; Fukuda, M. Handbook of Glycosyltransferases and Related Genes, 1st ed.; Springer: Tokyo, Japan, 2002. [Google Scholar]
- Barratt, J.; Smith, A.C.; Feehally, J. The pathogenic role of IgA1 O-linked glycosylation in the pathogenesis of IgA nephropathy. Nephrology 2007, 12, 275–284. [Google Scholar]
- Novak, J.; Julian, B.A.; Tomana, M.; Mestecky, J. IgA glycosylation and IgA immune complexes in the pathogenesis of IgA nephropathy. Semin. Nephrol. 2008, 28, 78–87. [Google Scholar]
- Mattu, T.S.; Pleass, R.J.; Willis, A.C.; Kilian, M.; Wormald, M.R.; Lellouch, A.C.; Rudd, P.M.; Woof, J.M.; Dwek, R.A. The glycosylation and structure of human serum IgA1, Fab, and Fc regions and the role of N-glycosylation on Fc alpha receptor interactions. J. Biol. Chem. 1998, 273, 2260–2272. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yagi, H.; Ohno, E.; Kondo, S.; Yoshida, A.; Kato, K. Development and Application of Multidimensional HPLC Mapping Method for O-linked Oligosaccharides. Biomolecules 2011, 1, 48-62. https://doi.org/10.3390/biom1010048
Yagi H, Ohno E, Kondo S, Yoshida A, Kato K. Development and Application of Multidimensional HPLC Mapping Method for O-linked Oligosaccharides. Biomolecules. 2011; 1(1):48-62. https://doi.org/10.3390/biom1010048
Chicago/Turabian StyleYagi, Hirokazu, Erina Ohno, Sachiko Kondo, Atsuhiro Yoshida, and Koichi Kato. 2011. "Development and Application of Multidimensional HPLC Mapping Method for O-linked Oligosaccharides" Biomolecules 1, no. 1: 48-62. https://doi.org/10.3390/biom1010048