
Role of Myofibrillar Protein Catabolism in Development of Glucocorticoid Myopathy: Aging and Functional Activity Aspects

Abstract
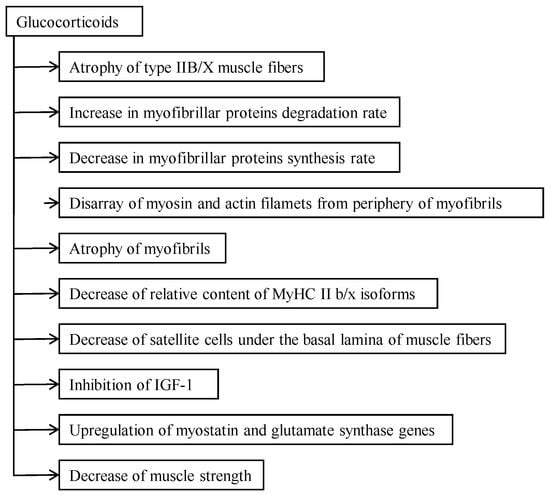
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Catabolic Action of Glucocorticoids
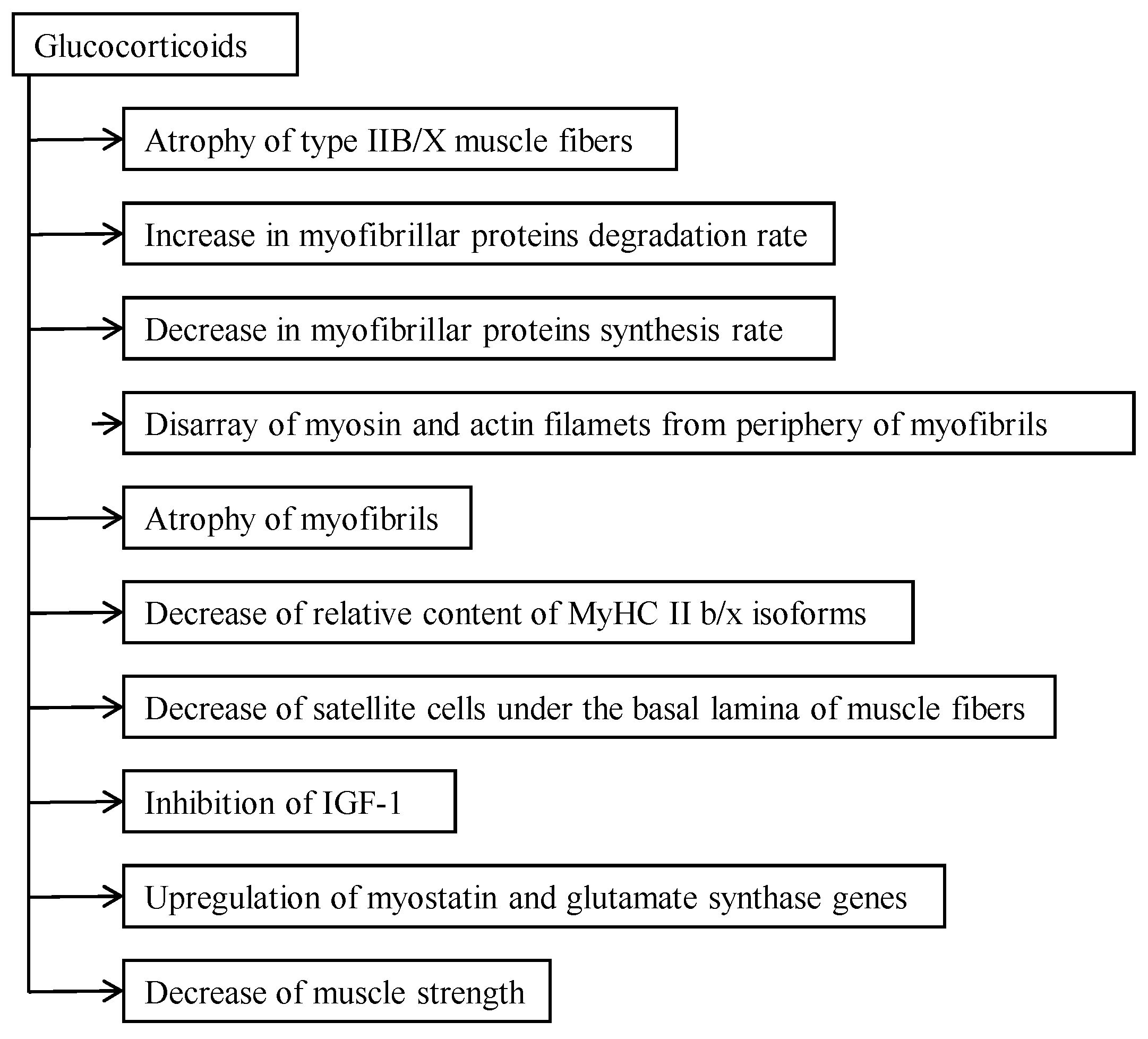
2.1. Relationship between Catabolic Action and Functional Activity of Muscle
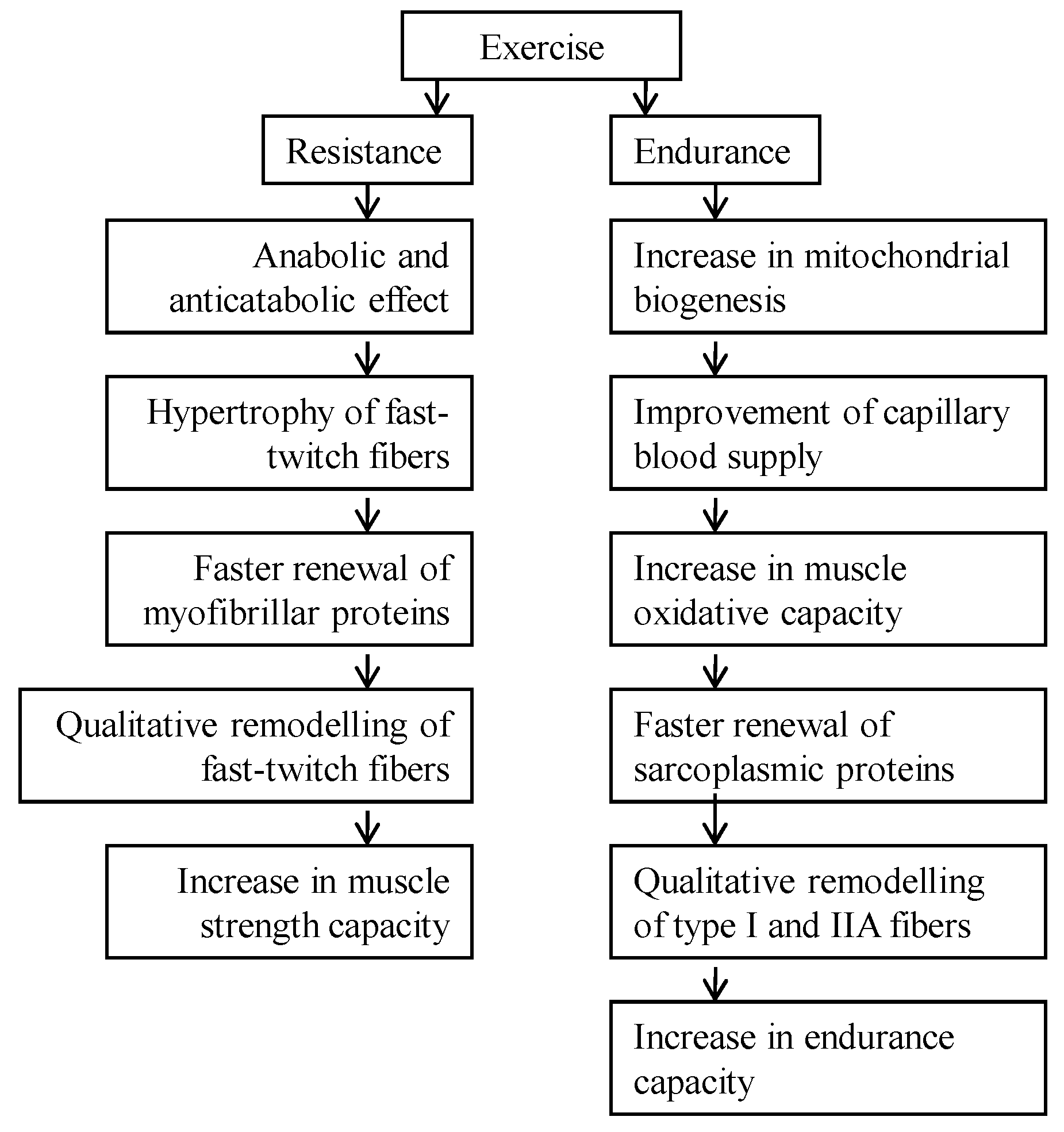
2.2. Aging Aspect of Catabolic Action of Glucocorticoids
3. Catabolic Action of Glucocorticoids in the Myofibrillar Compartment
4. Effect of Glucocorticoids on Muscle Strength and Motor Activity
4.1. Glucocorticoid-Caused Myopathy, Sarcopenia, and Cachexia
4.2. How to Prevent or Decelerate Skeletal Muscle Atrophy and Disability
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Seene, T. Turnover of skeletal muscle contractile proteins in glucocorticoid myopathy. J. Steroid Biochem. Mol. Biol. 1994, 50, 1–4. [Google Scholar] [CrossRef]
- Seene, T.; Umnova, M.; Kaasik, P. The exercise myopathy. In Overload, Performance Incompentence, and Regeneration in Sport; Lehmann, M., Foster, C., Gastmann, U., Keizer, H., Steinacker, J., Eds.; Kluwer Academic/Plenum Publishers: New York, NY, USA; Boston, MA, USA; Dordrecht, The Netherlands; London, UK; Moscow, Russia, 1999; pp. 119–130. [Google Scholar]
- Askari, A.; Vignos, P.J., Jr.; Moskowitz, R. Steroid myopathy in connective tissue disease. Am. J. Med. 1976, 61, 485–492. [Google Scholar] [CrossRef]
- Goldberg, A. Protein turnover in skeletal muscle in experimental corticosteroid myopathy. J. Biol. Chem. 1969, 244, 3217–3222. [Google Scholar] [PubMed]
- Tomas, F.; Munro, H.; Young, V. Effect of glucocorticoid administration on the rate of muscle protein breakdown in vivo in rats, as measured by urinary excretion on N-methylhistidine. Biochem. J. 1979, 178, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Kaasik, P.; Umnova, M.; Alev, K.; Selart, A.; Seene, T. Fine architectonics and protein turnover rate in myofibrils of glucocorticoid caused myopathic rats. J. Interdiscip. Histopathol. 2012, 1, 5–10. [Google Scholar] [CrossRef]
- Seene, T.; Viru, A. The catabolic effect of glucocorticoids on different types of skeletal muscle fibres and its dependence upon muscle activity and interaction with anabolic steroids. J. Steroid Biochem. 1982, 16, 349–352. [Google Scholar] [CrossRef]
- Tomas, F. Effect of corticosterone on myofibrillar protein turnover in diabetic rat as assessed by N-methylhistidine excretion. Biochem. J. 1982, 208, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Seene, T.; Alev, K. Effect of glucocorticoids on the turnover rate of actin and myosin heavy chains on different types of skeletal muscle fibres. J. Steroid Biochem. 1985, 22, 767–771. [Google Scholar] [CrossRef]
- Seene, T.; Umnova, M.; Alev, K.; Pehme, A. Effect of glucocorticoids on contractile apparatus of rat skeletal muscle. J. Steroid Biochem. 1988, 29, 313–317. [Google Scholar] [CrossRef]
- Seene, T.; Kaasik, P.; Pehme, A.; Alev, K.; Riso, E.M. The effect of glucocorticoids on the myosin heavy chain isoforms’ turnover in skeletal muscle. J. Steroid Biochem. Mol. Biol. 2003, 86, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Aru, M.; Alev, K.; Gapeyeva, H.; Vain, A.; Puhke, R.; Pehme, A.; Kaasik, P.; Selart, A.; Seene, T. Glucocorticoid-induced alterations in titin, nebulin, myosin heavy chain isoform content and viscosoelastic properties of rat skeletal muscle. Adv. Biol. Chem. 2013, 3, 70–75. [Google Scholar] [CrossRef]
- Holloszy, J.; Chen, M.; Cartee, G.; Young, J. Skeletal muscle atrophy in old rats: Defferential changes in three fiber types. Mech. Ageing Dev. 1991, 60, 199–213. [Google Scholar] [CrossRef]
- Attaix, D.; Mosoni, L.; Dardevet, D.; Combaret, L.; Patureau Mirand, P.; Grizard, J. Altered responses in skeletal muscle protein turnover during aging in anabolic and catabolic periods. Int. J. Biochem. Cell Biol. 2005, 37, 1962–1973. [Google Scholar] [CrossRef] [PubMed]
- Kaasik, P.; Umnova, M.; Pehme, A.; Alev, K.; Aru, M.; Selart, A.; Seene, T. Ageing and dexamethasone associated sarcopenia: Peculiarities of regeneration. J. Steroid Biochem. Mol. Biol. 2007, 105, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Kaasik, P.; Aru, M.; Alev, K.; Seene, T. Aging and regenerative capacity of skeletal muscle in rats. Curr. Aging Sci. 2012, 5, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Hickson, R.; Davis, J. Partial prevention of glucocorticoid induced muscle atrophy by endurance training. Am. J. Physiol. 1981, 241, E226–E232. [Google Scholar] [PubMed]
- Seene, T.; Kaasik, P. Role of exercise therapy in prevention of decline in aging muscle function: Glucocorticoid myopathy and unloading. J. Aging Res. 2012, 2012, 172492. [Google Scholar] [CrossRef] [PubMed]
- Smals, A.; Kloppenborg, P.; Benroad, T. Plasma testosterone profiles in cushing’s syndrome. J. Clin. Endocrinol. Metab. 1977, 45, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Kelly, F.; McGrath, J.; Goldspink, D.; Gullen, M. A morphological (biochemical) study on the action of corticosteroids on rat skeletal muscle. Muscle Nerve 1986, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Tada, O.; Higuchi, K.; Ohtsuka, A. Effects of cortiosterone on connection content and protein breakdown in rat skeletal muscle. Biosci. Biotechnol. Biochem. 2000, 64, 2686–2688. [Google Scholar] [CrossRef]
- Czerwinski, S.; Kurowski, T.; O’Neill, T.; Hickson, R. Initiating regular exercise protects against muscle atrophy from glucocorticoids. J. Appl. Physiol. 1987, 63, 1504–1510. [Google Scholar] [PubMed]
- Singleton, J.; Baker, B.; Thorburn, A. Dexamethasone inhibits insulin-like growth factor signaling and potentiates myoblast apoptosis. Endocrinology 2000, 14, 2945–2950. [Google Scholar] [CrossRef] [PubMed]
- Carballo-Jane, E.; Pandit, S.; Santoro, S.; Freund, C.; Luell, S.; Harris, G.; Forrest, M.; Sitlani, A. Skeletal muscle: a dual system to measure glucocorticoid—Dependent transactivation and transrepression of gene regulation. J. Steroid Biochem. Mol. Biol. 2004, 88, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Flack, K.; Davy, K.; Hulver, M.; Winett, R.; Friasrd, M.; Davy, B. Aging, resistance training, and diabetes prevention. J. Aging Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.; Tang, J.; Burd, N.; Rerecich, T.; Tarnopolsky, M.; Phillips, S. Differential stimulation of myofibrillar and sarcoplasmic protein synthesis with protein ingestion at rest and after resistance exercise. J. Physiol. 2009, 587, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.; Atherton, P.; Rennie, M.; Tarnopolsky, M.; Phillips, S. Resistance exercise enhances mTOR and MAPK signalling in human muscle over that seen at rest after bolus protein ingestion. Acta Physiol. (Oxf.) 2011, 201, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Fell, J.; Williams, D. The effect of aging on skeletal-muscle recovery from exercise: Possible implications for aging athletes. J. Aging Phys. Act. 2008, 16, 97–115. [Google Scholar] [PubMed]
- Brooks, S.; Faulkner, J. Contractile properties of skeletal muscles from young, adult, and aged mice. J. Physiol. 1988, 404, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, A.M.; Fidzianska, A.; Sculze, G.; Coper, H.; Ossowska, K.; Wolfart, S.; Hausmanova-Petrusewicz, I. Ultrastructural changes in the skeletal muscle of senile rats with significant age-dependent motor deficit. Basic Appl. Myol. 1998, 8, 185–190. [Google Scholar]
- Grounds, M. Age-associated changes in the response of skeletal muscle cells to exercise and regeneration. Ann. N. Y. Acad. Sci. 1998, 854, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.; Detkov, E.; Borisov, A.; Faulkner, J. Skeletal muscle regeneration in very old rats. J. Gerontol. A Biol. Sci. Med. Sci. 2001, 56, B224–B233. [Google Scholar] [CrossRef] [PubMed]
- Dardevet, D.; Sornet, C.; Taillandier, D.; Savary, I.; Attaix, D.; Grizard, J. Sensitivity and protein turnover response to glucocorticoids are different in skeletal muscle from adult and old rats. Lack of regulation of the ubiquitin-proteasome proteolytic pathway in ageing. J. Clin. Investig. 1995, 96, 2113–2119. [Google Scholar] [PubMed]
- Dardevet, D.; Sornet, C.; Savary, I.; Debras, E.; Patureau Mirand, P.; Grizard, J. Glucocorticoid effects on insulin and IGF-1-regulated muscle protein metabolism during aging. J. Endocrinol. 1998, 156, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Barani, A.; Durieux, A.; Sabido, O.; Freyssenet, D. Age-related changes in the mitotic and metabolic characteristics of muscle-derived cells. J. Appl. Physiol. 2003, 95, 2089–2098. [Google Scholar] [CrossRef] [PubMed]
- Machida, S.; Booth, F. Increased nuclear proteins in muscle satellite cells in aged animals as compared to young growing animals. Exp. Gerontol. 2004, 39, 1521–1525. [Google Scholar] [CrossRef] [PubMed]
- Hasty, P.; Campisi, J.; Hoeijmakers, J.; Van Steeg, H.; Vijg, J. Aging and genome maintenance: Lessons from the mouse? Science 2003, 299, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Ames, B. Alpha lipoic acid’s role in delaying the mitochondrial decay of aging. Ann. N. Y. Acad. Sci. 2004, 1019, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Seene, T.; Kaasik, P.; Riso, E.M. Review on aging, unloading and reloading: Changes in skeletal muscle quantity and quality. Arch. Gerontol. Geriatr. 2012, 54, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Seene, T.; Alev, K.; Kaasik, P.; Pehme, A. Changes in fast-twitch muscle oxidative capacity and myosin isoforms modulation during endurance training. J. Sports Med. Phys. Fitness 2007, 47, 124–132. [Google Scholar] [PubMed]
- Seene, T.; Kaasik, P.; Umnova, M. Structural rearrangements in contractile apparatus and resulting skeletal muscle remodelling: Effect of exercise training. J. Sports Med. Phys. Fitness 2009, 49, 410–423. [Google Scholar] [PubMed]
- Fernandez-Rodriques, E.; Stewart, P.; Cooper, M. The pituitary-adrenal axis and body composition. Pituitary 2009, 12, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Shultz, E.; Darr, K. The role of satellite cells in adaptive or induced fiber transformations. In The Dynamics State of Muscle Fibers; Pette, D., Ed.; Walter de Gruyter: Berlin, Germany, 1990; pp. 667–681. [Google Scholar]
- Järva, J.; Alev, K.; Seene, T. Myosin heavy chain composition in regulating skeletal muscle grafts. Basic Appl. Myol. 1997, 7, 137–141. [Google Scholar]
- Sandri, M. Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 2008, 23, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, N.; Yoshikawa, N.; Ito, N.; Maruyama, T.; Suzuki, Y.; Takeda, S.; Nakae, J.; Tagata, Y.; Nishitani, S.; Takehana, K.; et al. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab. 2011, 13, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, K.; Yamaguchi, A. Sarcopenia and cachexia: The adaptations of negative regulators of skeletal muscle mass. J. Cachexia Sarcopenia Muscle 2012, 3, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Evans, W.J.; Anker, S.D. Myopenia—A new unviversal term for muscle wasting. J. Cachexia Sarcopenia Muscle 2011, 2, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Von Haehling, S.; Anker, S.D. Treatment of cachexia: An overview of recent developments. J. Am. Med. Dir. Assoc. 2014, 15, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Coats, A.J.; Morley, J.E.; Rosano, G.; Bernabei, R.; von Haehling, S.; Kalantar-Zadeh, K. Muscle wasting disease: A proposal for a new disease classification. J. Cachexia Sarcopenia Muscle 2014, 5, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.C. Cancer cachexia and fat—Muscle physiology. N. Engl. J. Med. 2011, 365, 565–567. [Google Scholar] [CrossRef] [PubMed]
- Reznick, R.; Zong, H.; Li, J.; Morino, K.; Moore, I.; Yu, H.; Liu, Z.X.; Dong, J.; Mustard, K.; Hawley, S.; et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007, 5, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Winder, W.; Hardie, D. Inactivation of acetyl-CoA carboxylase and activation of AMP—Activated protein kinase in muscle during exercis. Am. J. Physiol. 1996, 270, E299–E304. [Google Scholar] [PubMed]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seene, T.; Kaasik, P. Role of Myofibrillar Protein Catabolism in Development of Glucocorticoid Myopathy: Aging and Functional Activity Aspects. Metabolites 2016, 6, 15. https://doi.org/10.3390/metabo6020015
Seene T, Kaasik P. Role of Myofibrillar Protein Catabolism in Development of Glucocorticoid Myopathy: Aging and Functional Activity Aspects. Metabolites. 2016; 6(2):15. https://doi.org/10.3390/metabo6020015
Chicago/Turabian StyleSeene, Teet, and Priit Kaasik. 2016. "Role of Myofibrillar Protein Catabolism in Development of Glucocorticoid Myopathy: Aging and Functional Activity Aspects" Metabolites 6, no. 2: 15. https://doi.org/10.3390/metabo6020015
APA StyleSeene, T., & Kaasik, P. (2016). Role of Myofibrillar Protein Catabolism in Development of Glucocorticoid Myopathy: Aging and Functional Activity Aspects. Metabolites, 6(2), 15. https://doi.org/10.3390/metabo6020015