Coordinating Metabolite Changes with Our Perception of Plant Abiotic Stress Responses: Emerging Views Revealed by Integrative—Omic Analyses

Abstract
:
1. Introduction
2. Transcriptional Regulation of Metabolic Networks
3. Metabolic Regulation of Transcriptional Networks
3.1. Metabolic DNA and RNA Structural Modifiers
3.2. Retrograde Signaling
3.3. Sugar Signaling

{kind=link}
| Abiotic stress | Soluble sugar | Sugar response | Gene expression | Gene | Gene model | Enzyme activity | Enzyme | Reference |
|---|---|---|---|---|---|---|---|---|
| Cold | gluc, fruc | increase | increase | FXK3 | At5g51830 | nd | nd | [88] |
| GOLS3 | At1g09350 | |||||||
| HXK2 | At2g19860 | |||||||
| RS5 | At5g40390 | |||||||
| SPS1 | At5g11110 | |||||||
| SPS1F | At5g20280 | |||||||
| decrease | SSR | At1g79440 | ||||||
| Nitrogen (N) deficiency | ∑sugar (suc + gluc + fruc) | increase | increase | TPP B | At1g78090 | increase | GlucoK | [89] [90] |
| PGM | At1g78050 | FUM | ||||||
| A/N-Inv D | At1g22650 | |||||||
| G6PDH | At1g24280 | |||||||
| PGDH | At1g64190 | |||||||
| ABI4 | At2g40220 | |||||||
| decrease | TPS3 | At1g17000 | decrease | AGPase | ||||
| CWINV4 | At2g36190 | NADP-GAPDH | ||||||
| NADP-IDH | ||||||||
| NR | ||||||||
| GS | ||||||||
| AspAT | ||||||||
| Potassium (K) deficiency | suc, gluc, fruc | increase | − | nd | nd | increase | FrucK | [91] |
| NADP-ME | ||||||||
| GS | ||||||||
| GOGAT | ||||||||
| decrease | NR | |||||||
| Phosphate (P) deficiency | suc, gluc, fruc | increase | increase | PPCK1 | At1g08650 | increase | cFBPase | [90] |
| PPCK2 | At3g04530 | SPS | ||||||
| BAM5 | At4g15210 | PEPCase | ||||||
| GWD3 | At4g24450 | GS | ||||||
| GBS1 | At1g32900 | GDH | ||||||
| GPT2 | At1g61800 | ShiDH | ||||||
| ADG2 | At5g19220 | |||||||
| APL3 | At4g39210 | |||||||
| SPS4 | At4g10120 | |||||||
| SPP1 | At1g51420 | |||||||
| SUS3 | At3g43190 | |||||||
| decrease | FLN1 | At3g54090 | decrease | PFP | ||||
| FLN2 | At1g69200 | |||||||
| Carbon availability | gluc | Increase (exogenous feeding) | increase | ABI4 | At2g40220 | nd | nd | [92] |
| ABI5 | At2g36270 | |||||||
| CTR1 | At5g03730 | |||||||
| Combined heat and drought | suc, tre, gluc, fruc | increase | decrease | AMY1 | At4g25000 | nd | nd | [93] |
| BAM5 | At4g15210 | |||||||
| C/VIF1 | At1g47960 | |||||||
| FLN1 | At3g54090 | |||||||
| G6PDH6 | At5g40760 | |||||||
| HXK2 | At2g19860 | |||||||
| PSL5 | At5g63840 | |||||||
| SPS1F | At5g20830 | |||||||
| SPS1 | At5g20280 | |||||||
| Heat | suc, gluc, fruc | increase | increase | NDB1 | At4g28220 | nd | nd | [93] |
| VHA-A | At1g78900 | |||||||
| GOLS1 | At2g47180 | |||||||
| A/N-INVC | At3g06500 | |||||||
| Drought | suc, gluc, fruc | increase | increase | APL3 | At4g39210 | nd | nd | [93] |
| SPS1 | At5g11110 | |||||||
| SUS1 | At5g20830 | |||||||
| G6PDH6 | At5g40760 | |||||||
| Drought | suc | increase | increase | CWINV1 | At3g13790 | nd | nd | [94,95] |
| CINV1 | At1g35580 | |||||||
| GDH1 | At5g18170 | |||||||
| GDH2 | At5g07440 | |||||||
| VAC-INV | At1g12240 | |||||||
| decrease | ADK1 | At3g09820 | ||||||
| GDH3 | At3g03910 | |||||||
| Anoxia | gluc, fruc | increase | increase | ADH1 | At1g77120 | nd | nd | [16] |
| SUS4 | At3g43190 | |||||||
| Salinity | suc, gluc | increase | increase | PYD4 | At3g08860 | nd | nd | [96] |
| BAM1 | At4g15210 | |||||||
| GPAT5 | At3g11430 | |||||||
| decrease | PDF1.2b | At2g26020 | ||||||
| PDF1.2 | At5g44420 |
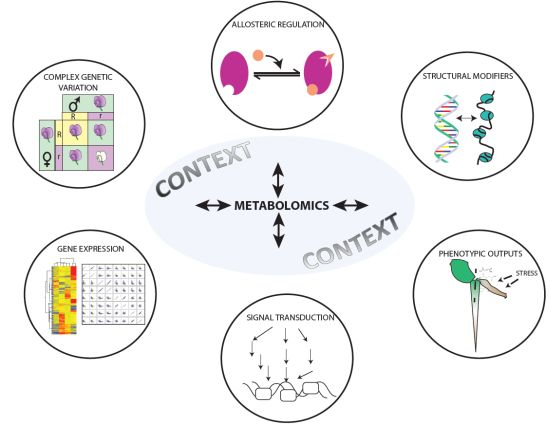
4. Integration with the Other—Omics Platforms: Phenomics, Genomics, Proteomics, and Metabolomics
5. Conclusions
Acknowledgements
Conflicts of Interest
References
- Bassel, G.W.; Lan, H.; Glaab, E.; Gibbs, D.J.; Gerjets, T.; Krasnogor, N.; Bonner, A.J.; Holdsworth, M.J.; Provart, N.J. Genome-wide network model capturing seed germination reveals coordinated regulation of plant cellular phase transitions. Proc. Natl. Acad. Sci. USA 2011, 108, 9709–9714. [Google Scholar] [CrossRef]
- Skirycz, A.; Vandenbroucke, K.; Clauw, P.; Maleux, K.; De Meyer, B.; Dhondt, S.; Pucci, A.; Gonzalez, N.; Hoeberichts, F.; Tognetti, V.B.; et al. Survival and growth of arabidopsis plants given limited water are not equal. Nat. Biotechnol. 2011, 29, 212–214. [Google Scholar] [CrossRef]
- Claeys, H.; Skirycz, A.; Maleux, K.; Inzé, D. Della signaling mediates stress-induced cell differentiation in arabidopsis leaves through modulation of anaphase-promoting complex/cyclosome activity. Plant Physiol. 2012, 159, 739–747. [Google Scholar] [CrossRef]
- Skirycz, A.; Claeys, H.; De Bodt, S.; Oikawa, A.; Shinoda, S.; Andriankaja, M.; Maleux, K.; Eloy, N.B.; Coppens, F.; Yoo, S.-D.; et al. Pause-and-stop: The effects of osmotic stress on cell proliferation during early leaf development in arabidopsis and a role for ethylene signaling in cell cycle arrest. Plant Cell. 2011, 23, 1876–1888. [Google Scholar] [CrossRef] [Green Version]
- Levey, S.; Wingler, A. Natural variation in the regulation of leaf senescence and relation to other traits in arabidopsis. Plant Cell. Environ. 2005, 28, 223–231. [Google Scholar] [CrossRef]
- Qin, F.; Kodaira, K.-S.; Maruyama, K.; Mizoi, J.; Tran, L.-S.P.; Fujita, Y.; Morimoto, K.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Spindly, a negative regulator of gibberellic acid signaling, is involved in the plant abiotic stress response. Plant Physiol. 2011, 157, 1900–1913. [Google Scholar] [CrossRef]
- Munns, R. Comparative physiology of salt and water stress. Plant Cell. Environ. 2002, 25, 239–250. [Google Scholar] [CrossRef]
- Wingler, A.; Roitsch, T. Metabolic regulation of leaf senescence: Interactions of sugar signalling with biotic and abiotic stress responses. Plant Biology 2008, 10, 50–62. [Google Scholar] [CrossRef]
- Kesari, R.; Lasky, J.R.; Villamor, J.G.; Des Marais, D.L.; Chen, Y.-J.C.; Liu, T.-W.; Lin, W.; Juenger, T.E.; Verslues, P.E. Intron-mediated alternative splicing of arabidopsis p5cs1 and its association with natural variation in proline and climate adaptation. Proc. Natl. Acad. Sci. USA 2012, 109, 9197–9202. [Google Scholar] [CrossRef]
- Yoshiba, Y.; Kiyosue, T.; Katagiri, T.; Ueda, H.; Mizoguchi, T.; Yamaguchi-Shinozaki, K.; Wada, K.; Harada, Y.; Shinozaki, K. Correlation between the induction of a gene for δ1-pyrroline-5-carboxylate synthetase and the accumulation of proline in arabidopsis thaliana under osmotic stress. Plant J. 1995, 7, 751–760. [Google Scholar]
- Waditee, R.; Bhuiyan, M.N.H.; Rai, V.; Aoki, K.; Tanaka, Y.; Hibino, T.; Suzuki, S.; Takano, J.; Jagendorf, A.T.; Takabe, T.; et al. Genes for direct methylation of glycine provide high levels of glycinebetaine and abiotic-stress tolerance in synechococcus and arabidopsis. Proc. Natl. Acad. Sci. USA 2005, 102, 1318–1323. [Google Scholar] [CrossRef]
- Ioannidis, N.E.; Cruz, J.A.; Kotzabasis, K.; Kramer, D.M. Evidence that putrescine modulates the higher plant photosynthetic proton circuit. PLoS one 2012, 7, e29864. [Google Scholar]
- Kaplan, F.; Kopka, J.; Haskell, D.W.; Zhao, W.; Schiller, K.C.; Gatzke, N.; Sung, D.Y.; Guy, C.L. Exploring the temperature-stress metabolome of arabidopsis. Plant Physiol. 2004, 136, 4159–4168. [Google Scholar] [CrossRef]
- Cuevas, J.C.; López-Cobollo, R.; Alcázar, R.; Zarza, X.; Koncz, C.; Altabella, T.; Salinas, J.; Tiburcio, A.F.; Ferrando, A. Putrescine is involved in arabidopsis freezing tolerance and cold acclimation by regulating abscisic acid levels in response to low temperature. Plant Physiol. 2008, 148, 1094–1105. [Google Scholar] [CrossRef]
- Renault, H.; El Amrani, A.; Berger, A.; Mouille, G.; Soubigou-Taconnat, L.; Bouchereau, A.; Deleu, C. Γ-aminobutyric acid transaminase deficiency impairs central carbon metabolism and leads to cell wall defects during salt stress in arabidopsis roots. Plant Cell. Environ. 2013, 36, 1009–1018. [Google Scholar] [CrossRef]
- Van Dongen, J.T.; Fröhlich, A.; Ramírez-Aguilar, S.J.; Schauer, N.; Fernie, A.R.; Erban, A.; Kopka, J.; Clark, J.; Langer, A.; Geigenberger, P. Transcript and metabolite profiling of the adaptive response to mild decreases in oxygen concentration in the roots of arabidopsis plants. Ann. Bot. 2009, 103, 269–280. [Google Scholar]
- Osuna, D.; Usadel, B.; Morcuende, R.; Gibon, Y.; Bläsing, O.E.; Höhne, M.; Günter, M.; Kamlage, B.; Trethewey, R.; Scheible, W.-R.; et al. Temporal responses of transcripts, enzyme activities and metabolites after adding sucrose to carbon-deprived arabidopsis seedlings. Plant J. 2007, 49, 463–491. [Google Scholar] [CrossRef]
- Raschke, M.; Boycheva, S.; Crèvecoeur, M.; Nunes-Nesi, A.; Witt, S.; Fernie, A.R.; Amrhein, N.; Fitzpatrick, T.B. Enhanced levels of vitamin b6 increase aerial organ size and positively affect stress tolerance in arabidopsis. Plant J. 2011, 66, 414–432. [Google Scholar] [CrossRef]
- Tunc-Ozdemir, M.; Miller, G.; Song, L.; Kim, J.; Sodek, A.; Koussevitzky, S.; Misra, A.N.; Mittler, R.; Shintani, D. Thiamin confers enhanced tolerance to oxidative stress in arabidopsis. Plant Physiol. 2009, 151, 421–432. [Google Scholar] [CrossRef]
- Miller, G.; Suzuki, N.; Rizhsky, L.; Hegie, A.; Koussevitzky, S.; Mittler, R. Double mutants deficient in cytosolic and thylakoid ascorbate peroxidase reveal a complex mode of interaction between reactive oxygen species, plant development, and response to abiotic stresses. Plant Physiol. 2007, 144, 1777–1785. [Google Scholar] [CrossRef]
- Szarka, A.; Tomasskovics, B.; Bánhegyi, G. The ascorbate-glutathione-α-tocopherol triad in abiotic stress response. Int. J. Mol. Sci. 2012, 13, 4458–4483. [Google Scholar] [CrossRef]
- Suzuki, N.; Koussevitzky, S.; Mittler, R.O.N.; Miller, G.A.D. Ros and redox signalling in the response of plants to abiotic stress. Plant Cell. Environ. 2012, 35, 259–270. [Google Scholar] [CrossRef]
- Urano, K.; Maruyama, K.; Ogata, Y.; Morishita, Y.; Takeda, M.; Sakurai, N.; Suzuki, H.; Saito, K.; Shibata, D.; Kobayashi, M.; et al. Characterization of the aba-regulated global responses to dehydration in arabidopsis by metabolomics. Plant J. 2009, 57, 1065–1078. [Google Scholar] [CrossRef]
- Saito, K.; Matsuda, F. Metabolomics for functional genomics, systems biology, and biotechnology. Annu Rev. Plant Biol 2010, 61, 463–489. [Google Scholar] [CrossRef]
- Fraire-Velázquez, S.; Balderas-Hernández, V.E. Abiotic stress in plants and metabolic responses. In Abiotic Stress—Plant Responses and Applications in Agriculture; Vahdati, K, Leslie, C., Eds.; Intechopen, 2013. [Google Scholar] [CrossRef]
- Obata, T.; Fernie, A. The use of metabolomics to dissect plant responses to abiotic stresses. Cell. Mol. Life Sci. 2012, 69, 3225–3243. [Google Scholar] [CrossRef]
- Kueger, S.; Steinhauser, D.; Willmitzer, L.; Giavalisco, P. High-resolution plant metabolomics: From mass spectral features to metabolites and from whole-cell analysis to subcellular metabolite distributions. Plant J. 2012, 70, 39–50. [Google Scholar] [CrossRef]
- Hur, M.; Campbell, A.A.; Almeida-de-Macedo, M.; Li, L.; Ransom, N.; Jose, A.; Crispin, M.; Nikolau, B.J.; Wurtele, E.S. A global approach to analysis and interpretation of metabolic data for plant natural product discovery. Nat. Prod. Rep. 2013, 30, 565–583. [Google Scholar] [CrossRef]
- Narsai, R.; Howell, K.A.; Carroll, A.; Ivanova, A.; Millar, A.H.; Whelan, J. Defining core metabolic and transcriptomic responses to oxygen availability in rice embryos and young seedlings. Plant Physiol. 2009, 151, 306–322. [Google Scholar] [CrossRef]
- Ralston-Hooper, K.; Jannasch, A.; Adamec, J.; Sepúlveda, M. The use of two-dimensional gas chromatography–time-of-flight mass spectrometry (gc× gc–tof-ms) for metabolomic analysis of polar metabolites. In Metabolic Profiling; 2011; Volume 708, pp. 205–211. [Google Scholar]
- Little, J.L. Artifacts in trimethylsilyl derivatization reactions and ways to avoid them. J. Chromatogr A 1999, 844, 1–22. [Google Scholar] [CrossRef]
- Halket, J.M.; Waterman, D.; Przyborowska, A.M.; Patel, R.K.P.; Fraser, P.D.; Bramley, P.M. Chemical derivatization and mass spectral libraries in metabolic profiling by gc/ms and lc/ms/ms. J. Exp. Bot. 2005, 56, 219–243. [Google Scholar]
- Golm metabolome Database (GMD). Available online: http://gmd.mpimp-golm.mpg.de/ (accessed on 27 June 2013).
- Giavalisco, P.; Li, Y.; Matthes, A.; Eckhardt, A.; Hubberten, H.-M.; Hesse, H.; Segu, S.; Hummel, J.; Köhl, K.; Willmitzer, L. Elemental formula annotation of polar and lipophilic metabolites using 13c, 15n and 34s isotope labelling, in combination with high-resolution mass spectrometry. Plant J. 2011, 68, 364–376. [Google Scholar] [CrossRef]
- Leng, J.; Wang, H.; Zhang, L.; Zhang, J.; Guo, Y. A highly sensitive isotope-coded derivatization method and its application for the mass spectrometric analysis of analytes containing the carboxyl group. Anal. Chim. Acta. 2013, 758, 114–121. [Google Scholar] [CrossRef]
- Gaquerel, E.; Kuhl, C.; Neumann, S. Computational annotation of plant metabolomics profiles via a novel network-assisted approach. Metabolomics 2013, 1–15. [Google Scholar]
- Metabolomic Tool Kit —GARnet. Available online: http://www.garnetcommunity.org.uk/resources/metabolomic-tool-kit/ (accessed on 27 June 2013).
- Pathway Activity Profiling (PAPi). Available online: http://www.4shared.com/file/s0uIYWIg/PAPi_10.html/ (accessed on 29 June 2013).
- Metabolites Biological Role. Available online: http://pdg.cnb.uam.es/mbrole/ (accessed on 26 July 2013).
- Fukushima, A.; Kusano, M.; Redestig, H.; Arita, M.; Saito, K. Metabolomic correlation-network modules in arabidopsis based on a graph-clustering approach. BMC Syst Biol 2011, 5, 1. [Google Scholar] [CrossRef]
- Saito, K.; Hirai, M.Y.; Yonekura-Sakakibara, K. Decoding genes with coexpression networks and metabolomics—“Majority report by precogs”. Trends Plant Sci. 2008, 13, 36–43. [Google Scholar] [CrossRef]
- Scholz, M.; Gatzek, S.; Sterling, A.; Fiehn, O.; Selbig, J. Metabolite fingerprinting: Detecting biological features by independent component analysis. Bioinformatics 2004, 20, 2447–2454. [Google Scholar] [CrossRef]
- Redestig, H.; Costa, I.G. Detection and interpretation of metabolite–transcript coresponses using combined profiling data. Bioinformatics 2011, 27, i357–i365. [Google Scholar] [CrossRef]
- Kamburov, A.; Cavill, R.; Ebbels, T.M.D.; Herwig, R.; Keun, H.C. Integrated pathway-level analysis of transcriptomics and metabolomics data with impala. Bioinformatics 2011, 27, 2917–2918. [Google Scholar] [CrossRef]
- IMPaLA: Integrated Molecular Pathway Level Analysis. Available online: http://impala.molgen.mpg.de/ (accessed on 26 June 2013).
- Painting omics data in biological pathways. Available online: http://www.paintomics.org/cgi-bin/main2.cgi/ (accessed on 29 June 2013).
- Sulpice, R.; Trenkamp, S.; Steinfath, M.; Usadel, B.; Gibon, Y.; Witucka-Wall, H.; Pyl, E.-T.; Tschoep, H.; Steinhauser, M.C.; Guenther, M.; et al. Network analysis of enzyme activities and metabolite levels and their relationship to biomass in a large panel of arabidopsis accessions. Plant Cell. 2010, 22, 2872–2893. [Google Scholar] [CrossRef]
- Cross, J.M.; von Korff, M.; Altmann, T.; Bartzetko, L.; Sulpice, R.; Gibon, Y.; Palacios, N.; Stitt, M. Variation of enzyme activities and metabolite levels in 24 arabidopsis accessions growing in carbon-limited conditions. Plant Physiol. 2006, 142, 1574–1588. [Google Scholar] [CrossRef]
- Gibon, Y.; Usadel, B.; Blaesing, O.; Kamlage, B.; Hoehne, M.; Trethewey, R.; Stitt, M. Integration of metabolite with transcript and enzyme activity profiling during diurnal cycles in arabidopsis rosettes. Genome Biol. 2006, 7, R76. [Google Scholar] [CrossRef]
- Hirai, M.Y.; Yano, M.; Goodenowe, D.B.; Kanaya, S.; Kimura, T.; Awazuhara, M.; Arita, M.; Fujiwara, T.; Saito, K. Integration of transcriptomics and metabolomics for understanding of global responses to nutritional stresses in arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2004, 101, 10205–10210. [Google Scholar] [CrossRef]
- Fernie, A.R.; Stitt, M. On the discordance of metabolomics with proteomics and transcriptomics: Coping with increasing complexity in logic, chemistry and network interactions. Plant Physiol. 2012, 158, 1139–1145. [Google Scholar] [CrossRef]
- Hannah, M.A.; Caldana, C.; Steinhauser, D.; Balbo, I.; Fernie, A.R.; Willmitzer, L. Combined transcript and metabolite profiling of arabidopsis grown under widely variant growth conditions facilitates the identification of novel metabolite-mediated regulation of gene expression. Plant Physiol. 2010, 152, 2120–2129. [Google Scholar] [CrossRef]
- Gouws, L.; Botes, E.; Wiese, A.J.; Trenkamp, S.; Torres-Jerez, I.; Tang, Y.; Hills, P.N.; Usadel, B.; Lloyd, J.R.; Fernie, A.; et al. The plant growth promoting substance, lumichrome, mimics starch and ethylene-associated symbiotic responses in lotus and tomato roots. Front. Plant Sci 2012. [Google Scholar] [CrossRef]
- Espinoza, C.; Degenkolbe, T.; Caldana, C.; Zuther, E.; Leisse, A.; Willmitzer, L.; Hincha, D.K.; Hannah, M.A. Interaction with diurnal and circadian regulation results in dynamic metabolic and transcriptional changes during cold acclimation in arabidopsis. PLoS One 2010, 5, e14101. [Google Scholar] [CrossRef]
- Ndamukong, I.; Jones, D.R.; Lapko, H.; Divecha, N.; Avramova, Z. Phosphatidylinositol 5-phosphate links dehydration stress to the activity of arabidopsis trithorax-like factor atx1. PLoS One 2010, 5, e13396. [Google Scholar]
- Saleh, A.; Alvarez-Venegas, R.; Yilmaz, M.; Le, O.; Hou, G.; Sadder, M.; Al-Abdallat, A.; Xia, Y.; Lu, G.; Ladunga, I.; et al. The highly similar arabidopsis homologs of trithorax atx1 and atx2 encode proteins with divergent biochemical functions. Plant Cell. 2008, 20, 568–579. [Google Scholar] [CrossRef]
- Pien, S.; Fleury, D.; Mylne, J.S.; Crevillen, P.; Inzé, D.; Avramova, Z.; Dean, C.; Grossniklaus, U. Arabidopsis trithorax1 dynamically regulates flowering locus c activation via histone 3 lysine 4 trimethylation. Plant Cell. 2008, 20, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Pical, C.; Westergren, T.; Dove, S.K.; Larsson, C.; Sommarin, M. Salinity and hyperosmotic stress induce rapid increases in phosphatidylinositol 4,5-bisphosphate, diacylglycerol pyrophosphate, and phosphatidylcholine in arabidopsis thaliana cells. J. Biol. Chem. 1999, 274, 38232–38240. [Google Scholar]
- Ndamukong, I.; Lapko, H.; Cerny, R.L.; Avramova, Z. A cytoplasm-specific activity encoded by the trithorax-like atx1 gene. Nucleic Acids Res. 2011, 39, 4709–4718. [Google Scholar] [CrossRef]
- Li, J.; Brader, G.; Palva, E.T. The wrky70 transcription factor: A node of convergence for jasmonate-mediated and salicylate-mediated signals in plant defense. Plant Cell. 2004, 16, 319–331. [Google Scholar] [CrossRef]
- Besseau, S.; Li, J.; Palva, E.T. Wrky54 and wrky70 co-operate as negative regulators of leaf senescence in arabidopsis thaliana. J. Exp. Bot. 2012, 63, 2667–2679. [Google Scholar] [CrossRef]
- Yao, Y.; Kovalchuk, I. Abiotic stress leads to somatic and heritable changes in homologous recombination frequency, point mutation frequency and microsatellite stability in arabidopsis plants. Mutat. Res.—Fund. Mol. M. 2011, 707, 61–66. [Google Scholar] [CrossRef]
- Weinberg, Z.; Wang, J.; Bogue, J.; Yang, J.; Corbino, K.; Moy, R.; Breaker, R. Comparative genomics reveals 104 candidate structured rnas from bacteria, archaea, and their metagenomes. Genome Biol. 2010, 11, R31. [Google Scholar] [CrossRef]
- Bocobza, S.; Adato, A.; Mandel, T.; Shapira, M.; Nudler, E.; Aharoni, A. Riboswitch-dependent gene regulation and its evolution in the plant kingdom. Genes Dev. 2007, 21, 2874–2879. [Google Scholar] [CrossRef]
- Wachter, A.; Tunc-Ozdemir, M.; Grove, B.C.; Green, P.J.; Shintani, D.K.; Breaker, R.R. Riboswitch control of gene expression in plants by splicing and alternative 3′ end processing of mrnas. Plant Cell. 2007, 19, 3437–3450. [Google Scholar] [CrossRef]
- Bocobza, S.E.; Malitsky, S.; Araújo, W.L.; Nunes-Nesi, A.; Meir, S.; Shapira, M.; Fernie, A.R.; Aharoni, A. Orchestration of thiamin biosynthesis and central metabolism by combined action of the thiamin pyrophosphate riboswitch and the circadian clock in arabidopsis. Plant Cell. 2013, 25, 288–307. [Google Scholar] [CrossRef]
- Rapala-Kozik, M.; Wolak, N.; Kujda, M.; Banas, A. The upregulation of thiamine (vitamin b1) biosynthesis in arabidopsis thaliana seedlings under salt and osmotic stress conditions is mediated by abscisic acid at the early stages of this stress response. BMC Plant Biol. 2012. [Google Scholar] [CrossRef]
- Zimmermann, P.; Hirsch-Hoffmann, M.; Hennig, L.; Gruissem, W. Genevestigator. Arabidopsis microarray database and analysis toolbox. Plant Physiol. 2004, 136, 2621–2632. [Google Scholar] [CrossRef]
- Leister, D. Retrograde signalling in plants: From simple to complex scenarios. Front. Plant Sci 2012. [Google Scholar] [CrossRef]
- Estavillo, G.M.; Chan, K.X.; Phua, S.Y.; Pogson, B.J. Reconsidering the nature and mode of action of metabolite retrograde signals from the chloroplast. Front. Plant Sci 2013. [Google Scholar] [CrossRef]
- Caldana, C.; Fernie, A.R.; Willmitzer, L.; Steinhauser, D. Unravelling retrograde signalling pathways: Finding candidate signalling molecules via metabolomics and systems biology driven approaches. Front. Plant Sci 2012. [Google Scholar] [CrossRef]
- Strand, A.; Asami, T.; Alonso, J.; Ecker, J.R.; Chory, J. Chloroplast to nucleus communication triggered by accumulation of mg-protoporphyrinix. Nature 2003, 421, 79–83. [Google Scholar] [CrossRef]
- Estavillo, G.M.; Crisp, P.A.; Pornsiriwong, W.; Wirtz, M.; Collinge, D.; Carrie, C.; Giraud, E.; Whelan, J.; David, P.; Javot, H.; et al. Evidence for a sal1-pap chloroplast retrograde pathway that functions in drought and high light signaling in arabidopsis. Plant Cell. 2011, 23, 3992–4012. [Google Scholar] [CrossRef]
- Xiao, Y.; Savchenko, T.; Baidoo, E.; Chehab, W.; Hayden, D.; Tolstikov, V.; Corwin, J.; Kliebenstein, D.; Keasling, J.; Dehesh, K. Retrograde signaling by the plastidial metabolite mecpp regulates expression of nuclear stress-response genes. Cell. 2012, 149, 1525–1535. [Google Scholar] [CrossRef]
- Baruah, A.; Šimková, K.; Hincha, D.K.; Apel, K.; Laloi, C. Modulation of 1o2-mediated retrograde signaling by the pleiotropic response locus 1 (prl1) protein, a central integrator of stress and energy signaling. Plant J. 2009, 60, 22–32. [Google Scholar] [CrossRef]
- Ramel, F.; Birtic, S.; Ginies, C.; Soubigou-Taconnat, L.; Triantaphylides, C.; Havaux, M. Carotenoid oxidation products are stress signals that mediate gene responses to singlet oxygen in plants. Proc. Natl. Acad. Sci. USA 2012, 109, 5535–5540. [Google Scholar]
- Ankele, E.; Kindgren, P.; Pesquet, E.; Strand, Å. In vivo visualization of mg-protoporphyrinix, a coordinator of photosynthetic gene expression in the nucleus and the chloroplast. Plant Cell. 2007, 19, 1964–1979. [Google Scholar] [CrossRef]
- Moulin, M.; McCormac, A.C.; Terry, M.J.; Smith, A.G. Tetrapyrrole profiling in arabidopsis seedlings reveals that retrograde plastid nuclear signaling is not due to mg-protoporphyrin ix accumulation. Proc. Natl. Acad. Sci. USA 2008, 105, 15178–15183. [Google Scholar]
- Mochizuki, N.; Tanaka, R.; Tanaka, A.; Masuda, T.; Nagatani, A. The steady-state level of mg-protoporphyrin ix is not a determinant of plastid-to-nucleus signaling in arabidopsis. Proc. Natl. Acad. Sci. USA 2008, 105, 15184–15189. [Google Scholar] [CrossRef]
- Woodson, J.D.; Perez-Ruiz, J.M.; Chory, J. Heme synthesis by plastid ferrochelatase i regulates nuclear gene expression in plants. Curr. Biol. 2011, 21, 897–903. [Google Scholar] [CrossRef]
- Duncan, O.; Taylor, N.L.; Carrie, C.; Eubel, H.; Kubiszewski-Jakubiak, S.; Zhang, B.; Narsai, R.; Millar, A.H.; Whelan, J. Multiple lines of evidence localize signaling, morphology, and lipid biosynthesis machinery to the mitochondrial outer membrane of arabidopsis. Plant Physiol. 2011, 157, 1093–1113. [Google Scholar] [CrossRef]
- Duncan, O.; van der Merwe, M.J.; Daley, D.O.; Whelan, J. The outer mitochondrial membrane in higher plants. Trends Plant Sci. 2013, 18, 207–217. [Google Scholar] [CrossRef]
- Giegé, P.; Heazlewood, J.L.; Roessner-Tunali, U.; Millar, A.H.; Fernie, A.R.; Leaver, C.J.; Sweetlove, L.J. Enzymes of glycolysis are functionally associated with the mitochondrion in arabidopsis cells. Plant Cell. 2003, 15, 2140–2151. [Google Scholar] [CrossRef]
- Graham, J.W.A.; Williams, T.C.R.; Morgan, M.; Fernie, A.R.; Ratcliffe, R.G.; Sweetlove, L.J. Glycolytic enzymes associate dynamically with mitochondria in response to respiratory demand and support substrate channeling. Plant Cell. 2007, 19, 3723–3738. [Google Scholar] [CrossRef]
- Cho, Y.-H.; Yoo, S.-D.; Sheen, J. Regulatory functions of nuclear hexokinase1 complex in glucose signaling. Cell. 2006, 127, 579–589. [Google Scholar] [CrossRef]
- Moore, B.; Zhou, L.; Rolland, F.; Hall, Q.; Cheng, W.-H.; Liu, Y.-X.; Hwang, I.; Jones, T.; Sheen, J. Role of the arabidopsis glucose sensor hxk1 in nutrient, light, and hormonal signaling. Science 2003, 300, 332–336. [Google Scholar] [CrossRef]
- Cho, Y.-H.; Sheen, J.; Yoo, S.-D. Low glucose uncouples hexokinase1-dependent sugar signaling from stress and defense hormone abscisic acid and c2h4 responses in arabidopsis. Plant Physiol. 2010, 152, 1180–1182. [Google Scholar] [CrossRef]
- Kaplan, F.; Kopka, J.; Sung, D.Y.; Zhao, W.; Popp, M.; Porat, R.; Guy, C.L. Transcript and metabolite profiling during cold acclimation of arabidopsis reveals an intricate relationship of cold-regulated gene expression with modifications in metabolite content. Plant J. 2007, 50, 967–981. [Google Scholar] [CrossRef]
- Scheible, W.-R.; Morcuende, R.; Czechowski, T.; Fritz, C.; Osuna, D.; Palacios-Rojas, N.; Schindelasch, D.; Thimm, O.; Udvardi, M.K.; Stitt, M. Genome-wide reprogramming of primary and secondary metabolism, protein synthesis, cellular growth processes, and the regulatory infrastructure of arabidopsis in response to nitrogen. Plant Physiol. 2004, 136, 2483–2499. [Google Scholar] [CrossRef]
- Morcuende, R.; Bari, R.; Gibon, Y.; Zheng, W.; Pant, B.D.; Blaesing, O.; Usadel, B.; Czechowski, T.; Udvardi, M.K.; Stitt, M.; et al. Genome-wide reprogramming of metabolism and regulatory networks of arabidopsis in response to phosphorus. Plant Cell. Environ. 2007, 30, 85–112. [Google Scholar] [CrossRef]
- Armengaud, P.; Sulpice, R.; Miller, A.J.; Stitt, M.; Amtmann, A.; Gibon, Y. Multilevel analysis of primary metabolism provides new insights into the role of potassium nutrition for glycolysis and nitrogen assimilation in arabidopsis roots. Plant Physiol. 2009, 150, 772–785. [Google Scholar] [CrossRef]
- Arroyo, A.; Bossi, F.; Finkelstein, R.R.; León, P. Three genes that affect sugar sensing (abscisic acid insensitive 4, abscisic acid insensitive 5, and constitutive triple response 1) are differentially regulated by glucose in arabidopsis. Plant Physiol. 2003, 133, 231–242. [Google Scholar] [CrossRef]
- Rizhsky, L.; Liang, H.; Shuman, J.; Shulaev, V.; Davletova, S.; Mittler, R. When defense pathways collide. The response of arabidopsis to a combination of drought and heat stress. Plant Physiol. 2004, 134, 1683–1696. [Google Scholar] [CrossRef]
- Hummel, I.; Pantin, F.; Sulpice, R.; Piques, M.; Rolland, G.; Dauzat, M.; Christophe, A.; Pervent, M.; Bouteillé, M.; Stitt, M.; et al. Arabidopsis plants acclimate to water deficit at low cost through changes of carbon usage: An integrated perspective using growth, metabolite, enzyme, and gene expression analysis. Plant Physiol. 2010, 154, 357–372. [Google Scholar] [CrossRef]
- Van Houtte, H.; Vandesteene, L.; Lopez-Galvis, L.; Lemmens, L.; Kissel, E.; Carpentier, S.; Feil, R.; Avonce, N.; Beeckman, T.; Lunn, J.; et al. Over-expression of the trehalase gene attre1 leads to increased drought stress tolerance in arabidopsis and is involved in aba-induced stomatal closure. Plant Physiol. 2013, 161, 1158–1171. [Google Scholar] [CrossRef]
- Lippold, F.; Sanchez, D.H.; Musialak, M.; Schlereth, A.; Scheible, W.-R.; Hincha, D.K.; Udvardi, M.K. Atmyb41 regulates transcriptional and metabolic responses to osmotic stress in arabidopsis. Plant Physiol. 2009, 149, 1761–1772. [Google Scholar] [CrossRef]
- Baena-Gonzalez, E.; Rolland, F.; Thevelein, J.M.; Sheen, J. A central integrator of transcription networks in plant stress and energy signalling. Nature 2007, 448, 938–942. [Google Scholar] [CrossRef]
- Jossier, M.; Bouly, J.-P.; Meimoun, P.; Arjmand, A.; Lessard, P.; Hawley, S.; Grahame Hardie, D.; Thomas, M. Snrk1 (snf1-related kinase 1) has a central role in sugar and aba signalling in Arabidopsis thaliana. Plant J. 2009, 59, 316–328. [Google Scholar] [CrossRef]
- Toroser, D.; Plaut, Z.; Huber, S.C. Regulation of a plant snf1-related protein kinase by glucose-6-phosphate. Plant Physiol. 2000, 123, 403–412. [Google Scholar] [CrossRef]
- Zhang, Y.; Primavesi, L.F.; Jhurreea, D.; Andralojc, P.J.; Mitchell, R.A.C.; Powers, S.J.; Schluepmann, H.; Delatte, T.; Wingler, A.; Paul, M.J. Inhibition of snf1-related protein kinase1 activity and regulation of metabolic pathways by trehalose-6-phosphate. Plant Physiol. 2009, 149, 1860–1871. [Google Scholar] [CrossRef]
- Paul, M.J.; Jhurreea, D.; Zhang, Y.; Primavesi, L.F.; Delatte, T.; Schluepmann, H.; Wingler, A. Up-regulation of biosynthetic processes associated with growth by trehalose 6-phosphate. Plant Signal. Behav 2010, 5, 386–392. [Google Scholar] [CrossRef]
- Fragoso, S.; Espíndola, L.; Páez-Valencia, J.; Gamboa, A.; Camacho, Y.; Martínez-Barajas, E.; Coello, P. Snrk1 isoforms akin10 and akin11 are differentially regulated in arabidopsis plants under phosphate starvation. Plant Physiol. 2009, 149, 1906–1916. [Google Scholar]
- Kolbe, A.; Tiessen, A.; Schluepmann, H.; Paul, M.; Ulrich, S.; Geigenberger, P. Trehalose 6-phosphate regulates starch synthesis via posttranslational redox activation of adp-glucose pyrophosphorylase. Proc. Natl. Acad. Sci. USA 2005, 102, 11118–11123. [Google Scholar] [CrossRef]
- Delatte, T.L.; Sedijani, P.; Kondou, Y.; Matsui, M.; de Jong, G.J.; Somsen, G.W.; Wiese-Klinkenberg, A.; Primavesi, L.F.; Paul, M.J.; Schluepmann, H. Growth arrest by trehalose-6-phosphate: An astonishing case of primary metabolite control over growth by way of the snrk1 signaling pathway. Plant Physiol. 2011, 157, 160–174. [Google Scholar] [CrossRef]
- Rahmani, F.; Hummel, M.; Schuurmans, J.; Wiese-Klinkenberg, A.; Smeekens, S.; Hanson, J. Sucrose control of translation mediated by an upstream open reading frame-encoded peptide. Plant Physiol. 2009, 150, 1356–1367. [Google Scholar] [CrossRef]
- Wahl, V.; Ponnu, J.; Schlereth, A.; Arrivault, S.; Langenecker, T.; Franke, A.; Feil, R.; Lunn, J.E.; Stitt, M.; Schmid, M. Regulation of flowering by trehalose-6-phosphate signaling in arabidopsis thaliana. Science 2013, 339, 704–707. [Google Scholar] [CrossRef]
- Cho, Y.-H.; Hong, J.-W.; Kim, E.-C.; Yoo, S.-D. Regulatory functions of snrk1 in stress-responsive gene expression and in plant growth and development. Plant Physiol. 2012, 158, 1955–1964. [Google Scholar] [CrossRef]
- Ng, S.; Giraud, E.; Duncan, O.; Law, S.R.; Wang, Y.; Xu, L.; Narsai, R.; Carrie, C.; Walker, H.; Day, D.A.; et al. Cyclin-dependent kinase e1 (cdke1) provides a cellular switch in plants between growth and stress responses. J. Biol. Chem. 2013, 288, 3449–3459. [Google Scholar] [CrossRef]
- Nargang, F.E.; Adames, K.; Rüb, C.; Cheung, S.; Easton, N.; Nargang, C.E.; Chae, M.S. Identification of genes required for alternative oxidase production in the neurospora crassa gene knockout library. G3 (Bethesda) 2012, 2, 1345–1356. [Google Scholar] [CrossRef]
- Giraud, E.; Van Aken, O.; Ho, L.H.M.; Whelan, J. The transcription factor abi4 is a regulator of mitochondrial retrograde expression of alternative oxidase1a. Plant Physiol. 2009, 150, 1286–1296. [Google Scholar] [CrossRef]
- Koussevitzky, S.; Nott, A.; Mockler, T.C.; Hong, F.; Sachetto-Martins, G.; Surpin, M.; Lim, J.; Mittler, R.; Chory, J. Signals from chloroplasts converge to regulate nuclear gene expression. Science 2007, 316, 715–719. [Google Scholar] [CrossRef]
- Giraud, E.; Ho, L.H.M.; Clifton, R.; Carroll, A.; Estavillo, G.; Tan, Y.-F.; Howell, K.A.; Ivanova, A.; Pogson, B.J.; Millar, A.H.; et al. The absence of alternative oxidase1a in arabidopsis results in acute sensitivity to combined light and drought stress. Plant Physiol. 2008, 147, 595–610. [Google Scholar] [CrossRef]
- Shkolnik-Inbar, D.; Adler, G.; Bar-Zvi, D. Abi4 downregulates expression of the sodium transporter hkt1;1 in arabidopsis roots and affects salt tolerance. Plant J. 2013, 73, 993–1005. [Google Scholar] [CrossRef]
- Van Aken, O.; Whelan, J. Comparison of transcriptional changes to chloroplast and mitochondrial perturbations reveals common and specific responses in arabidopsis. Front. Plant Sci 2012. [Google Scholar] [CrossRef]
- Cho, Y.-H.; Yoo, S.-D. Signaling role of fructose mediated by fins1/fbp in arabidopsis thaliana. PLoS Genet. 2011, 7, e1001263. [Google Scholar] [CrossRef]
- Koßmann, J.; Sonnewald, U.; Willmitzer, L. Reduction of the chloroplastic fructose-1,6-bisphosphatase in transgenic potato plants impairs photosynthesis and plant growth. Plant J. 1994, 6, 637–650. [Google Scholar]
- Zrenner, R.; Krause, K.-P.; Apel, P.; Sonnewald, U. Reduction of the cytosolic fructose-1,6-bisphosphatase in transgenic potato plants limits photosynthetic sucrose biosynthesis with no impact on plant growth and tuber yield. Plant J. 1996, 9, 671–681. [Google Scholar]
- Arsova, B.; Hoja, U.; Wimmelbacher, M.; Greiner, E.; Üstün, Ş.; Melzer, M.; Petersen, K.; Lein, W.; Börnke, F. Plastidial thioredoxin z interacts with two fructokinase-like proteins in a thiol-dependent manner: Evidence for an essential role in chloroplast development in arabidopsis and nicotiana benthamiana. Plant Cell. 2010, 22, 1498–1515. [Google Scholar] [CrossRef]
- Deprost, D.; Yao, L.; Sormani, R.; Moreau, M.; Leterreux, G.; Nicolai, M.; Bedu, M.; Robaglia, C.; Meyer, C. The arabidopsis tor kinase links plant growth, yield, stress resistance and mrna translation. EMBO Rep. 2007, 8, 864–870. [Google Scholar] [CrossRef]
- Mahfouz, M.M.; Kim, S.; Delauney, A.J.; Verma, D.P.S. Arabidopsis target of rapamycin interacts with raptor, which regulates the activity of s6 kinase in response to osmotic stress signals. Plant Cell. 2006, 18, 477–490. [Google Scholar] [CrossRef]
- Moreau, M.; Azzopardi, M.; Clément, G.; Dobrenel, T.; Marchive, C.; Renne, C.; Martin-Magniette, M.-L.; Taconnat, L.; Renou, J.-P.; Robaglia, C.; et al. Mutations in the arabidopsis homolog of lst8/gβl, a partner of the target of rapamycin kinase, impair plant growth, flowering, and metabolic adaptation to long days. Plant Cell. 2012, 24, 463–481. [Google Scholar] [CrossRef] [Green Version]
- Jazwinski, S.M. The retrograde response: When mitochondrial quality control is not enough. Biochim. Biophys. Acta 2013, 1833, 400–409. [Google Scholar] [CrossRef]
- Warren, C.; Aranda, I.; Cano, F.J. Metabolomics demonstrates divergent responses of two eucalyptus species to water stress. Metabolomics 2012, 8, 186–200. [Google Scholar] [CrossRef] [Green Version]
- Yobi, A.; Wone, B.W.M.; Xu, W.; Alexander, D.C.; Guo, L.; Ryals, J.A.; Oliver, M.J.; Cushman, J.C. Comparative metabolic profiling between desiccation-sensitive and desiccation-tolerant species of selaginella reveals insights into the resurrection trait. Plant J. 2012, 72, 983–999. [Google Scholar]
- Sanchez, D.H.; Pieckenstain, F.L.; Szymanski, J.; Erban, A.; Bromke, M.; Hannah, M.A.; Kraemer, U.; Kopka, J.; Udvardi, M.K. Comparative functional genomics of salt stress in related model and cultivated plants identifies and overcomes limitations to translational genomics. PLoS One 2011, 6, e17094. [Google Scholar] [CrossRef]
- Sanchez, D.H.; Schwabe, F.; Erban, A.; Udvardi, M.K.; Kopka, J. Comparative metabolomics of drought acclimation in model and forage legumes. Plant Cell. Environ. 2012, 35, 136–149. [Google Scholar] [CrossRef]
- Widodo; Patterson, J.H.; Newbigin, E.; Tester, M.; Bacic, A.; Roessner, U. Metabolic responses to salt stress of barley (hordeum vulgare l.) cultivars, sahara and clipper, which differ in salinity tolerance. J. Exp. Bot. 2009, 60, 4089–4103. [Google Scholar] [CrossRef]
- Witt, S.; Galicia, L.; Lisec, J.; Cairns, J.; Tiessen, A.; Araus, J.L.; Palacios-Rojas, N.; Fernie, A.R. Metabolic and phenotypic responses of greenhouse-grown maize hybrids to experimentally controlled drought stress. Mol. Plant 2012, 5, 401–417. [Google Scholar] [CrossRef]
- Stitt, M.; Sulpice, R.; Keurentjes, J. Metabolic networks: How to identify key components in the regulation of metabolism and growth. Plant Physiol. 2010, 152, 428–444. [Google Scholar] [CrossRef]
- Riedelsheimer, C.; Lisec, J.; Czedik-Eysenberg, A.; Sulpice, R.; Flis, A.; Grieder, C.; Altmann, T.; Stitt, M.; Willmitzer, L.; Melchinger, A.E. Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 8872–8877. [Google Scholar] [CrossRef]
- Sulpice, R.; Nikoloski, Z.; Tschoep, H.; Antonio, C.; Kleessen, S.; Larhlimi, A.; Selbig, J.; Ishihara, H.; Gibon, Y.; Fernie, A.R.; et al. Impact of the carbon and nitrogen supply on relationships and connectivity between metabolism and biomass in a broad panel of arabidopsis accessions. Plant Physiol. 2013, 162, 347–363. [Google Scholar] [CrossRef]
- Degenkolbe, T.; Giavalisco, P.; Zuther, E.; Seiwert, B.; Hincha, D.K.; Willmitzer, L. Differential remodeling of the lipidome during cold acclimation in natural accessions of arabidopsis thaliana. Plant J. 2012, 72, 972–982. [Google Scholar]
- Lindsley, J.E.; Rutter, J. Whence cometh the allosterome? Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 10533–10535. [Google Scholar] [CrossRef]
- Fernie, A.R.; Morgan, J.A. Analysis of metabolic flux using dynamic labelling and metabolic modelling. Plant Cell. Environ. 2013, 36, 1738–1750. [Google Scholar] [CrossRef]
- Baxter, C.; Redestig, H.; Schauer, N.; Repsilber, D.; Patil, K.; Nielsen, J.; Selbig, J.; Liu, J.; Fernie, A.; Sweetlove, L. The metabolic response of heterotrophic arabidopsis cells to oxidative stress. Plant Physiol. 2007, 143, 312–325. [Google Scholar]
- Lehmann, M.; Laxa, M.; Sweetlove, L.; Fernie, A.; Obata, T. Metabolic recovery of arabidopsis thaliana roots following cessation of oxidative stress. Metabolomics 2012, 8, 143–153. [Google Scholar] [CrossRef]
- Giavalisco, P.; Köhl, K.; Hummel, J.; Seiwert, B.; Willmitzer, L. 13c isotope-labeled metabolomes allowing for improved compound annotation and relative quantification in liquid chromatography-mass spectrometry-based metabolomic research. Anal. Chem. 2009, 81, 6546–6551. [Google Scholar] [CrossRef]
- Nakabayashi, R.; Saito, K. Metabolomics for unknown plant metabolites. Anal. Bioanal Chem 2013, 405, 5005–5011. [Google Scholar] [CrossRef]
- Nakabayashi, R.; Sawada, Y.; Yamada, Y.; Suzuki, M.; Hirai, M.Y.; Sakurai, T.; Saito, K. Combination of liquid chromatography–fourier transform ion cyclotron resonance-mass spectrometry with 13c-labeling for chemical assignment of sulfur-containing metabolites in onion bulbs. Anal. Chem. 2013, 85, 1310–1315. [Google Scholar] [CrossRef]
- Bocobza, S.E.; Willmitzer, L.; Raikhel, N.; Aharoni, A. Discovery of new modules in metabolic biology using chemometabolomics. Plant Physiol. 2012, 160, 1160–1163. [Google Scholar] [CrossRef]
- Tohge, T.; Ramos, M.S.; Nunes-Nesi, A.; Mutwil, M.; Giavalisco, P.; Steinhauser, D.; Schellenberg, M.; Willmitzer, L.; Persson, S.; Martinoia, E.; et al. Toward the storage metabolome: Profiling the barley vacuole. Plant Physiol. 2011, 157, 1469–1482. [Google Scholar] [CrossRef] [Green Version]
- Moussaieff, A.; Rogachev, I.; Brodsky, L.; Malitsky, S.; Toal, T.W.; Belcher, H.; Yativ, M.; Brady, S.M.; Benfey, P.N.; Aharoni, A. High-resolution metabolic mapping of cell types in plant roots. Proc. Natl. Acad. Sci. USA 2013, 110, E1232–E1241. [Google Scholar] [CrossRef]
- Long, T.A.; Tsukagoshi, H.; Busch, W.; Lahner, B.; Salt, D.E.; Benfey, P.N. The bhlh transcription factor popeye regulates response to iron deficiency in arabidopsis roots. Plant Cell. 2010, 22, 2219–2236. [Google Scholar] [CrossRef]
- Gifford, M.L.; Dean, A.; Gutierrez, R.A.; Coruzzi, G.M.; Birnbaum, K.D. Cell-specific nitrogen responses mediate developmental plasticity. Proc. Natl. Acad. Sci. USA 2008, 105, 803–808. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Radomiljac, J.D.; Whelan, J.; Van der Merwe, M. Coordinating Metabolite Changes with Our Perception of Plant Abiotic Stress Responses: Emerging Views Revealed by Integrative—Omic Analyses. Metabolites 2013, 3, 761-786. https://doi.org/10.3390/metabo3030761
Radomiljac JD, Whelan J, Van der Merwe M. Coordinating Metabolite Changes with Our Perception of Plant Abiotic Stress Responses: Emerging Views Revealed by Integrative—Omic Analyses. Metabolites. 2013; 3(3):761-786. https://doi.org/10.3390/metabo3030761
Chicago/Turabian StyleRadomiljac, Jordan D., James Whelan, and Margaretha Van der Merwe. 2013. "Coordinating Metabolite Changes with Our Perception of Plant Abiotic Stress Responses: Emerging Views Revealed by Integrative—Omic Analyses" Metabolites 3, no. 3: 761-786. https://doi.org/10.3390/metabo3030761