
Oxidative Stress and Human Skin Connective Tissue Aging


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
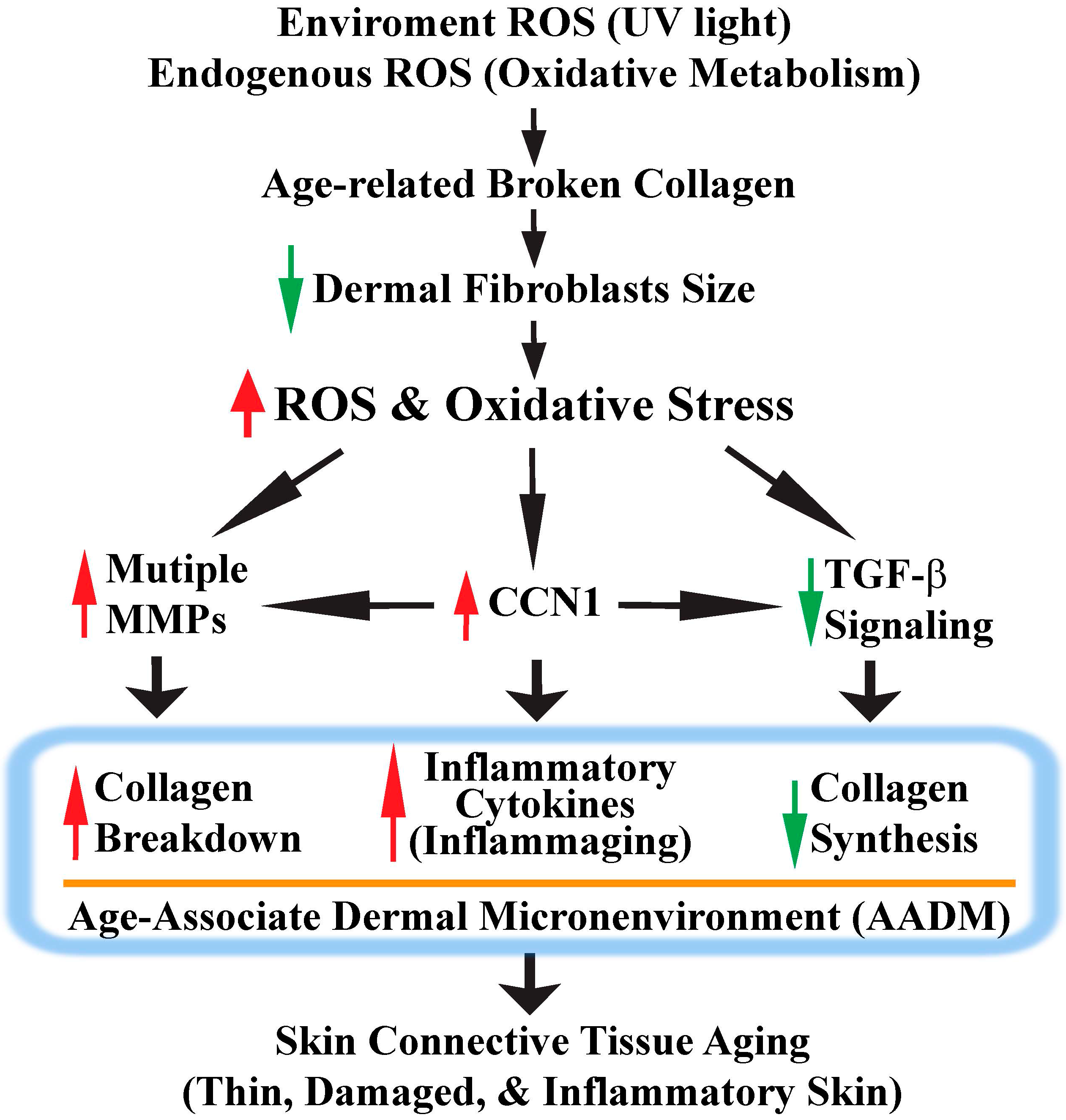
:1. Introduction
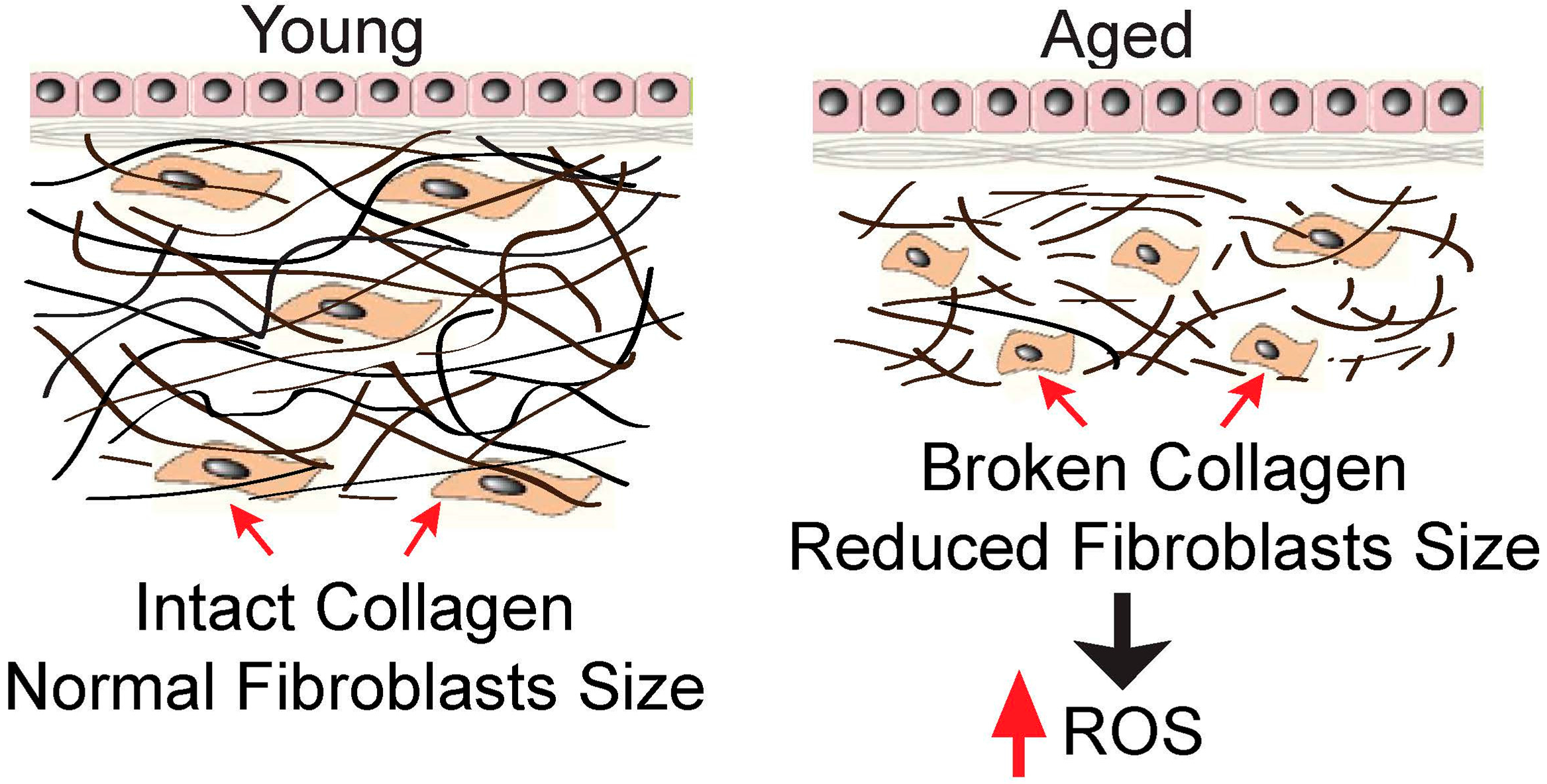
2. Collagen Fragmentation Collapses Dermal Fibroblasts and Increases Intracellular ROS Generation
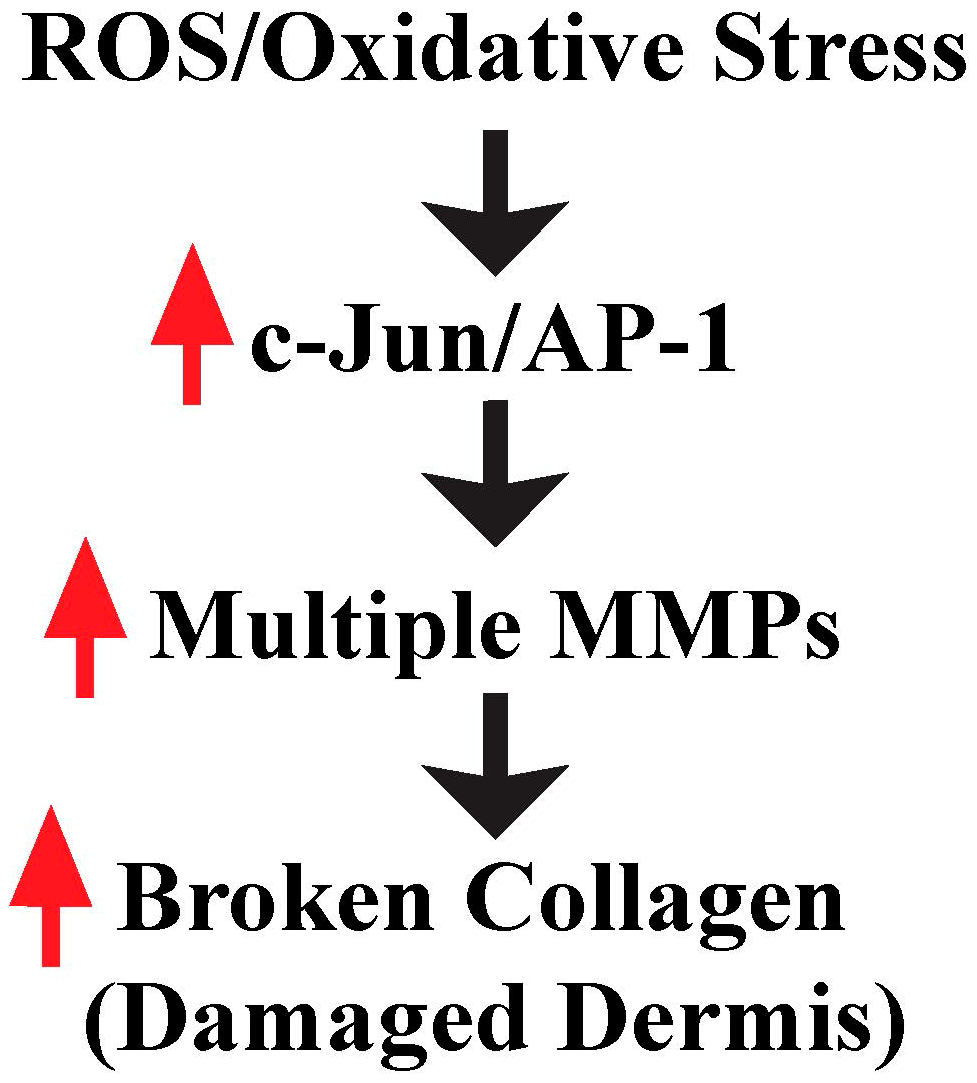
3. ROS/Oxidative Stress Contributes to Damaged Dermis by Induction of Multiple MMPs
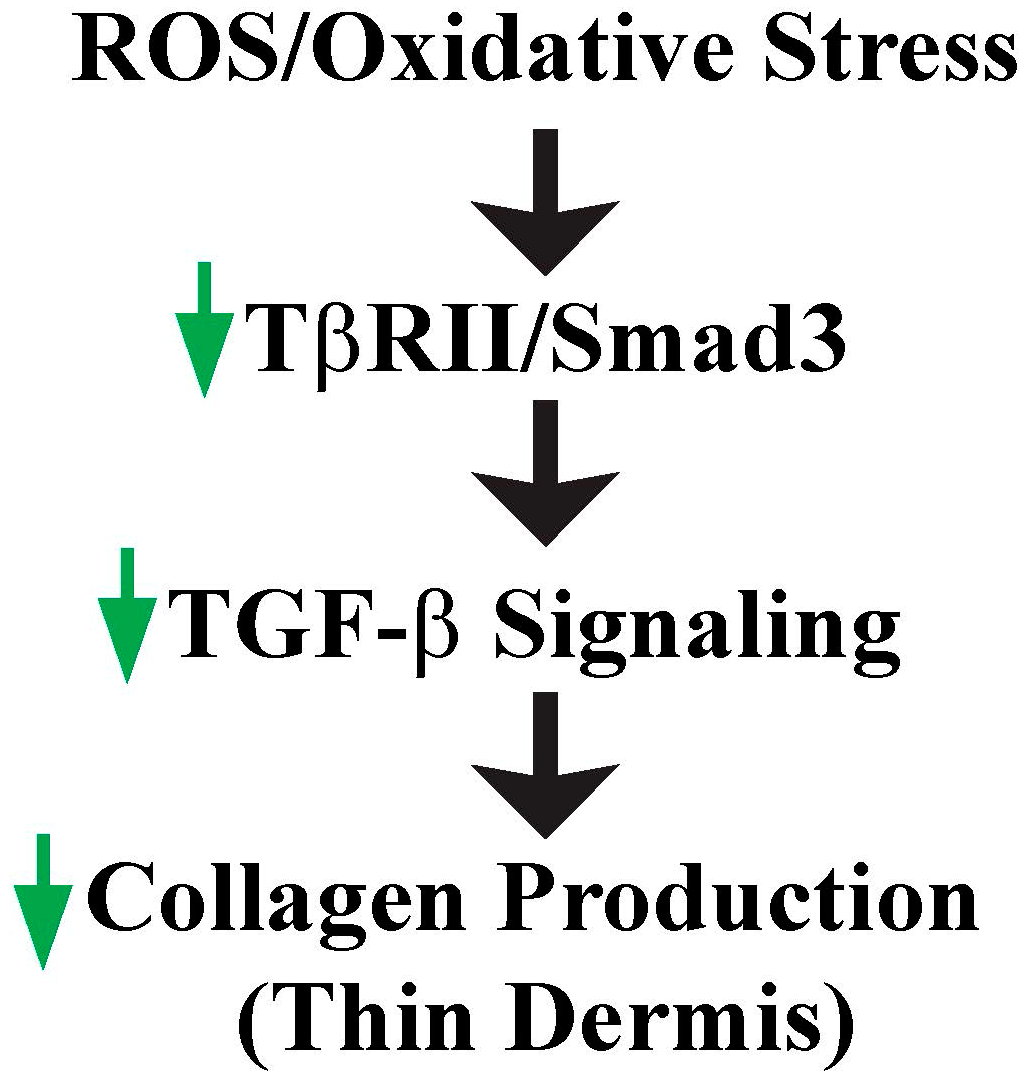
4. ROS/Oxidative Stress Contributes to Thin Dermis by Inhibition of TGF-β Signaling
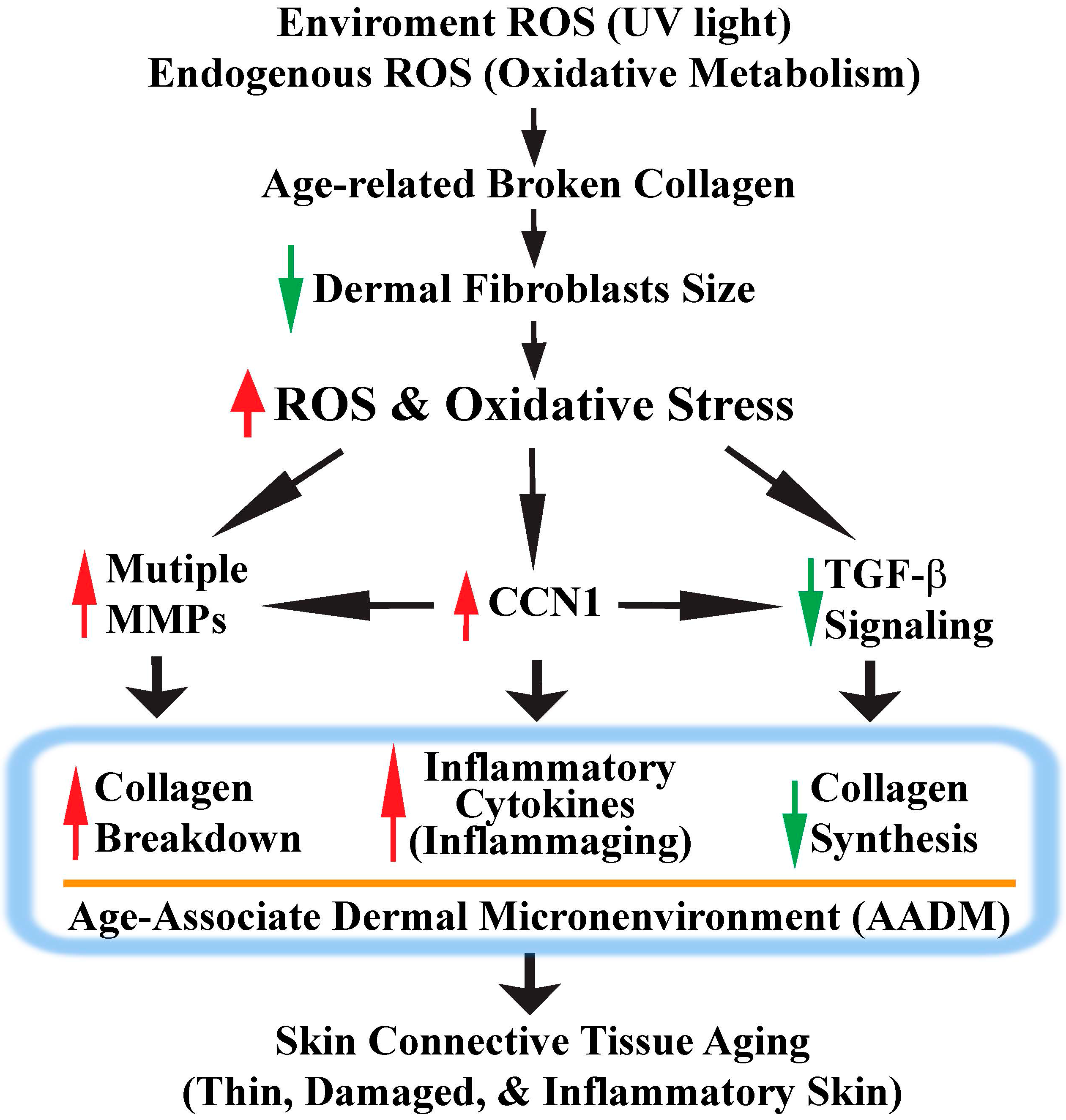
5. CCN1 Functions as a Critical Mediator of Oxidative Stress-Induced Skin Connective Tissue Aging
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Quan, T.; Fisher, G.J. Role of age-associated alterations of the dermal extracellular matrix microenvironment in human skin aging: A mini-review. Gerontology 2015, 61, 427–434. [Google Scholar] [CrossRef]
- Yaar, M.; Eller, M.S.; Gilchrest, B.A. Fifty years of skin aging. J. Investig. Dermatol. Symp. Proc. 2002, 7, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H. Photoaging in Asians. Photodermatol. Photoimmunol. Photomed. 2003, 19, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Wlaschek, M.; Tantcheva-Poor, I.; Naderi, L.; Ma, W.; Schneider, L.A.; Razi-Wolf, Z.; Schuller, J.; Scharffetter-Kochanek, K. Solar UV irradiation and dermal photoaging. J. Photochem. Photobiol. B 2001, 63, 41–51. [Google Scholar] [CrossRef]
- Uitto, J.; Bernstein, E.F. Molecular mechanisms of cutaneous aging: Connective tissue alterations in the dermis. J. Investig. Dermatol. Symp. Proc. 1998, 3, 41–44. [Google Scholar] [PubMed]
- Fisher, G.J.; Wang, Z.Q.; Datta, S.C.; Varani, J.; Kang, S.; Voorhees, J.J. Pathophysiology of premature skin aging induced by ultraviolet light. N. Engl. J. Med. 1997, 337, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.J.; Varani, J.; Voorhees, J.J. Looking older: Fibroblast collapse and therapeutic implications. Arch. Dermatol. 2008, 144, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Lavker, R.M. Structural alterations in exposed and unexposed aged skin. J. Investig. Dermatol. 1979, 73, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Jacob, M.P. Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomed. Pharmacother. 2003, 57, 195–202. [Google Scholar] [CrossRef]
- Cheresh, D.A.; Stupack, D.G. Regulation of angiogenesis: Apoptotic cues from the ECM. Oncogene 2008, 27, 6285–6298. [Google Scholar] [CrossRef] [PubMed]
- Eaglstein, W.H. Wound healing and aging. Clin. Geriatr. Med. 1989, 5, 183–188. [Google Scholar] [PubMed]
- Valencia, I.C.; Falabella, A.; Kirsner, R.S.; Eaglstein, W.H. Chronic venous insufficiency and venous leg ulceration. J. Am. Acad. Dermatol. 2001, 44, 401–424. [Google Scholar] [CrossRef] [PubMed]
- Achyut, B.R.; Bader, D.A.; Robles, A.I.; Wangsa, D.; Harris, C.C.; Ried, T.; Yang, L. Inflammation-mediated genetic and epigenetic alterations drive cancer development in the neighboring epithelium upon stromal abrogation of TGF-β signaling. PLoS Genet. 2013, 9, e1003251. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. TGF-β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004, 303, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Mittapalli, V.R.; Madl, J.; Loffek, S.; Kiritsi, D.; Kern, J.S.; Romer, W.; Nystrom, A.; Bruckner-Tuderman, L. Injury-driven stiffening of the dermis expedites skin carcinoma progression. Cancer Res. 2016, 76, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The aging process. Proc. Natl. Acad. Sci. USA 1981, 78, 7124–7128. [Google Scholar] [CrossRef] [PubMed]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Fisher, G.J.; Quan, T.; Purohit, T.; Shao, Y.; Cho, M.K.; He, T.; Varani, J.; Kang, S.; Voorhees, J.J. Collagen fragmentation promotes oxidative stress and elevates matrix metalloproteinase-1 in fibroblasts in aged human skin. Am. J. Pathol. 2009, 174, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.J.; Kang, S.; Varani, J.; Bata-Csorgo, Z.; Wan, Y.; Datta, S.; Voorhees, J.J. Mechanisms of photoaging and chronological skin aging. Arch. Dermatol. 2002, 138, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Valacchi, G.; Sticozzi, C.; Pecorelli, A.; Cervellati, F.; Cervellati, C.; Maioli, E. Cutaneous responses to environmental stressors. Ann. N. Y. Acad. Sci. 2012, 1271, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Vierkotter, A.; Krutmann, J. Environmental influences on skin aging and ethnic-specific manifestations. Dermatoendocrinol 2012, 4, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Robichaud, P.; Quan, T. Oxidative stress and CCN1 protein in human skin connective tissue aging. AIMS Mol. Sci. 2016, 3, 269–279. [Google Scholar] [CrossRef]
- Fisher, G.J.; Shao, Y.; He, T.; Qin, Z.; Perry, D.; Voorhees, J.J.; Quan, T. Reduction of fibroblast size/mechanical force down-regulates TGF-β type ii receptor: Implications for human skin aging. Aging Cell 2016, 15, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Varani, J.; Schuger, L.; Dame, M.K.; Leonard, C.; Fligiel, S.E.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Reduced fibroblast interaction with intact collagen as a mechanism for depressed collagen synthesis in photodamaged skin. J. Investig. Dermatol. 2004, 122, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Wang, F.; Shao, Y.; Rittie, L.; Xia, W.; Orringer, J.S.; Voorhees, J.J.; Fisher, G.J. Enhancing structural support of the dermal microenvironment activates fibroblasts, endothelial cells, and keratinocytes in aged human skin in vivo. J. Investig. Dermatol. 2013, 133, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Alenghat, F.J.; Nauli, S.M.; Kolb, R.; Zhou, J.; Ingber, D.E. Global cytoskeletal control of mechanotransduction in kidney epithelial cells. Exp. Cell Res. 2004, 301, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Cellular mechanotransduction: Putting all the pieces together again. FASEB J. 2006, 20, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Silver, F.H.; Siperko, L.M.; Seehra, G.P. Mechanobiology of force transduction in dermal tissue. Skin Res. Technol. 2003, 9, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Butler, J.P.; Ingber, D.E. Mechanotransduction across the cell surface and through the cytoskeleton. Science 1993, 260, 1124–1127. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.; Cho, M.K.; Perry, D.; Quan, T. Age-associated reduction of cell spreading induces mitochondrial DNA common deletion by oxidative stress in human skin dermal fibroblasts: Implication for human skin connective tissue aging. J. Biomed. Sci. 2015, 22, 62. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Quan, T.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Oxidative exposure impairs TGF-β pathway via reduction of type II receptor and SMAD3 in human skin fibroblasts. Age 2014, 36, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Robichaud, P.; He, T.; Fisher, G.J.; Voorhees, J.J.; Quan, T. Oxidant exposure induces cysteine-rich protein 61 (CCN1) via c-Jun/AP-1 to reduce collagen expression in human dermal fibroblasts. PLoS ONE 2014, 9, e115402. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Fisher, G.J.; Quan, T. Cysteine-rich protein 61 (CCN1) domain-specific stimulation of matrix metalloproteinase-1 expression through αVβ3 integrin in human skin fibroblasts. J. Biol. Chem. 2013, 288, 12386–12394. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Qin, Z.; Xia, W.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Matrix-degrading metalloproteinases in photoaging. J. Investig. Dermatol. Symp. Proc. 2009, 14, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; He, T.; Shao, Y.; Lin, L.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Elevated cysteine-rich 61 mediates aberrant collagen homeostasis in chronologically aged and photoaged human skin. Am. J. Pathol. 2006, 169, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Qin, Z.; Robichaud, P.; Voorhees, J.J.; Fisher, G.J. CCN1 contributes to skin connective tissue aging by inducing age-associated secretory phenotype in human skin dermal fibroblasts. J. Cell Commun. Signal. 2011, 5, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Little, E.; Quan, H.; Qin, Z.; Voorhees, J.J.; Fisher, G.J. Elevated matrix metalloproteinases and collagen fragmentation in photodamaged human skin: Impact of altered extracellular matrix microenvironment on dermal fibroblast function. J. Investig. Dermatol. 2013, 133, 1362–1366. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Shao, Y.; He, T.; Voorhees, J.J.; Fisher, G.J. Reduced expression of connective tissue growth factor (CTGF/CCN2) mediates collagen loss in chronologically aged human skin. J. Investig. Dermatol. 2010, 130, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Okubo, T.; Voorhees, J.J.; Fisher, G.J.; Quan, T. Elevated cysteine-rich protein 61 (CCN1) promotes skin aging via upregulation of IL-1β in chronically sun-exposed human skin. Age 2014, 36, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Bergfeld, W.F. The aging skin. Int. J. Fertil. Women’s Med. 1997, 42, 57–66. [Google Scholar]
- Smith, J.G., Jr.; Davidson, E.A.; Sams, W.M., Jr.; Clark, R.D. Alterations in human dermal connective tissue with age and chronic sun damage. J. Investig. Dermatol. 1962, 39, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Varani, J.; Warner, R.L.; Gharaee-Kermani, M.; Phan, S.H.; Kang, S.; Chung, J.H.; Wang, Z.Q.; Datta, S.C.; Fisher, G.J.; Voorhees, J.J. Vitamin A antagonizes decreased cell growth and elevated collagen-degrading matrix metalloproteinases and stimulates collagen accumulation in naturally aged human skin. J. Investig. Dermatol. 2000, 114, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Kohrmann, A.; Kammerer, U.; Kapp, M.; Dietl, J.; Anacker, J. Expression of matrix metalloproteinases (MMPs) in primary human breast cancer and breast cancer cell lines: New findings and review of the literature. BMC Cancer 2009, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, L.; Cho, H.; Xie, X.; Eliceiri, G.L. Additional MDA-MB-231 breast cancer cell matrix metalloproteinases promote invasiveness. J. Cell Physiol. 2008, 216, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Brenneisen, P.; Sies, H.; Scharffetter-Kochanek, K. Ultraviolet-B irradiation and matrix metalloproteinases: From induction via signaling to initial events. Ann. N. Y. Acad. Sci. 2002, 973, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.J.; Datta, S.C.; Talwar, H.S.; Wang, Z.Q.; Varani, J.; Kang, S.; Voorhees, J.J. Molecular basis of sun-induced premature skin ageing and retinoid antagonism. Nature 1996, 379, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Chakraborti, S.; Mandal, M.; Das, S.; Mandal, A.; Chakraborti, T. Regulation of matrix metalloproteinases: An overview. Mol. Cell Biochem. 2003, 253, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Benbow, U.; Brinckerhoff, C.E. The AP-1 site and MMP gene regulation: What is all the fuss about? Matrix Biol. 1997, 15, 519–526. [Google Scholar] [CrossRef]
- Gutman, A.; Wasylyk, B. The collagenase gene promoter contains a TPA and oncogene-responsive unit encompassing the PEA3 and AP-1 binding sites. EMBO J. 1990, 9, 2241–2246. [Google Scholar] [PubMed]
- Angel, P.; Karin, M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1991, 1072, 129–157. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H.; Kang, S.; Varani, J.; Lin, J.; Fisher, G.J.; Voorhees, J.J. Decreased extracellular-signal-regulated kinase and increased stress-activated map kinase activities in aged human skin in vivo. J. Investig. Dermatol. 2000, 115, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.H.; Rhie, G.E.; Kim, Y.K.; Park, C.H.; Cho, K.H.; Kim, K.H.; Eun, H.C.; Chung, J.H. H2O2 accumulation by catalase reduction changes map kinase signaling in aged human skin in vivo. J. Investig. Dermatol. 2005, 125, 221–229. [Google Scholar] [PubMed]
- Ignotz, R.A.; Massague, J. Transforming growth factor β stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 1986, 261, 4337–4345. [Google Scholar] [PubMed]
- Patil, A.S.; Sable, R.B.; Kothari, R.M. An update on transforming growth factor-β (TGF-β): Sources, types, functions and clinical applicability for cartilage/bone healing. J. Cell Physiol. 2011, 226, 3094–3103. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Rosenbloom, J.; Jimenez, S.A. Transforming growth factor β (TGF β) causes a persistent increase in steady-state amounts of type I and type III collagen and fibronectin mRNAs in normal human dermal fibroblasts. Biochem. J. 1987, 247, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; He, T.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Connective tissue growth factor: Expression in human skin in vivo and inhibition by ultraviolet irradiation. J. Investig. Dermatol. 2002, 118, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.C.; Young, D.A.; Waters, J.G.; Rowan, A.D.; Chantry, A.; Edwards, D.R.; Clark, I.M. The comparative role of activator protein 1 and smad factors in the regulation of Timp-1 and MMP-1 gene expression by transforming growth factor-β1. J. Biol. Chem. 2003, 278, 10304–10313. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. The transforming growth factor-β family. Annu. Rev. Cell Biol. 1990, 6, 597–641. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. How cells read TGF-β signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; He, T.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation alters transforming growth factor β/smad pathway in human skin in vivo. J. Investig. Dermatol. 2002, 119, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; He, T.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation blocks cellular responses to transforming growth factor-β by down-regulating its type-II receptor and inducing Smad7. J. Biol. Chem. 2001, 276, 26349–26356. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; He, T.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Solar ultraviolet irradiation reduces collagen in photoaged human skin by blocking transforming growth factor-β type II receptor/Smad signaling. Am. J. Pathol. 2004, 165, 741–751. [Google Scholar] [CrossRef]
- Lau, L.F.; Lam, S.C. The CCN family of angiogenic regulators: The integrin connection. Exp. Cell Res. 1999, 248, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Planque, N.; Perbal, B. A structural approach to the role of CCN (CYR61/CTGF/NOV) proteins in tumourigenesis. Cancer Cell Int. 2003, 3, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perbal, B. CCN proteins: Multifunctional signalling regulators. Lancet 2004, 363, 62–64. [Google Scholar] [CrossRef]
- Leask, A.; Abraham, D.J. All in the CCN family: Essential matricellular signaling modulators emerge from the bunker. J. Cell Sci. 2006, 119, 4803–4810. [Google Scholar] [CrossRef] [PubMed]
- Perbal, B.; Brigstock, D.R.; Lau, L.F. Report on the second international workshop on the CCN family of genes. Mol. Pathol. 2003, 56, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Lau, L.F. Functions and mechanisms of action of CCN matricellular proteins. Int. J. Biochem. Cell Biol. 2009, 41, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Kireeva, M.L.; Mo, F.E.; Yang, G.P.; Lau, L.F. Cyr61, a product of a growth factor-inducible immediate-early gene, promotes cell proliferation, migration, and adhesion. Mol. Cell Biol. 1996, 16, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Chen, N.; Lau, L.F. The angiogenic factors Cyr61 and connective tissue growth factor induce adhesive signaling in primary human skin fibroblasts. J. Biol. Chem. 2001, 276, 10443–10452. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.E.; Muntean, A.G.; Chen, C.C.; Stolz, D.B.; Watkins, S.C.; Lau, L.F. Cyr61 (CCN1) is essential for placental development and vascular integrity. Mol. Cell Biol. 2002, 22, 8709–8720. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Qin, Z.; Xu, Y.; He, T.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation induces Cyr61/CCN1, a mediator of collagen homeostasis, through activation of transcription factor AP-1 in human skin fibroblasts. J. Investig. Dermatol. 2010, 130, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Shin, S.; Qin, Z.; Fisher, G.J. Expression of CCN family of genes in human skin in vivo and alterations by solar-simulated ultraviolet irradiation. J. Cell Commun. Signal. 2009, 3, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Qin, Z.; Voorhees, J.J.; Fisher, G.J. Cysteine-rich protein 61 (CCN1) mediates replicative senescence-associated aberrant collagen homeostasis in human skin fibroblasts. J. Cell. Biochem. 2012, 113, 3011–3018. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Mo, F.E.; Lau, L.F. The angiogenic factor Cyr61 activates a genetic program for wound healing in human skin fibroblasts. J. Biol. Chem. 2001, 276, 47329–47337. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.; Chen, C.C.; Lau, L.F. Matricellular protein CCN1 activates a proinflammatory genetic program in murine macrophages. J. Immunol. 2010, 184, 3223–3232. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 945–963. [Google Scholar] [CrossRef] [PubMed]
- Daynes, R.A.; Araneo, B.A.; Ershler, W.B.; Maloney, C.; Li, G.Z.; Ryu, S.Y. Altered regulation of IL-6 production with normal aging. Possible linkage to the age-associated decline in dehydroepiandrosterone and its sulfated derivative. J. Immunol. 1993, 150, 5219–5230. [Google Scholar] [PubMed]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Maggio, M.; Guralnik, J.M.; Longo, D.L.; Ferrucci, L. Interleukin-6 in aging and chronic disease: A magnificent pathway. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 575–584. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, Y.; Quan, T. Oxidative Stress and Human Skin Connective Tissue Aging. Cosmetics 2016, 3, 28. https://doi.org/10.3390/cosmetics3030028
Tu Y, Quan T. Oxidative Stress and Human Skin Connective Tissue Aging. Cosmetics. 2016; 3(3):28. https://doi.org/10.3390/cosmetics3030028
Chicago/Turabian StyleTu, Yidong, and Taihao Quan. 2016. "Oxidative Stress and Human Skin Connective Tissue Aging" Cosmetics 3, no. 3: 28. https://doi.org/10.3390/cosmetics3030028