
Ribosome Assembly as Antimicrobial Target

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- (1)
- Mutations of membrane components affect the permeability barrier and, alternatively, transport proteins are affected, shifting the import:export ratio towards the export.
- (2)
- The antibiotic target is altered by blocking binding or modifying the binding site, causing insensitivity to the drug. Mutation rates for types 1 and 2 are in the range of 10−6 to 10−8, i.e., one bacterium out of 106 to 108 is resistant to the respective drug.
- (3)
- Plasmids coding for enzymes that modify (acetylation, phosphorylation of adenylation) or degrade the antibiotic [3].
- (4)
- (5)
- Rare mechanisms are dilution (overproduction) of the target molecule or activation of alternative pathways. Both are known for trimethoprim that inhibits dihydrofolate reductase [16].
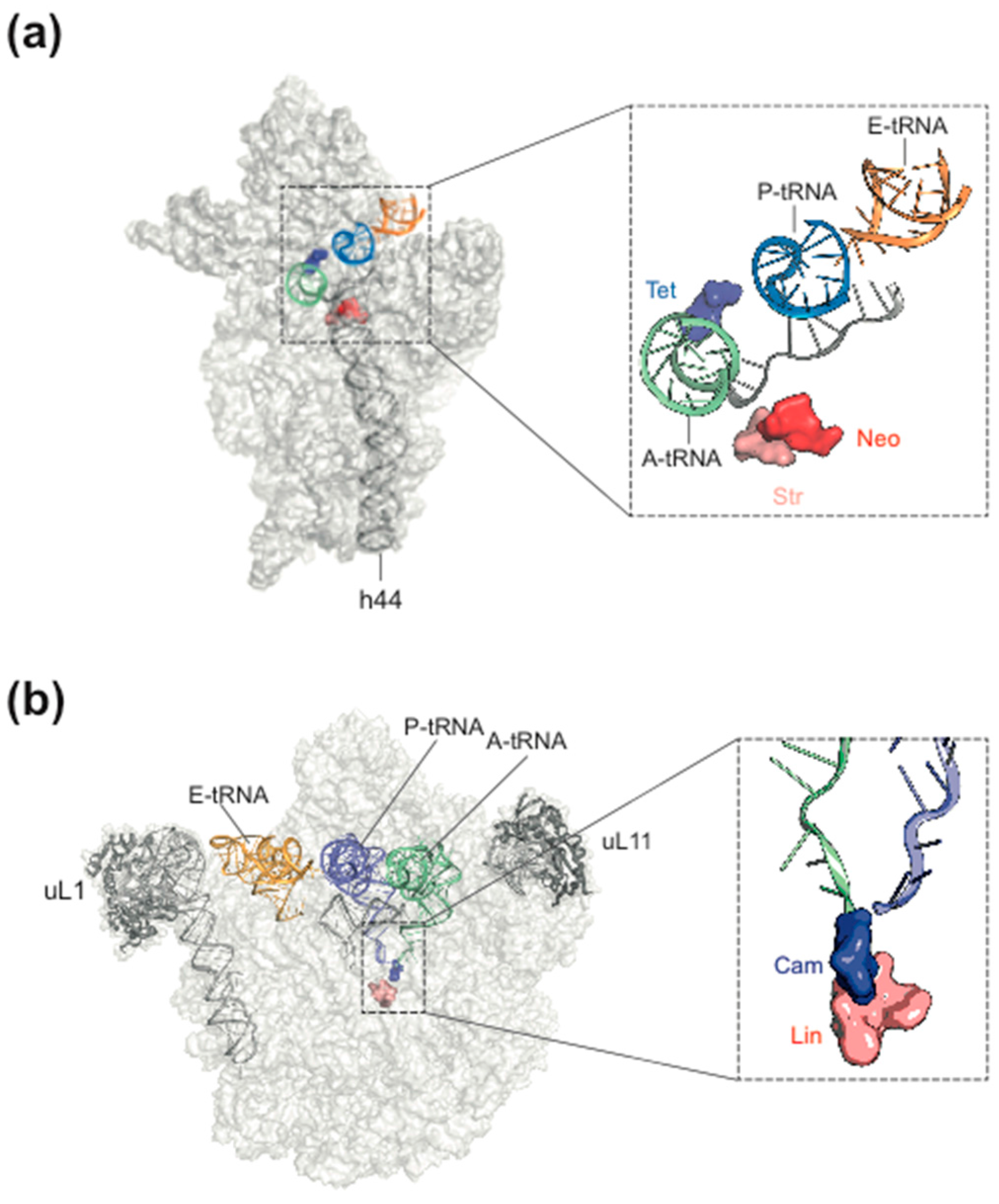
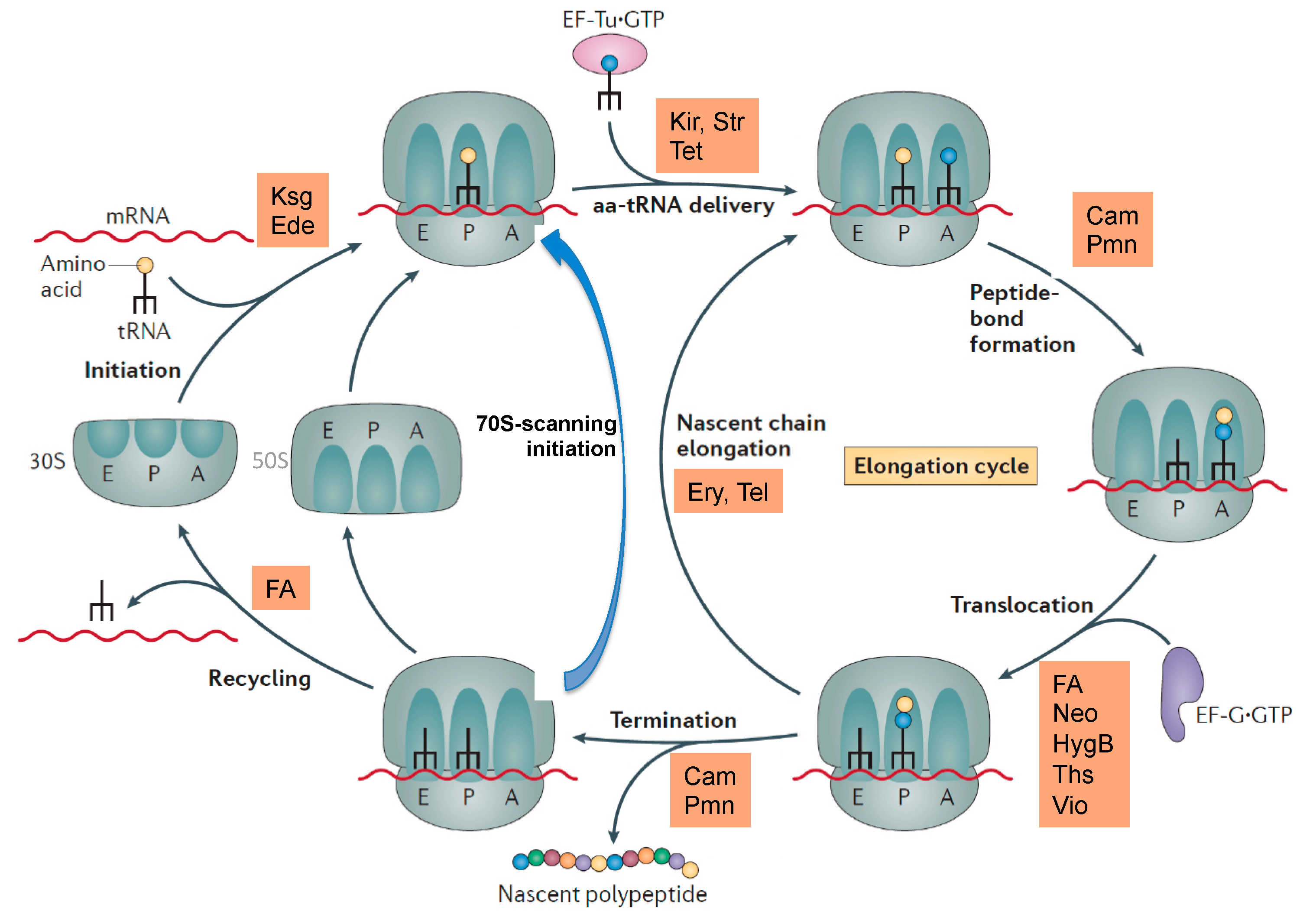
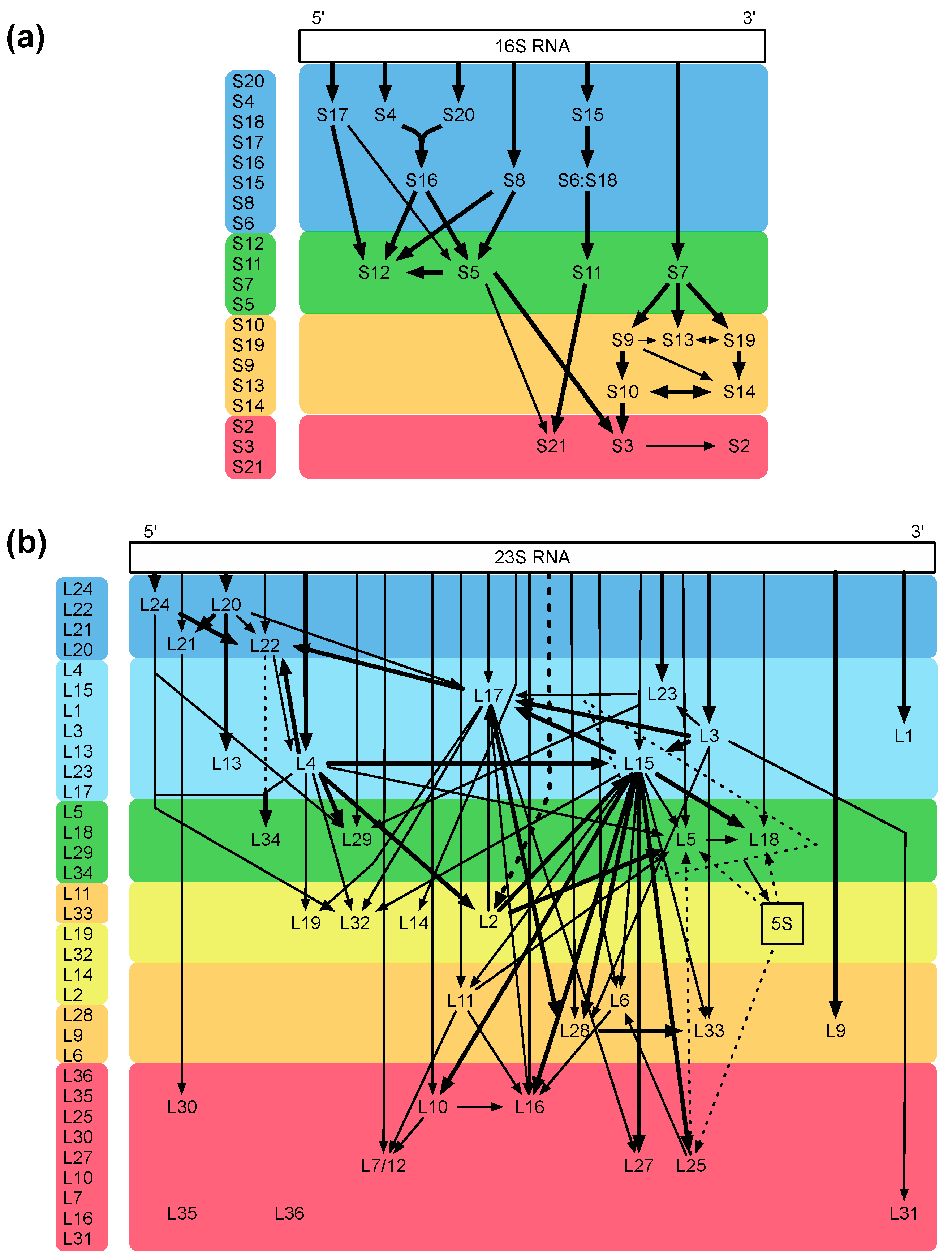
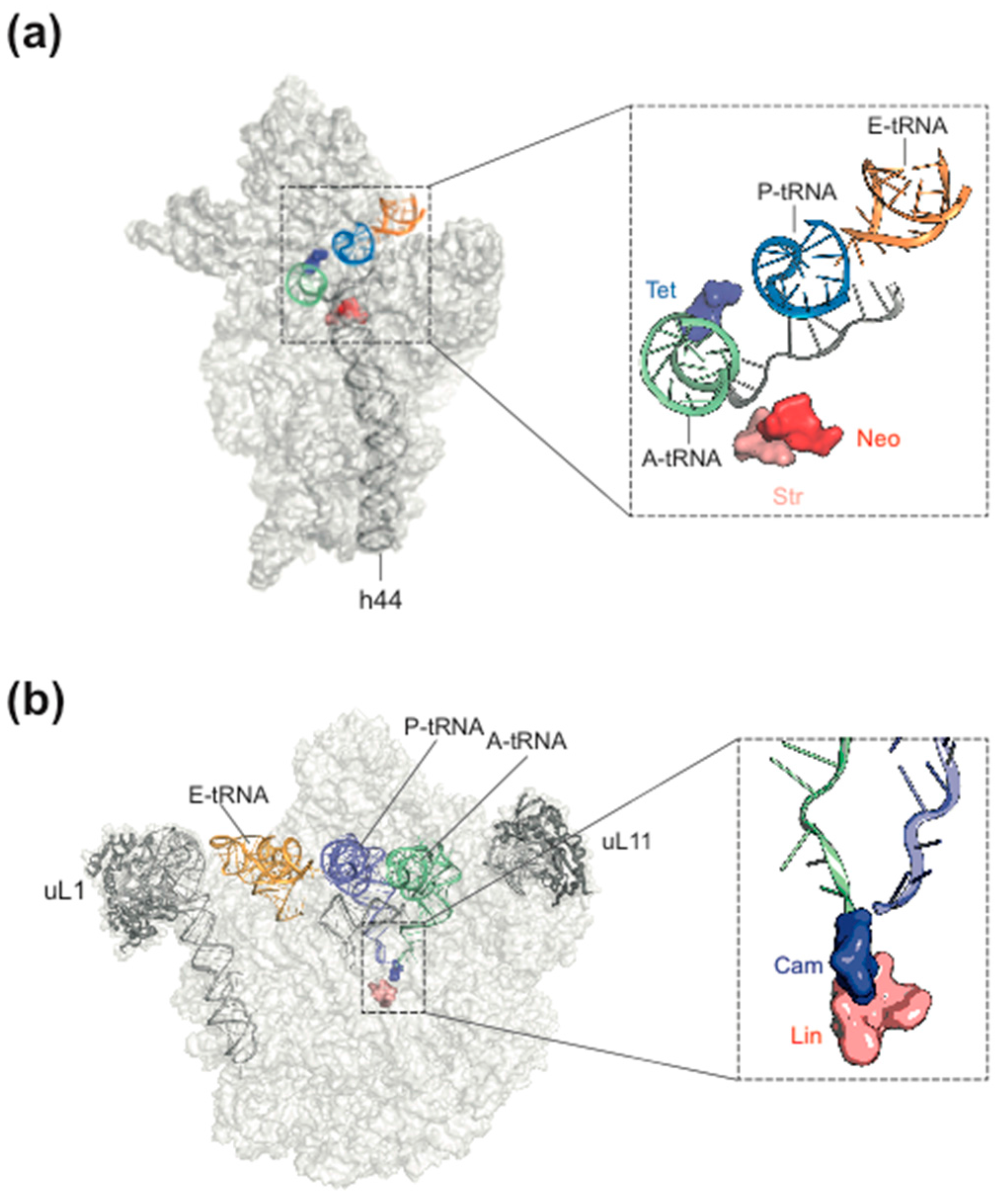
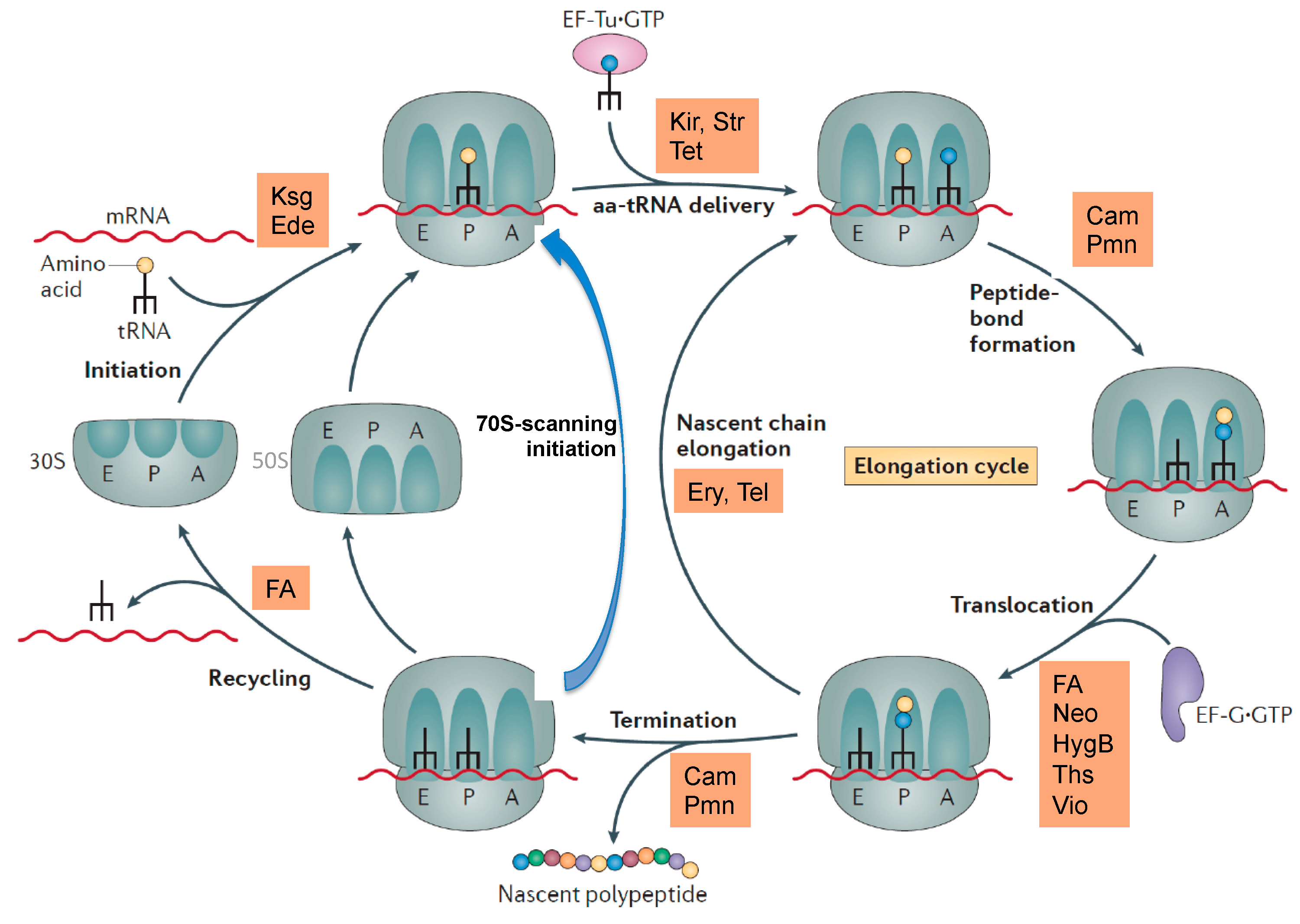
2. Ribosome Assembly as Attractive Target for New Antimicrobials
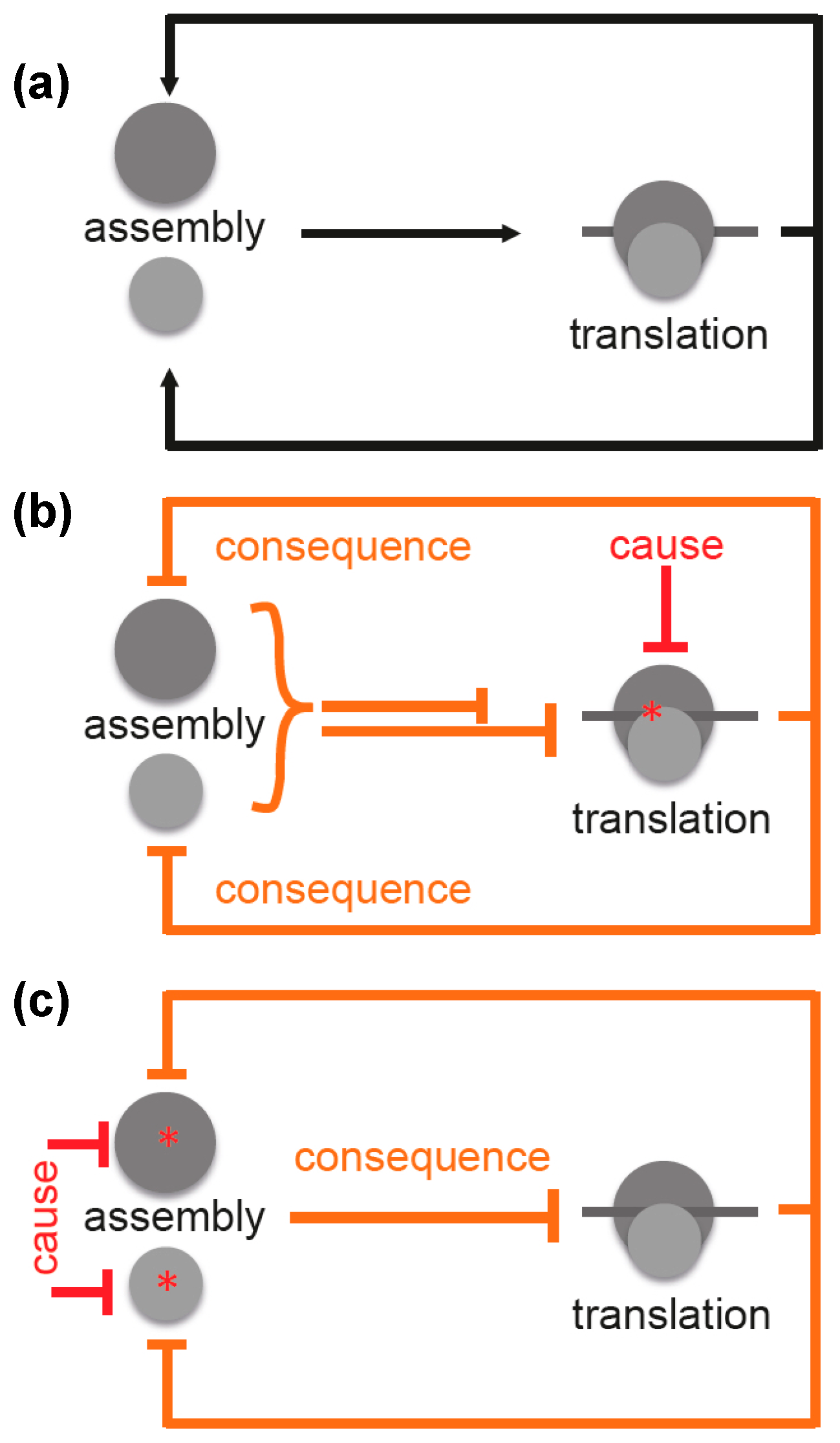
3. Ribosome Assembly and Translation Are Coupled in Bacteria
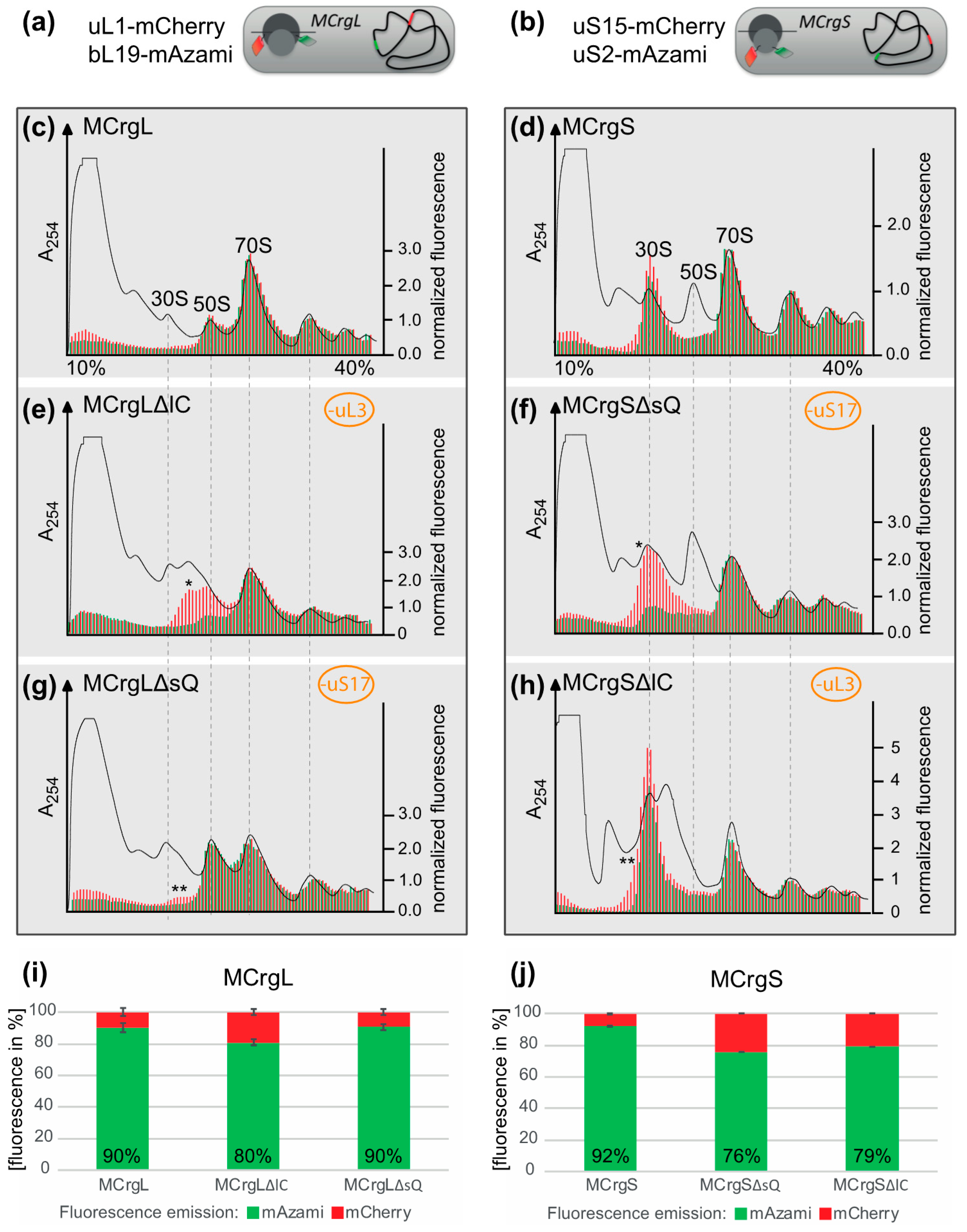
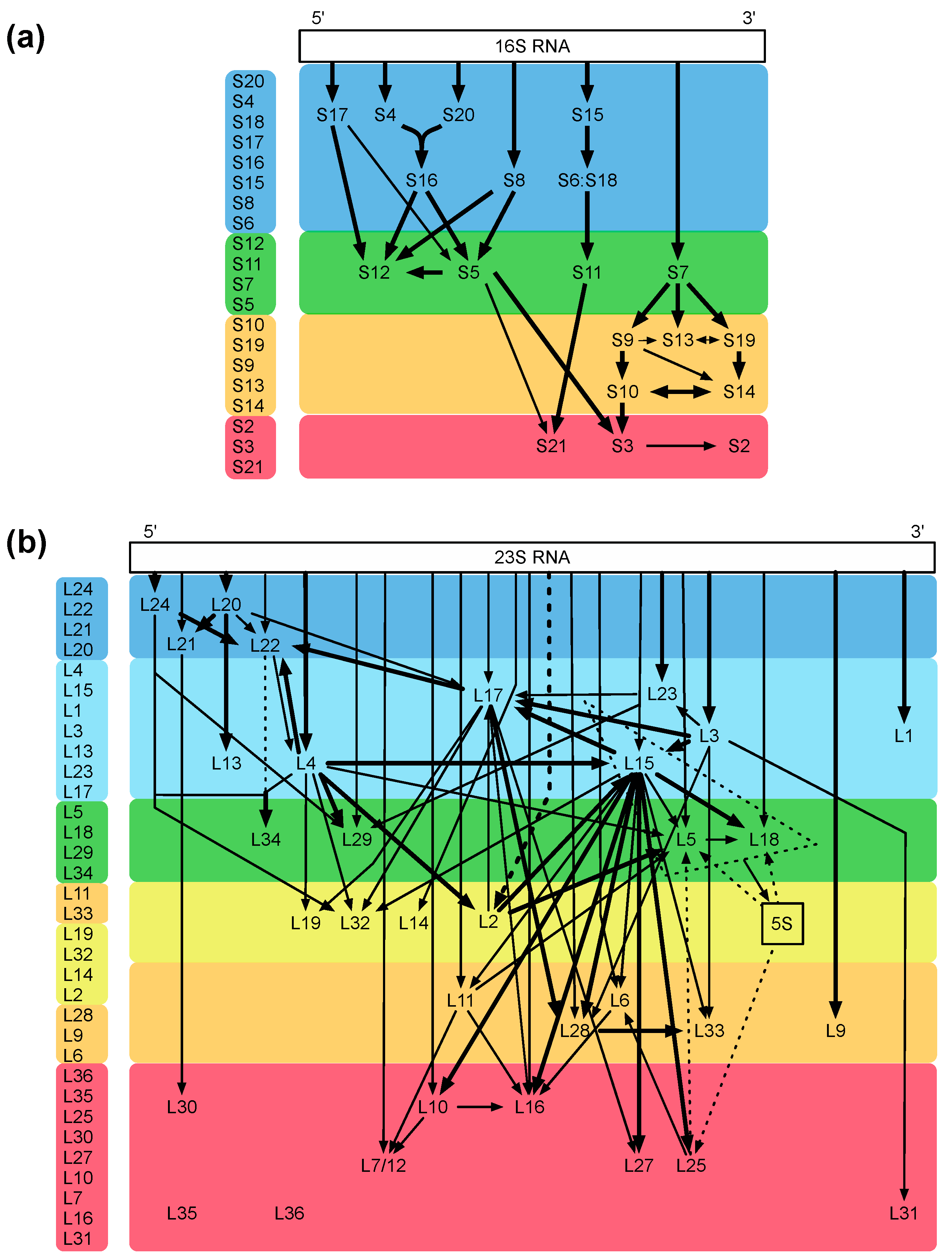
4. Specific Readouts for Assembly Are Needed
5. Materials and Methods
5.1. Media, Buffers, Antibodies and Antibiotics
5.2. Plasmids and Bacterial Strains
5.3. λ-Red Recombineering
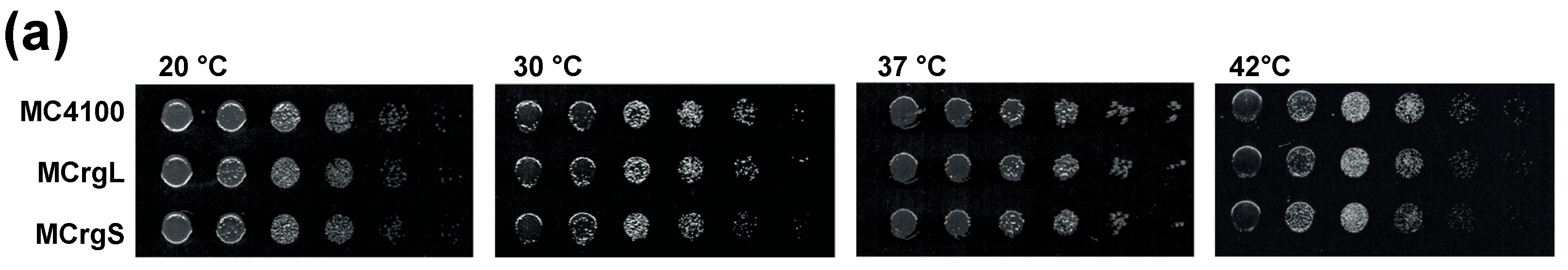
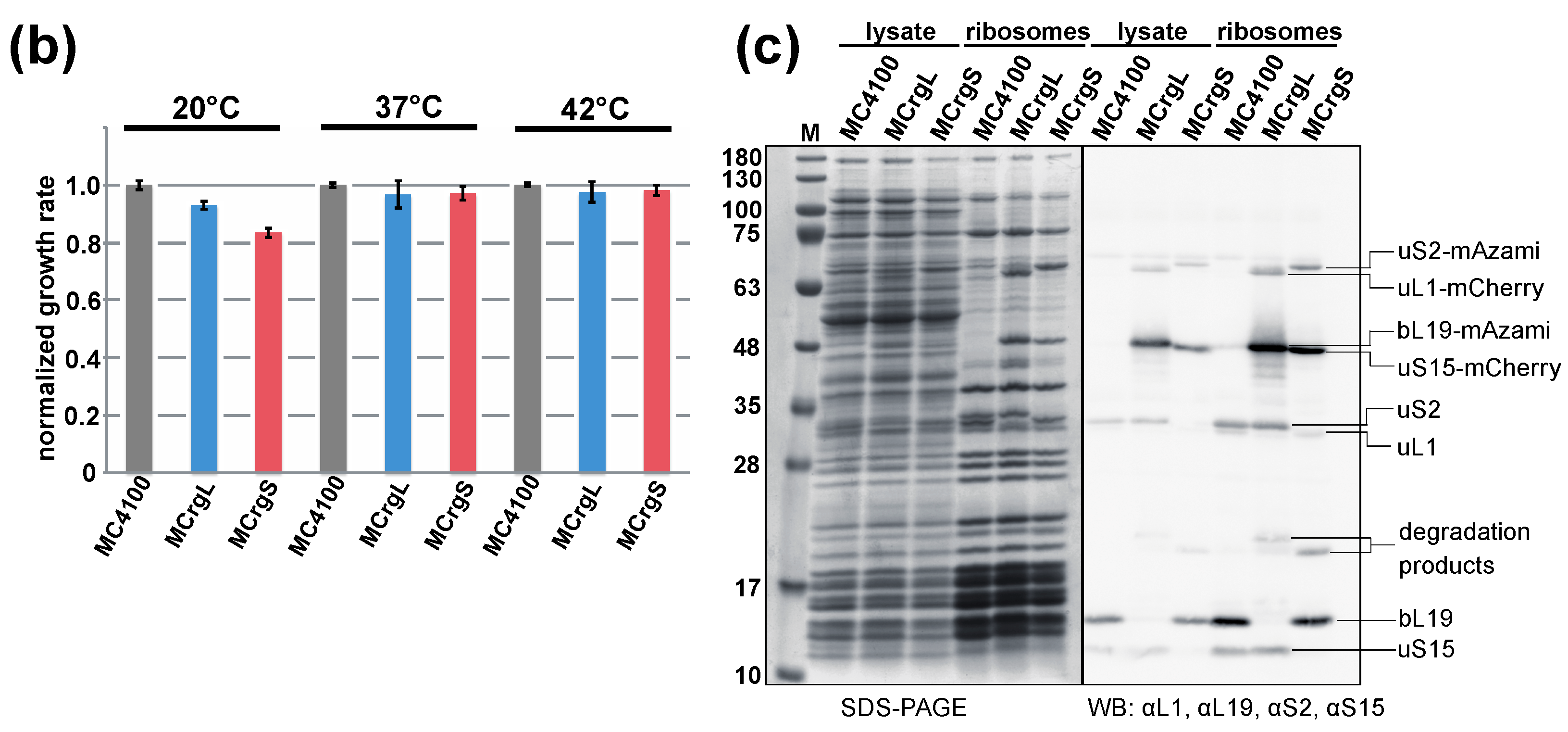
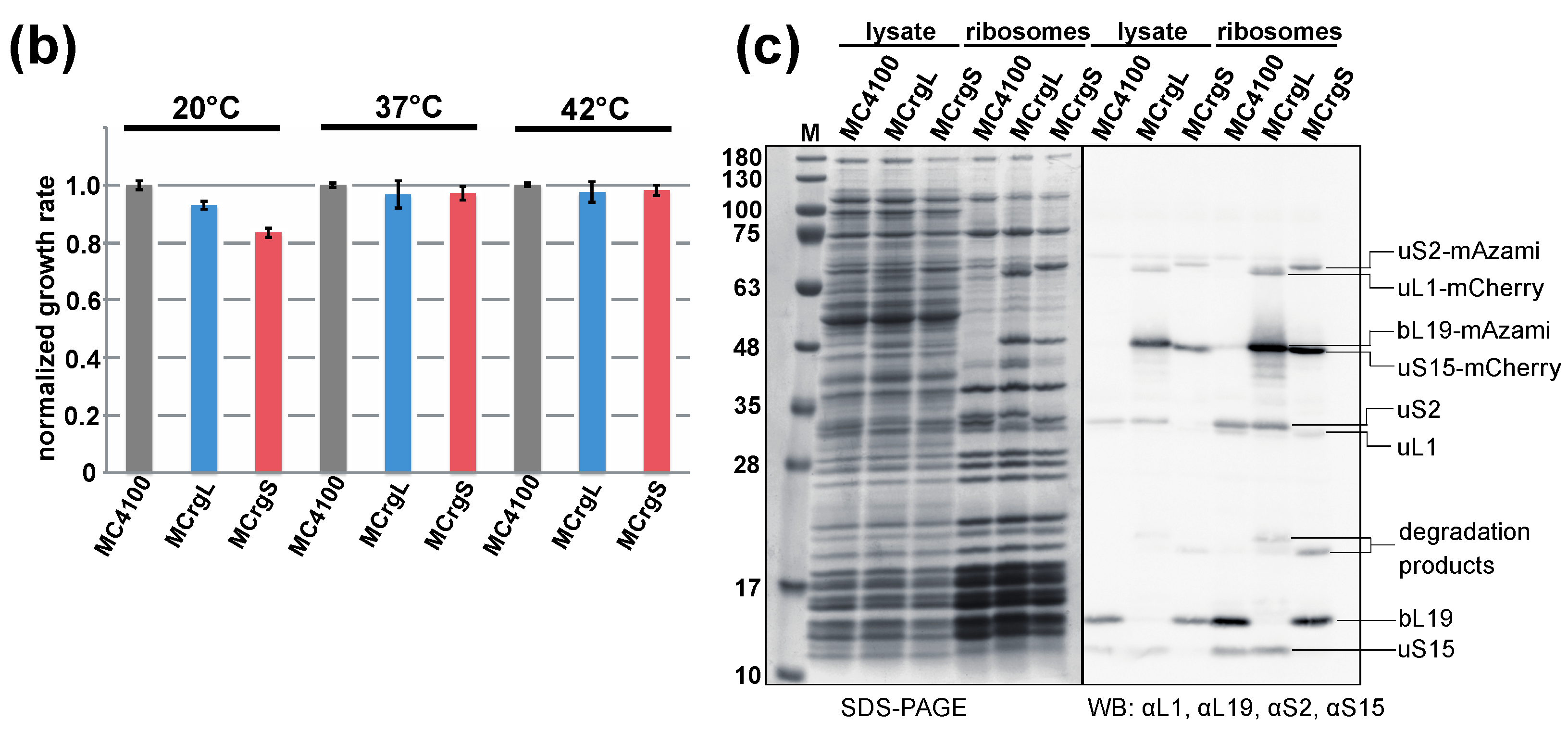
5.4. Cell Growth Analyses
5.5. Purification of Ribosomes by Sucrose Cushion Centrifugation
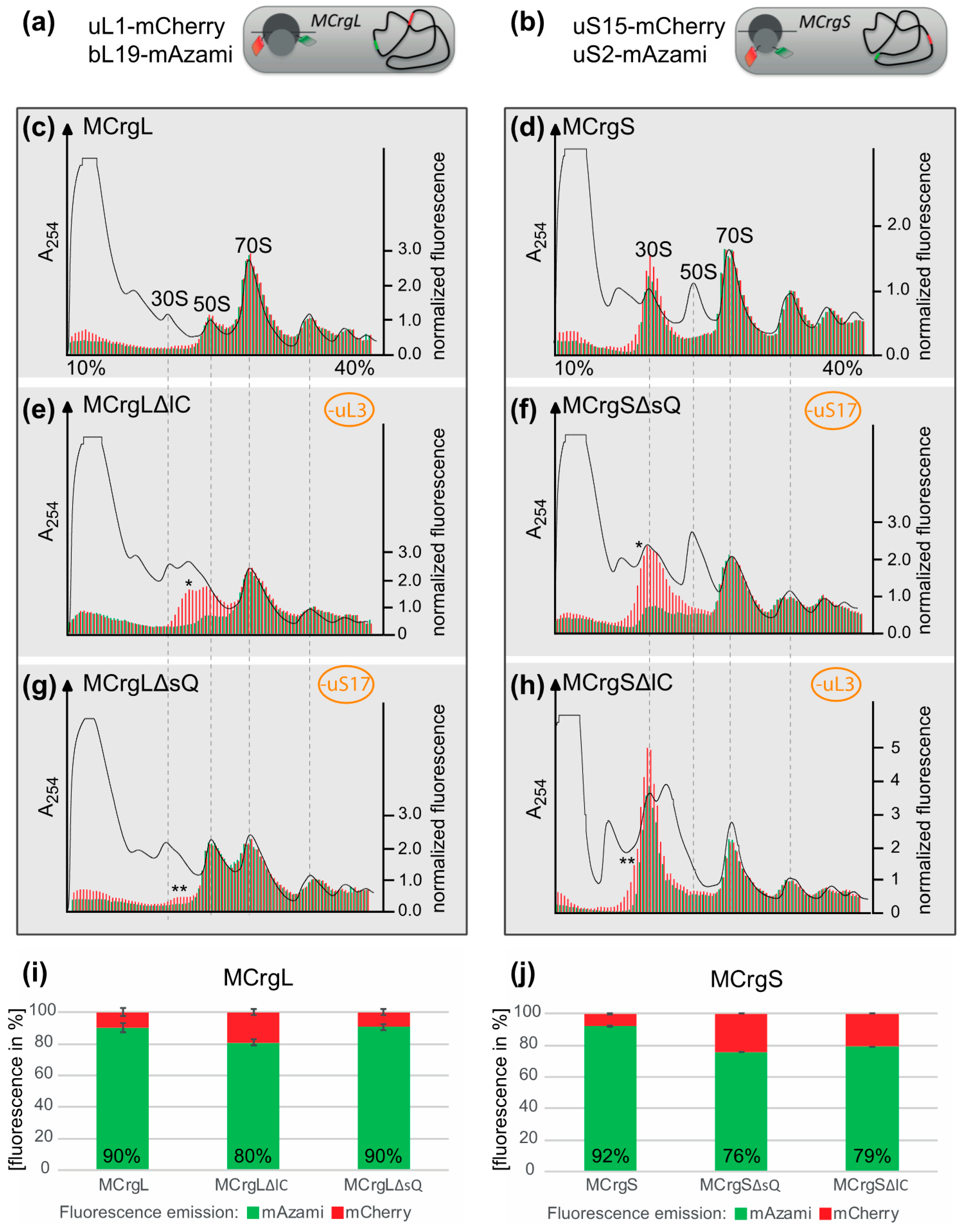
5.6. Sucrose Gradient Centrifugation
5.7. Polysome Analysis and Fluorometric Analysis of the Sucrose Fractions
6. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Da | molecular mass in Dalton |
| EGFP | Enhanced green fluorescent protein |
| LB | Lysogeny broth (medium) |
| LB agar | LB and agar containing breeding grounds |
| rRNA | ribosomal RNA |
| PTC | peptidyl transferase center |
| tRNA | transfer RNA |
| TRIS | Tris(hydroxymethyl)-aminomethan |
| aa-tRNA | aminoacyl tRNA |
| GTP | guanosin triphosphate |
| GDP | guanosin diphosphate |
| EF-Tu | elongation factor thermo-unstable |
| EF-G | elongation factor G |
| FA | fusidic acid |
| mAzami | Monomeric green fluorescent protein |
| mCherry | Monomeric red fluorescent protein |
| OD600 | Optical density at 600 nm wavelength |
| S.D. | standard deviation |
| SDS PAGE | Sodium dodecyl sulfate poly acrylamide gel electrophoresis |
References
- Cundliffe, E.; Demain, A.L. Avoidance of suicide in antibiotic-producing microbes. J. Ind. Microbiol. Biotechnol. 2010, 37, 643–672. [Google Scholar] [CrossRef] [PubMed]
- Nierhaus, K.H.; Wittmann, H.G. Ribosomal function and its inhibition by antibiotics in prokaryotes. Naturwissenschaften 1980, 67, 234–250. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Nierhaus, K.H.; Wilson, D.N. Protein Synthesis and Ribosome Structure: Translating the Genome; WILEY-VCH Verlag GmbH&Co.: Weinheim, Germay, 2004; p. 579. [Google Scholar]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Tenson, T.; Mankin, A. Antibiotics and the ribosome. Mol. Microbiol. 2006, 59, 1664–1677. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 2014, 12, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, D. Inhibitors of protein biosynthesis. Mol. Biol. Biochem. Biophys. 1979, 30. Available online: http://link.springer.com/book/10.1007/978-3-642-81309-2 (accessed on 23 May 2016). [Google Scholar]
- Cundliffe, E. Antibiotic inhibitors of ribosome function. In The Molecular Basis of Antibiotic Action; Gale, E.F., Cundliffe, E., Reynolds, P.E., Richmond, M.H., Waring, M.J., Eds.; John Willey & Sons: London, UK; New York, NY, USA; Sydney, Australia; Toronto, ON, Canada, 1981; pp. 402–545. [Google Scholar]
- Polacek, N.; Mankin, A.S. The ribosomal peptidyl transferase center: Structure, function, evolution, inhibition. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 285–311. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Wittek, D.; Gupta, R.; Qin, B.; Ueda, T.; Krause, R.; Yamamoto, K.; Albrecht, R.; Pech, M.; Nierhaus, K.H. 70S-scanning initiation is a novel and frequent initiation mode of ribosomal translation in bacteria. Proc. Natl. Acad. Sci. USA 2016, 113, E1180–E1189. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Connell, S.R.; Trieber, C.A.; Dinos, G.P.; Einfeldt, E.; Taylor, D.E.; Nierhaus, K.H. Mechanism of Tet(O)-mediated tetracycline resistance. EMBO J. 2003, 22, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Connell, S.R.; Tracz, D.M.; Nierhaus, K.H.; Taylor, D.E. Ribosomal protection proteins and their mechanism of tetracycline resistance. Antimicrob. Agents Chemother. 2003, 47, 3675–3681. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, F.; Starosta, A.L.; Arenz, S.; Sohmen, D.; Donhofer, A.; Wilson, D.N. Tetracycline antibiotics and resistance mechanisms. Biol. Chem. 2014, 395, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Huovinen, P. Trimethoprim resistance. Antimicrob. Agents Chemother. 1987, 31, 1451–1456. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Farha, M.A.; Brown, E.D. Unconventional screening approaches for antibiotic discovery. Ann. N. Y. Acad. Sci. 2015, 1354, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Maguire, B.A. Inhibition of bacterial ribosome assembly: A suitable drug target? Microbiol. Mol. Biol. Rev. 2009, 73, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Traub, P.; Nomura, M. Structure and function of E. coli ribosomes. V. Reconstitution of functionally active 30S ribosomal particles from RNA and proteins. Proc. Natl. Acad. Sci. USA 1968, 59, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Nierhaus, K.H.; Dohme, F. Total reconstitution of functionally active 50S ribosomal subunits from Escherichia coli. Proc. Natl. Acad. Sci. USA 1974, 71, 4713–4717. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, V.; Nierhaus, K.H. Initiator proteins for the assembly of the 50S subunit from E. coli ribosomes. Proc. Natl. Acad. Sci. USA 1982, 79, 7238–7242. [Google Scholar] [CrossRef] [PubMed]
- Spillmann, S.; Dohme, F.; Nierhaus, K.H. Assembly in vitro of the 50S subunit from Escherichia coli ribosomes: Proteins essential for the first heat dependent conformational change. J. Mol. Biol. 1977, 115, 513–523. [Google Scholar] [CrossRef]
- Nowotny, V.; Nierhaus, K.H. Protein l20 from the large subunit of Escherichia coli ribosomes is an assembly protein. J. Mol. Biol. 1980, 137, 391–399. [Google Scholar] [CrossRef]
- Held, W.A.; Ballou, B.; Mizushima, S.; Nomura, M. Assembly mapping of 30S ribosomal proteins from E. coli. Further studies. J. Biol. Chem. 1974, 249, 3103–3111. [Google Scholar] [PubMed]
- Herold, M.; Nierhaus, K.H. Incorporation of six additional proteins to complete the assembly map of the 50 S subunit from Escherichia coli ribosomes. J. Biol. Chem. 1987, 262, 8826–8833. [Google Scholar] [PubMed]
- Nierhaus, K.H. The assembly of prokaryotic ribosomes. Biochimie 1991, 73, 739–755. [Google Scholar] [CrossRef]
- Chen, S.S.; Williamson, J.R. Characterization of the ribosome biogenesis landscape in E. coli using quantitative mass spectrometry. J. Mol. Biol. 2013, 425, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Kurland, C.G.; Nomura, M.; Watson, J.D. The physical properties of the chloromycetin particles. J. Mol. Biol. 1962, 4, 388–394. [Google Scholar] [CrossRef]
- MacKenzie, F.M.; Gould, I.M. The post-antibiotic effect. J. Antimicrob. Chemother. 1993, 32, 519–537. [Google Scholar] [CrossRef] [PubMed]
- Bogenhagen, D.F.; Martin, D.W.; Koller, A. Initial steps in RNA processing and ribosome assembly occur at mitochondrial DNA nucleoids. Cell Metab. 2014, 19, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Yunis, A.A. Chloramphenicol toxicity: 25 years of research. Am. J. Med. 1989, 87, 44N–48N. [Google Scholar] [PubMed]
- Kuter, D.J.; Tillotson, G.S. Hematologic effects of antimicrobials: Focus on the oxazolidinone linezolid. Pharmacotherapy 2001, 21, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Bottger, E.C. Mutant A1555G mitochondrial 12S rRNA and aminoglycoside susceptibility. Antimicrob. Agents Chemother. 2010, 54, 3073–3075. [Google Scholar] [CrossRef] [PubMed]
- Takebe, Y.; Miura, A.; Bedwell, D.M.; Tam, M.; Nomura, M. Increased expression of ribosomal genes during inhibition of ribosome assembly in Escherichia coli. J. Mol. Biol. 1985, 184, 23–30. [Google Scholar] [CrossRef]
- Champney, W.S.; Burdine, R. 50S ribosomal subunit synthesis and translation are equivalent targets for erythromycin inhibition in staphylococcus aureus. Antimicrob. Agents Chemother. 1996, 40, 1301–1303. [Google Scholar] [PubMed]
- Siibak, T.; Peil, L.; Xiong, L.; Mankin, A.; Remme, J.; Tenson, T. Erythromycin- and chloramphenicol-induced ribosomal assembly defects are secondary effects of protein synthesis inhibition. Antimicrob. Agents Chemother. 2009, 53, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Siibak, T.; Peil, L.; Donhofer, A.; Tats, A.; Remm, M.; Wilson, D.N.; Tenson, T.; Remme, J. Antibiotic-induced ribosomal assembly defects result from changes in the synthesis of ribosomal proteins. Mol. Microbiol. 2011, 80, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Kannan, K.; Vazquez-Laslop, N.; Mankin, A.S. Selective protein synthesis by ribosomes with a drug-obstructed exit tunnel. Cell 2012, 151, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.; Champney, W.S. 30S ribosomal subunit assembly is a target for inhibition by aminoglycosides in Escherichia coli. Antimicrob. Agents Chemother. 2002, 46, 1546–1549. [Google Scholar] [CrossRef] [PubMed]
- Nikolay, R.; Schloemer, R.; Schmidt, S.; Mueller, S.; Heubach, A.; Deuerling, E. Validation of a fluorescence-based screening concept to identify ribosome assembly defects in Escherichia coli. Nucleic Acids Res. 2014, 42, e100. [Google Scholar] [CrossRef] [PubMed]
- Nikolay, R.; Schloemer, R.; Mueller, S.; Deuerling, E. Fluorescence-based monitoring of ribosome assembly landscapes. BMC Mol. Biol. 2015, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Herzog, A.; Yaguchi, M.; Cabezon, T.; Corchuelo, M.C.; Petre, J.; Bollen, A. A missense mutation in the gene coding for ribosomal protein S17 (rpsQ) leading to ribosomal assembly defectivity in Escherichia coli. Mol. Gen. Genet. 1979, 171, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Lhoest, J.; Colson, C. Cold-sensitive ribosome assembly in an Escherichia coli mutant lacking a single methyl group in ribosomal protein l3. Eur. J. Biochem. 1981, 121, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Ellis, H.M.; Lee, E.C.; Jenkins, N.A.; Copeland, N.G.; Court, D.L. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. USA 2000, 97, 5978–5983. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolay, R.; Schmidt, S.; Schlömer, R.; Deuerling, E.; Nierhaus, K.H. Ribosome Assembly as Antimicrobial Target. Antibiotics 2016, 5, 18. https://doi.org/10.3390/antibiotics5020018
Nikolay R, Schmidt S, Schlömer R, Deuerling E, Nierhaus KH. Ribosome Assembly as Antimicrobial Target. Antibiotics. 2016; 5(2):18. https://doi.org/10.3390/antibiotics5020018
Chicago/Turabian StyleNikolay, Rainer, Sabine Schmidt, Renate Schlömer, Elke Deuerling, and Knud H. Nierhaus. 2016. "Ribosome Assembly as Antimicrobial Target" Antibiotics 5, no. 2: 18. https://doi.org/10.3390/antibiotics5020018