Synthesis and Biological Evaluation of a New Polymeric Conjugate and Nanocarrier with Osteotropic Properties



Abstract
:1. Introduction
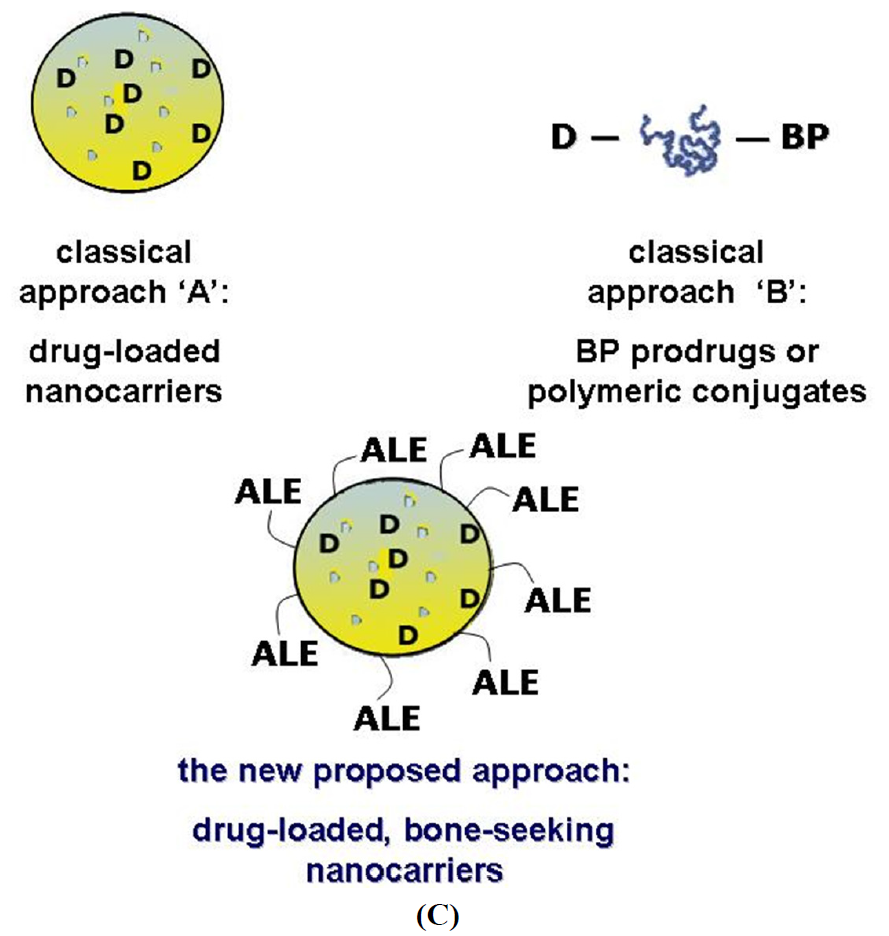
1.1. Approaches for Drug Targeting to Bone
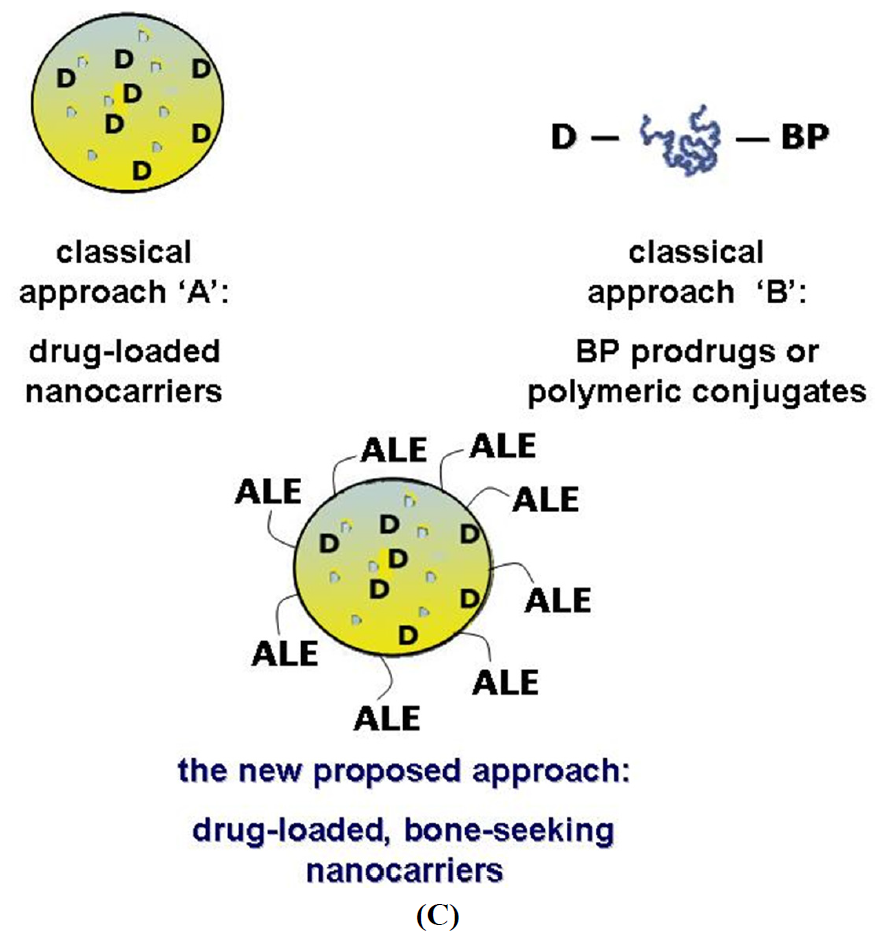
1.2. A New Targeting Strategy
2. Experimental Section
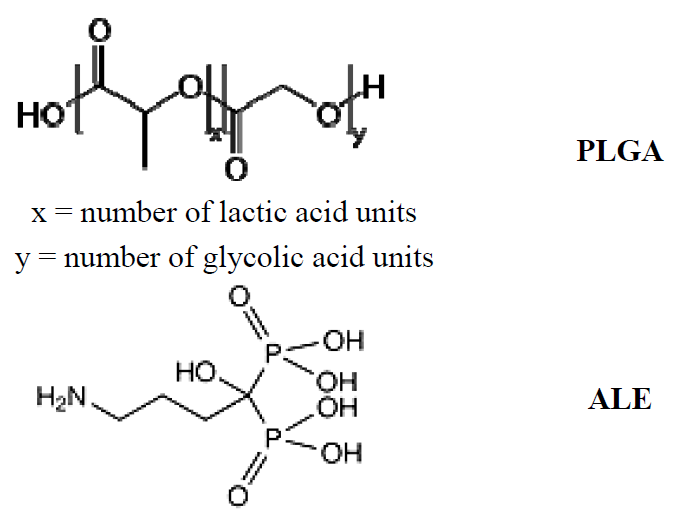
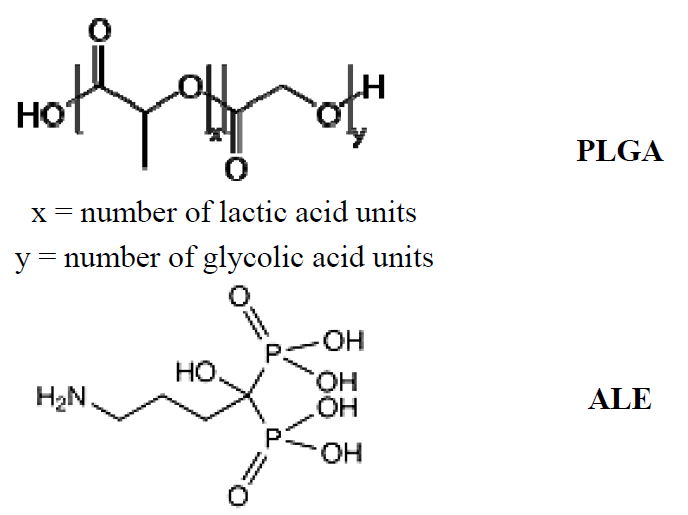
2.1. Synthesis and Characterization of the PLGA-ALE Conjugate
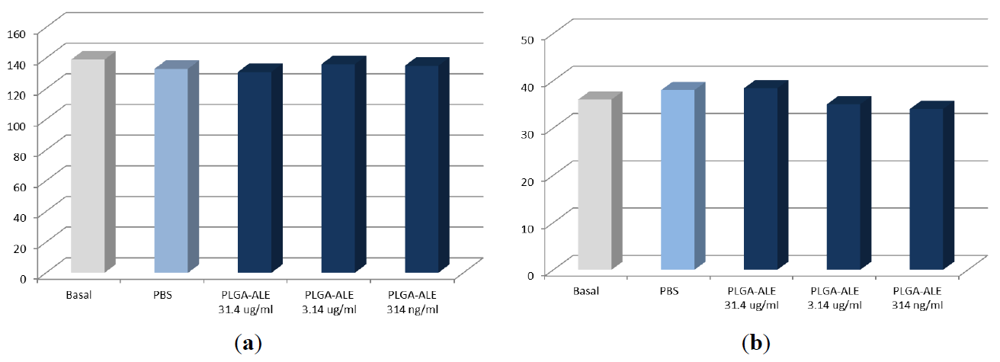
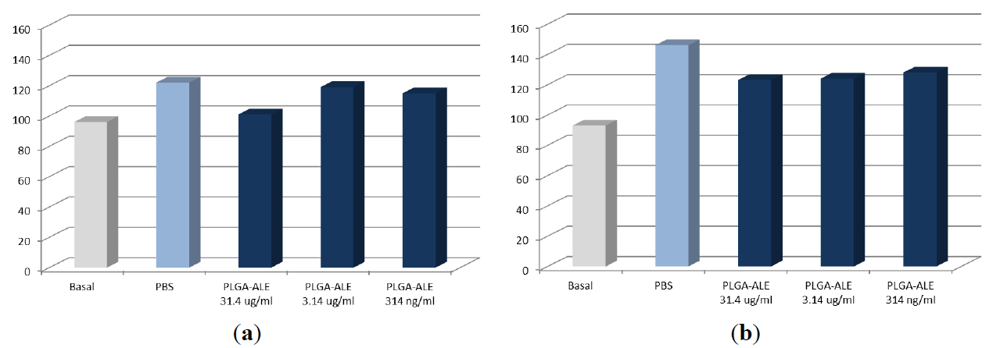
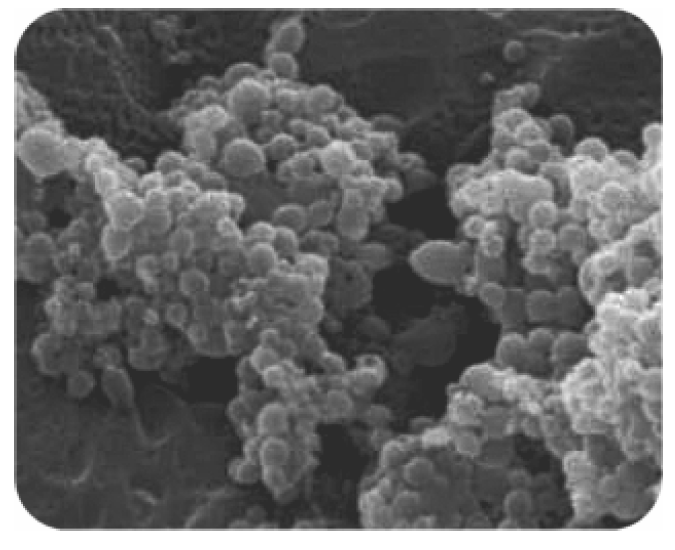
2.2. PLGA-ALE Nanoparticles

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
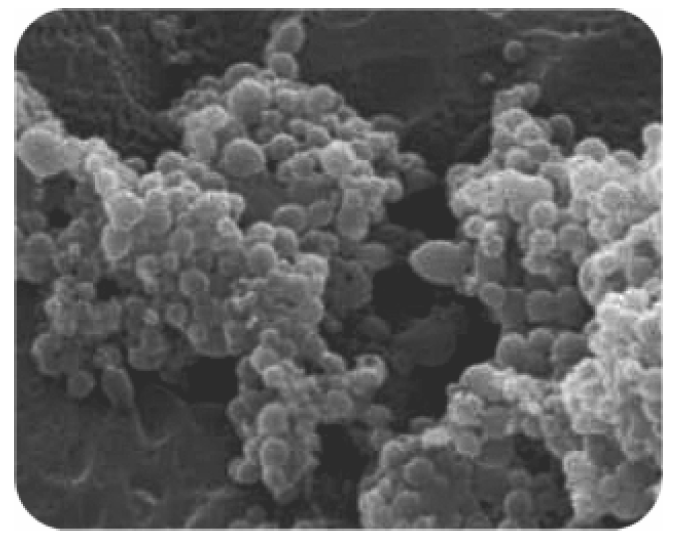
{kind=link}
| Production method | Zeta Potential (mV) | Mean Particle Size (nm) | PDI |
|---|---|---|---|
| Solvent evaporation (organic phase) | |||
| Acetone | −37.6 ± 4.1 | 188.0 ± 21.3 | 0.258 |
| DMSO | −38.9 ± 3.7 | 286.9 ± 9.60 | 0.156 |
| Acetone/DMSO (1:1, v/v) | −37.2 ± 5.0 | 198.7 ± 17.3 | 0.348 |
| Dialysis | −39.3 ± 4.7 | 438.6 ± 11.1 | 0.033 |
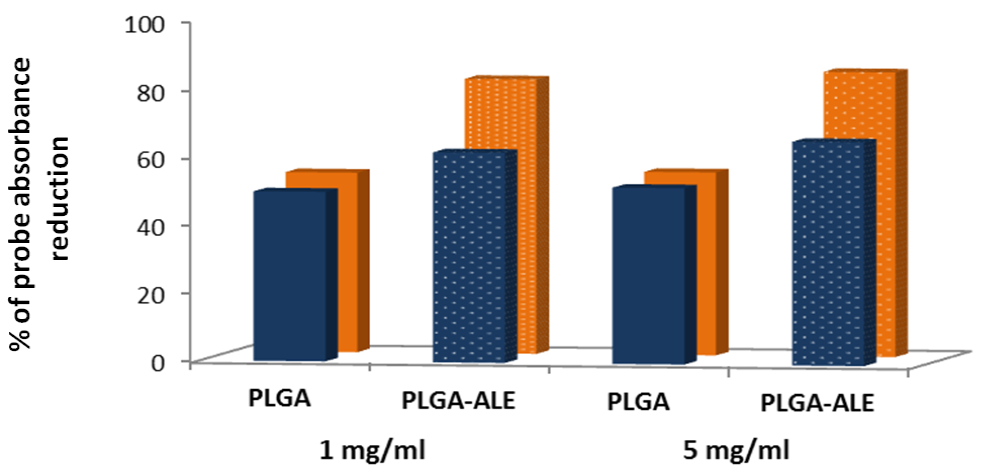
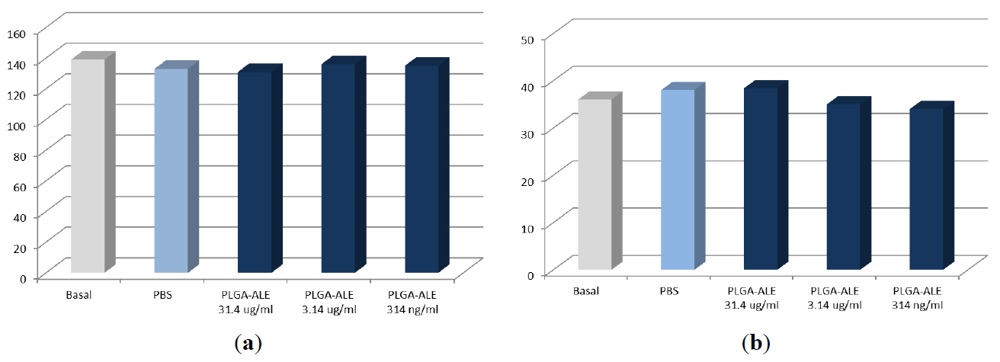
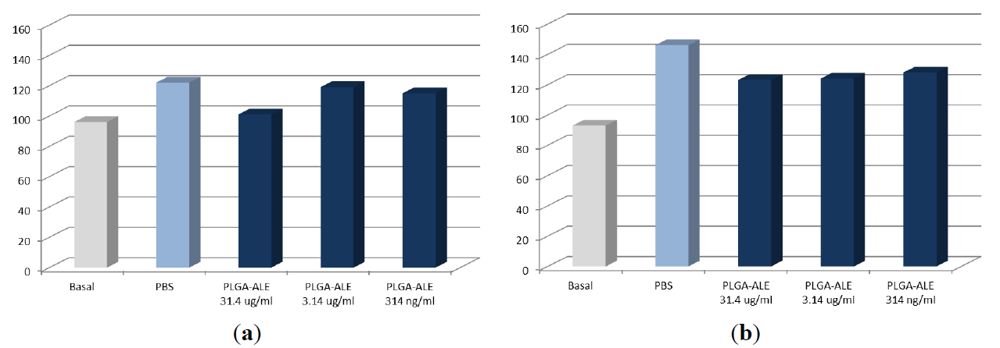
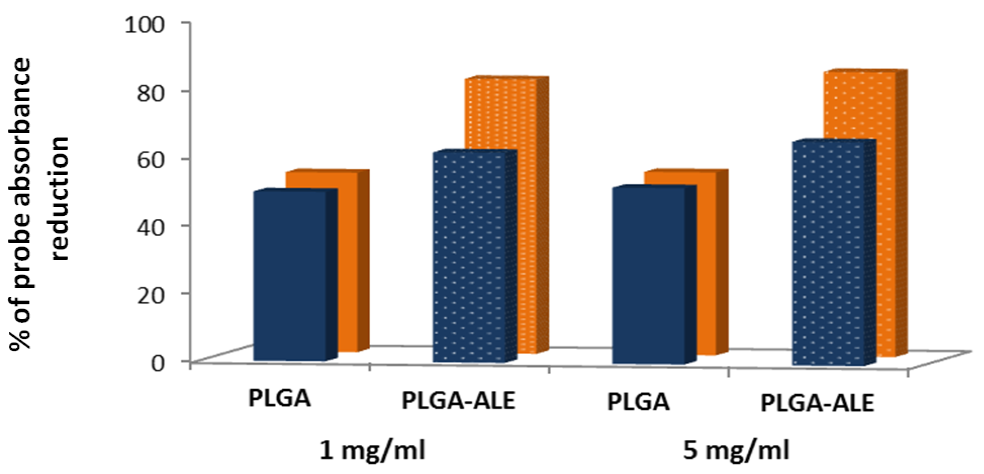
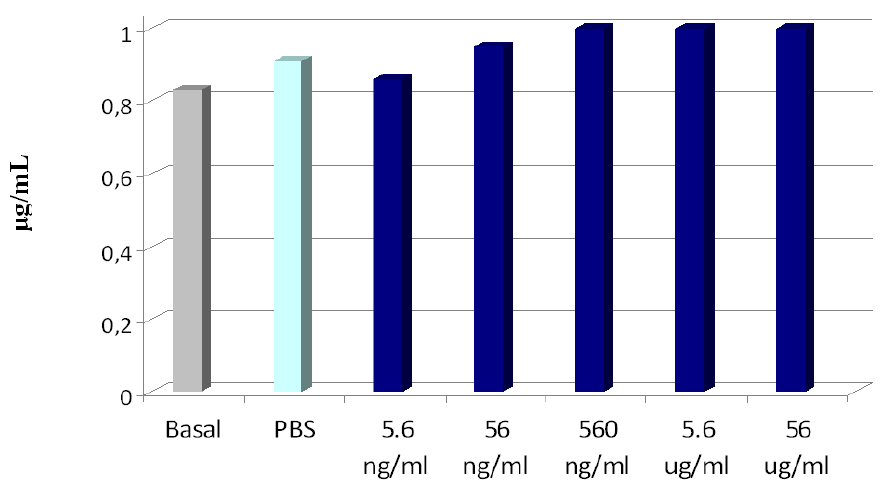
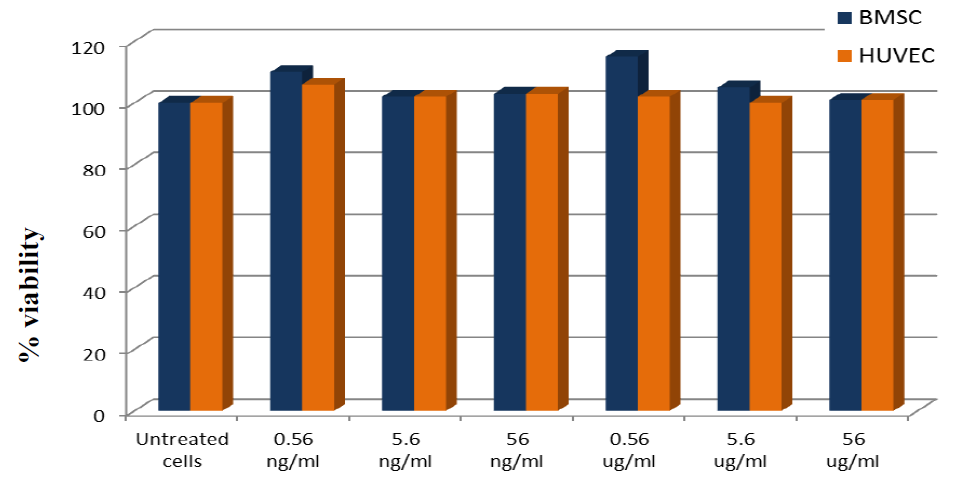
2.3. Biocompatibility Studies on PLGA-ALE NP
| Sample | NP concentration | % hemolysis |
|---|---|---|
| PLGA-ALE NP | 0.56 ng/mL | 0 ± 0.155 |
| 5.6 ng/mL | 0.167 ± 0.191 | |
| 56 ng/mL | 0.356 ± 0.309 | |
| 0.56 μg/mL | 0 ± 0.272 | |
| 5.6 μg/mL | 0.289 ± 0.320 | |
| 56 μg/mL | 0.001 ± 0.165 | |
| Saponin | - | 132.7 ± 2.74 |
| PBS | - | 0 |
| Distilled water | - | 100 |
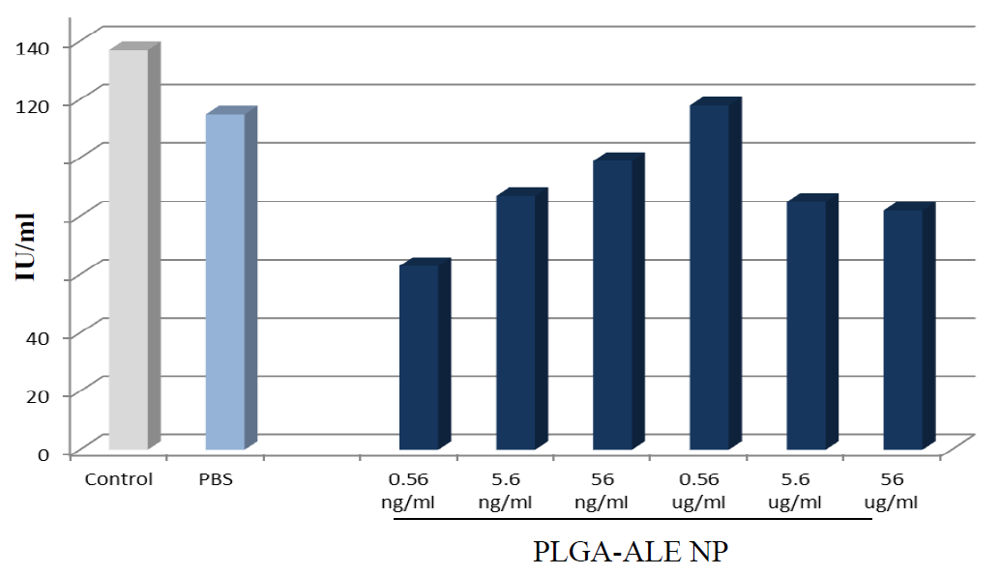
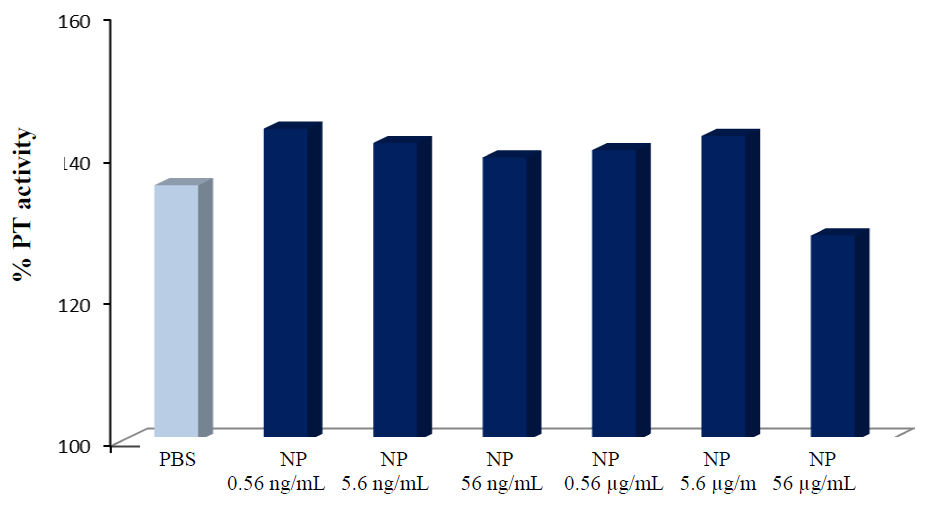

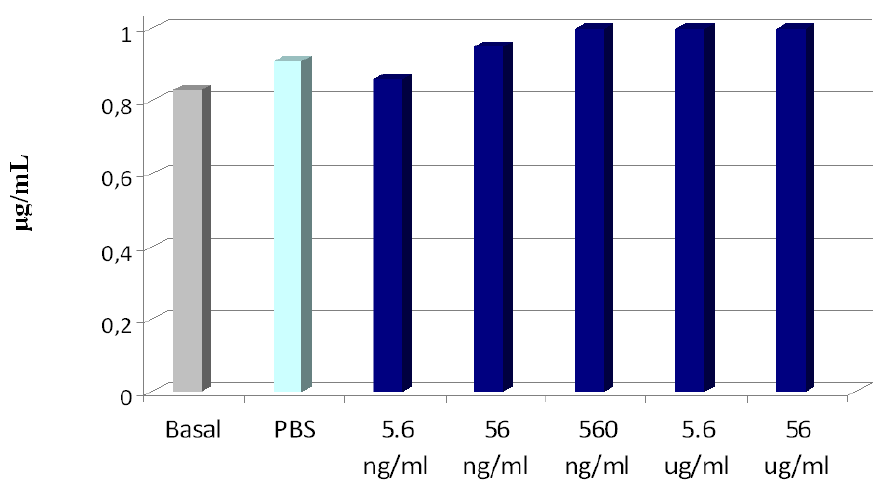

3. DOX-Loaded Nanoparticles
3.1. Preparation and Characterization of the NP [38]
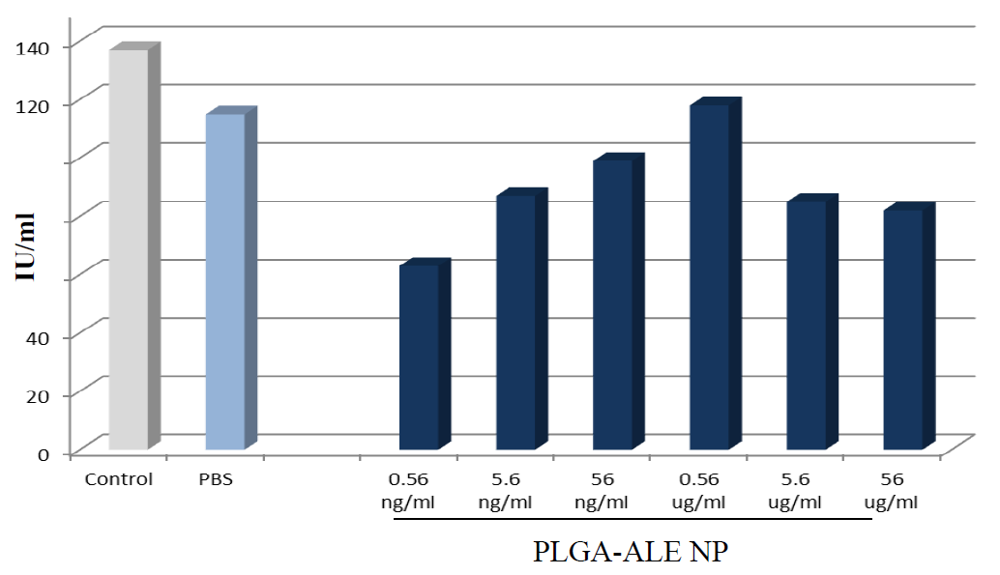
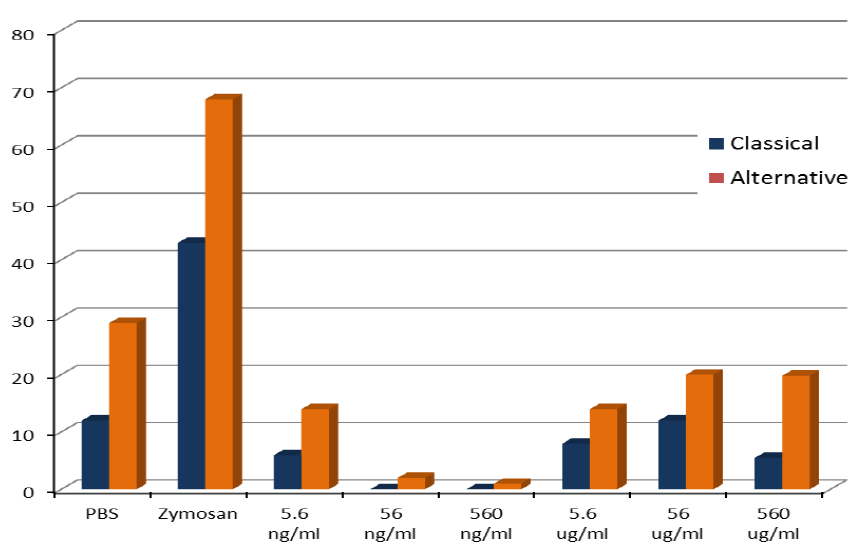
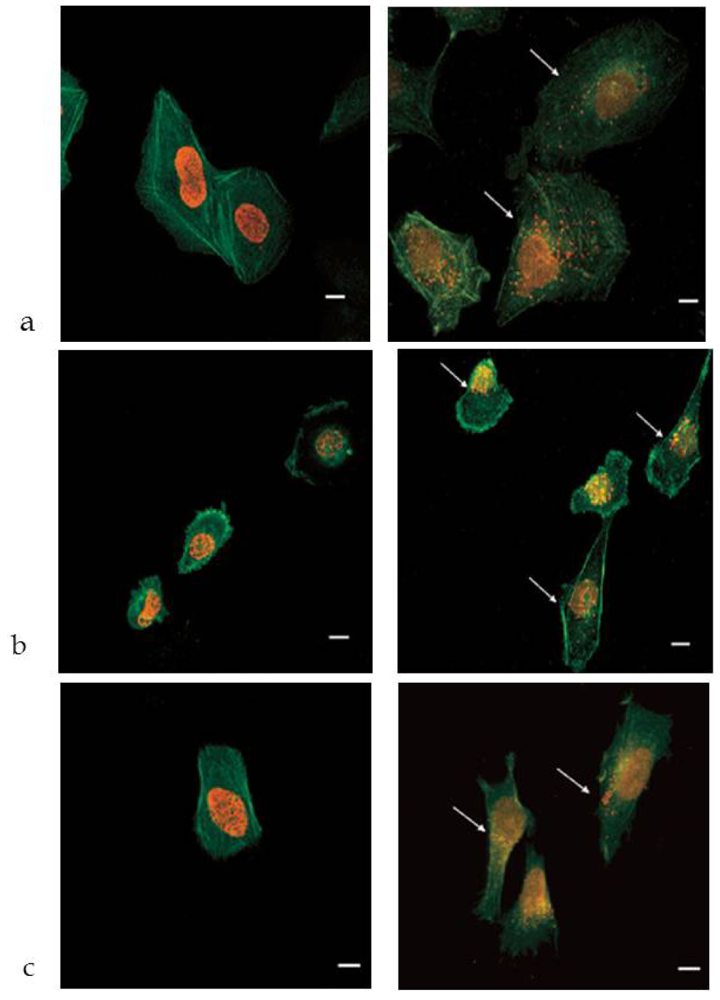
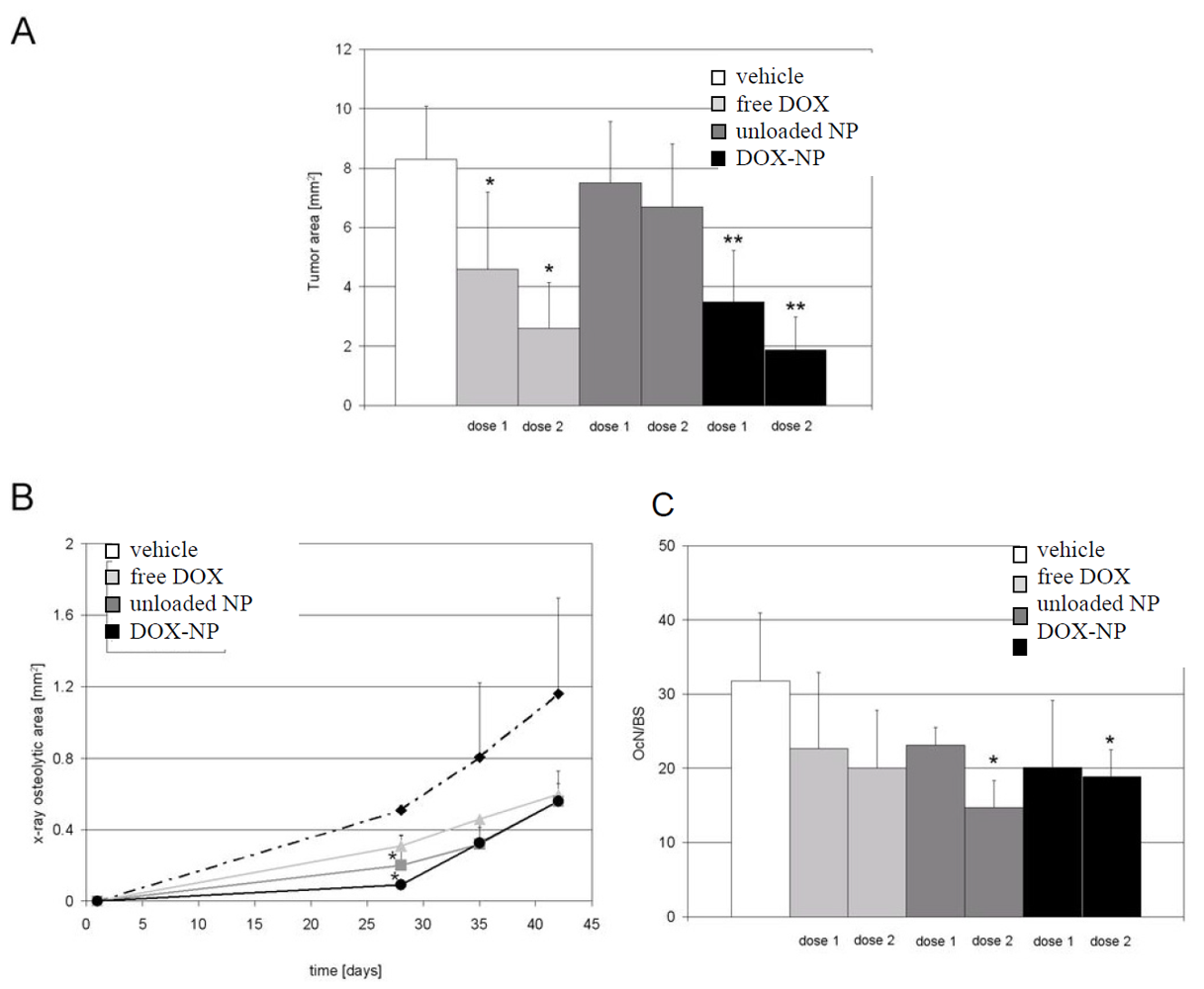
3.2. In Vitro Studies
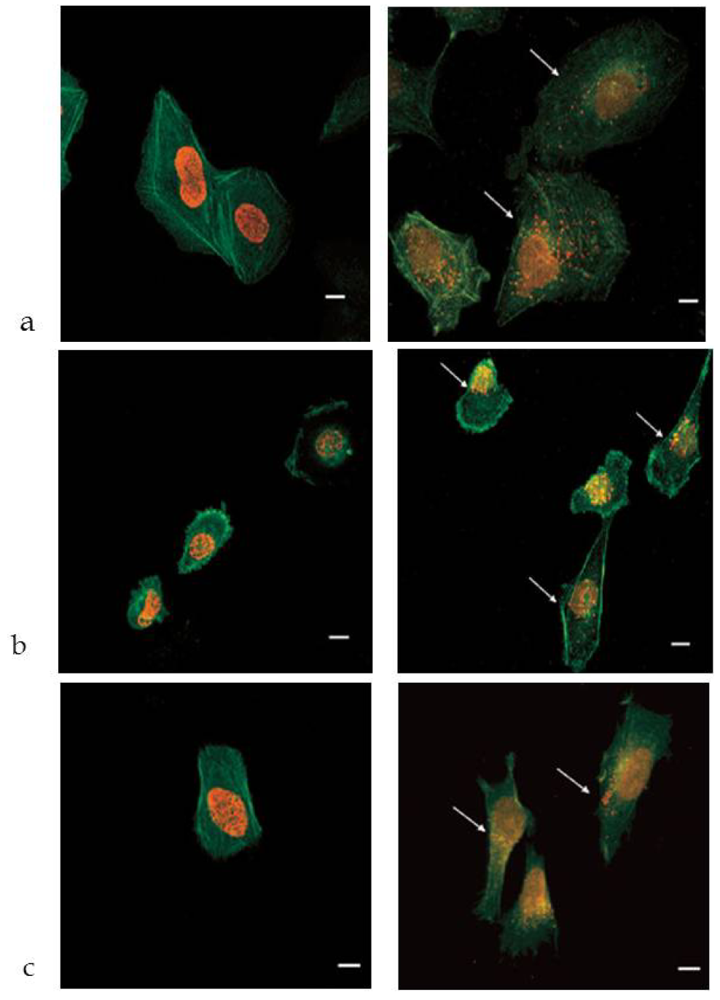
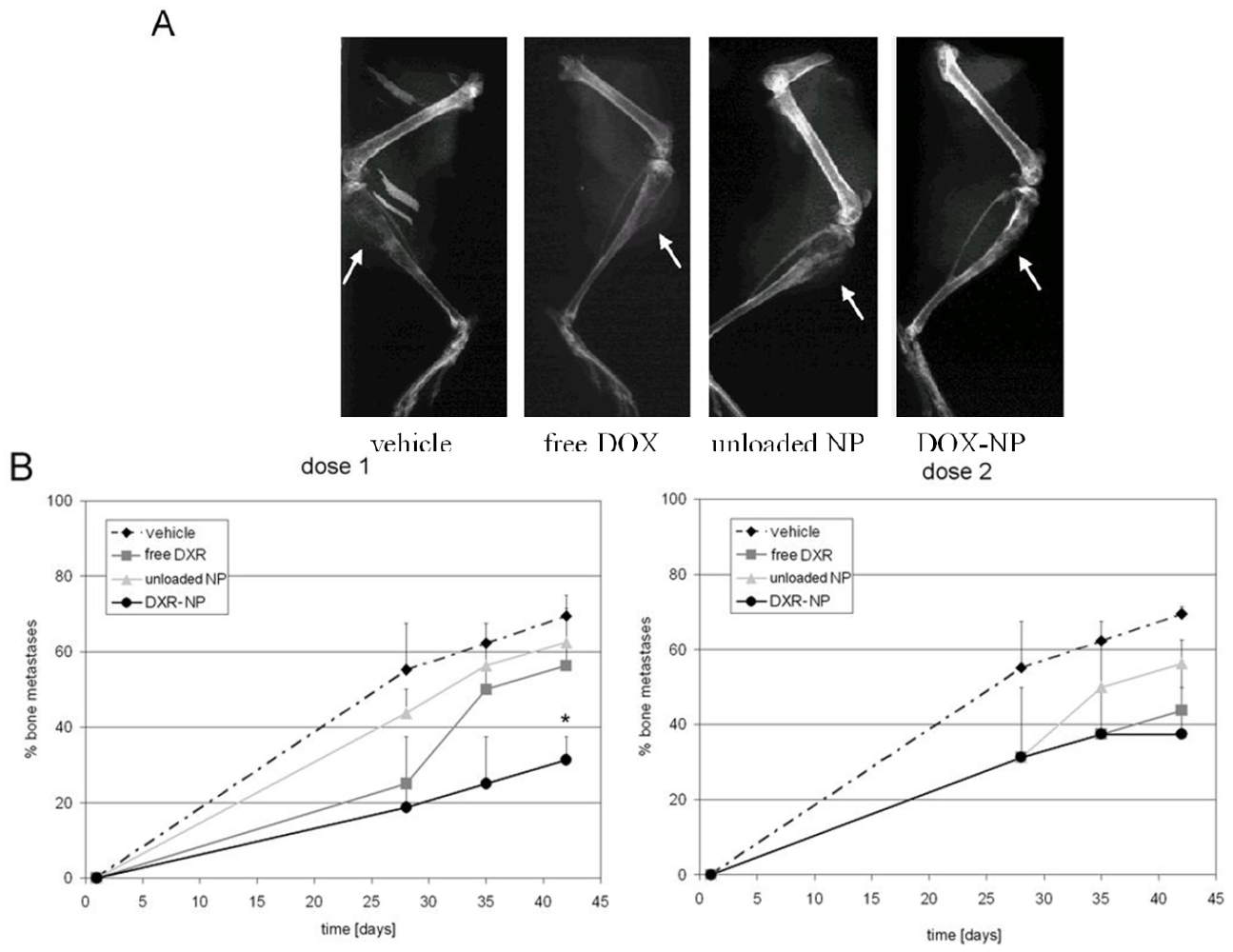
3.3. In Vivo Studies
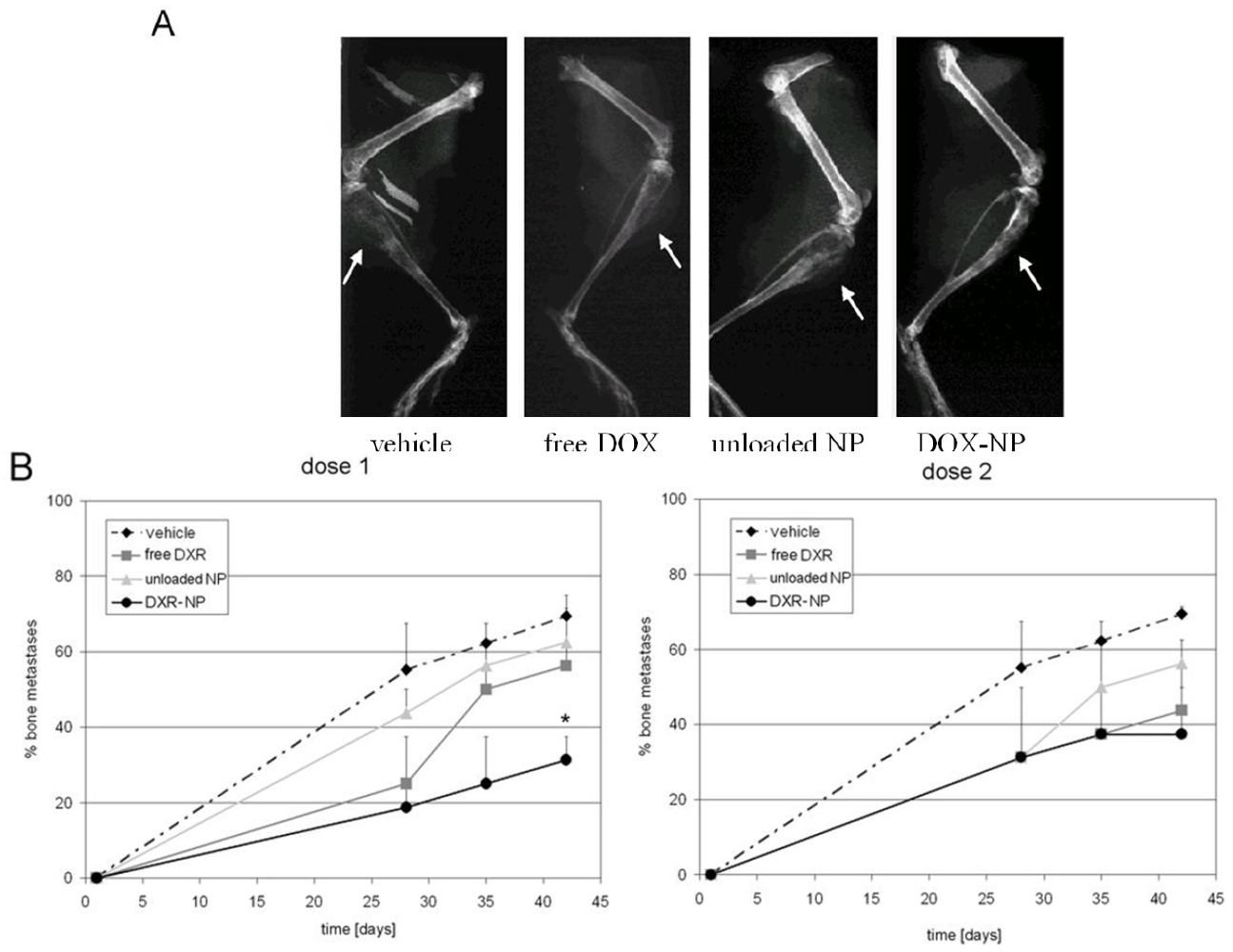
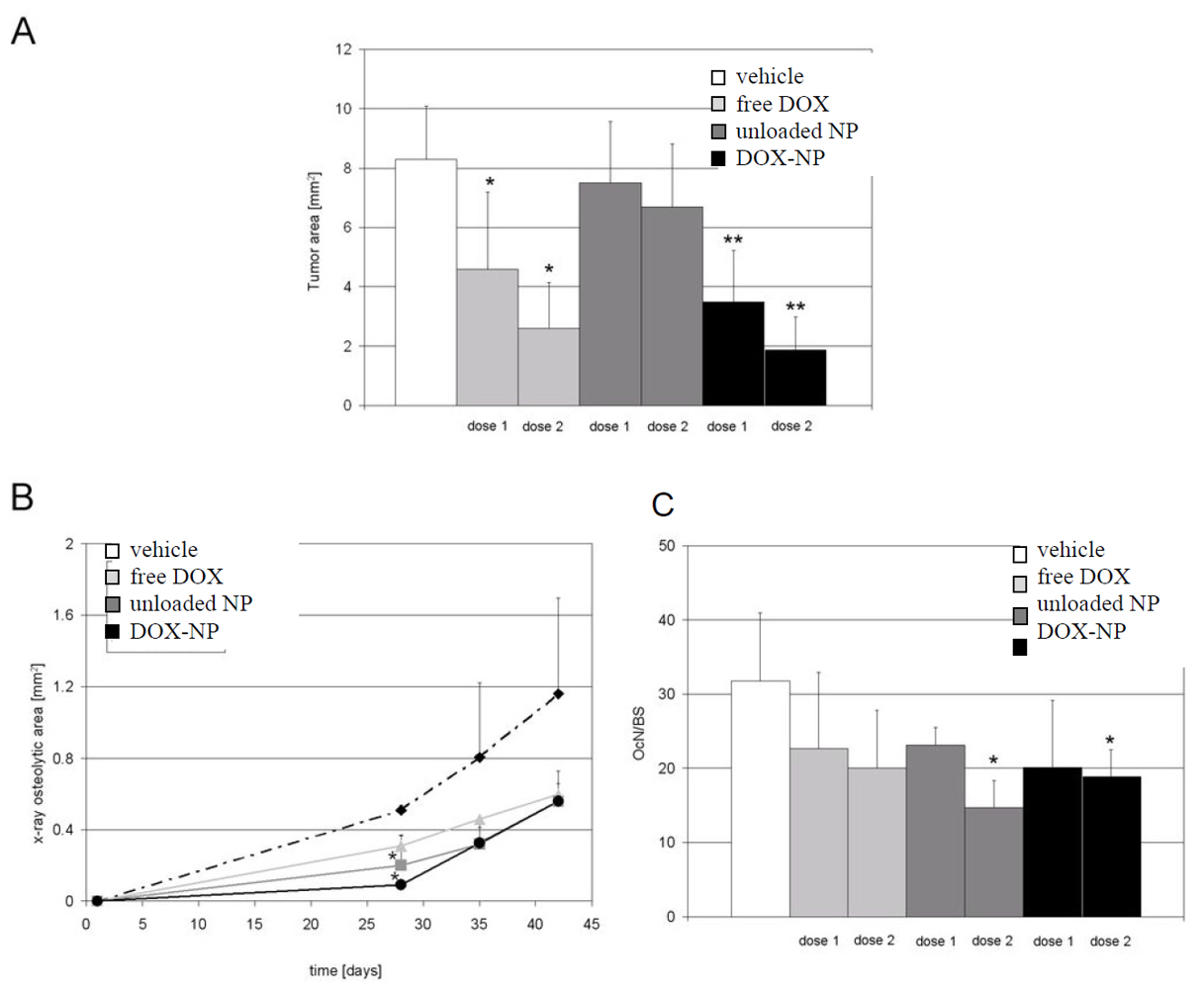
4. Conclusions
Acknowledgments
References
- Basile, L.; Pignatello, R.; Passirani, C. Active targeting strategies for anticancer drug nanocarriers. Curr. Drug Deliv. 2012, in press. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmark of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Roodman, G.D. Mechanisms of bone metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef]
- Coleman, R.E. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176. [Google Scholar] [CrossRef]
- Taube, T.; Elomaa, I.; Blomqvist, C.; Beneton, M.N.C.; Kanis, J.A. Histomorphometric evidence for osteoclast-mediated bone resorption in metastatic breast cancer. Bone 1994, 15, 161–166. [Google Scholar] [CrossRef]
- Boyde, A.; Maconnachie, E.; Reid, S.A.; Delling, G.; Mundy, G.R. Scanning electron microscopy in bone pathology: Review of methods, potential and applications. Scan. Electron. Microsc. 1986, 4, 1537–1554. [Google Scholar]
- Guise, T.A.; Mohammad, K.S.; Clines, G.; Stebbins, E.G.; Wong, D.H.; Higgins, L.S.; Vessella, R.; Corey, E.; Padalecki, S.; Suva, L.; Chirgwin, J.M. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin. Cancer Res. 2006, 12, 6213s–6216s. [Google Scholar] [CrossRef]
- Houston, S.J.; Rubens, R.D. The systemic treatment of bone metastases. Clin. Orthop. 1995, 312, 95–104. [Google Scholar]
- Lin, A.; Ray, M.E. Targeted and systemic radiotherapy in the treatment of bone metastasis. Cancer Metastasis. Rev. 2006, 25, 669–675. [Google Scholar] [CrossRef]
- Iyer, A.K.; Khaled, G.; Fang, J.; Maeda, H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov. Today 2006, 11, 812–818. [Google Scholar] [CrossRef]
- Hirsjärvi, S.; Passirani, C.; Benoit, J.P. Passive and active tumor targeting with nanocarriers. Curr. Drug Discov. Technol. 2011, 8, 188–196. [Google Scholar] [CrossRef]
- Hirabayashi, H.; Fujisaki, J. Bone-specific drug delivery systems: Approaches via chemical modification of bone-seeking agents. Clin. Pharmacokinet. 2003, 42, 1319–1330. [Google Scholar] [CrossRef]
- Wang, D.; Miller, S.C.; Kopeckova, P.; Kopecek, J. Bone-targeting macromolecular therapeutics. Adv. Drug Del. Rev. 2005, 57, 1049–1076. [Google Scholar] [CrossRef]
- Kratz, F. Bisphosphonate-prodrugs. Patent No. WO/2011/023367, 3 March 2011. [Google Scholar]
- Masarachia, P.; Weinreb, M.; Balena, R.; Rodan, G.A. Comparison of the distribution of 3H-alendronate and 3H-etidronate in rat and mouse bones. Bone 1996, 19, 281–290. [Google Scholar] [CrossRef]
- Coleman, R.E. Metastatic bone disease: Clinical features, pathophysiology, and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176. [Google Scholar] [CrossRef]
- Clézardin, P.; Ebetino, F.H.; Fournier, P.G.J. Bisphosphonates and cancer-induced bone disease: Beyond their antiresorptive activity. Cancer Res. 2005, 65, 4971–4974. [Google Scholar] [CrossRef]
- Koul, H.K.; Koul, S.; Meacham, R.B. New role for an established drug? Bisphosphonates as potential anticancer agents. Prostate Cancer Prostatic Dis. 2012, in press. [Google Scholar]
- Fujisaki, J.; Tokunaga, Y.; Takahashi, T.; Hirose, T.; Shimojo, F.; Kagayama, A.; Hata, T. Osteotropic drug delivery system (ODDS) based on bisphosphonic prodrug. I: Synthesis and in vivo characterization of osteotropic carboxyfluorescein. J. Drug Target 1995, 3, 273–282. [Google Scholar] [CrossRef]
- Wong, R.K.S.; Wiffen, P.J. Bisphosphonates for the relief of pain secondary to bone metastases. Cochrane Database Syst. Rev. 2002, 2. [Google Scholar] [CrossRef]
- Zhang, S.; Gangal, G.; Uludağ, H. ‘Magic bullets’ for bone diseases: Progress in rational design of bone-seeking medicinal agents. Chem. Soc. Rev. 2007, 36, 507–531. [Google Scholar] [CrossRef]
- Bauss, F.; Esswein, A.; Reiff, K.; Sponer, G.; Müller-Beckmann, B. Effect of 17beta-estradiol-bisphosphonate conjugates, potential bone-seeking estrogen pro-drugs, on 17beta-estradiol serum kinetics and bone mass in rats. Calcif. Tissue Int. 1996, 59, 168–173. [Google Scholar] [CrossRef]
- Niemi, R.; Vepsalainen, J.; Taipale, H.; Jairvinen, T. Bisphosphonate prodrugs: Synthesis and in vitro evaluation of novel acyloxyalkyl esters of clodronic acid. J Med Chem 1999, 2, 5053–5058. [Google Scholar]
- MBC Pharma’s Solutions; MBC Pharma: Aurora, CO, USA. Available online: http://www.mbcpharma.com/html/technology.html (last accessed on January 12, 2012).
- Wright, J.E.; Gittens, S.A.; Bansal, G.; Kitov, P.I.; Sindrey, D.; Kucharski, C.; Uludağ, H. A comparison of mineral affinity of bisphosphonate-protein conjugates constructed with disulfide and thioether linkages. Biomaterials 2006, 27, 769–784. [Google Scholar] [CrossRef]
- Ogawa, K.; Mukai, T.; Inoue, Y.; Ono, M.; Saji, H. Development of a novel 99mTc-chelate conjugated bisphosphonate with high affinity for bone as a bone scintigraphic agent. J. Nucl. Med. 2006, 47, 2042–2047. [Google Scholar]
- Tanaka, K.S.; Houghton, T.J.; Kang, T.; Dietrich, E.; Delorme, D.; Ferreira, S.S.; Caron, L.; Viens, F.; Arhin, F.F.; Sarmiento, I.; et al. Bisphosphonated fluoroquinolone esters as osteotropic prodrugs for the prevention of osteomyelitis. Bioorg. Med. Chem. 2008, 16, 9217–9229. [Google Scholar] [CrossRef]
- Tanaka, K.S.; Dietrich, E.; Ciblat, S.; Métayer, C.; Arhin, F.F.; Sarmiento, I.; Moeck, G.; Parr, T.R., Jr.; Far, A.R. Synthesis and in vitro evaluation of bisphosphonated glycopeptide prodrugs for the treatment of osteomyelitis. Bioorg. Med. Chem. Lett. 2010, 20, 1355–1359. [Google Scholar] [CrossRef]
- McPherson, J.C.; Runner, R.; Buxton, T.B.; Hartmann, J.F.; Farcasiu, D.; Bereczki, I.; Rőth, E.; Tollas, S.; Ostorházi, E.; Rozgonyi, F.; Herczegh, P. Synthesis of osteotropic hydroxybisphosphonate derivatives of fluoroquinolone antibacterials. Eur. J. Med. Chem. 2012, 47C, 615–618. [Google Scholar]
- Wang, D.; Miller, S.; Sima, M.; Kopeckova, P.; Kopecek, J. Synthesis and evaluation of water-soluble polymeric bone-targeted drug delivery systems. Bioconj. Chem. 2003, 14, 853–859. [Google Scholar] [CrossRef]
- Satchi-Fainaro, R.; Miller, K.; Shabat, D.; Erez, R. Conjugates of a polymer, a bisphosphonate and an anti-angiogenesis agent and uses thereof in the treatment and monitoring of bone related diseases. US Patent 20110142764, 16 June 2011. [Google Scholar]
- Miller, K.; Erez, R.; Segal, E.; Shabat, D.; Satchi-Fainaro, R. Targeting bone metastases with a bispecific anticancer and antiangiogenic polymer-alendronate-taxane conjugate. Angew. Chem. Int. Ed. Engl. 2009, 48, 2949–2954. [Google Scholar] [CrossRef]
- Bala, I.; Hariharan, S.; Kumar, M.N. PLGA nanoparticles in drug delivery: The state of the art. Crit. Rev. Ther. Drug Carr. Syst. 2004, 21, 387–422. [Google Scholar] [CrossRef]
- Drugs.com: Alendronate. Available online: http://www.drugs.com/pro/alendronate.html (accessed on 12 January 2012).
- DeRuiter, J.; Clark, R. Bisphosphonates: Calcium antiresorptive agents. Revision Spring 2002. Available online: http://www.duc.auburn.edu/~deruija/endo_bisphos.pdf (accessed on July 2011).
- Pignatello, R.; Cenni, E.; Miceli, D.; Fotia, C.; Salerno, M.; Granchi, D.; Avnet, S.; Sarpietro, M.G.; Castelli, F.; Baldini, N. A novel biomaterial for osteotropic drug nanocarriers. Synthesis and biocompatibility evaluation of a PLGA-alendronate conjugate. Nanomedicine 2009, 4, 161–175. [Google Scholar] [CrossRef]
- Cenni, E.; Granchi, D.; Avnet, S.; Fotia, C.; Salerno, M.; Micieli, D.; Sarpietro, M.G.; Pignatello, R.; Castelli, F.; Baldini, N. Biocompatibility of poly(D,L-lactide-co-glycolide) nanoparticles conjugated with alendronate. Biomaterials 2008, 29, 1400–1411. [Google Scholar] [CrossRef]
- Salerno, M.; Cenni, E.; Fotia, C.; Avnet, S.; Granchi, D.; Castelli, F.; Micieli, D.; Pignatello, R.; Capulli, M.; Rucci, N.; et al. Bone-targeted doxorubicin-loaded nanoparticles as a tool for the treatment of skeletal metastases. Curr. Cancer Drug Targets 2010, 10, 649–659. [Google Scholar] [CrossRef]
- Choi, S.W.; Kim, J.H. Design of surface-modified poly(D,L-lactide-co-glycolide) nanoparticles for targeted drug delivery to bone. J. Control. Release 2007, 122, 24–30. [Google Scholar] [CrossRef]
- Oberdorster, G.; Maynard, A.; Donaldson, K.; Castranova, V.; Fitzpatrick, J.; Ausman, K.; Carter, J.; Karn, B.; Kreyling, W.; Lai, D.; The ILSI Research Foundation/Risk Science Institute Nanomaterial Toxicity Screening Working Group; et al. Principles for characterizing the potential human health effects from exposure to nanomaterials: Elements for a screening strategy. Part. Fibre Toxicol. 2005, 2. [Google Scholar] [CrossRef]
- Borm, P.J.A.; Robbins, D.; Haubold, S.; Kuhlbusch, T.; Fissan, H.; Donaldson, K.; Schins, R.; Stone, V.; Kreyling, W.; Lademann, J.; et al. The potential risk of nanomaterials: A review carried out for ECETOC. Part. Fibre Toxicol. 2006, 3. [Google Scholar] [CrossRef]
- Cenni, E.; Avnet, S.; Granchi, D.; Fotia, C.; Salerno, M.; Micieli, D.; Sarpietro, M.G.; Pignatello, R.; Castelli, F.; Baldini, N. The effect of poly(D,L-lactide-co-glycolide) nanoparticles conjugated with alendronate on human osteoclast precursors. J. Mat. Sci. Polym. Med. 2011, 7. [Google Scholar] [CrossRef]
- Drugs.com: Doxil. Available online: http://www.drugs.com/pro/doxil.html (accessed on 12 January 2012).
- Andreopoulou, E.; Gaiotti, D.; Kim, E.; Downey, A.; Mirchandani, D.; Hamilton, A.; Jacobs, A.; Curtin, J.; Muggia, F. Pegylated liposomal doxorubicin HCL (PLD; Caelyx/Doxil®): Experience with long-term maintenance in responding patients with recurrent epithelial ovarian cancer. Ann. Oncol. 2007, 18, 716–721. [Google Scholar]
- Momparler, R.L.; Karon, M.; Siegel, S.E.; Avila, F. Effect of adriamycin on DNA, RNA, and protein synthesis in cell-free systems and intact cells. Cancer Res. 1976, 36, 2891–2895. [Google Scholar]
- Panyam, J.; Zhou, W.Z.; Prabha, S.; Sahoo, S.K.; Labhasetwar, V. Rapid endo-lysosomal escape of poly(D,L-lactide-co-glycolide) nanoparticles: Implications for drug and gene delivery. FASEB J. 2002, 16, 1217–1226. [Google Scholar] [CrossRef]
- Vasir, J.K.; Labhasetwar, V. Biodegradable nanoparticles for cytosolic delivery of therapeutics. Adv. Drug Deliv. Rev. 2007, 59, 718–728. [Google Scholar] [CrossRef]
- Clézardin, P.; Ebetino, F.H.; Fournier, P.G.J. Bisphosphonates and cancer-induced bone disease: Beyond their antiresorptive activity. Cancer Res. 2005, 65, 4971–4974. [Google Scholar] [CrossRef]
- Santini, D.; Schiavon, G.; Angeletti, S.; Vincenzi, B.; Gasparro, S.; Grilli, C.; La Cesa, A.; Virzí, V.; Leoni, V.; Budillon, A.; et al. Last generation of amino-bisphosphonates (N-BPs) and cancer angio-genesis: A new role for these drugs? Recent Pat. Anticancer Drug Discov. 2006, 1, 383–396. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pignatello, R.; Sarpietro, M.G.; Castelli, F. Synthesis and Biological Evaluation of a New Polymeric Conjugate and Nanocarrier with Osteotropic Properties. J. Funct. Biomater. 2012, 3, 79-99. https://doi.org/10.3390/jfb3010079
Pignatello R, Sarpietro MG, Castelli F. Synthesis and Biological Evaluation of a New Polymeric Conjugate and Nanocarrier with Osteotropic Properties. Journal of Functional Biomaterials. 2012; 3(1):79-99. https://doi.org/10.3390/jfb3010079
Chicago/Turabian StylePignatello, Rosario, Maria Grazia Sarpietro, and Francesco Castelli. 2012. "Synthesis and Biological Evaluation of a New Polymeric Conjugate and Nanocarrier with Osteotropic Properties" Journal of Functional Biomaterials 3, no. 1: 79-99. https://doi.org/10.3390/jfb3010079
APA StylePignatello, R., Sarpietro, M. G., & Castelli, F. (2012). Synthesis and Biological Evaluation of a New Polymeric Conjugate and Nanocarrier with Osteotropic Properties. Journal of Functional Biomaterials, 3(1), 79-99. https://doi.org/10.3390/jfb3010079