
Predictions of Physicochemical Properties of Ionic Liquids with DFT

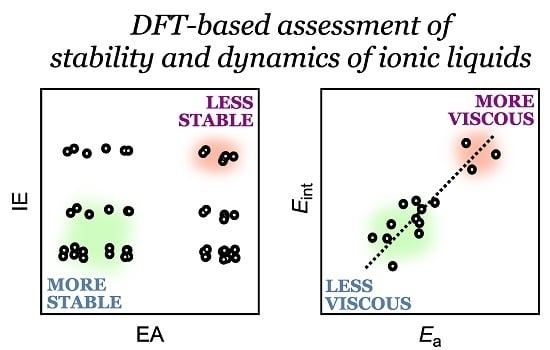
Abstract
:
1. Introduction
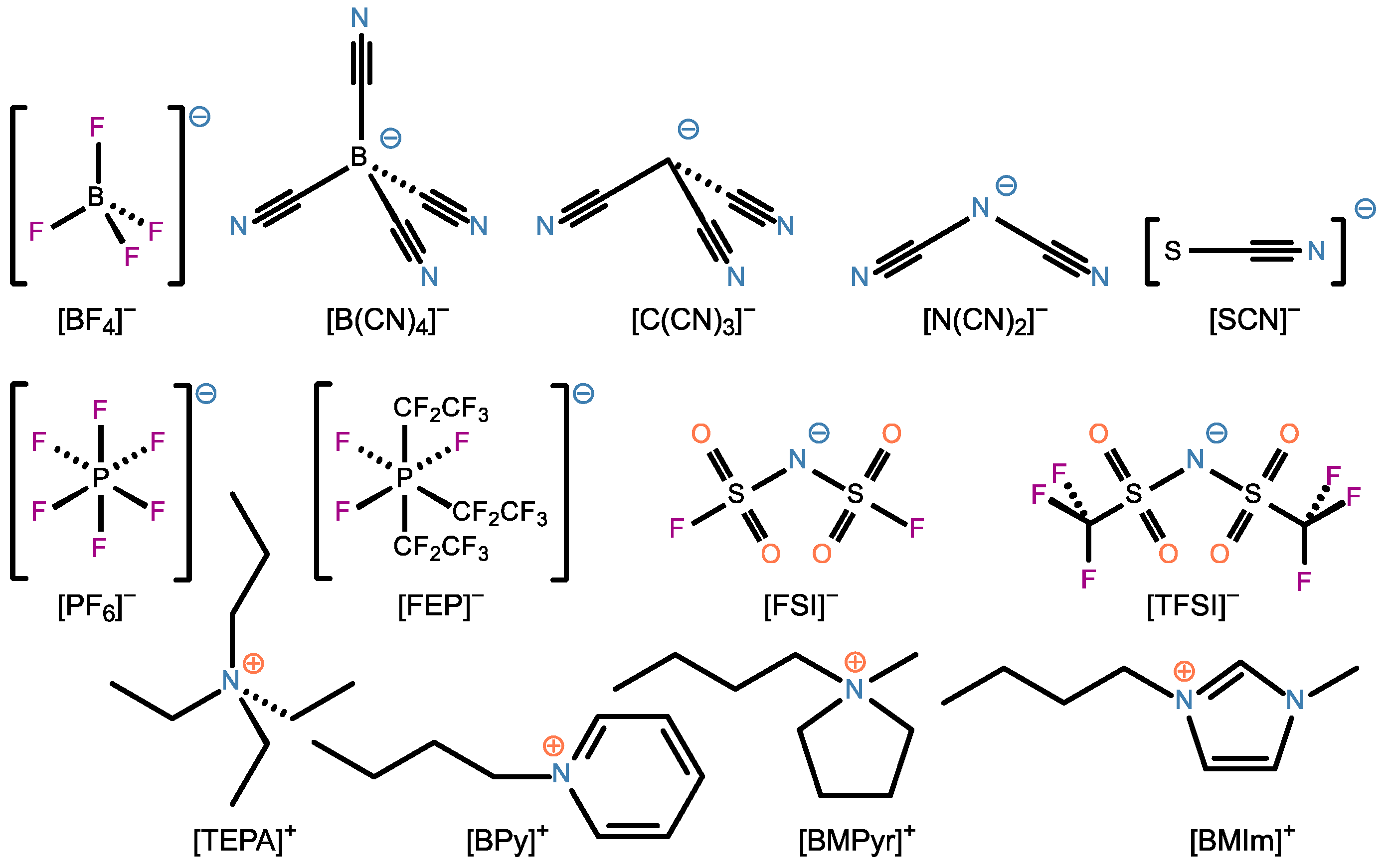
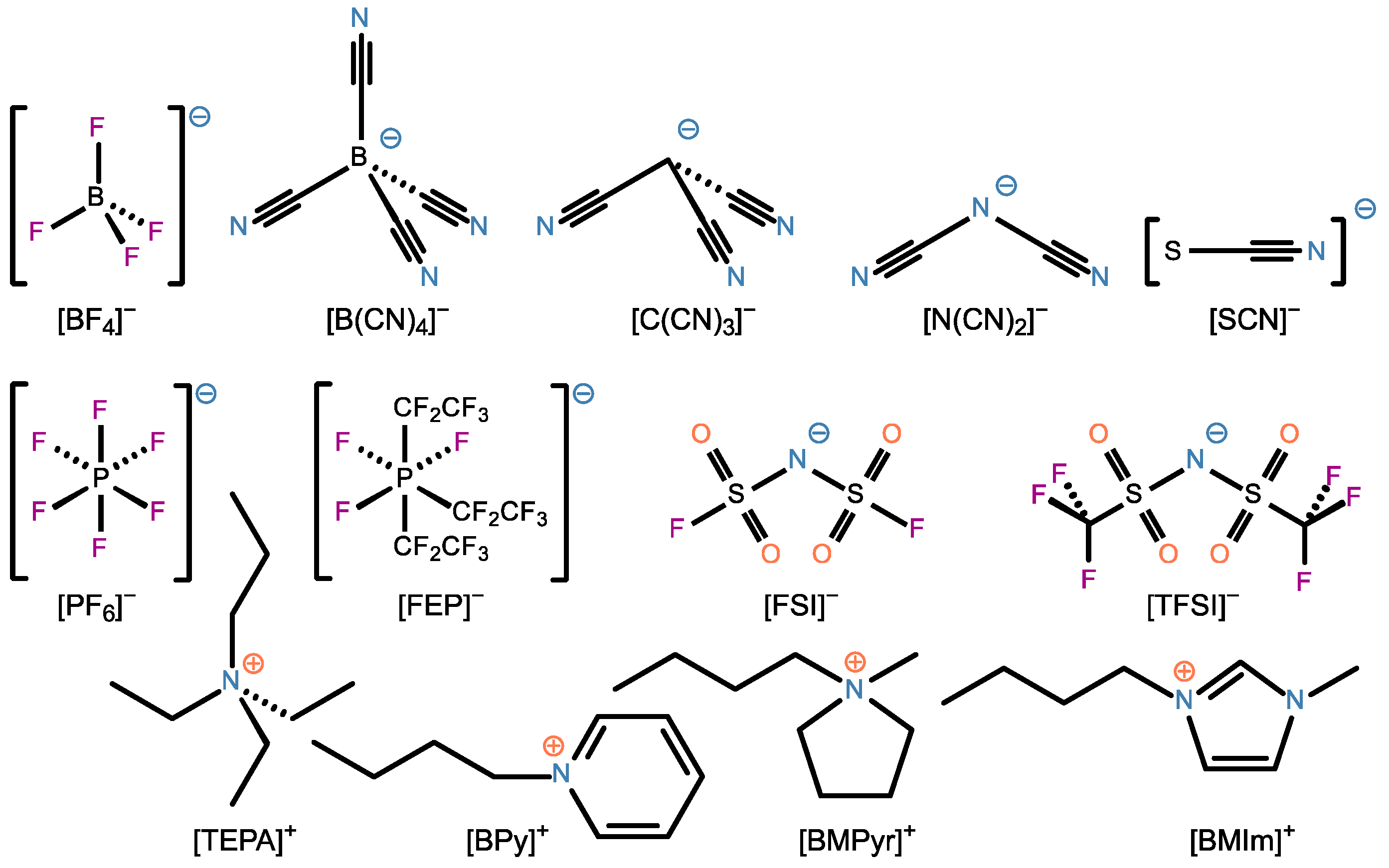
2. Materials and Methods
3. Results
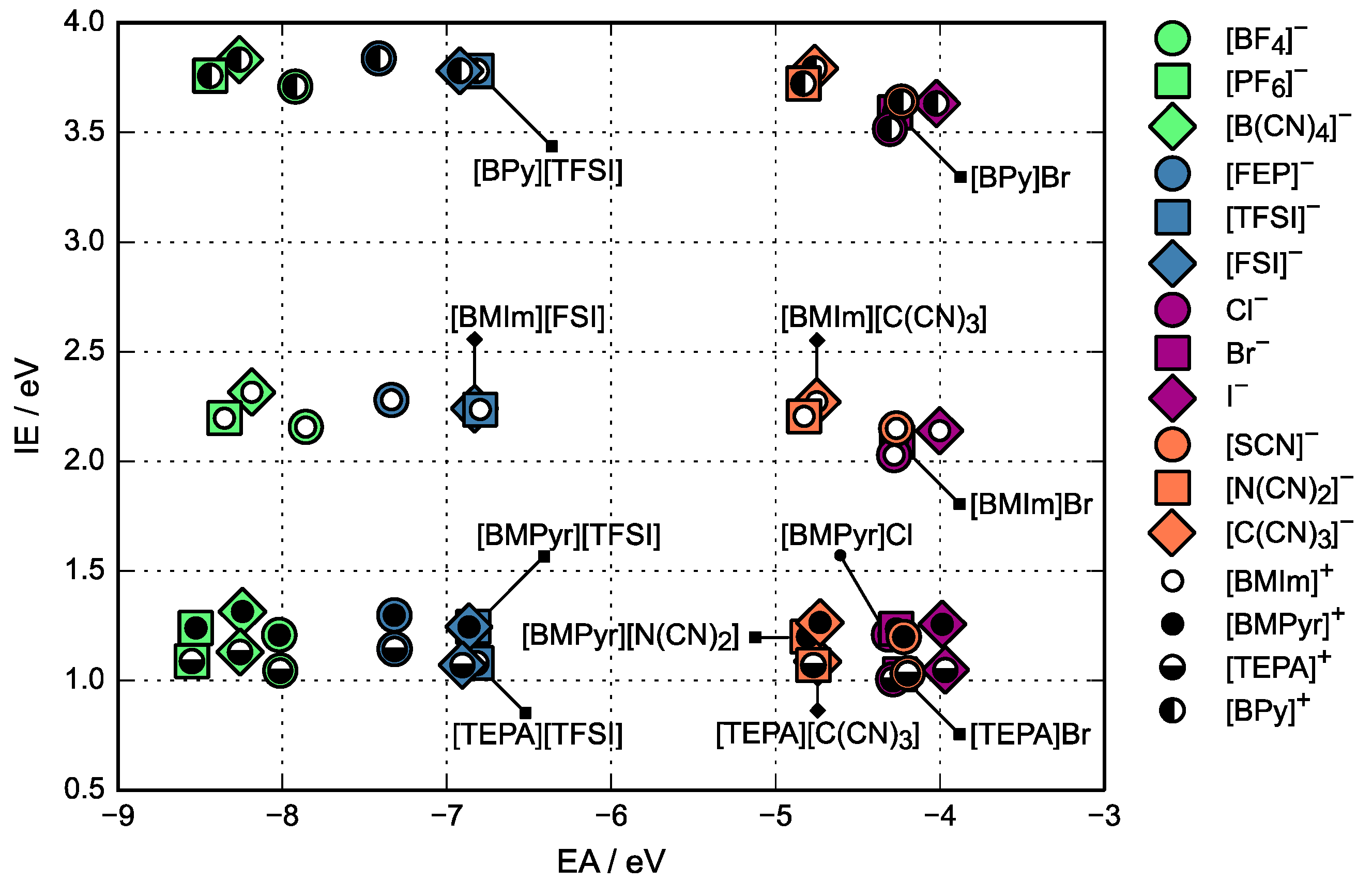
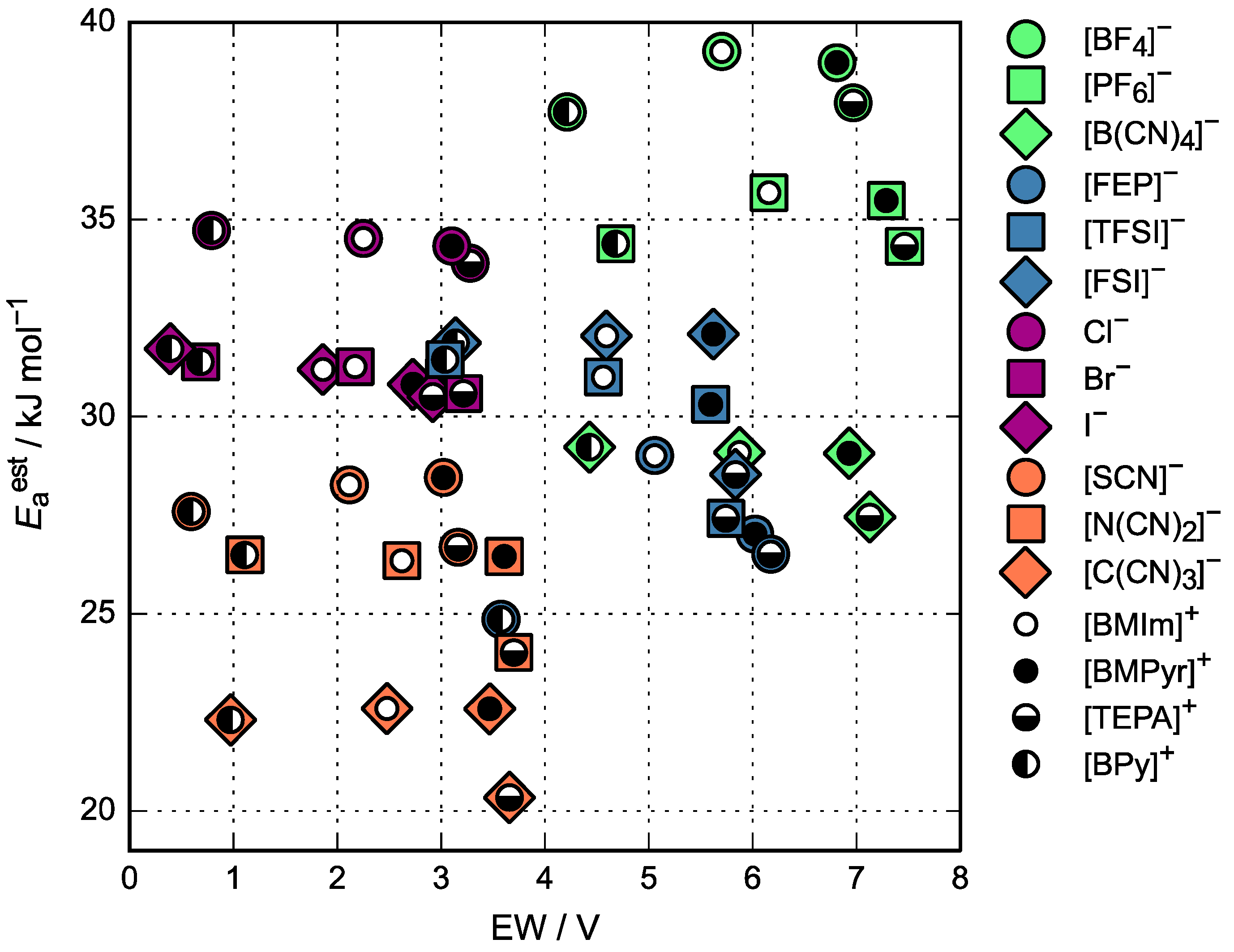
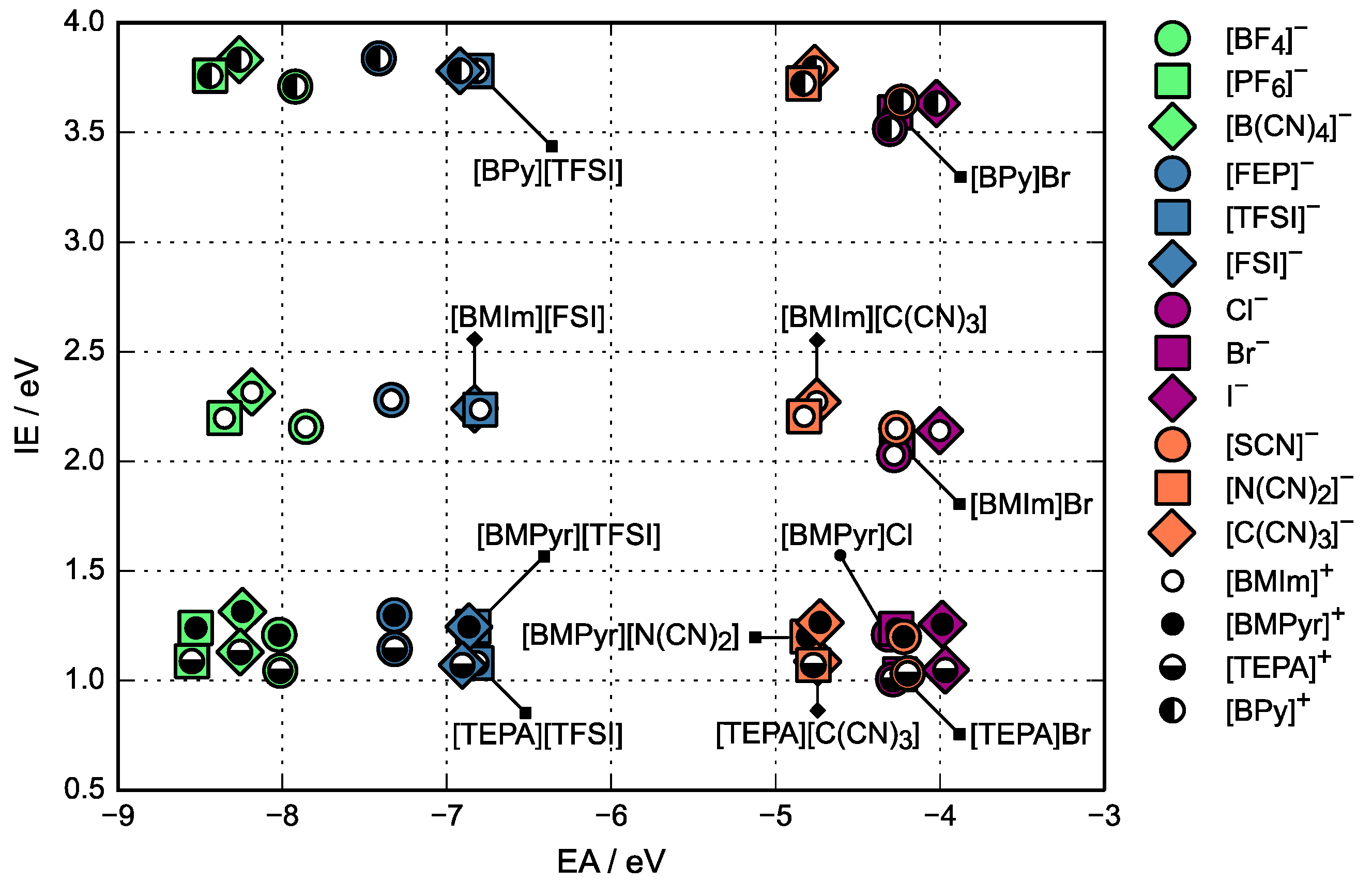
3.1. Relative Electrochemical Stability
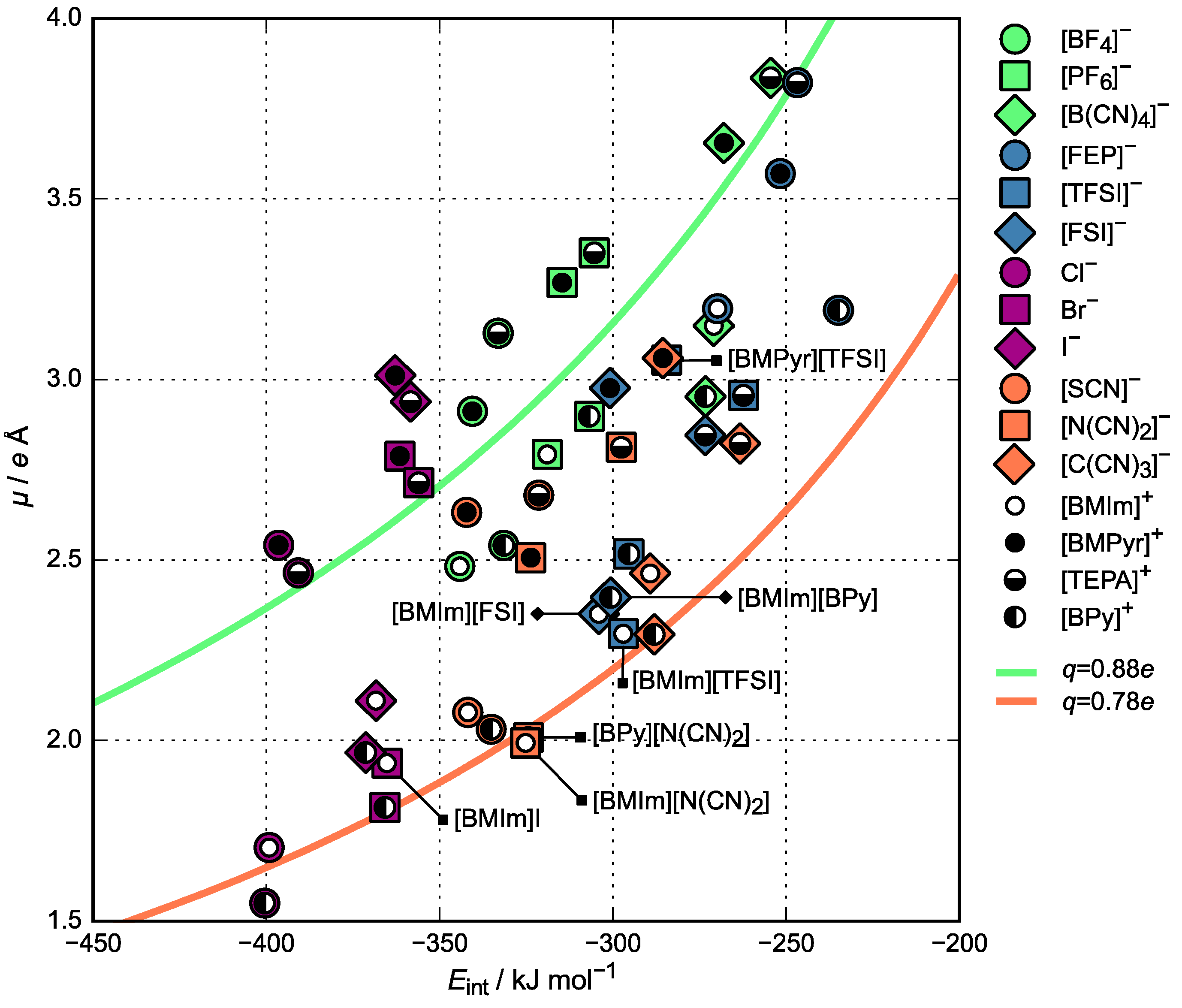
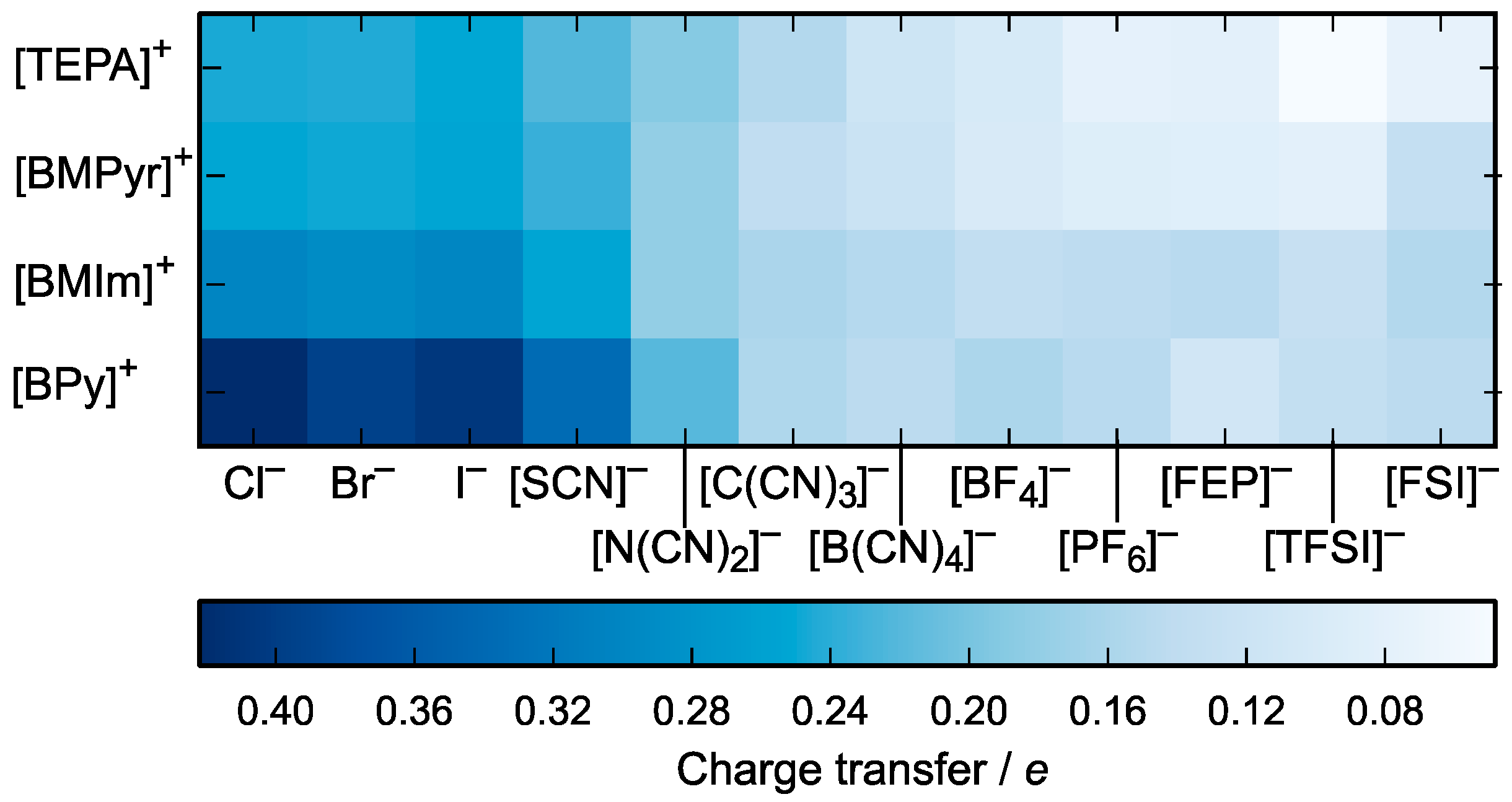
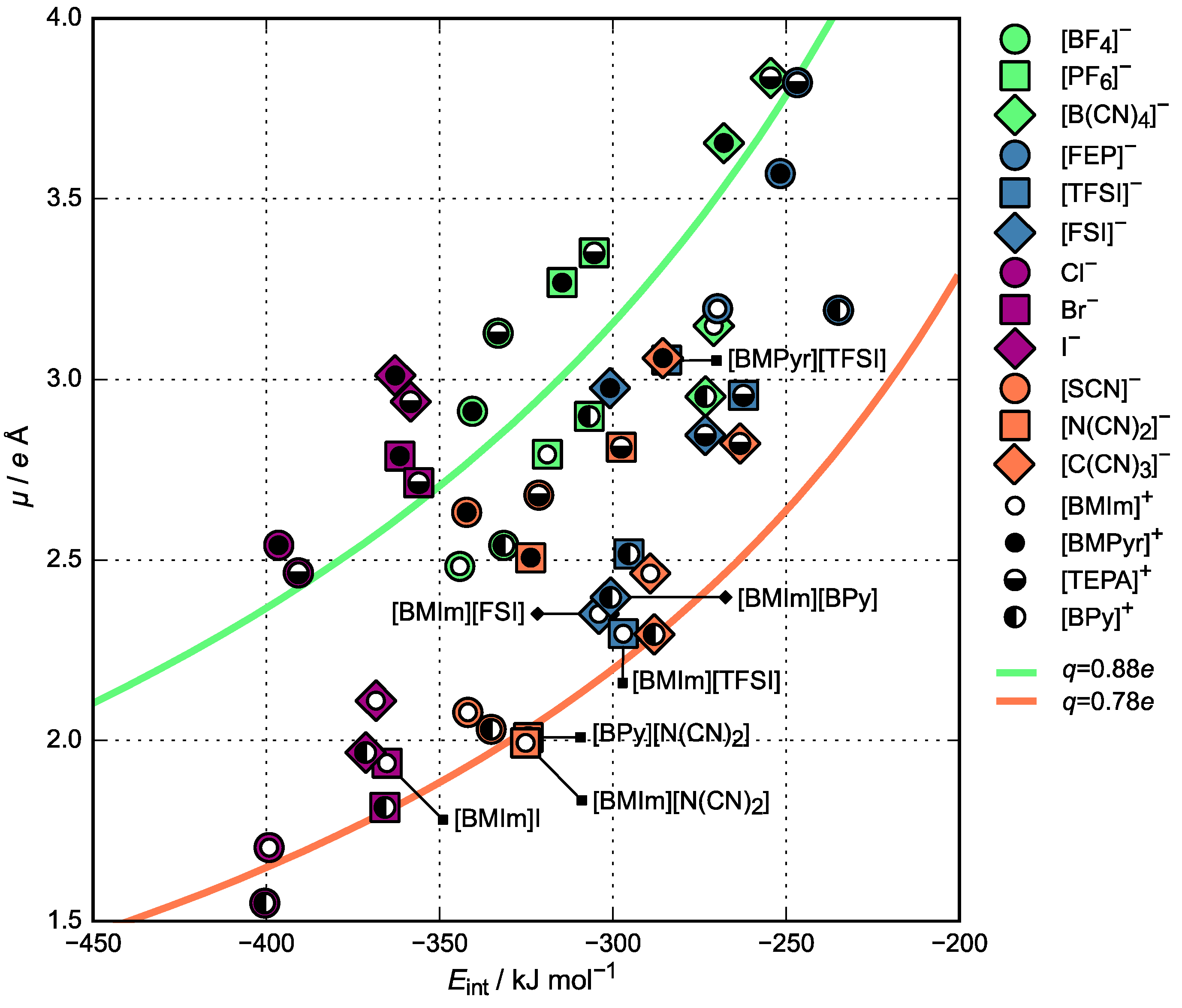
3.2. Charge Density Distribution
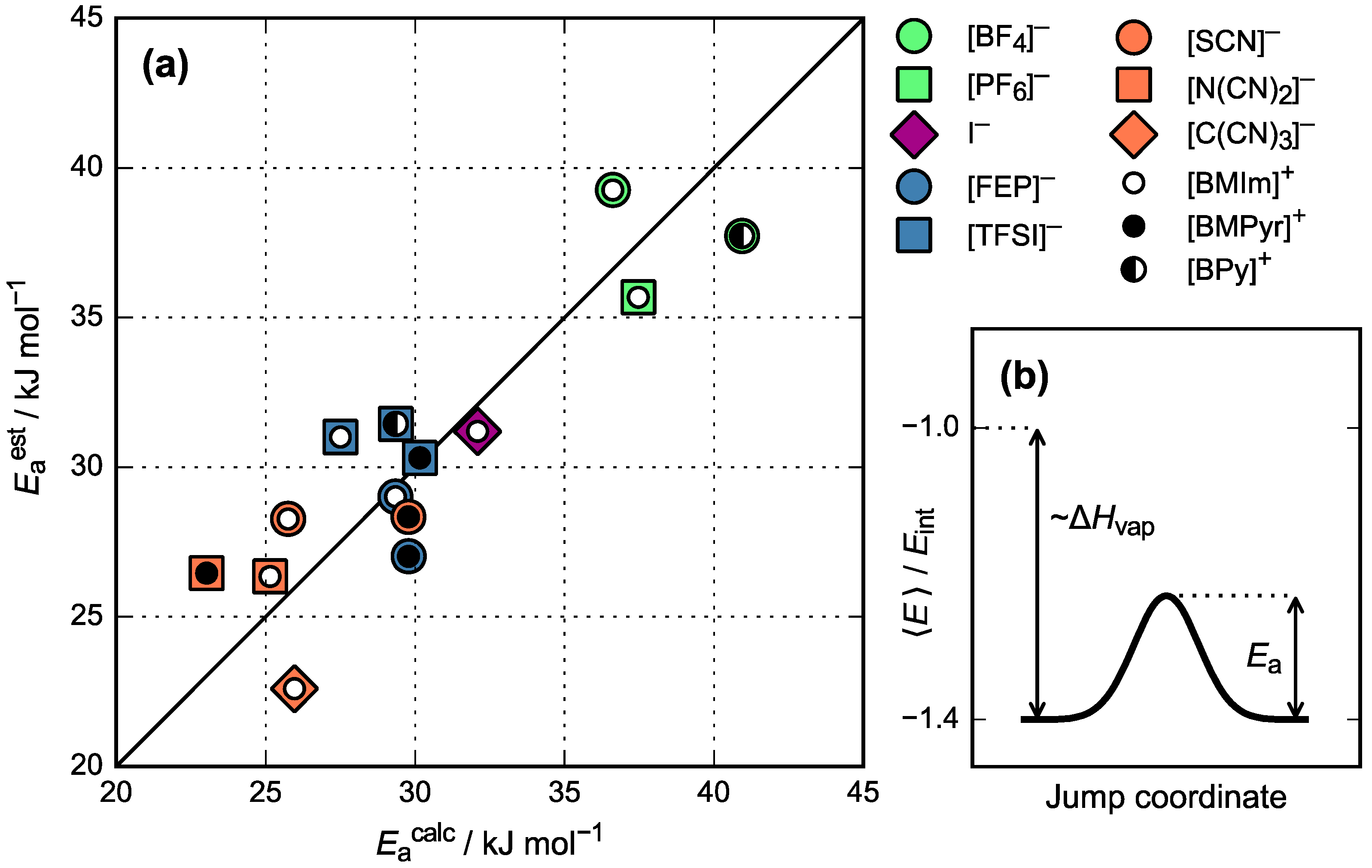
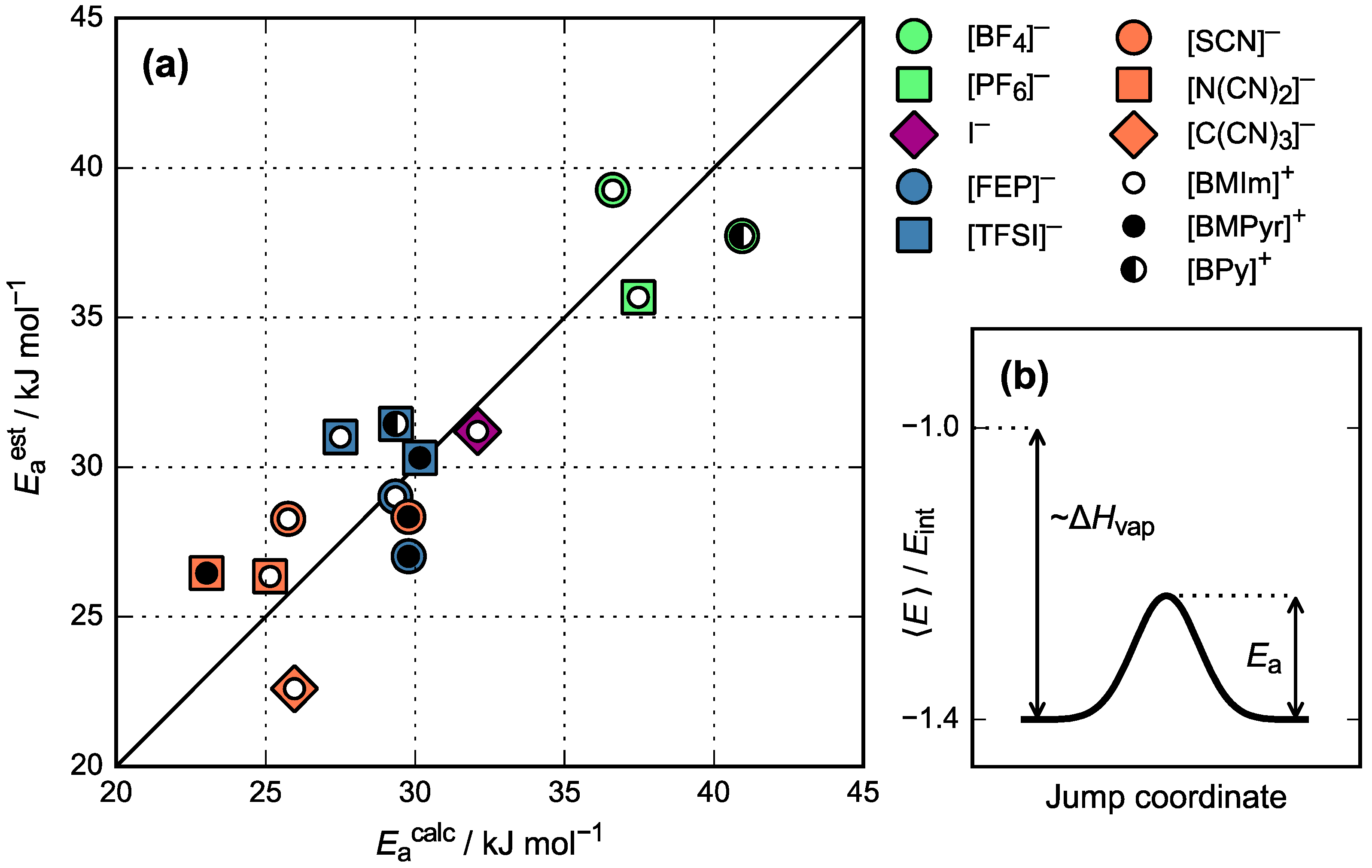
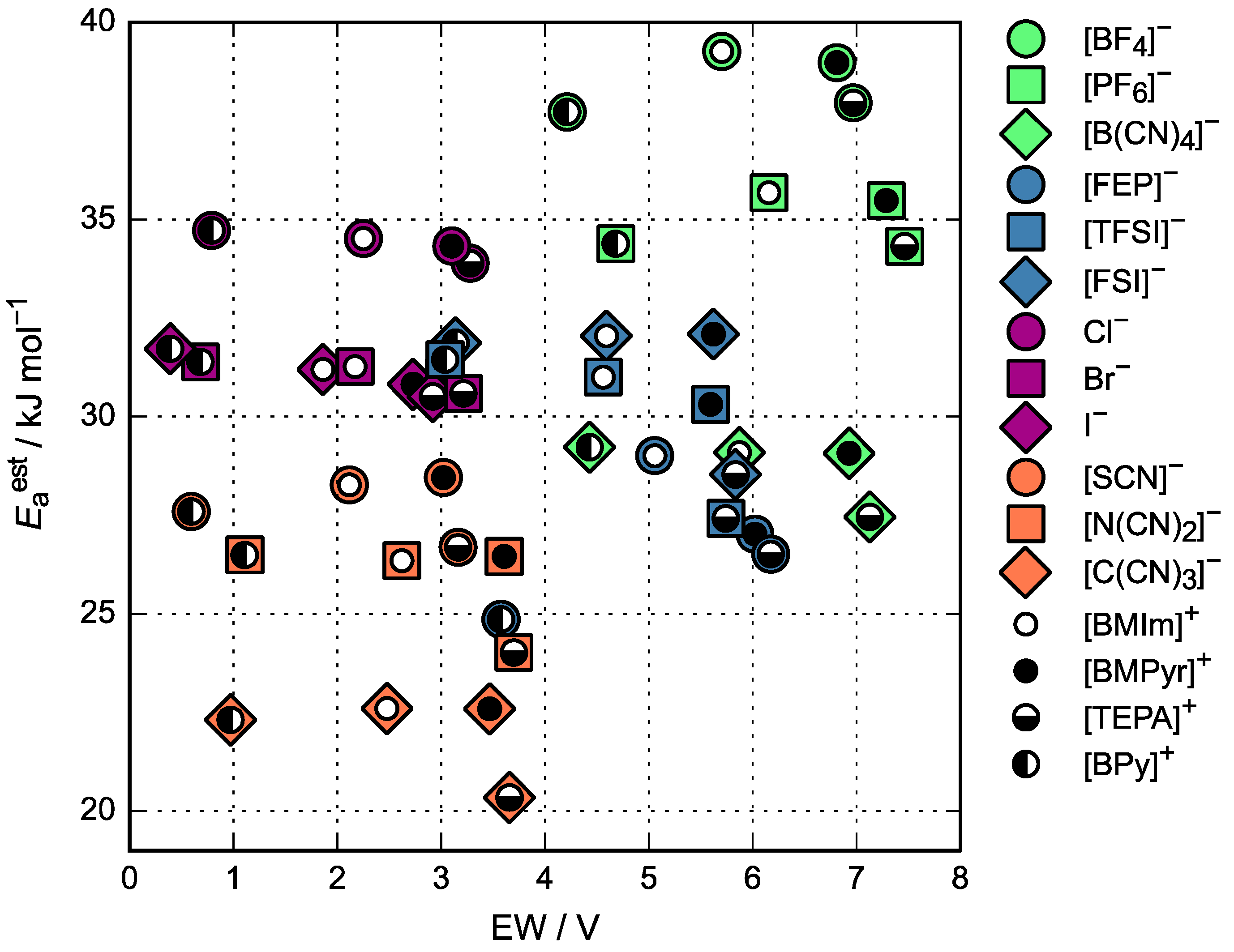
3.3. Activation Energy of Viscosity
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
| DFT | Density functional theory |
| BLYP | Becke’s 88 exchange functional and Lee, Yang and Parr’s correlation functional |
| Def2-TZVP | Ahlrics’ style triple-zeta basis set |
| D3 | Grimme’s dispersion correction |
| gCP | Grimme’s counterpoise correction |
| B3LYP | Hybrid Becke’s three parameter exchange functional and Lee, Yang and Parr’s correlation functional |
| ΔSCF | method to estimate electron affinities and ionization energies |
| EA | Electron Affinity |
| IE | Ionization Energy |
| Eint | Interaction energy of an ionic pair |
| CHELPG | Charges from Electrostatic Potentials using a Grid based method |
| ΔHvap | Heat of vaporization |
| Vm | Molar volume |
| D | Self-diffusion coefficient |
Appendix A
Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ionic Liquid | T Range/K | Max. η/Pa·s | Eacalc/kJ·mol−1 |
|---|---|---|---|
| [BMIm]BF4 | 278.15−338.15 | 0.36 [45] | 36.62 |
| [BMIm]PF6 | 288.15−373.15 | 0.533 [45] | 37.47 |
| [BMIm]TFSI | 278.15−373.15 | 0.143 [45] | 27.50 |
| [BMIm]I | 353.15−388.15 | 0.0365 [46] | 32.09 |
| [BMIm]N(CN)2 | 293.15−343.15 | 0.03679 [47] | 25.15 |
| [BMIm]SCN | 313.15−353.15 | 0.0322 [48] | 25.75 |
| [BMIm]C(CN)3 | 298.15−338.15 | 0.02784 [49] | 25.96 |
| [BMIm]FAP | 293.15−373.15 | 0.1003 [50] | 29.34 |
| [BMPyr]SCN | 298.15−348.15 | 0.109 [51] | 29.84 |
| [BMPyr]N(CN)2 | 293.15−343.15 | 0.0413 [52] | 23.02 |
| [BMPyr]FAP | 290−365 | 0.355 [53] | 29.78 |
| [BMPyr]TFSI | 283.15−353.15 | 0.168 [54] | 30.16 |
| [BPy]BF4 | 298.15−343.15 | 0.145 [55] | 40.94 |
| [BPy]TFSI | 303.15−328.15 | 0.04915 [56] | 29.36 |
References
- Fedorov, M.V.; Kornyshev, A.A. Ionic Liquids at Electrified Interfaces. Chem. Rev. 2014, 114, 2978–3036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacFarlane, D.R.; Tachikawa, N.; Forsyth, M.; Pringle, J.M.; Howlett, P.C.; Elliott, G.D.; Davis, J.H.; Watanabe, M.; Simon, P.; Angell, C.A. Energy applications of ionic liquids. Energy Environ. Sci. 2014, 7, 232–250. [Google Scholar] [CrossRef] [Green Version]
- Korth, M. Large-scale virtual high-throughput screening for the identification of new battery electrolyte solvents: Evaluation of electronic structure theory methods. Phys. Chem. Chem. Phys. 2014, 16, 7919–7926. [Google Scholar] [CrossRef] [PubMed]
- Borodin, O.; Olguin, M.; Spear, C.E.; Leiter, K.W.; Knap, J. Towards high throughput screening of electrochemical stability of battery electrolytes. Nanotechnology 2015, 26, 354003. [Google Scholar] [CrossRef] [PubMed]
- Schütter, C.; Husch, T.; Korth, M.; Balducci, A. Toward New Solvents for EDLCs: From Computational Screening to Electrochemical Validation. J. Phys. Chem. C 2015, 119, 13413–13424. [Google Scholar] [CrossRef]
- Husch, T.; Yilmazer, N.D.; Balducci, A.; Korth, M. Large-scale virtual high-throughput screening for the identification of new battery electrolyte solvents: Computing infrastructure and collective properties. Phys. Chem. Chem. Phys. 2015, 17, 3394–3401. [Google Scholar] [CrossRef] [PubMed]
- Husch, T.; Korth, M. Charting the known chemical space for non-aqueous lithium-air battery electrolyte solvents. Phys. Chem. Chem. Phys. 2015, 17, 22596–22603. [Google Scholar] [CrossRef] [PubMed]
- Husch, T.; Korth, M. How to estimate solid-electrolyte-interphase features when screening electrolyte materials. Phys. Chem. Chem. Phys. 2015, 17, 22799–22808. [Google Scholar] [CrossRef] [PubMed]
- Borodin, O.; Olguin, M.; Spear, C.; Leiter, K.; Knap, J.; Yushin, G.; Childs, A.; Xu, K. Challenges with Quantum Chemistry-Based Screening of Electrochemical Stability of Lithium Battery Electrolytes. ECS Trans. 2015, 69, 113–123. [Google Scholar] [CrossRef]
- Qu, X.; Jain, A.; Rajput, N.N.; Cheng, L.; Zhang, Y.; Ong, S.P.; Brafman, M.; Maginn, E.; Curtiss, L.A.; Persson, K.A. The Electrolyte Genome project: A big data approach in battery materials discovery. Comput. Mater. Sci. 2015, 103, 56–67. [Google Scholar] [CrossRef]
- Cheng, L.; Assary, R.S.; Qu, X.; Jain, A.; Ong, S.P.; Rajput, N.N.; Persson, K.; Curtiss, L.A. Accelerating Electrolyte Discovery for Energy Storage with High-Throughput Screening. J. Phys. Chem. Lett. 2015, 6, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Zahn, S.; MacFarlane, D.; Izgorodina, E.I. Assessment of Kohn-Sham Density Functional Theory and Moller-Plesset Perturbation Theory for Ionic Liquids. Phys. Chem. Chem. Phys. 2013, 15, 13664–13675. [Google Scholar] [CrossRef] [PubMed]
- Lage-Estebanez, I.; Ruzanov, A.; de la Vega, J.M.G.; Fedorov, M.V.; Ivaništšev, V.B. Self-interaction error in DFT-based modelling of ionic liquids. Phys. Chem. Chem. Phys. 2016, 18, 2175–2182. [Google Scholar] [CrossRef] [PubMed]
- Ivaništšev, V.; Kirchner, K. NaRIBaS: A Scripting Framework for Computational Modelling of Nanomaterials and Room Temperature Ionic Liquids in Bulk and Slab. 2015. Available online: www.github.com/vladislavivanistsev/NaRIBaS (accessed on 15 July 2016).
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Density functional theory with London dispersion corrections. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Kruse, H.; Grimme, S. A geometrical correction for the inter- and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems. J. Chem. Phys. 2012, 136, 154101. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Ong, S.P.; Andreussi, O.; Wu, Y.; Marzari, N.; Ceder, G. Electrochemical Windows of Room-Temperature Ionic Liquids from Molecular Dynamics and Density Functional Theory Calculations. Chem. Mater. 2011, 23, 2979–2986. [Google Scholar] [CrossRef]
- Pandian, S.; Raju, S.G.; Hariharan, K.S.; Kolake, S.M.; Park, D.-H.; Lee, M.-J. Functionalized ionic liquids as electrolytes for lithium-ion batteries. J. Power Sources 2015, 286, 204–209. [Google Scholar] [CrossRef]
- Ivaništšev, V.; O’Connor, S.; Fedorov, M.V. Poly(a)morphic portrait of the electrical double layer in ionic liquids. Electrochem. Commun. 2014, 48, 61–64. [Google Scholar] [CrossRef]
- Galiński, M.; Lewandowski, A.; Stępniak, I. Ionic liquids as electrolytes. Electrochim. Acta 2006, 51, 5567–5580. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Kirchner, B.; Malberg, F.; Firaha, D.S.; Hollóczki, O. Ion pairing in ionic liquids. J. Phys. Condens. Matter 2015, 27, 463002. [Google Scholar] [CrossRef] [PubMed]
- Schröder, C. Comparing reduced partial charge models with polarizable simulations of ionic liquids. Phys. Chem. Chem. Phys. 2012, 14, 3089–3102. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, S. Factors Controlling the Diffusion of Ions in Ionic Liquids. ChemPhysChem 2012, 13, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Maginn, E.J. Direct Correlation between Ionic Liquid Transport Properties and Ion Pair Lifetimes: A Molecular Dynamics Study. J. Phys. Chem. Lett. 2015, 6, 700–705. [Google Scholar] [CrossRef] [PubMed]
- De Riva, J.; Ferro, V.R.; del Olmo, L.; Ruiz, E.; Lopez, R.; Palomar, J. Statistical Refinement and Fitting of Experimental Viscosity-to-Temperature Data in Ionic Liquids. Ind. Eng. Chem. Res. 2014, 53, 10475–10484. [Google Scholar] [CrossRef]
- Batista, M.L.; Coutinho, J.A.; Gomes, J.R. Prediction of Ionic Liquids Properties through Molecular Dynamics Simulations. Curr. Phys. Chem. 2014, 4, 151–172. [Google Scholar] [CrossRef]
- Borodin, O. Relation between Heat of Vaporization, Ion Transport, Molar Volume, and Cation- Anion Binding Energy for Ionic Liquids. J. Phys. Chem. B 2009, 113, 12353–12357. [Google Scholar] [CrossRef] [PubMed]
- Borodin, O.; Vatamanu, J.; Smith, G. Bulk and Interfacial Behavior of Ionic Liquids from Molecular Dynamics Simulations. ECS Trans. 2010, 33, 583–599. [Google Scholar]
- Bernard, U.L.; Izgorodina, E.I.; MacFarlane, D.R. New Insights into the Relationship between Ion-Pair Binding Energy and Thermodynamic and Transport Properties of Ionic Liquids. J. Phys. Chem. C 2010, 114, 20472–20478. [Google Scholar] [CrossRef]
- Hunt, P.A. Why does a reduction in hydrogen bonding lead to an increase in viscosity for the 1-butyl-2, 3-dimethyl-imidazolium-based ionic liquids? J. Phys. Chem. B 2007, 111, 4844–4853. [Google Scholar] [CrossRef] [PubMed]
- Varela, L.M.; Carrete, J.; García, M.; Rodríguez, J.R.; Gallego, L.J.; Turmine, M.; Cabeza, O. Pseudolattice theory of ionic liquids. In Ionic Liquids: Theory, Properties, New Approaches; Kokorin, A., Ed.; InTech: Shanghai, China, 2011; pp. 347–366. [Google Scholar]
- Izgorodina, E.I.; Bernard, U.L.; Dean, P.M.; Pringle, J.M.; MacFarlane, D.R. The Madelung Constant of Organic Salts. Cryst. Growth Des. 2009, 9, 4834–4839. [Google Scholar] [CrossRef]
- Sangoro, J.R.; Iacob, C.; Serghei, A.; Friedrich, C.; Kremer, F. Universal scaling of charge transport in glass-forming ionic liquids. Phys. Chem. Chem. Phys. 2009, 11, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Ivanova, A.S.; Couchot-Vore, D.; Garrett-Roe, S. Ultrafast structure and dynamics in ionic liquids: 2D-IR spectroscopy probes the molecular origin of viscosity. J. Phys. Chem. Lett. 2014, 5, 1541–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kincaid, J.F.; Eyring, H.; Stearn, A.E. The Theory of Absolute Reaction Rates and its Application to Viscosity and Diffusion in the Liquid State. Chem. Rev. 1941, 28, 301–365. [Google Scholar] [CrossRef]
- Cohen, E. Fifty years of kinetic theory. Phys. Stat. Mech. Its Appl. 1993, 194, 229–257. [Google Scholar] [CrossRef]
- Madden, P.A.; Wilson, M. Covalent effects in ionic liquids. J. Phys. Condens. Matter 2000, 12, A95. [Google Scholar] [CrossRef]
- Salgado, J.; Regueira, T.; Lugo, L.; Vijande, J.; Fernández, J.; García, J. Density and viscosity of three (2,2,2-trifluoroethanol + 1-butyl-3-methylimidazolium) ionic liquid binary systems. J. Chem. Thermodyn. 2014, 70, 101–110. [Google Scholar] [CrossRef]
- Ghatee, M.H.; Zare, M.; Moosavi, F.; Zolghadr, A.R. Temperature-Dependent Density and Viscosity of the Ionic Liquids 1-Alkyl-3-methylimidazolium Iodides: Experiment and Molecular Dynamics Simulation. J. Chem. Eng. Data 2010, 55, 3084–3088. [Google Scholar] [CrossRef]
- Seoane, R.G.; Corderí, S.; Gómez, E.; Calvar, N.; González, E.J.; Macedo, E.A.; Domínguez, Á. Temperature Dependence and Structural Influence on the Thermophysical Properties of Eleven Commercial Ionic Liquids. Ind. Eng. Chem. Res. 2012, 51, 2492–2504. [Google Scholar] [CrossRef]
- Larriba, M.; Navarro, P.; García, J.; Rodríguez, F. Selective extraction of toluene from n-heptane using [emim][SCN] and [bmim][SCN] ionic liquids as solvents. J. Chem. Thermodyn. 2014, 79, 266–271. [Google Scholar] [CrossRef]
- Zubeir, L.F.; Romanos, G.E.; Weggemans, W.M.A.; Iliev, B.; Schubert, T.J.S.; Kroon, M.C. Solubility and Diffusivity of CO2 in the Ionic Liquid 1-Butyl-3-methylimidazolium Tricyanomethanide within a Large Pressure Range (0.01 MPa to 10 MPa). J. Chem. Eng. Data 2015, 60, 1544–1562. [Google Scholar] [CrossRef]
- Almantariotis, D.; Stevanovic, S.; Fandiño, O.; Pensado, A.S.; Padua, A. a. H.; Coxam, J.-Y.; Costa Gomes, M.F. Absorption of carbon dioxide, nitrous oxide, ethane and nitrogen by 1-alkyl-3-methylimidazolium (C(n)mim, n = 2,4,6) tris(pentafluoroethyl)trifluorophosphate ionic liquids (eFAP). J. Phys. Chem. B 2012, 116, 7728–7738. [Google Scholar] [CrossRef] [PubMed]
- Domańska, U.; Królikowska, M. Density and Viscosity of Binary Mixtures of Thiocyanate Ionic Liquids + Water as a Function of Temperature. J. Solut. Chem. 2012, 41, 1422–1445. [Google Scholar] [CrossRef] [PubMed]
- González, E.J.; González, B.; Macedo, E.A. Thermophysical Properties of the Pure Ionic Liquid 1-Butyl-1-methylpyrrolidinium Dicyanamide and Its Binary Mixtures with Alcohols. J. Chem. Eng. Data 2013, 58, 1440–1448. [Google Scholar] [CrossRef]
- Fletcher, S.I.; Sillars, F.B.; Hudson, N.E.; Hall, P.J. Physical Properties of Selected Ionic Liquids for Use as Electrolytes and Other Industrial Applications. J. Chem. Eng. Data 2010, 55, 778–782. [Google Scholar] [CrossRef]
- Tokuda, H.; Tsuzuki, S.; Susan, M.A.B.H.; Hayamizu, K.; Watanabe, M. How Ionic Are Room-Temperature Ionic Liquids? An Indicator of the Physicochemical Properties. J. Phys. Chem. B 2006, 110, 19593–19600. [Google Scholar] [CrossRef] [PubMed]
- Khupse, N.D.; Kumar, A. Dramatic Change in Viscosities of Pure Ionic Liquids upon Addition of Molecular Solvents. J. Solut. Chem. 2009, 38, 589–600. [Google Scholar] [CrossRef]
- Oliveira, F.S.; Freire, M.G.; Carvalho, P.J.; Coutinho, J.A.P.; Lopes, J.N.C.; Rebelo, L.P.N.; Marrucho, I.M. Structural and Positional Isomerism Influence in the Physical Properties of Pyridinium NTf2-Based Ionic Liquids: Pure and Water-Saturated Mixtures. J. Chem. Eng. Data 2010, 55, 4514–4520. [Google Scholar] [CrossRef]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karu, K.; Ruzanov, A.; Ers, H.; Ivaništšev, V.; Lage-Estebanez, I.; García de la Vega, J.M. Predictions of Physicochemical Properties of Ionic Liquids with DFT. Computation 2016, 4, 25. https://doi.org/10.3390/computation4030025
Karu K, Ruzanov A, Ers H, Ivaništšev V, Lage-Estebanez I, García de la Vega JM. Predictions of Physicochemical Properties of Ionic Liquids with DFT. Computation. 2016; 4(3):25. https://doi.org/10.3390/computation4030025
Chicago/Turabian StyleKaru, Karl, Anton Ruzanov, Heigo Ers, Vladislav Ivaništšev, Isabel Lage-Estebanez, and José M. García de la Vega. 2016. "Predictions of Physicochemical Properties of Ionic Liquids with DFT" Computation 4, no. 3: 25. https://doi.org/10.3390/computation4030025