Plasma Membranes Modified by Plasma Treatment or Deposition as Solid Electrolytes for Potential Application in Solid Alkaline Fuel Cells

Abstract
:1. Introduction
2. Results and Discussion
2.1. Morphological and Structural Properties
2.1.1. SEM Observations


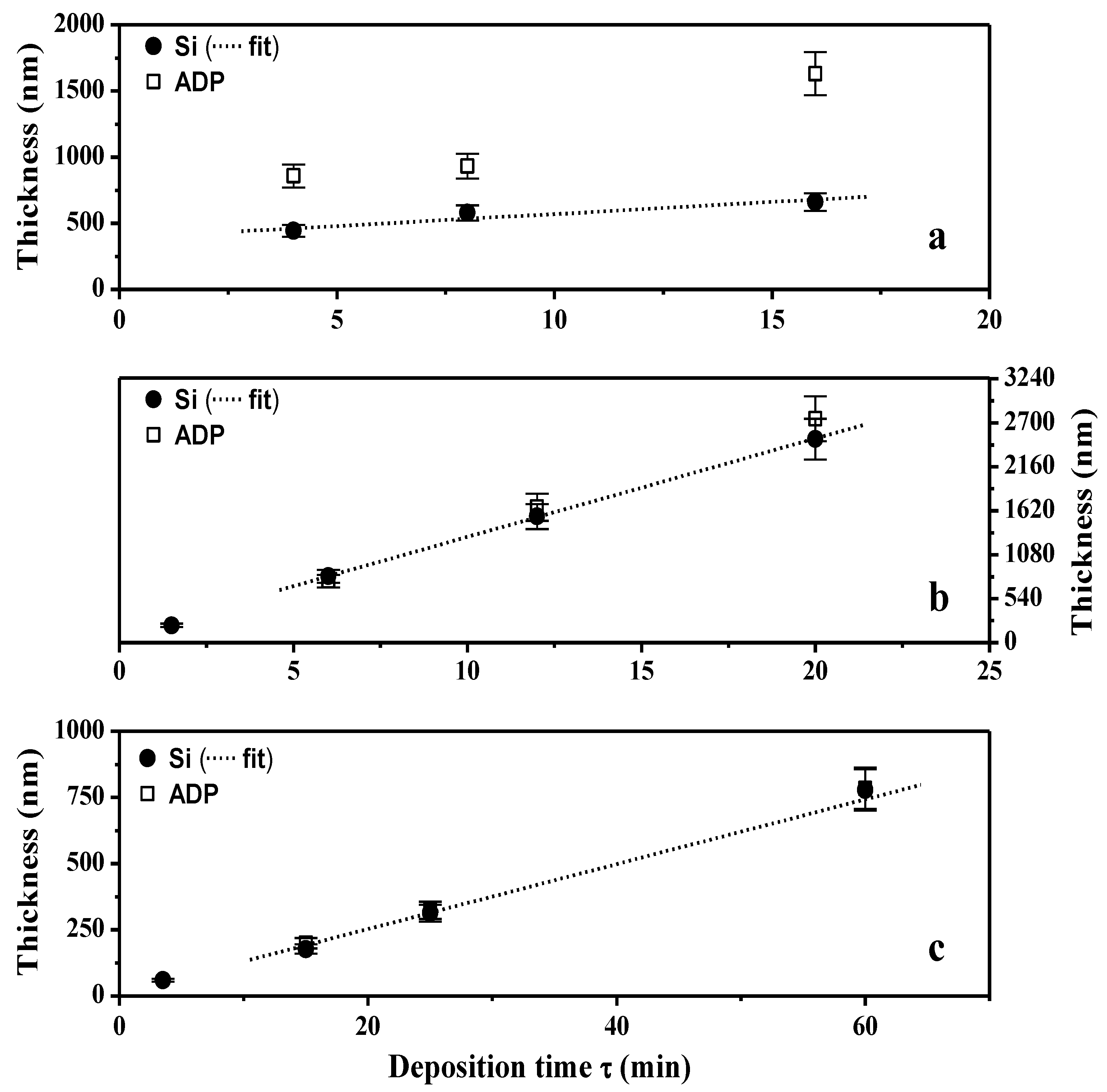
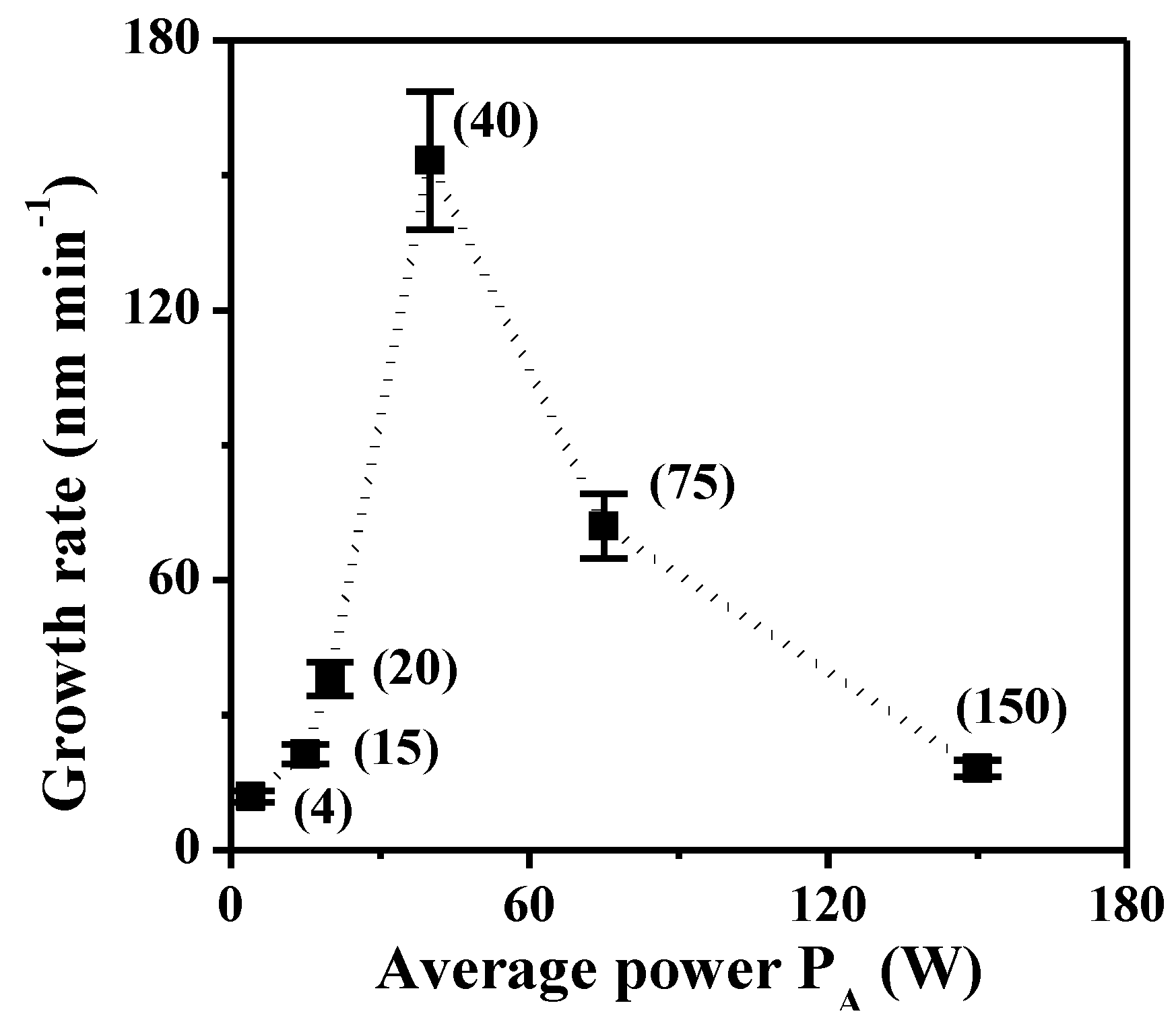
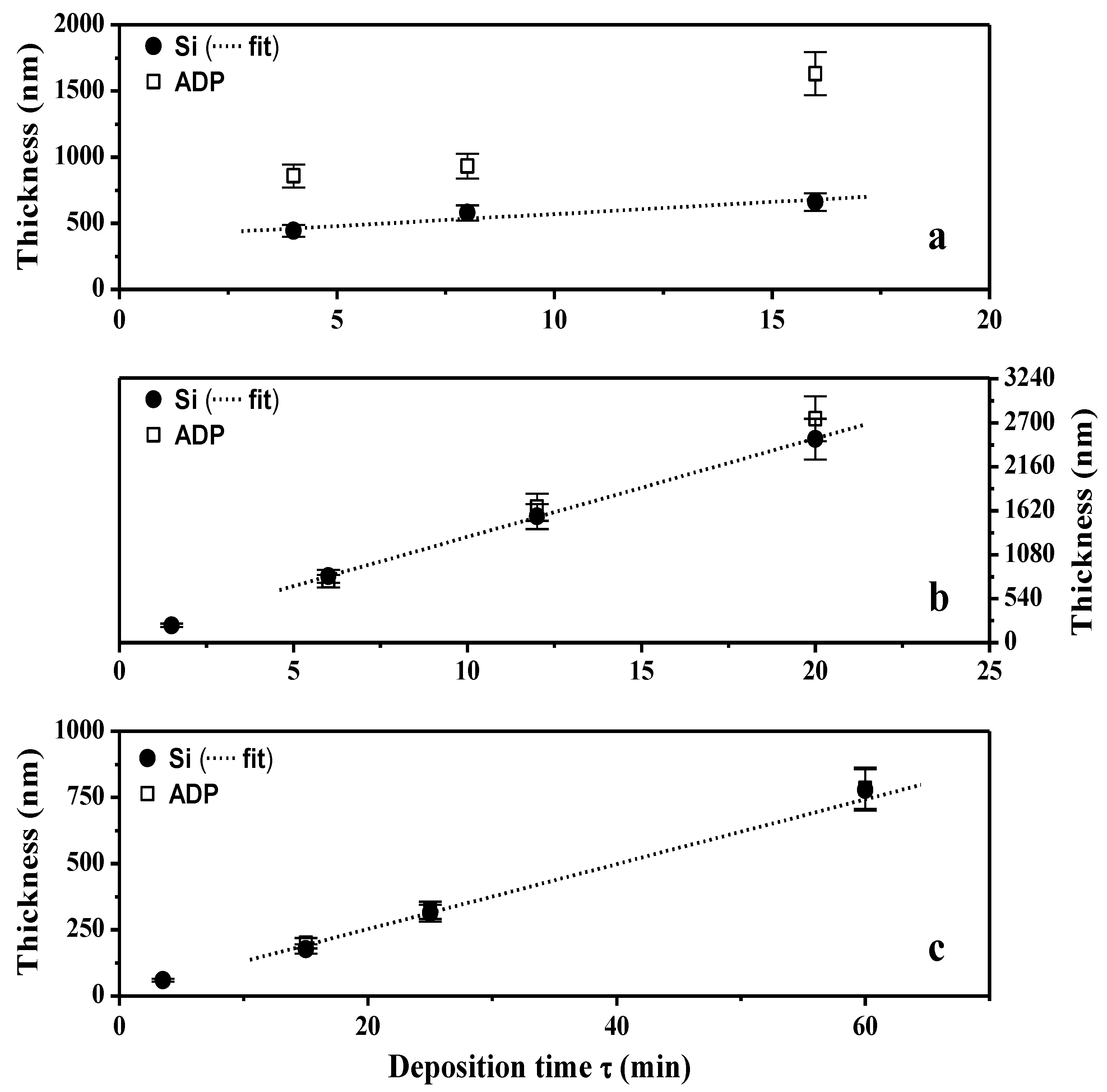
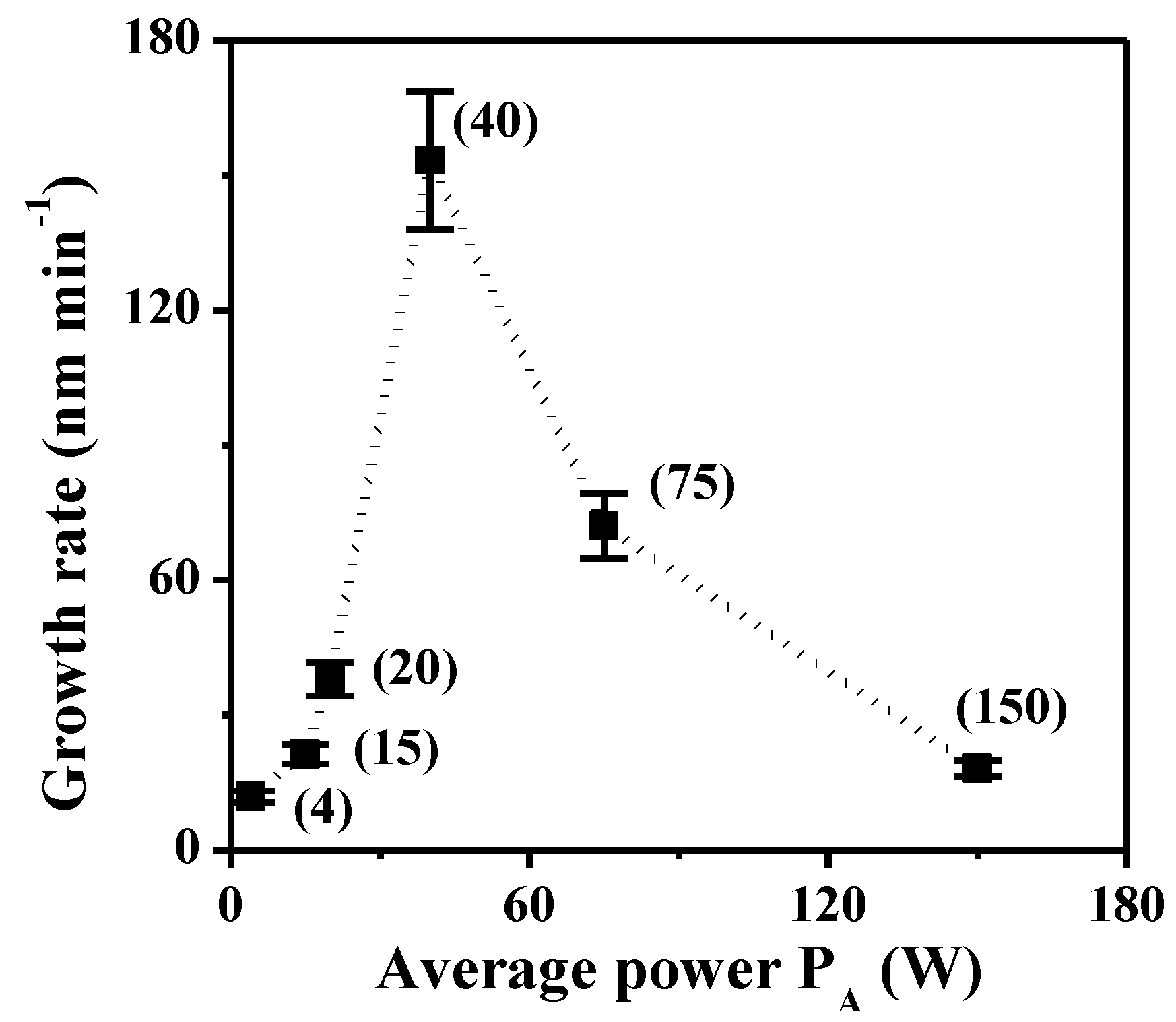
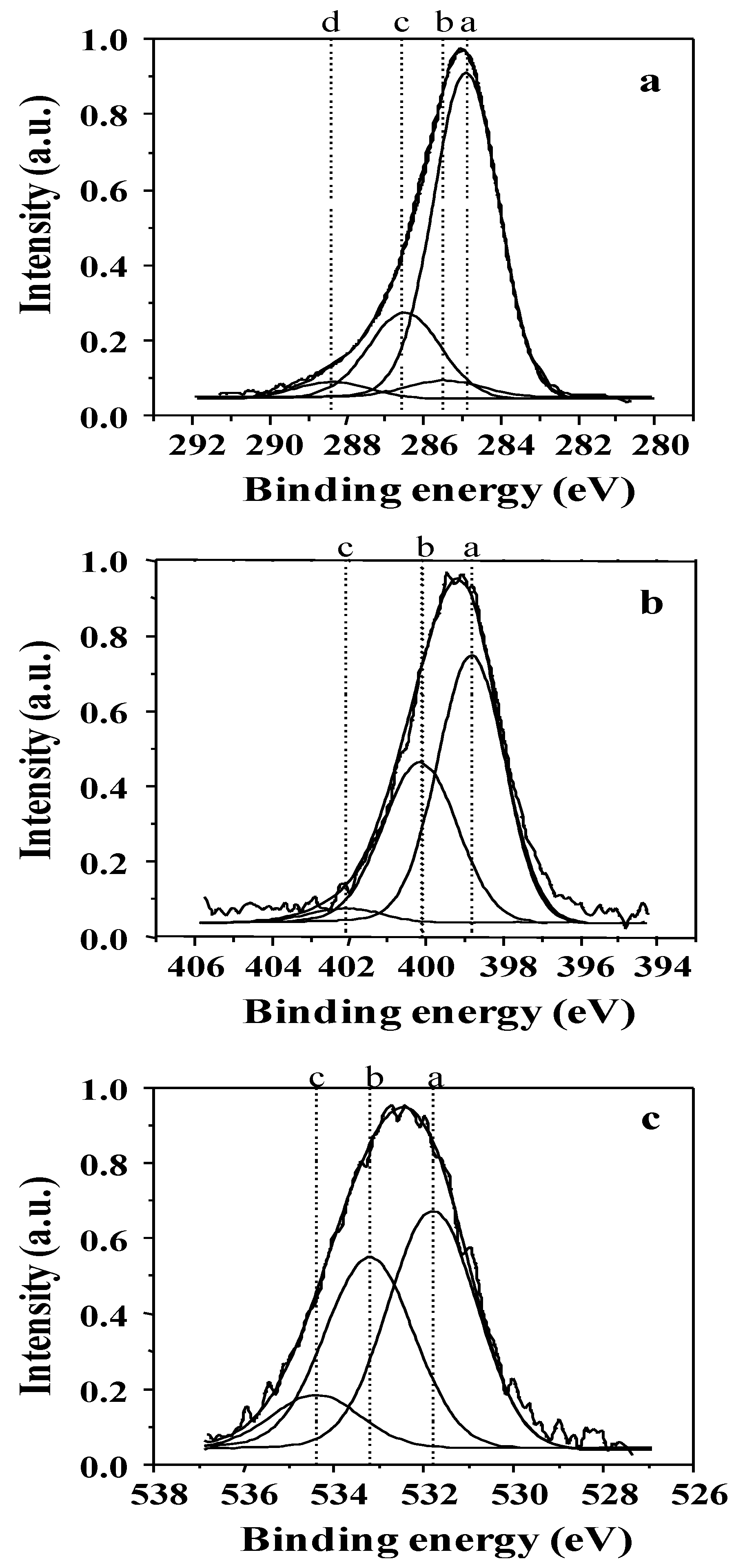
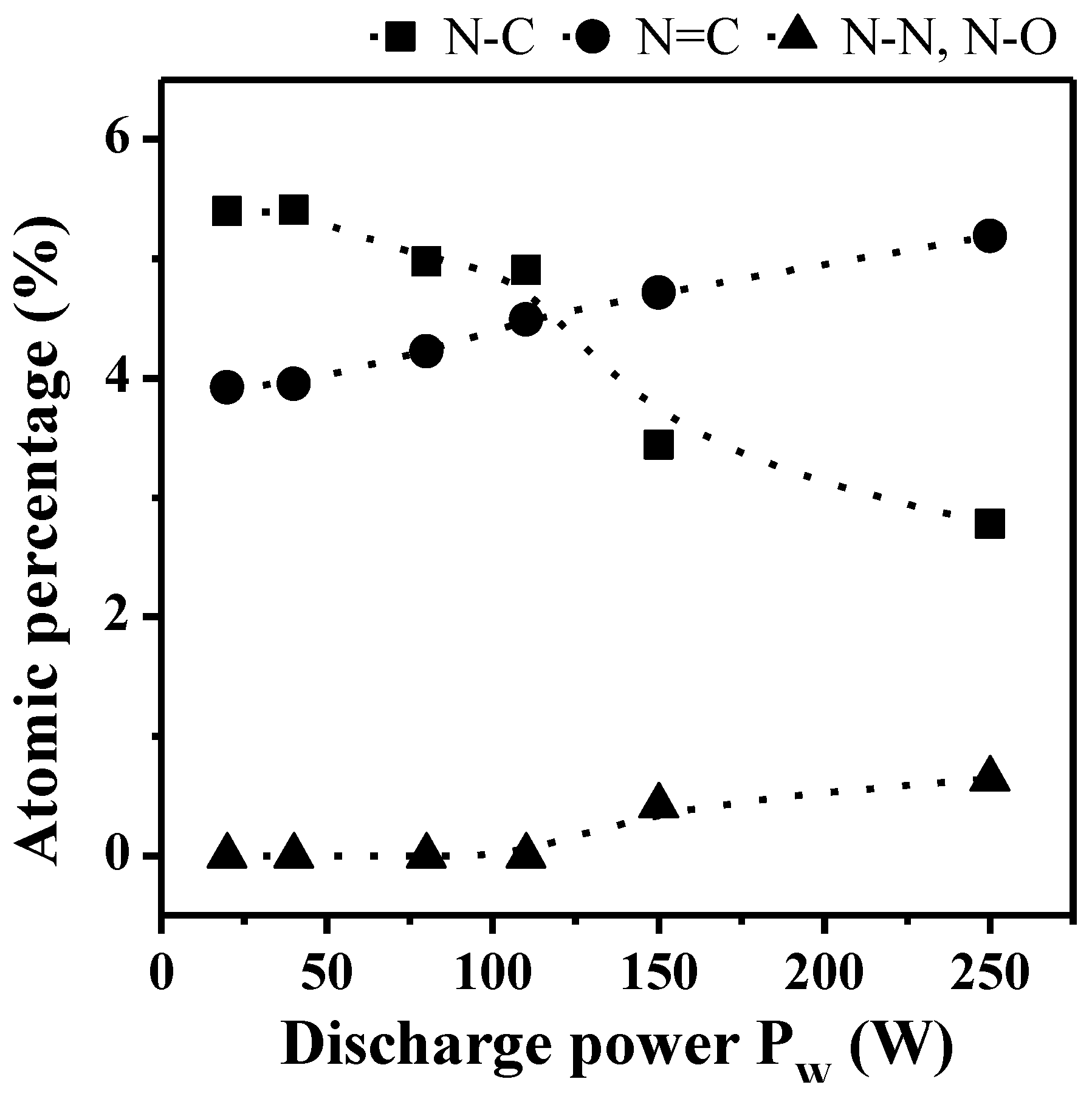
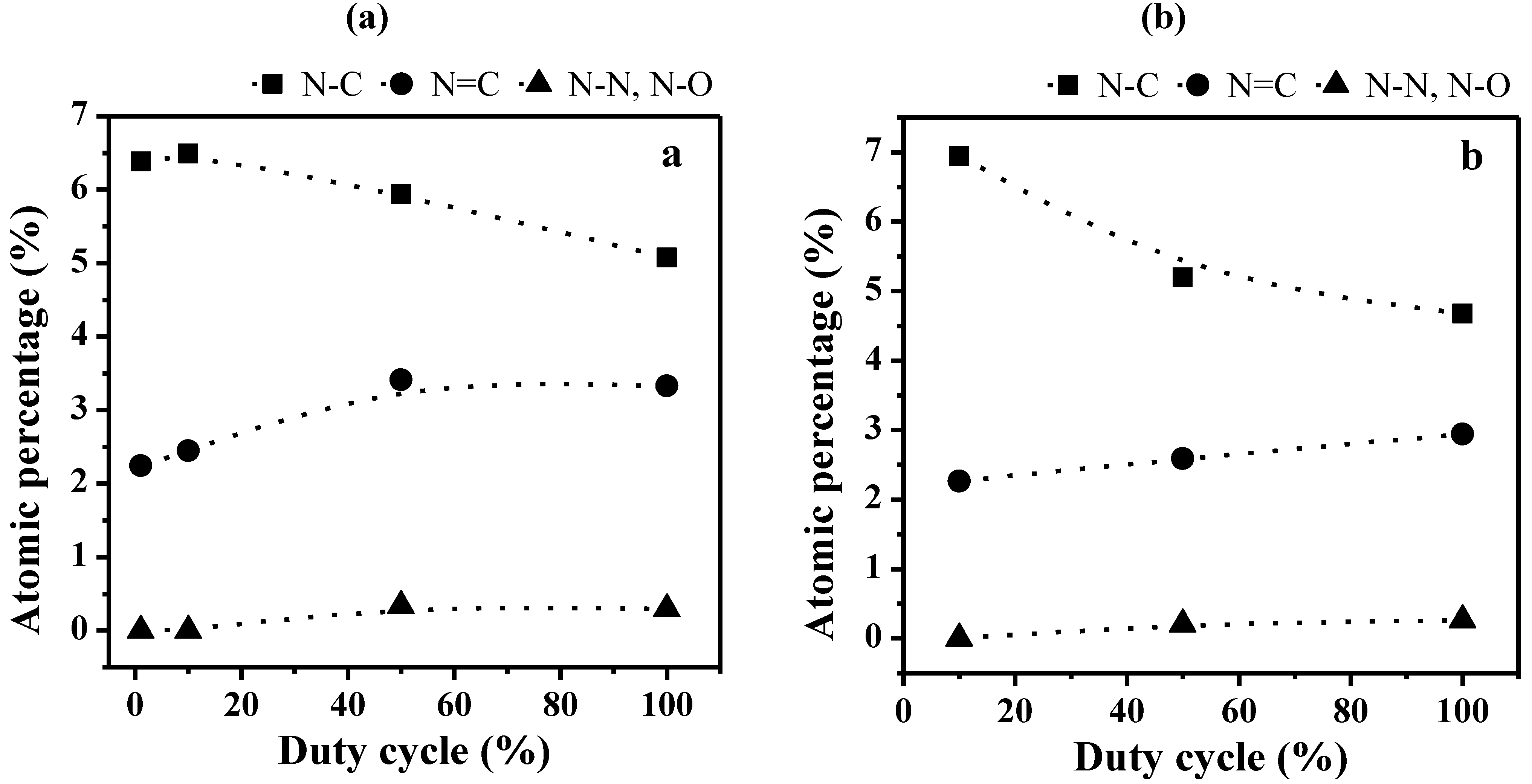
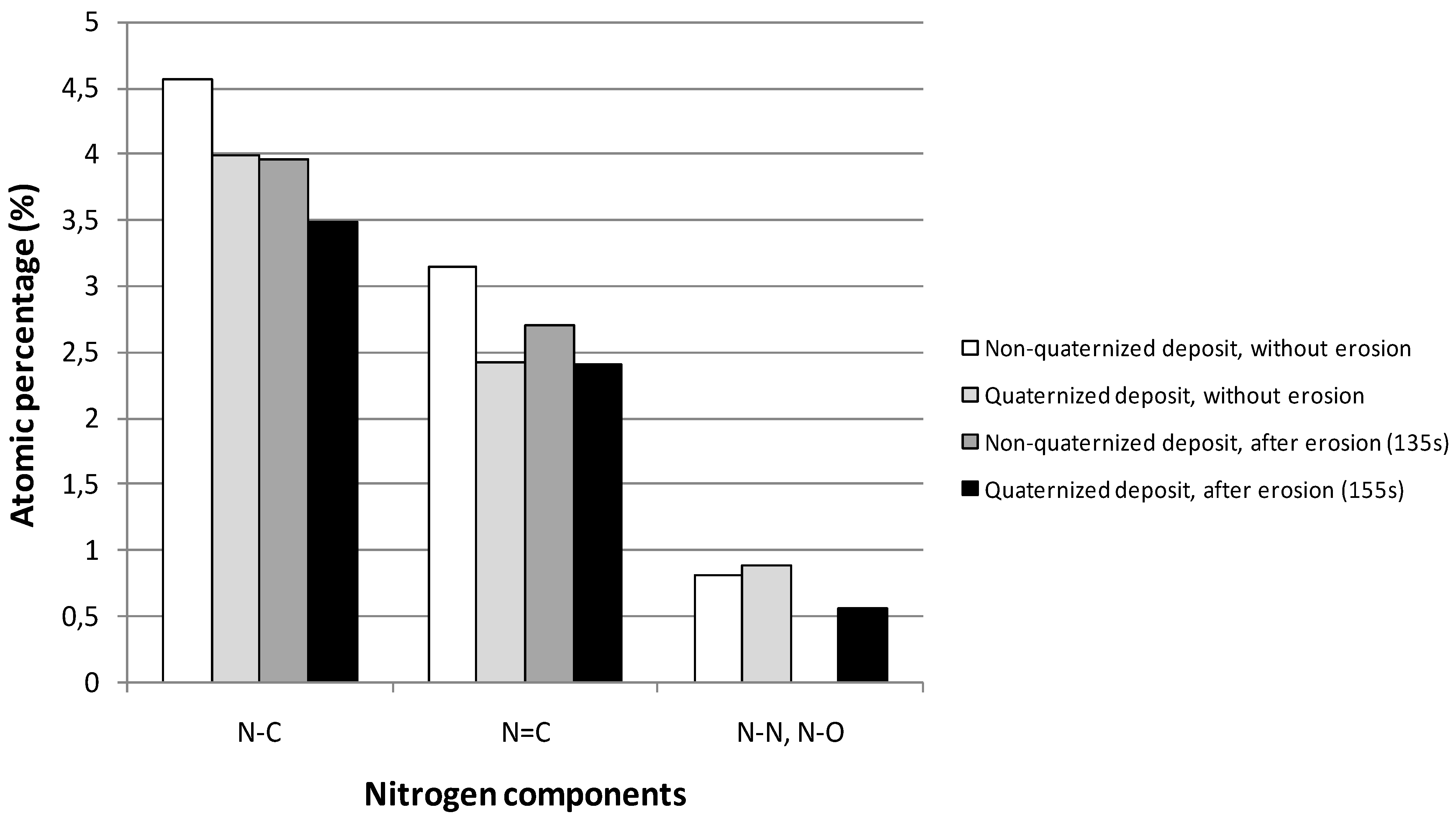
2.1.2. XPS Analyses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Photoelectron | Peak | Bonding energy (eV) | FWHM (eV) | Assignment | References |
|---|---|---|---|---|---|
| C1s | a | 284.8 ± 0.1 | 2.1 | C–C(–C), C–H | [32,33,34] |
| b | 285.5 ± 0.1 | 2.2 | sp2 C=N, C–C(–O), C–C(–N) | [35,36] | |
| c | 286.6 ± 0.1 | 2.2 | C–N, C–O (OCN) | [33,35] | |
| d | 288.4 ± 0.1 | 2.3 | C=O | [32,35,36] | |
| N1s | a | 398.6 ± 0.1 | 2.2 | sp3 N–C | [37,38,39] |
| b | 400.1 ± 0.1 | 2.2 | sp2 N=C | [40] | |
| c | 402.1 ± 0.1 | 2.3 | N–N, N–O | [39,41,42] | |
| O1s | a | 531.8 ± 0.1 | 2.2 | O=C | [32,43] |
| b | 533.2 ± 0.1 | 2.2 | O–C | [35,36] | |
| c | 534.5 ± 0.1 | 2.3 | O–H | [32,43] |
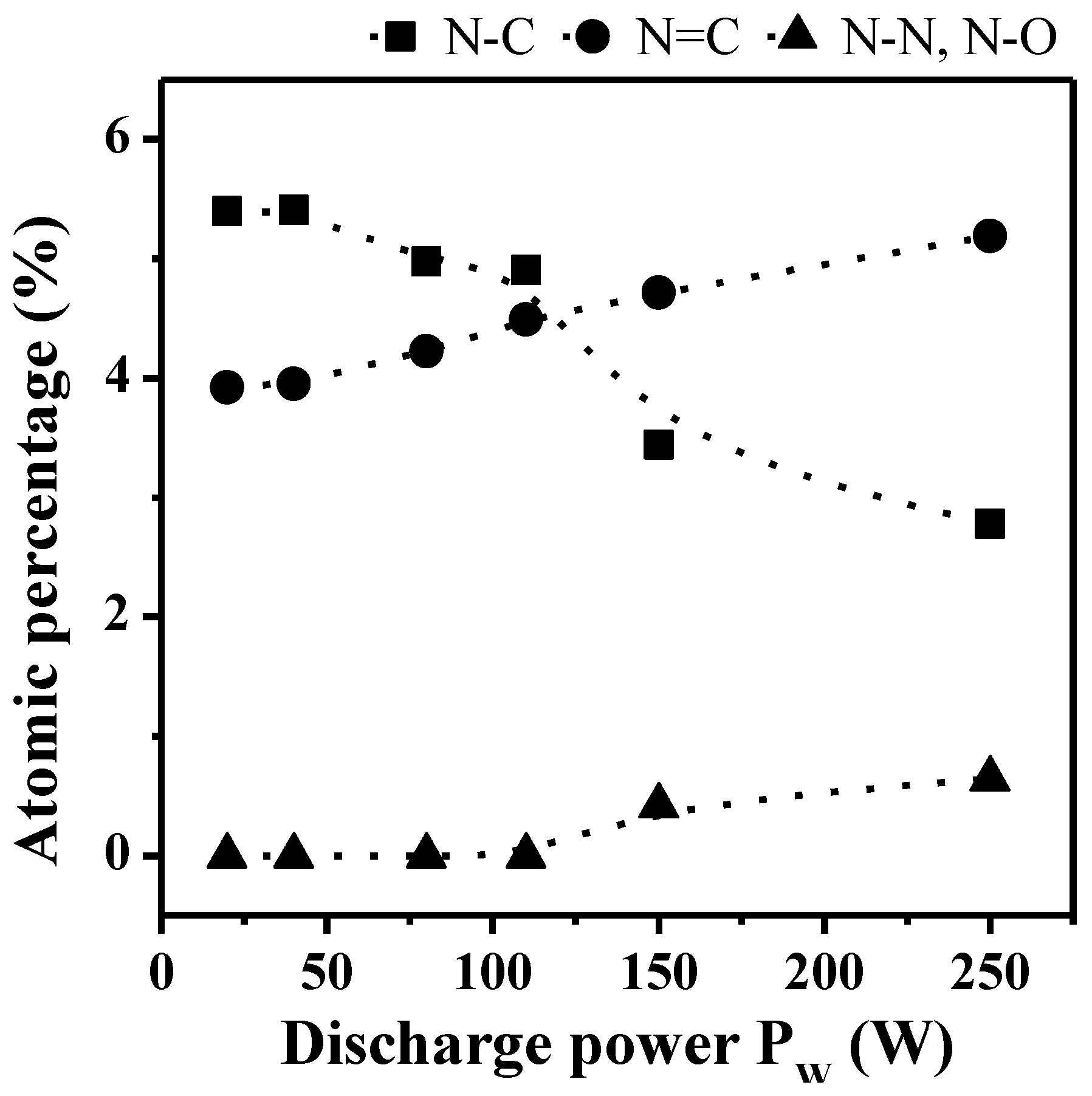
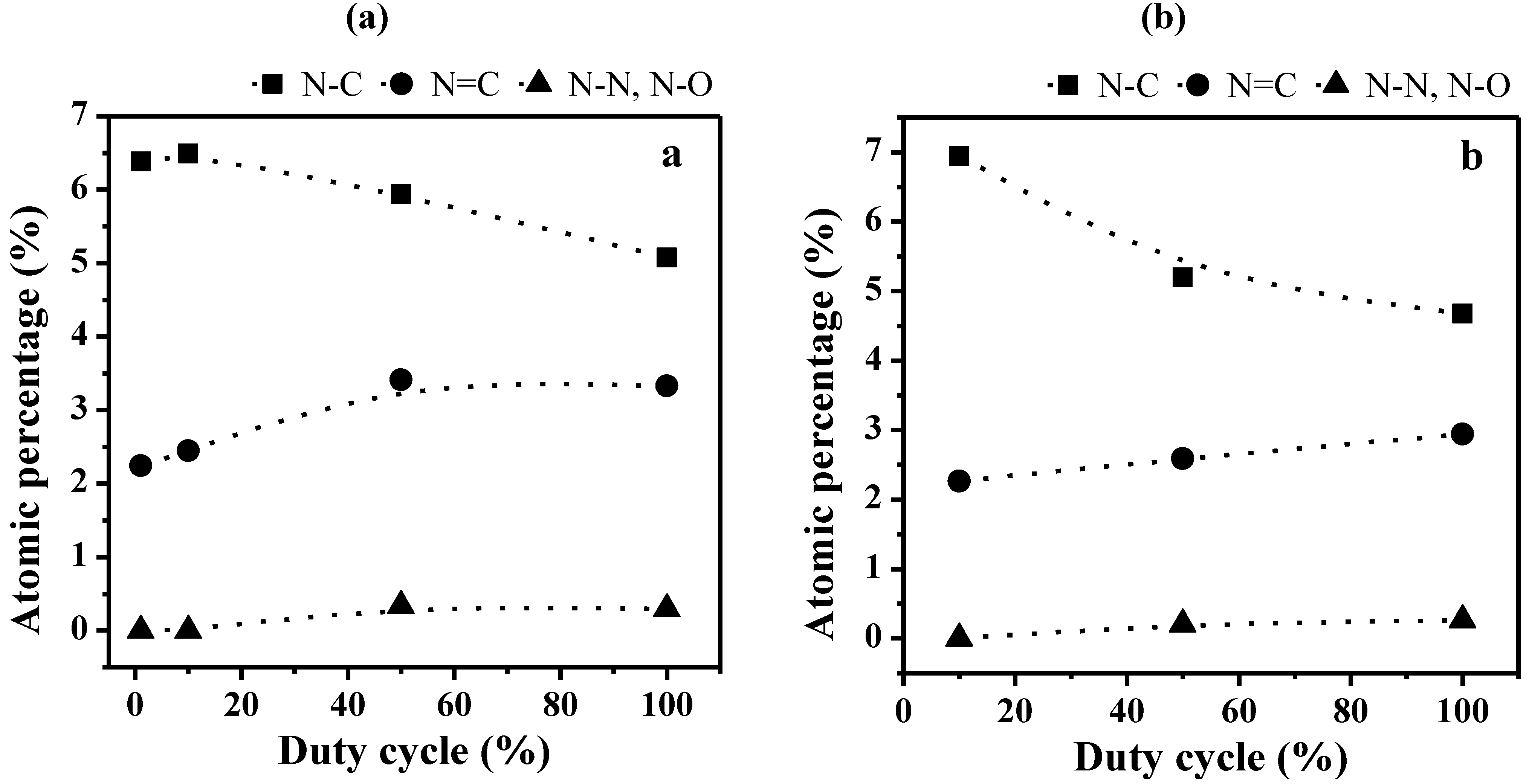
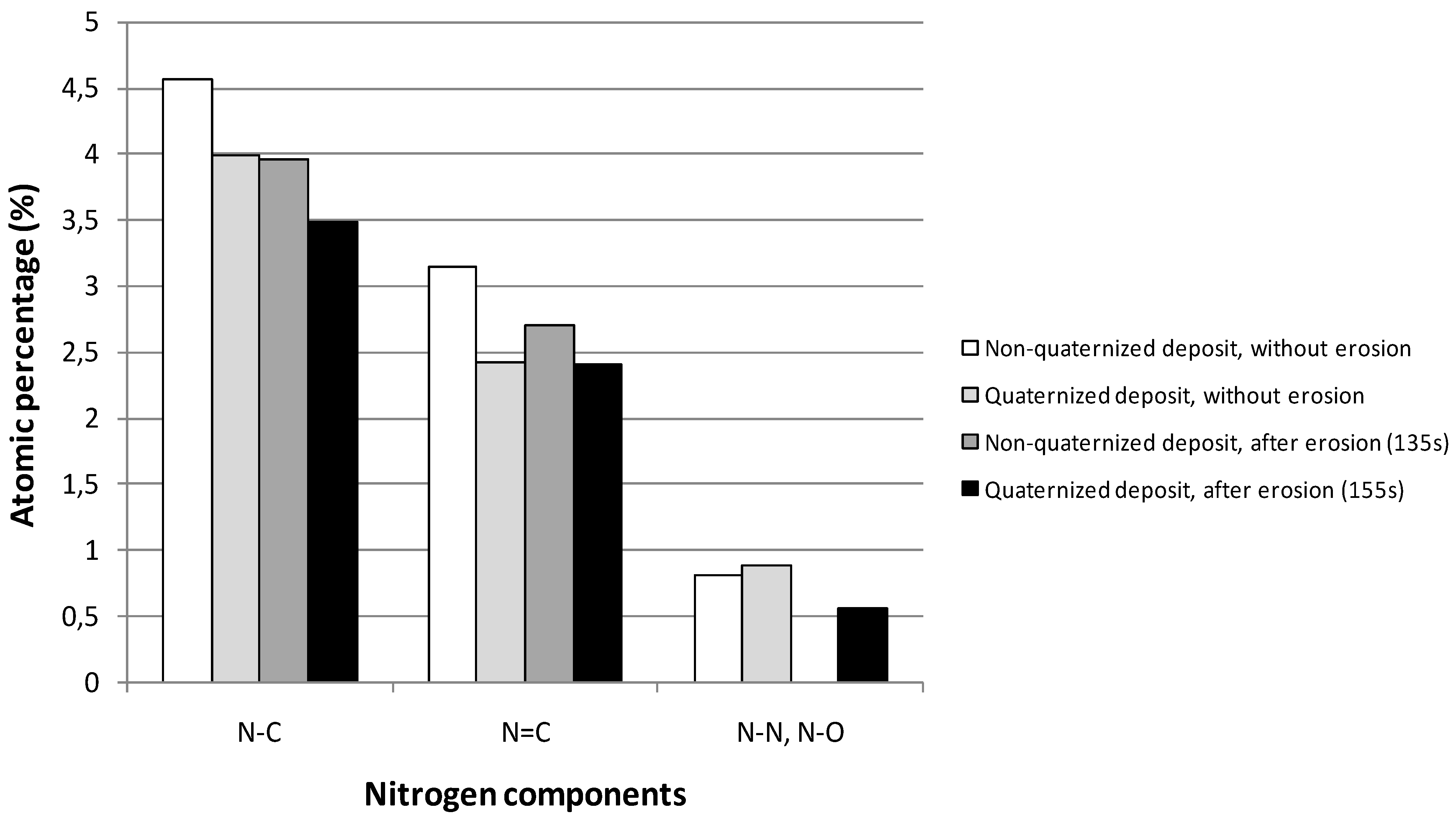
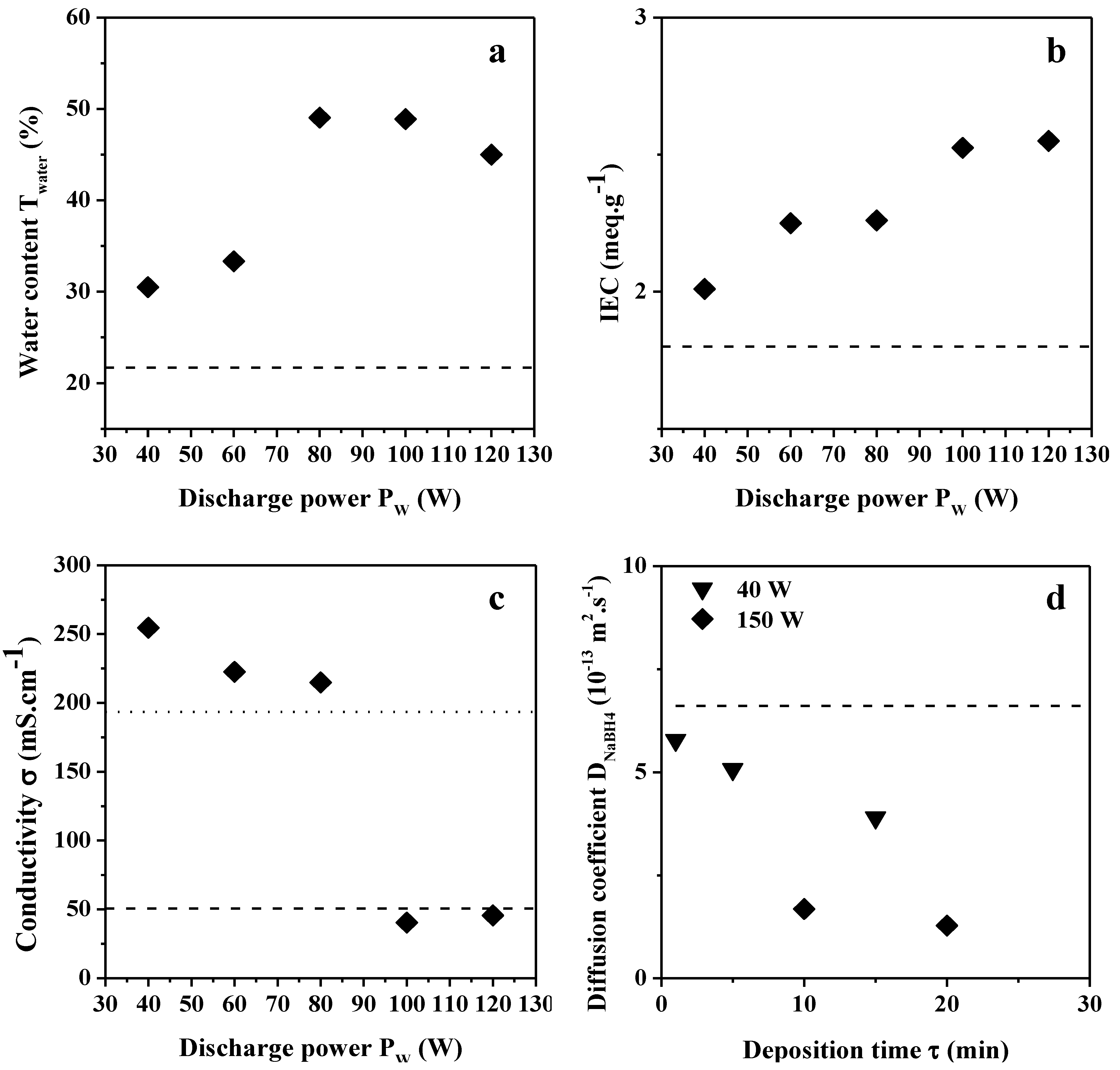
2.2. Transport Properties
2.2.1. Water Content, IEC and Hydroxide Conductivity
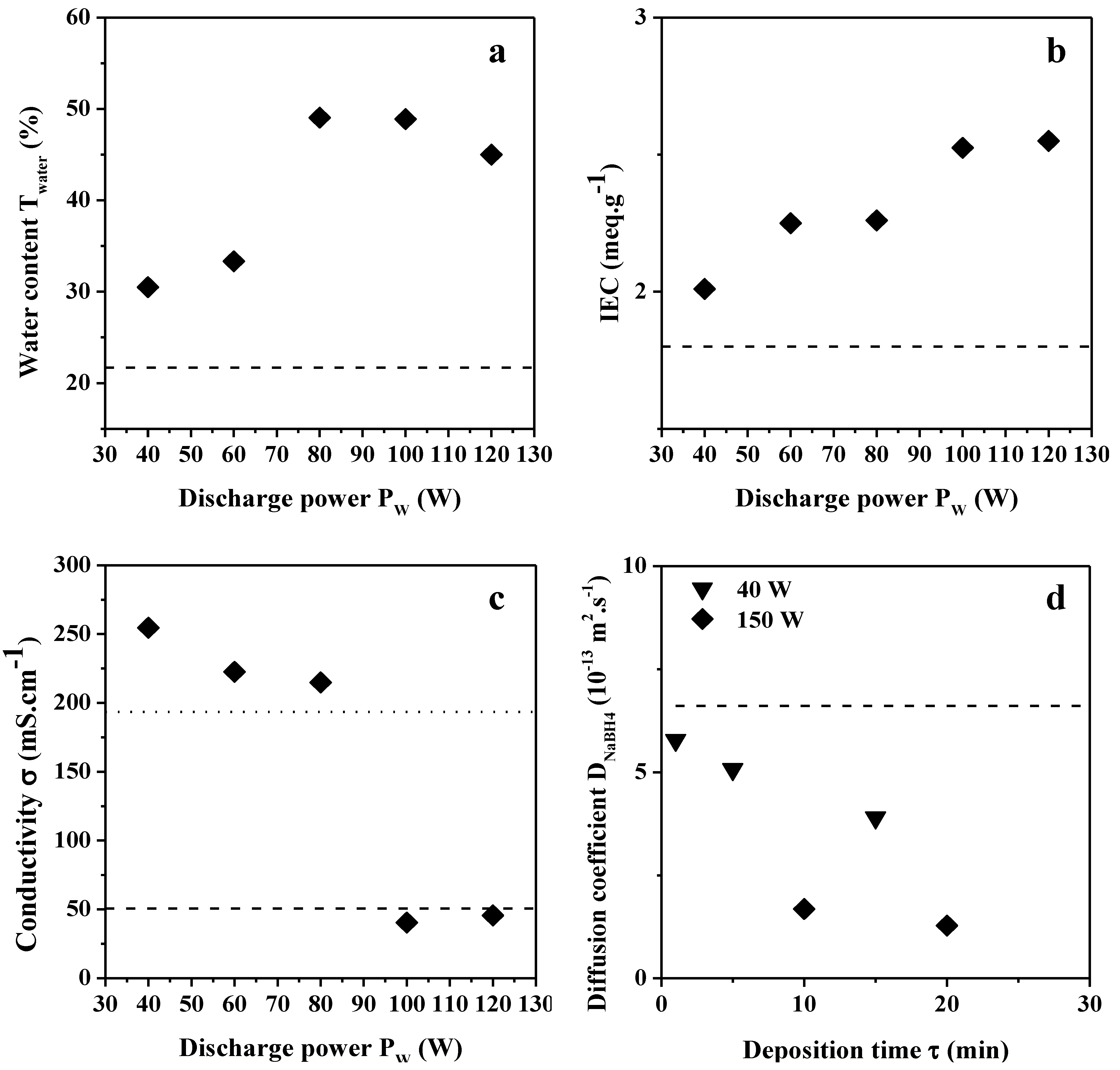
2.2.2. NaBH4 and Glycerol Permeability
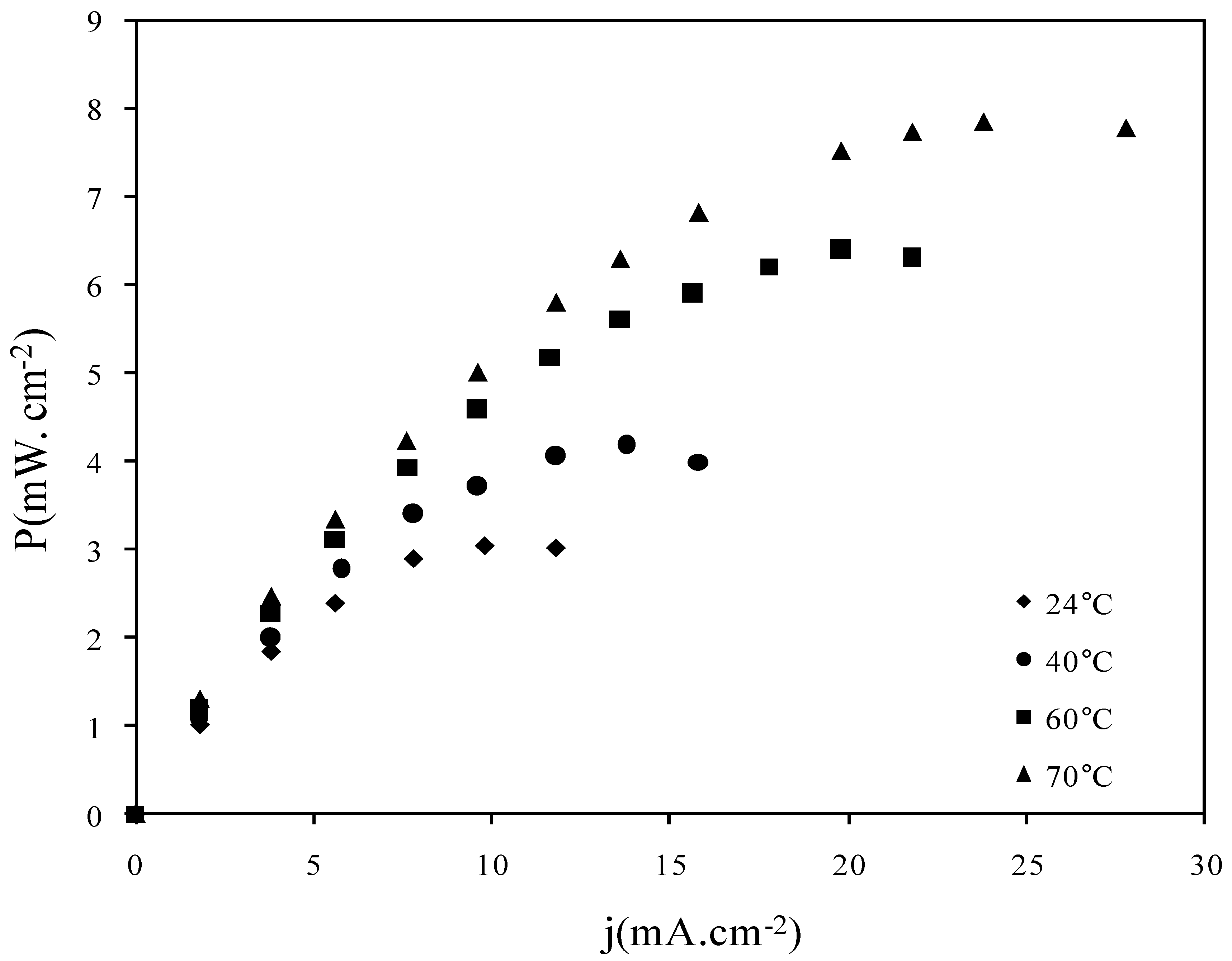
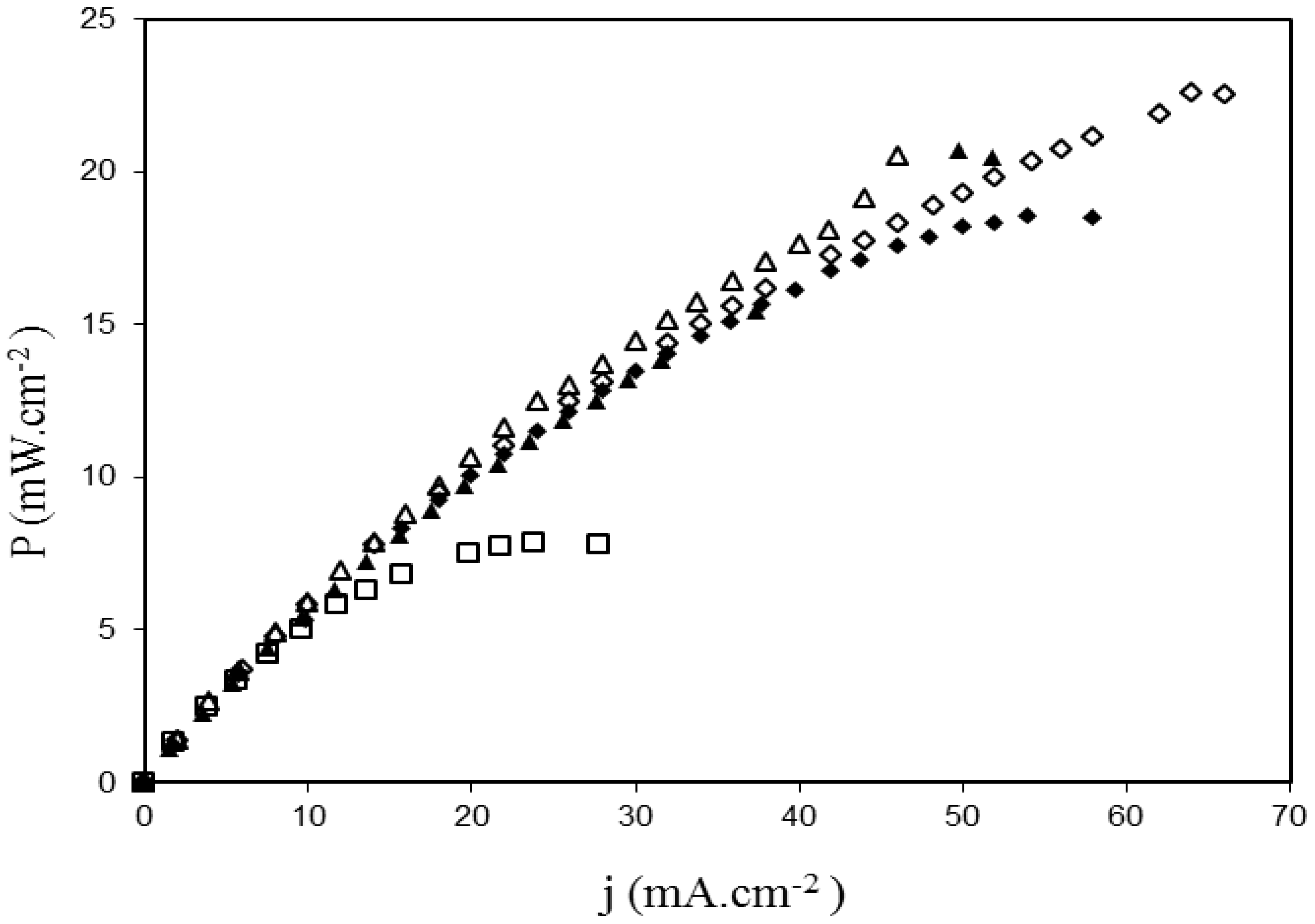
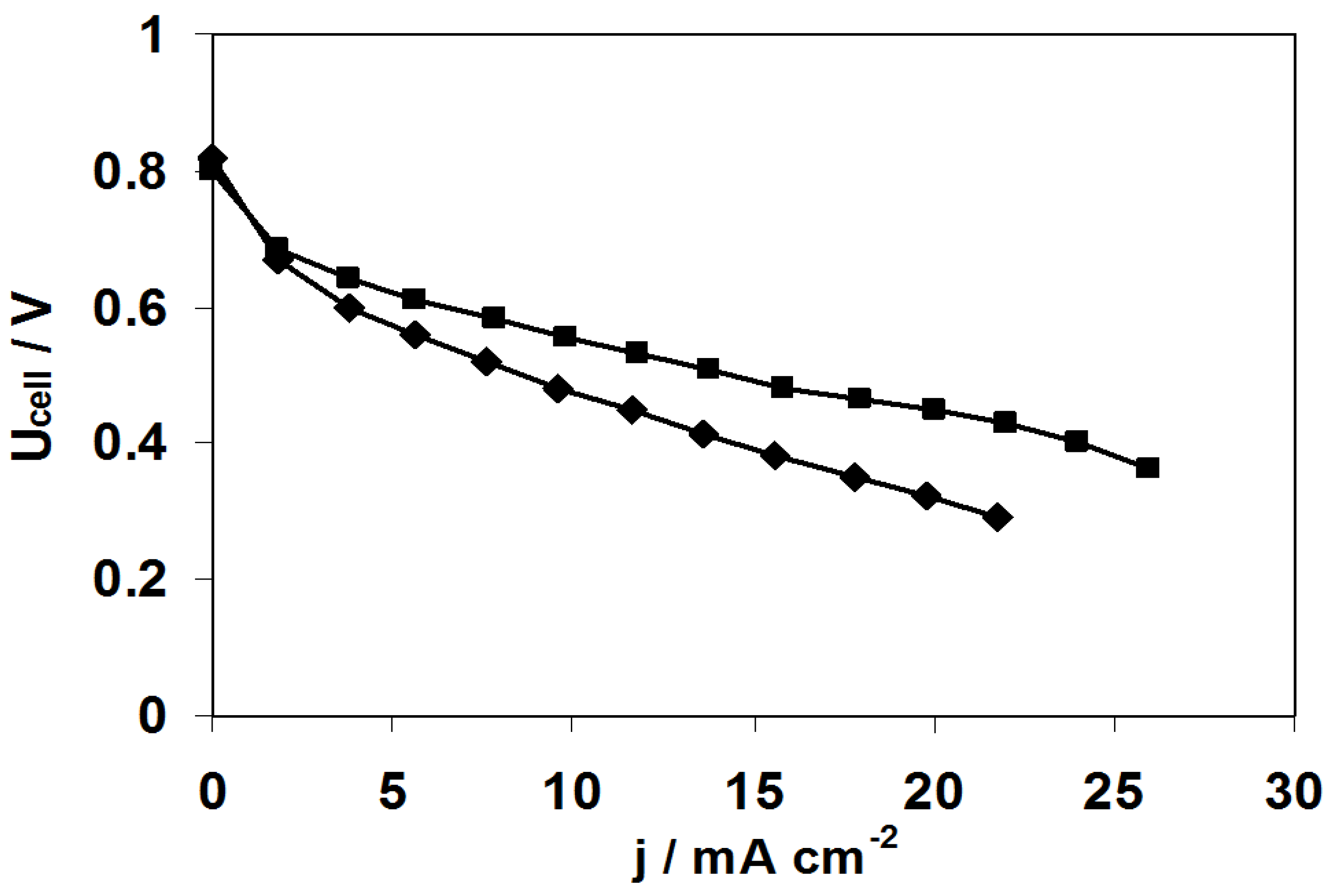
2.3. Fuel Cell Tests

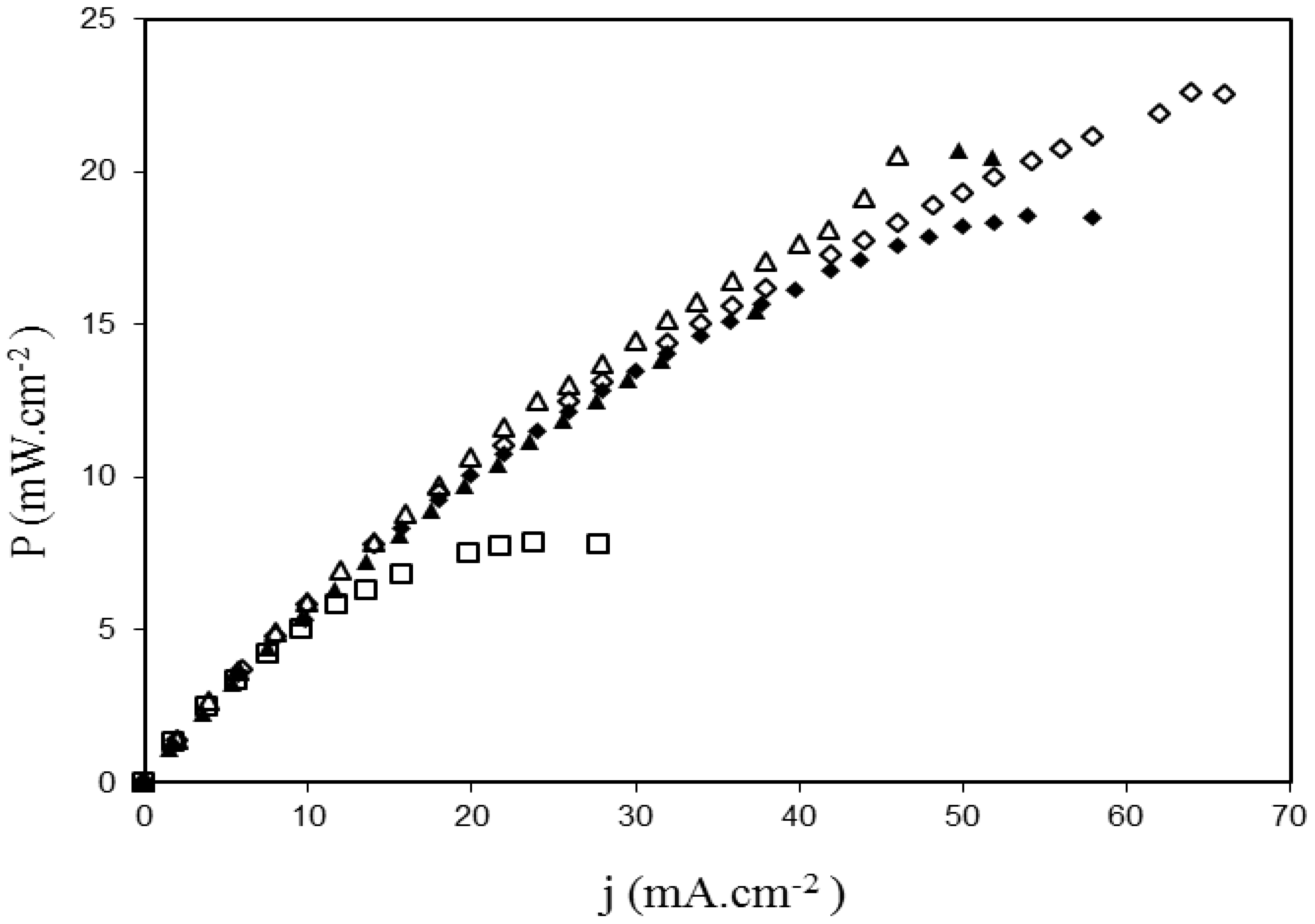
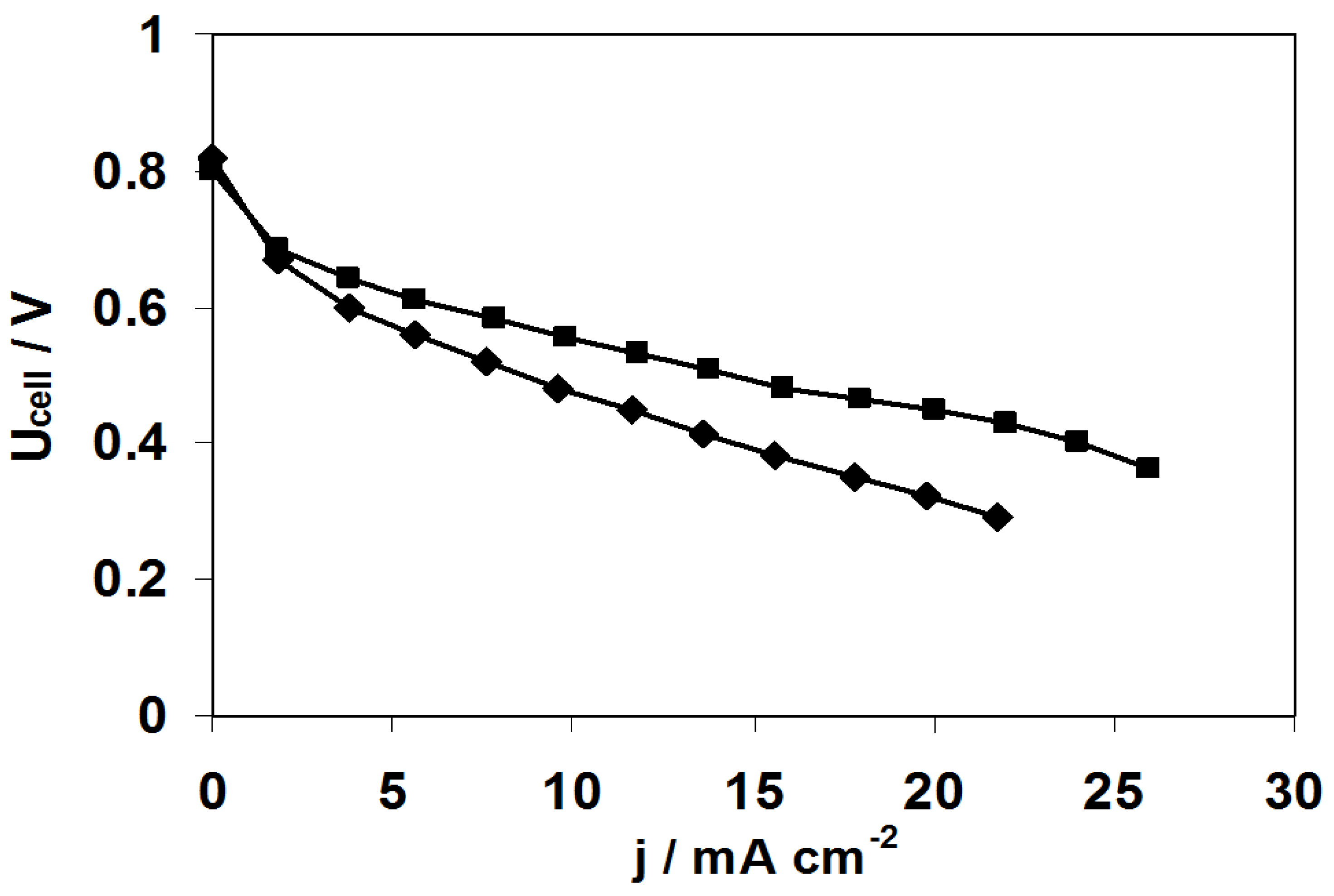
3. Experimental Section
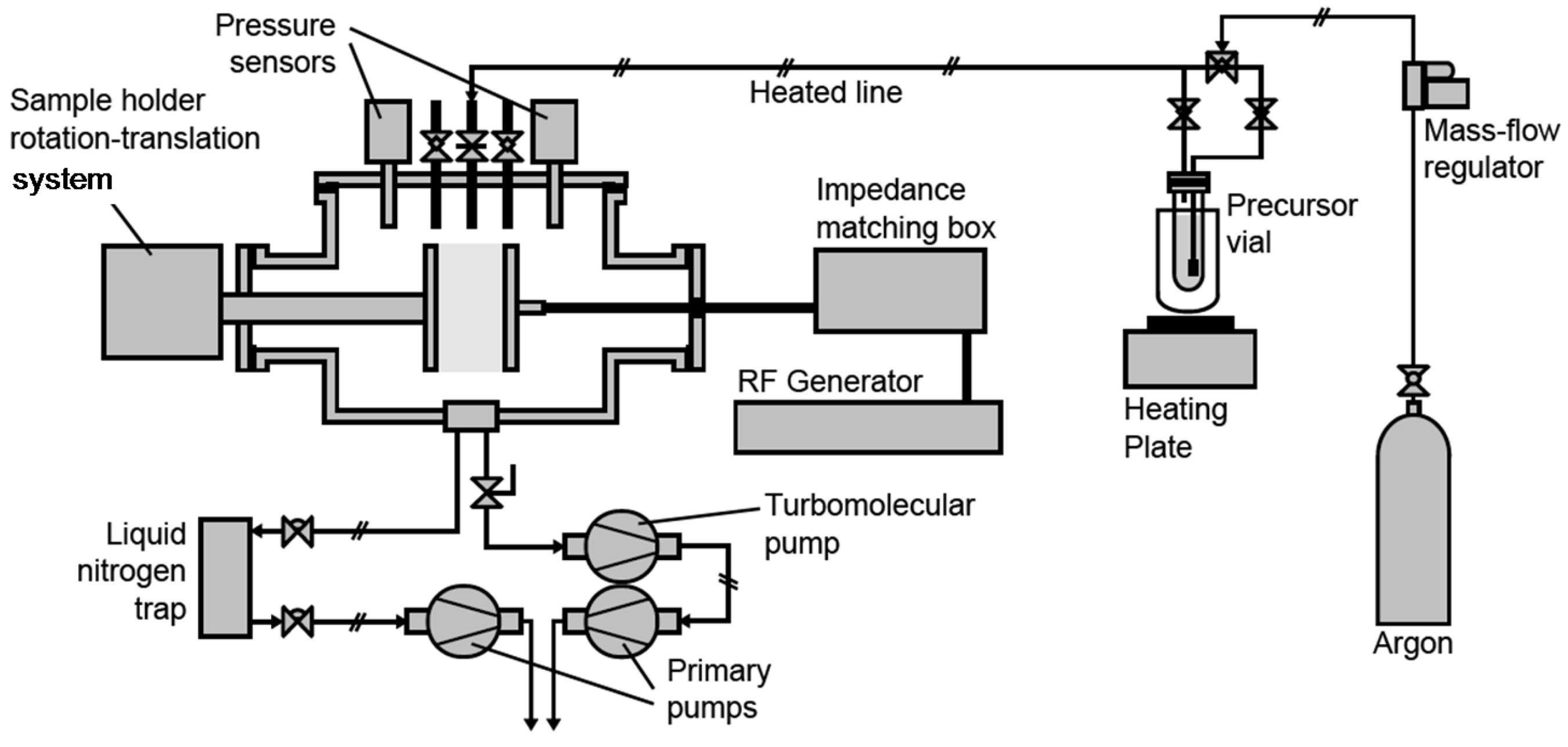
3.1. Plasma Process Equipment
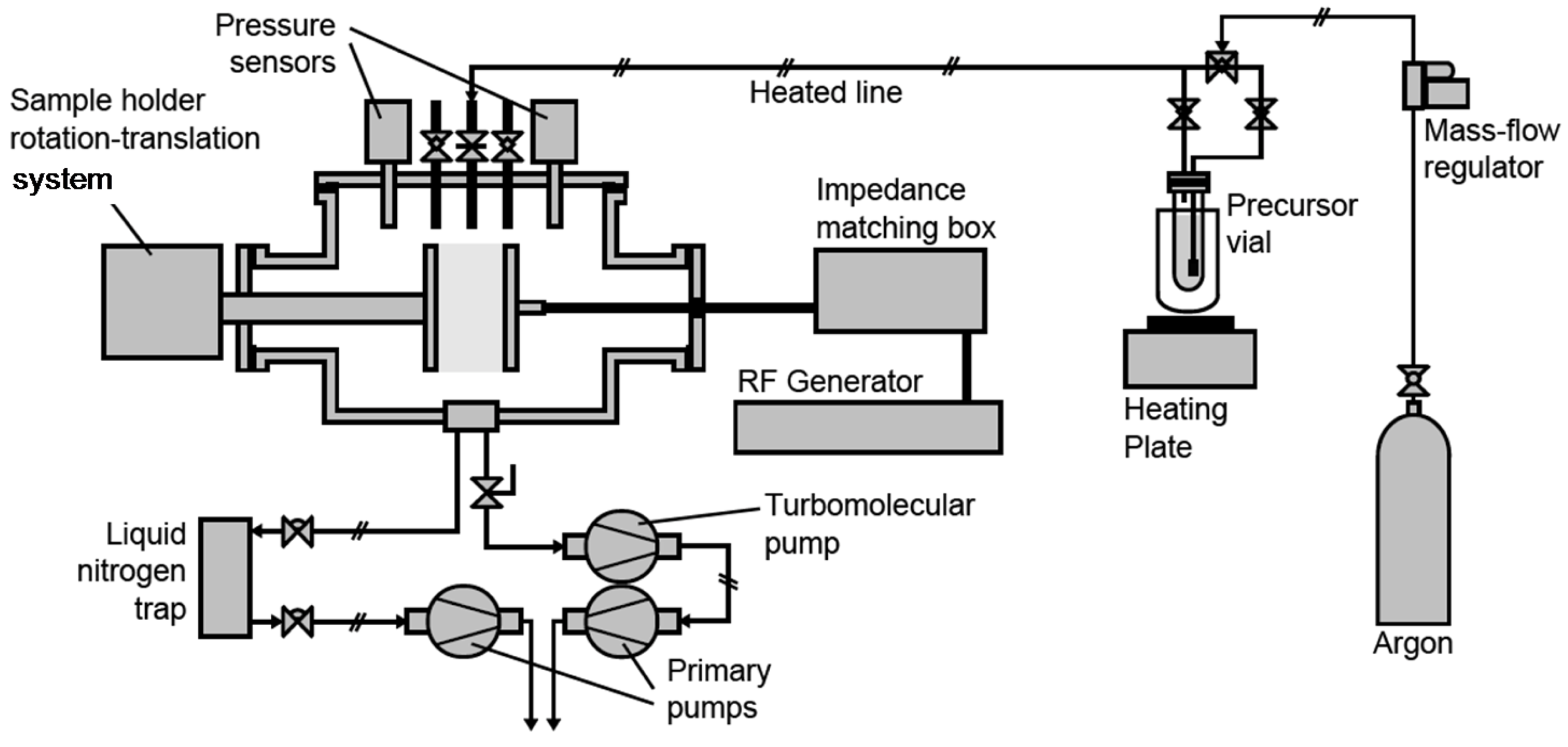
3.2. Plasma Process Procedure
3.3. Membranes Characterization
3.4. Membrane-Electrodes Assemblies Preparation and Fuel Cell Tests
4. Conclusions
Acknowledgments
References
- Alberti, G.; Casciola, M. Composite membranes for medium-temperature PEM fuel cells. Rev. Mater. Res. 2003, 33, 129–154. [Google Scholar] [CrossRef]
- Vetter, S.; Ruffmann, B.; Buder, I.; Nunes, S.P. Proton conductive membranes of sulfonated poly(ether ketone ketone). J. Membr. Sci. 2005, 260, 181–188. [Google Scholar] [CrossRef]
- Morse, J.D. Micro-fuel cell power sources. Int. J. Energy Res. 2007, 31, 576–602. [Google Scholar] [CrossRef]
- Wan, N.; Wang, C.; Mao, Z. Titanium substrate based micro-PEMFC operating under ambient conditions. Electrochem. Commun. 2007, 9, 511–516. [Google Scholar] [CrossRef]
- Rozière, J.; Jones, D.J. Advances in the development of inorganic-organic membranes for fuel cell applications. Adv. Polym. Sci. 2008, 215, 219–264. [Google Scholar]
- Smith, B.; Sridhar, S.; Khan, A.A. Solid polymer electrolyte membranes for fuel cell applications. J. Membr. Sci. 2005, 259, 10–26. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Hu, L.; Zhuang, L.; Lu, J.; Xu, B. A feasibility analysis for alkaline membrane direct methanol fuel cell: Thermodynamic disadvantages versus kinetic advantages. Electrochem. Commun. 2003, 5, 662–666. [Google Scholar] [CrossRef]
- Yang, C.C. A feasibility analysis for alkaline membrane direct methanol fuel cell: thermodynamic disadvantages versus kinetic advantages. Int. J. Hydrog. Energy 2004, 29, 135–143. [Google Scholar] [CrossRef]
- Demarconnay, L.; Coutanceau, C.; Léger, J.-M. Electroreduction of dioxygen (ORR) in alkaline medium on Ag/C and Pt/C nanostructured catalysts—Effect of the presence of methanol. Electrochim. Acta 2004, 49, 4513–4521. [Google Scholar] [CrossRef]
- Demarconnay, L.; Coutanceau, C.; Léger, J.M. Study of the oxygen electroreduction at nanostructured PtBi catalysts in alkaline medium. Electrochim. Acta 2008, 53, 3232–3241. [Google Scholar] [CrossRef]
- Simoes, M.; Baranton, S.; Coutanceau, C. Electro-oxidation of glycerol at Pd based nano-catalysts for an application in alkaline fuel cells for chemicals and energy cogeneration. Appl. Catal. B Environ. 2010, 93, 354–362. [Google Scholar] [CrossRef]
- Ilie, A.; Simoes, M.; Baranton, S.; Coutanceau, C.; Martemianov, S. Influence of operational parameters and of catalytic materials on electrical performance of direct glycerol solid alkaline membrane fuel cells. J. Power Sources 2011, 196, 4965–4971. [Google Scholar]
- Molina-Concha, B.; Chatenet, M.; Coutanceau, C.; Hahn, F. In situ infrared (FTIR) study of the borohydride oxidation reaction. Electrochem. Commun. 2009, 11, 223–226. [Google Scholar] [CrossRef]
- Jamard, R.; Salomon, J.; Martinent-Beaumont, A.; Coutanceau, C. Life time test in direct borohydride fuel cell system. J. Power Sources 2009, 193, 779–787. [Google Scholar] [CrossRef]
- Yu, E.H.; Scott, K. Development of direct methanol alkaline fuel cells using anion exchange membranes. J. Power Sources 2004, 137, 248–256. [Google Scholar] [CrossRef]
- Adams, L.A.; Poynton, S.D.; Tamain, C.; Slade, R.C.T.; Varcoe, J.R. A carbon dioxide tolerant aqueous-electrolyte-free anion-exchange membrane alkaline fuel cell. ChemSusChem 2008, 1, 79–81. [Google Scholar] [CrossRef] [Green Version]
- Jamard, R.; Latour, A.; Salomon, J.; Capron, P.; Martinent-Beaumont, A. Study of fuel efficiency in a direct borohydride fuel cell. J. Power Sources 2008, 176, 287–292. [Google Scholar] [CrossRef]
- Bae, I.S.; Cho, S.H.; Lee, S.B.; Kim, Y.; Boo, J.H. Growth of plasma-polymerized thin films by PECVD method and study on their surface and optical characteristics. Surf. Coat. Technol. 2005, 193, 142–146. [Google Scholar] [CrossRef]
- Schieda, M.; Roualdès, S.; Durand, J.; Martinent, A.; Marsacq, D. Plasma-polymerized thin films as new membranes for miniature solid alkaline fuel cells. Desalination 2006, 199, 286–288. [Google Scholar] [CrossRef]
- Matsuoka, K.; Chiba, S.; Iriyama, Y.; Abe, T.; Matsuoka, M.; Kikuchi, K.; Ogumi, Z. Preparation of anion-exchange membrane by plasma polymerization and its use in alkaline fuel cells. Thin Solid Films 2008, 516, 3309–3313. [Google Scholar] [CrossRef]
- Hu, J.; Meng, Y.; Zhang, C.; Fang, S. Plasma-polymerized alkaline anion-exchange membrane: Synthesis and structure characterization. Thin Solid Films 2011, 519, 2155–2162. [Google Scholar]
- Choi, W.C.; Kim, J.D.; Woo, S.I.; Fang, S. Modification of proton conducting membrane for reducing methanol crossover in a direct-methanol fuel cell. J. Power Sources 2001, 96, 411–414. [Google Scholar] [CrossRef]
- Lue, S.J.; Shih, T.S.; Wei, T.C. Plasma modification on a Nafion membrane for direct methanol fuel cell applications. Korean J. Chem. Eng. 2006, 23, 441–446. [Google Scholar] [CrossRef]
- Bae, B.; Kim, D.; Kim, H.J.; Lim, T.H.; Oh, I.H.; Ha, H.Y. Surface characterization of Argon-plasma-modified perfluorosulfonic acid membranes. J. Phys. Chem. B 2006, 110, 4240–4246. [Google Scholar]
- Ramdutt, D.; Charles, C.; Hudspeth, J.; Ladewig, B.; Gengenbach, T.; Boswell, R.; Dicks, A.; Brault, P. Low energy plasma treatment of Nafion membranes for PEM fuel cells. J. Power Sources 2007, 165, 41–48. [Google Scholar] [CrossRef]
- Zhang, C.; Hu, J.; Cong, J.; Zhao, Y.; Shen, W.; Toyoda, H.; Nagatsu, M.; Meng, Y. Pulsed plasma-polymerized alkaline anion-exchange membranes for potential application in direct alcohol fuel cells. J. Power Sources 2011, 196, 5386–5393. [Google Scholar] [CrossRef]
- Yasuda, H. Plasma Polymerization; Academic Press: Orlando, FL, USA, 1985. [Google Scholar]
- D’agostino, R. Plasma Deposition, Treatment, and Etching of Polymers; Academic Press: San Diego, CA, USA, 1990. [Google Scholar]
- Roualdès, S.; Schieda, M.; Durivault, L.; Guesmi, I.; Gérardin, E.; Durand, J. Ion-exchange plasma membranes for fuel cells on a micrometer scale. Chem. Vap. Deposition 2007, 13, 361–369. [Google Scholar] [CrossRef]
- Yasuda, H.; Hirotsu, T. Critical evaluation of conditions of plasma polymerization. J. Polym. Sci. Part A Polym. Chem. 1978, 16, 743–759. [Google Scholar]
- Yasuda, H.K. Plasma Polymerization and Plasma Interactions with Polymeric Materials; John Wiley & Sons: New York, NY, USA, 1990. [Google Scholar]
- Gerenser, L.J. An X-ray photoemission spectroscopy study of chemical interactions at silver/plasma modified polyethylene interfaces: Correlations with adhesion. J. Vac. Sci. Technol. A 1988, 6, 2897–2903. [Google Scholar] [CrossRef]
- Liu, C.H.; Huang, C. Structural changes of boron carbide induced by Zr incorporation. J. Mater. Sci. 2000, 35, 387–390. [Google Scholar] [CrossRef]
- Grigonis, A.; Silinskas, M.; Kopustinskas, V. Investigation of ion irradiation effects in a-SixC1−x:H thin films. Vacuum 2002, 68, 257–261. [Google Scholar] [CrossRef]
- Briggs, D.; Seah, M.P. Practical Surface Analysis: Auger and X-ray Photoelectron Spectroscopy, 2nd ed; John Wiley & Sons: Chichester, UK, 1990. [Google Scholar]
- Ektessabi, A.M.; Yamaguchi, K. Changes in chemical states of PET films due to low and high energy oxygen ion beam. Thin Solid Films 2000, 377, 793–797. [Google Scholar] [CrossRef]
- Barber, M.; Connor, J.A.; Guest, M.F.; Hillier, I.H.; Schwarz, M.; Stacey, M. Bonding in some donor-acceptor complexes involving boron trifluoride: Study by means of ESCA and molecular orbital calculations. J. Chem. Soc. Faraday Trans. 1973, 2, 551–558. [Google Scholar]
- Mansour, A.; Ugolini, D. Photoelectron-spectroscopy study of amorphous a-CNx:H. Phys. Rev. B 1993, 47, 10201–10209. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Cardinaud, C.; Turban, G. Spectroscopic determination of the structure of amorphous nitrogenated carbon films. J. Appl. Phys. 1998, 83, 4491–4500. [Google Scholar] [CrossRef]
- He, Z.; Carter, G.; Colligon, J.S. Ion-assisted deposition of CN and SiCN film. Thin Solid Films 1996, 283, 90–96. [Google Scholar] [CrossRef]
- Hao, J.; Xu, T.; Liu, W. Effect of deposition pressure on microstructure and properties of hydrogenated carbon nitride films prepared by DC-RF-PECVD. J. Non-Cryst. Solids 2005, 351, 3671–3676. [Google Scholar]
- Hao, J.; Xu, T.; Liu, W. Preparation and characterization of hydrogenated carbon nitride films synthesized by dual DC-RF plasma system. Mater. Sci. Eng. A 2005, 408, 297–302. [Google Scholar] [CrossRef]
- Avila, A.; Montero, I.; Galan, L.; Ripalda, J.M.; Levy, R. Behavior of oxygen doped SiC thin films: An X-ray photoelectron spectroscopy study. J. Appl. Phys. 2001, 89, 212–216. [Google Scholar]
- Li, S.D.; Wang, C.C.; Chen, C.Y. Preparation and characterization of a novel bipolar membrane by plasma-induced polymerization. J. Membr. Sci. 2008, 318, 429–434. [Google Scholar] [CrossRef]
- Roualdes, S.; Topala, I.; Mahdjoub, H.; Rouessac, V.; Sistat, P.; Durand, J. Sulfonated polystyrene-type plasma-polymerized membranes for miniature DMFC. J. Power Sources 2006, 158, 1270–1281. [Google Scholar] [CrossRef]
- Lide, D.R. CRC Handbook of Chemistry and Physics, 88th ed; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Kadjo, A.J.J.; Garnier, J.P.; Maye, J.P.; Relot, F.; Martemianov, S. Performance and instabilities of proton exchange membrane fuel cells. Russ. J. Electrochem. 2006, 42, 467–475. [Google Scholar] [CrossRef]
- Kadjo, A.J.J.; Brault, P.; Caillard, A.; Coutanceau, C.; Garnier, J.P.; Martemianov, S. Improvement of proton exchange membrane fuel cell electrical performance by optimization of operating parameters and electrodes preparation. J. Power Sources 2007, 172, 613–622. [Google Scholar] [CrossRef]
- Bograchev, D.; Gueguen, M.; Grandidier, J.C.; Martemianov, S. Stress and plastic deformation of MEA in fuel cells: Stresses generated during cell assembly. J. Power Sources 2008, 180, 393–401. [Google Scholar] [CrossRef]
- Bograchev, D.; Gueguen, M.; Grandidier, J.C.; Martemianov, S. Stress and plastic deformation of MEA in running fuel cell. Int. J. Hydrog. Energy 2008, 33, 5703–5717. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Reinholdt, M.; Ilie, A.; Roualdès, S.; Frugier, J.; Schieda, M.; Coutanceau, C.; Martemianov, S.; Flaud, V.; Beche, E.; Durand, J. Plasma Membranes Modified by Plasma Treatment or Deposition as Solid Electrolytes for Potential Application in Solid Alkaline Fuel Cells. Membranes 2012, 2, 529-552. https://doi.org/10.3390/membranes2030529
Reinholdt M, Ilie A, Roualdès S, Frugier J, Schieda M, Coutanceau C, Martemianov S, Flaud V, Beche E, Durand J. Plasma Membranes Modified by Plasma Treatment or Deposition as Solid Electrolytes for Potential Application in Solid Alkaline Fuel Cells. Membranes. 2012; 2(3):529-552. https://doi.org/10.3390/membranes2030529
Chicago/Turabian StyleReinholdt, Marc, Alina Ilie, Stéphanie Roualdès, Jérémy Frugier, Mauricio Schieda, Christophe Coutanceau, Serguei Martemianov, Valérie Flaud, Eric Beche, and Jean Durand. 2012. "Plasma Membranes Modified by Plasma Treatment or Deposition as Solid Electrolytes for Potential Application in Solid Alkaline Fuel Cells" Membranes 2, no. 3: 529-552. https://doi.org/10.3390/membranes2030529