
An Archaeosome-Adjuvanted Vaccine and Checkpoint Inhibitor Therapy Combination Significantly Enhances Protection from Murine Melanoma

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Vaccine Delivery Systems and Route of Immunization
2.2. Mouse Strains and Adoptive Cell Transfer
2.3. Tumor Model (B16-OVA, Melanoma)
2.4. Assessment of In Vivo Cytolytic Activity
2.5. Detection of OVA- Specific CD8+ T Cells
2.6. Checkpoint Inhibitor Combination Therapy
3. Results
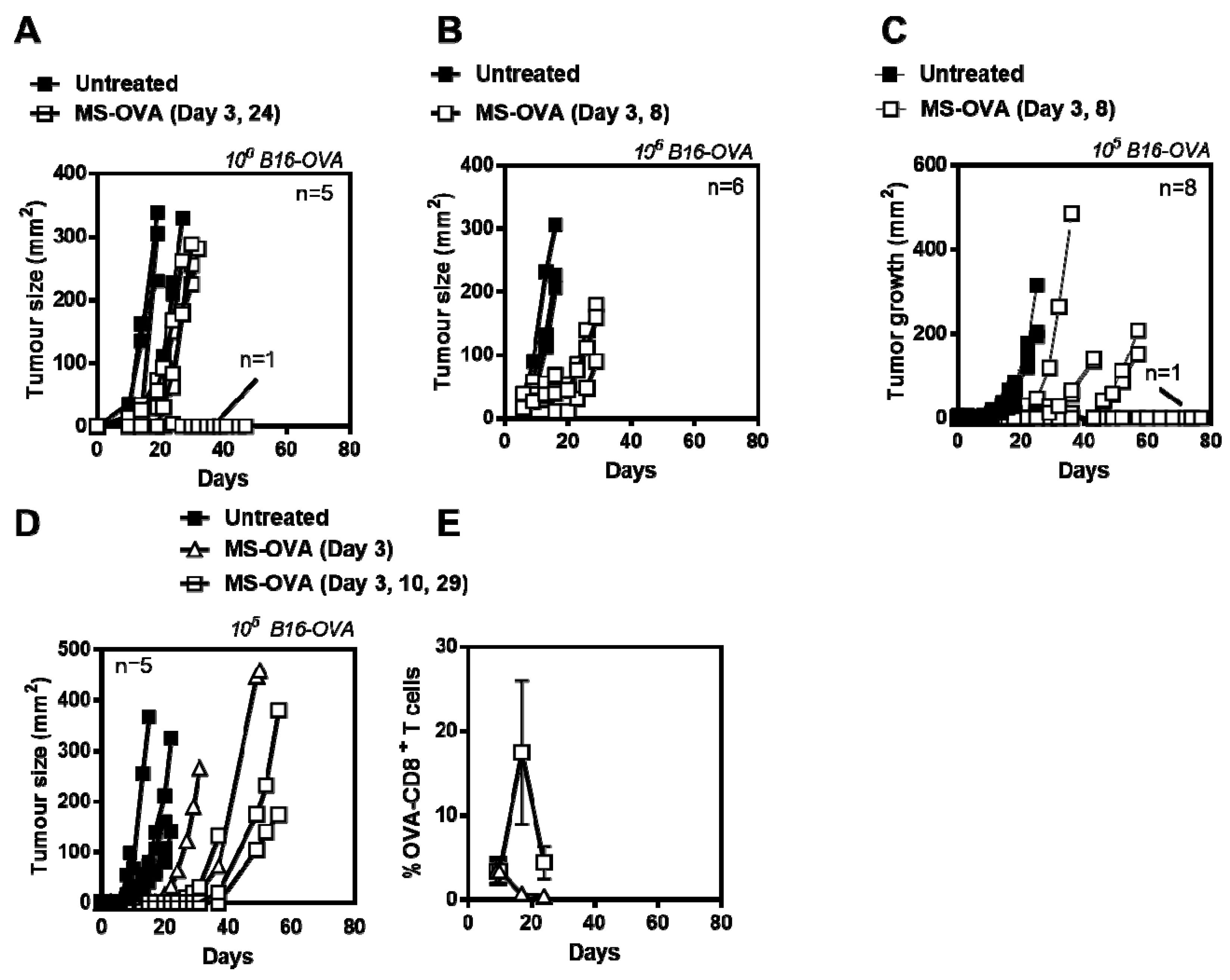
3.1. MS-OVA Therapy Against B16-OVA Melanoma Induces OVA-CD8+ T Cells and Modest Tumor Protection
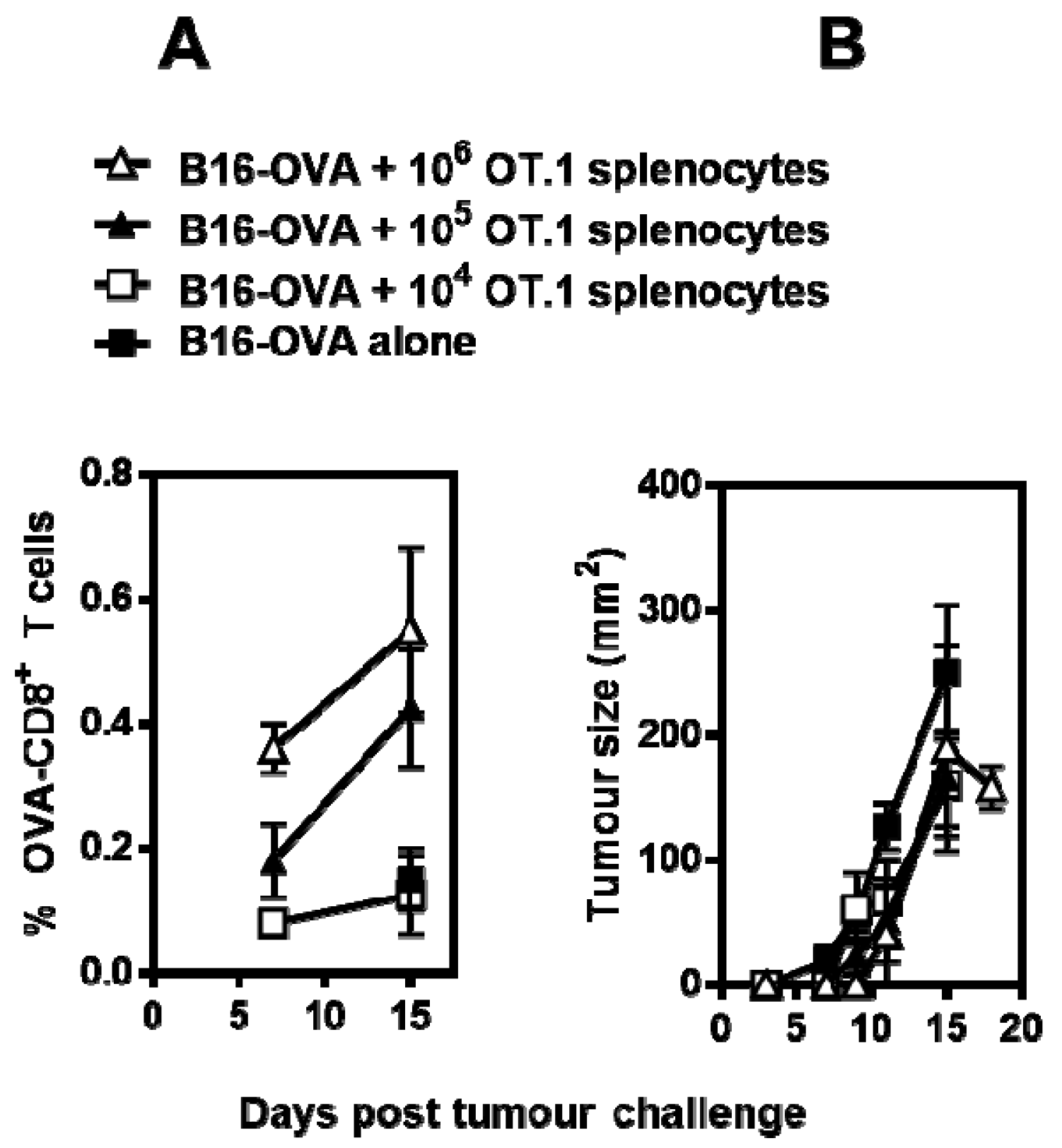
3.2. OVA-CD8+ T Cell Cytotoxicity Is Impaired in Mice Bearing Solid B16-OVA Tumors
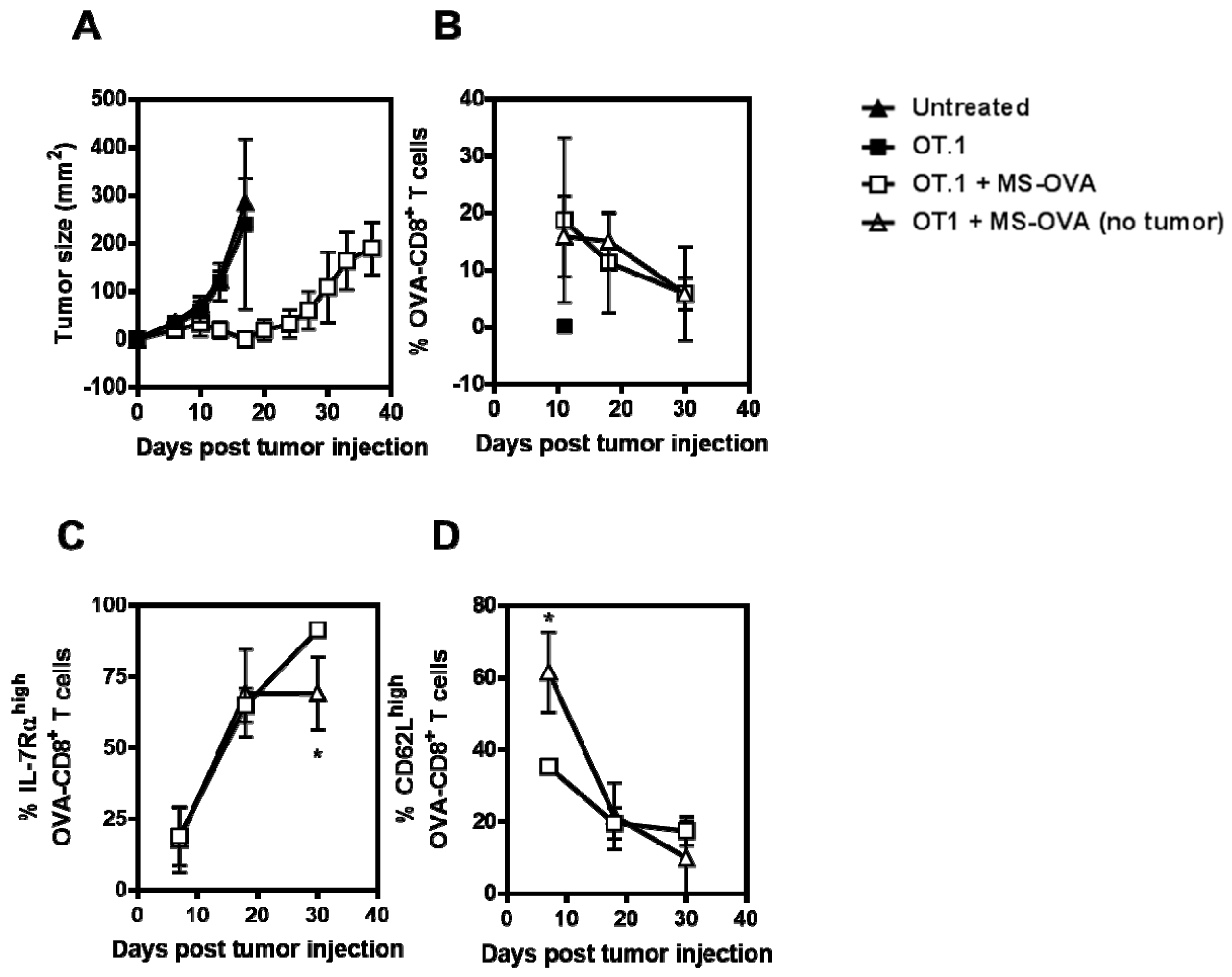
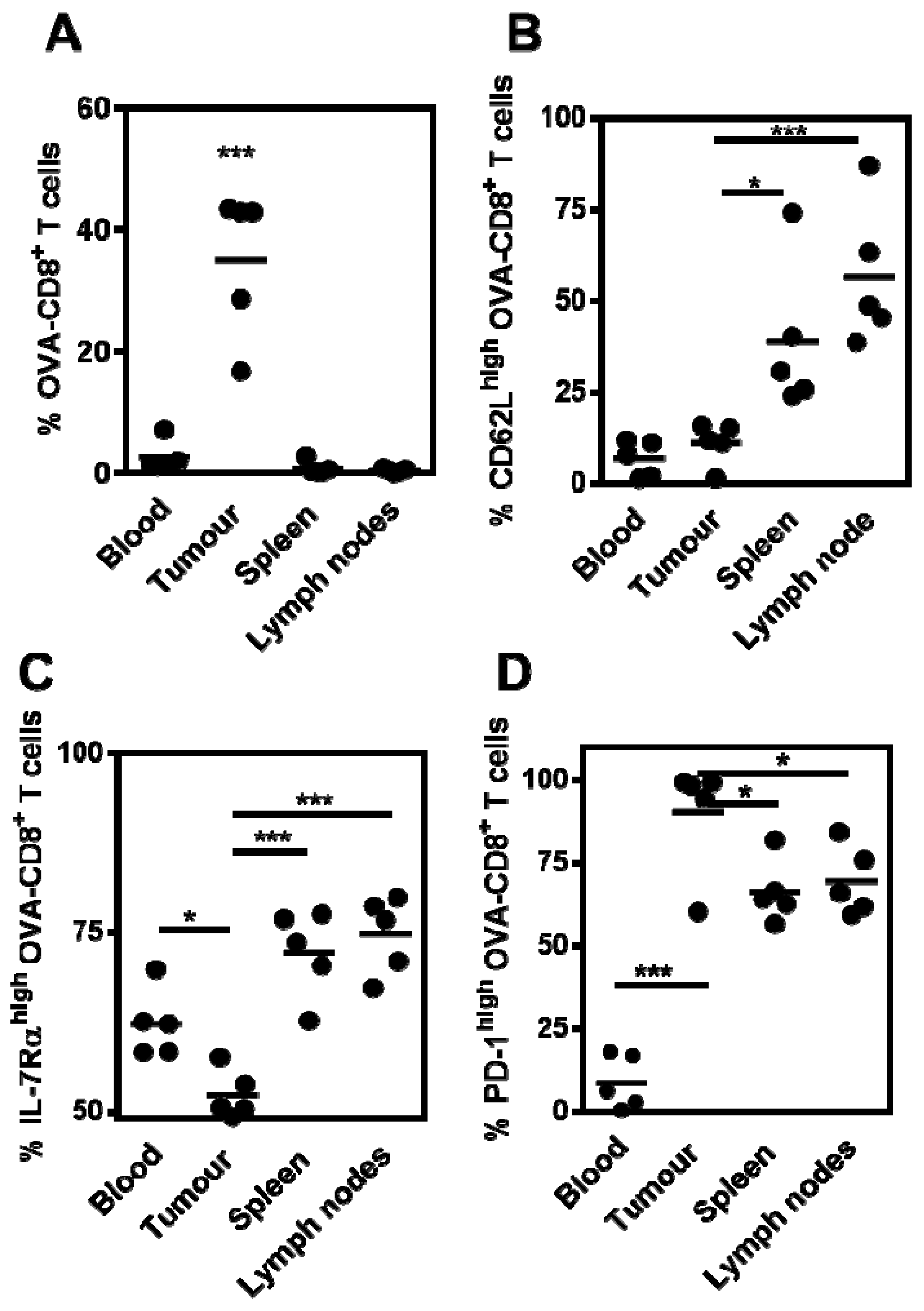
3.3. Phenotype and Frequency of Responding OVA-CD8+ T Cells
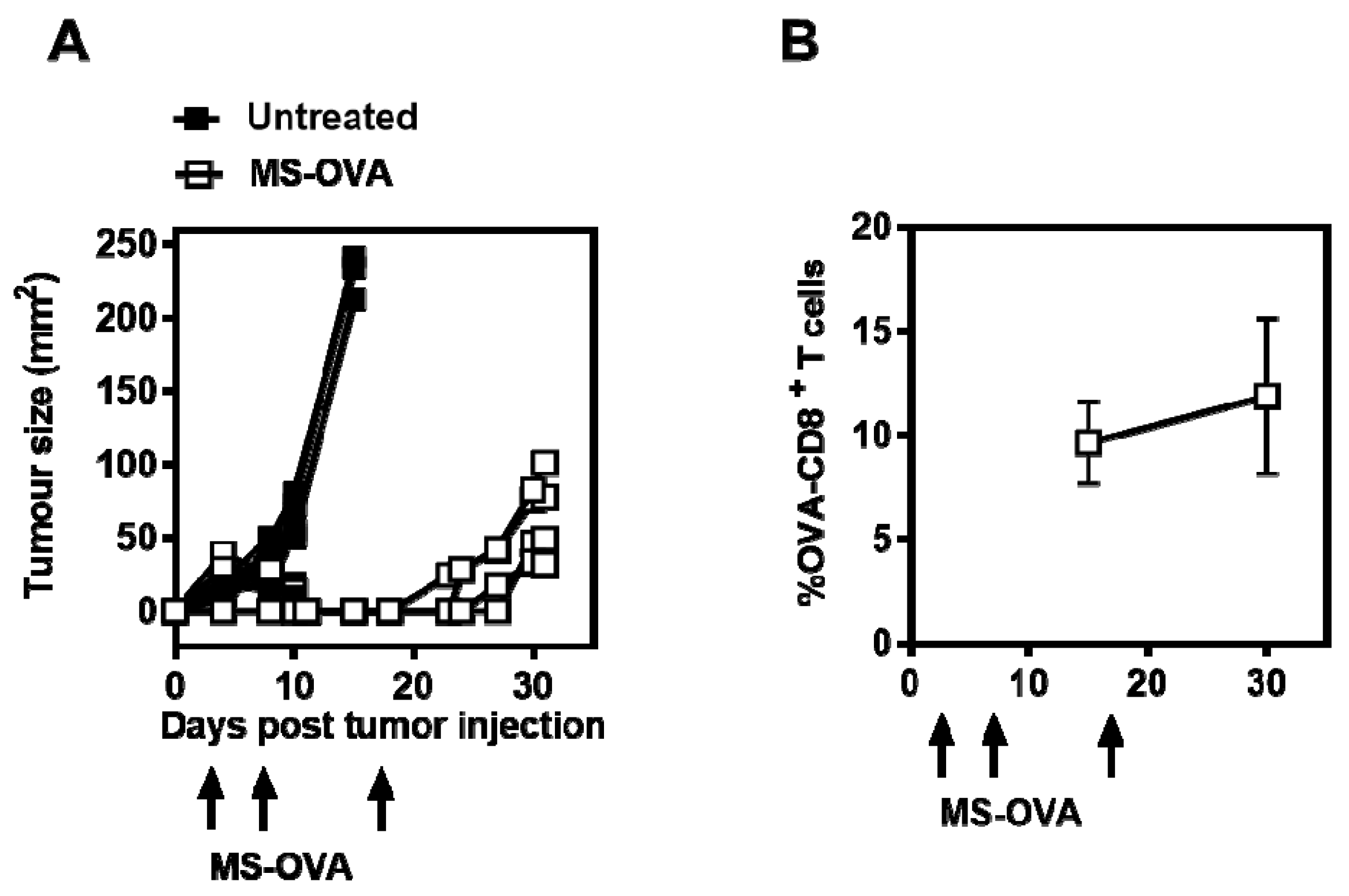
3.4. Tumor-Infiltrating CD8+ T Cells Express PD-1 Following Therapeutic MS-OVA Treatment
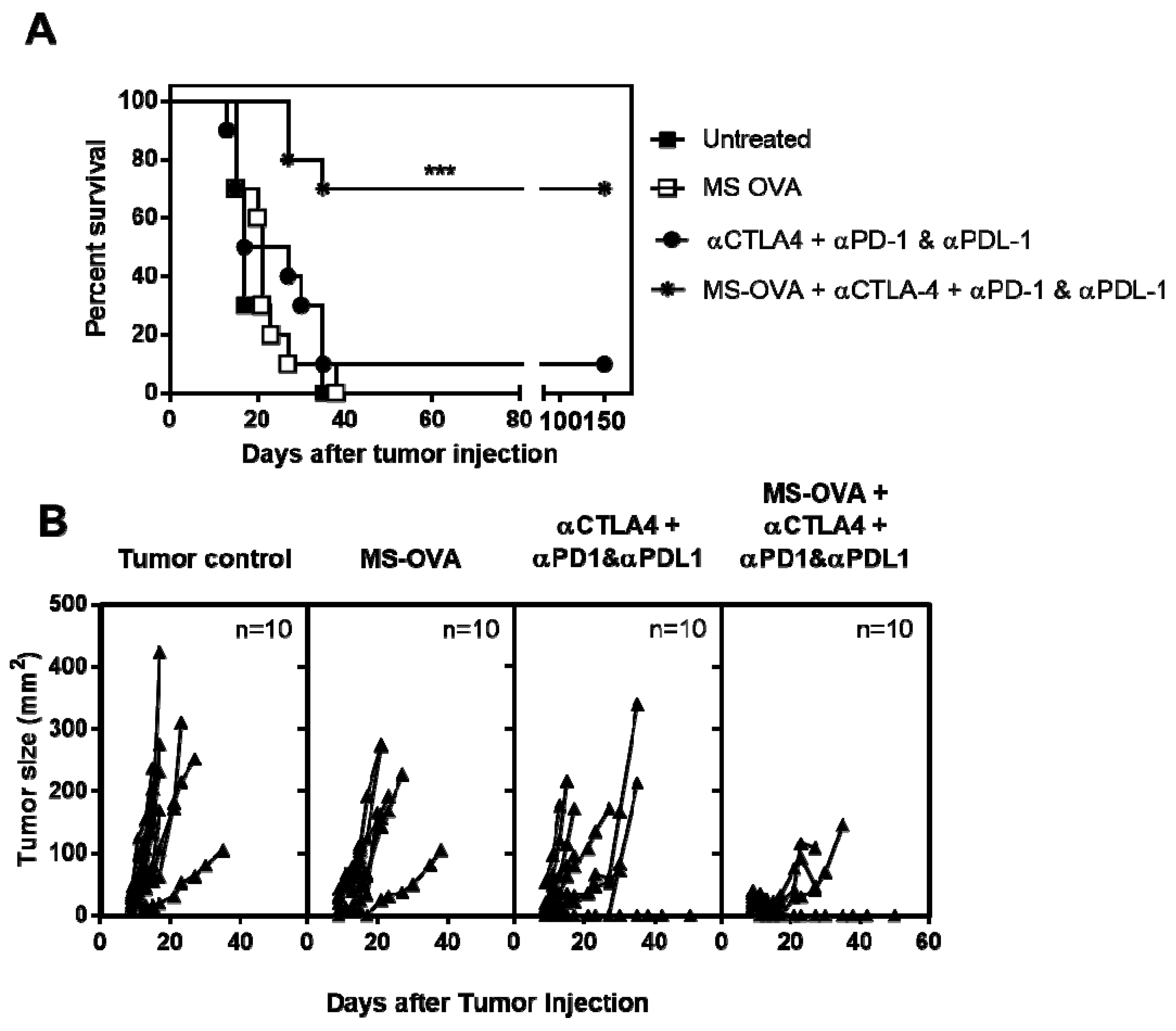
3.5. Combination Checkpoint Inhibitor Therapy with MS-OVA
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix
References
- Hamm, C.; Petrella, T.; Verma, S. Biochemotherapy for the treatment of metastatic malignant melanoma: A systematic review. Cancer Treat. Rev. 2008, 34, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final Version of 2009 AJCC Melanoma Staging and Classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef] [PubMed]
- Sznol, M.; Kluger, H.M.; Callahan, M.K.; Postow, M.A.; Gordon, R.A.; Segal, N.H.; Rizvi, N.; Lesokhin, A.; Atkins, M.B.; Kirkwood, J.; et al. Survival, response duration, and activity by BRAF mutation (MT) status of nivolumab (NIVO, anti-PD-1, BMS-936558, ONO-4538) and ipilimumab (IPI) concurrent therapy in advanced melanoma (MEL). J. Clin. Oncol. 2014, 32, LBA9003. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Redman, J.M.; Gibney, G.T.; Atkins, M.B. Advances in immunotherapy for melanoma. BMC Med. 2016, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Maia, M.C.; Hansen, A.R. A comprehensive review of immunotherapies in prostate cancer. Crit. Rev. Oncol. Hematol. 2017, 113, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Brustugun, O.T.; Sprauten, M.; Helland, Å. Real-world data on nivolumab treatment of non-small cell lung cancer. Acta Oncol. Stockh. Swed. 2017, 56, 438–440. [Google Scholar] [CrossRef] [PubMed]
- Zibelman, M.; Ramamurthy, C.; Plimack, E.R. Emerging role of immunotherapy in urothelial carcinoma-Advanced disease. Urol. Oncol. 2016, 34, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Hude, I.; Sasse, S.; Engert, A.; Bröckelmann, P.J. The emerging role of immune checkpoint inhibition in malignant lymphoma. Haematologica 2017, 102, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Haq, K.; Jia, Y.; Krishnan, L. Archaeal lipid vaccine adjuvants for induction of cell-mediated immunity. Expert Rev. Vaccines 2016, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, L.; Dicaire, C.J.; Patel, G.B.; Sprott, G.D. Archaeosome vaccine adjuvants induce strong humoral, cell-mediated, and memory responses: Comparison to conventional liposomes and alum. Infect. Immun. 2000, 68, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, L.; Sad, S.; Patel, G.B.; Sprott, G.D. Archaeosomes induce long-term CD8+ cytotoxic T cell response to entrapped soluble protein by the exogenous cytosolic pathway, in the absence of CD4+ T cell help. J. Immunol. 2000, 165, 5177–5185. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, L.; Deschatelets, L.; Stark, F.C.; Gurnani, K.; Sprott, G.D. Archaeosome adjuvant overcomes tolerance to tumor-associated melanoma antigens inducing protective CD8 T cell responses. Clin. Dev. Immunol. 2010, 2010, 578432. [Google Scholar] [CrossRef] [PubMed]
- Stark, F.C.; McCluskie, M.J.; Krishnan, L. Homologous Prime-Boost Vaccination with OVA Entrapped in Self-Adjuvanting Archaeosomes Induces High Numbers of OVA-Specific CD8+ T Cells that Protect Against Subcutaneous B16-OVA Melanoma. Vaccines 2016, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Klebanoff, C.A.; Palmer, D.C.; Wrzesinski, C.; Kerstann, K.; Yu, Z.; Finkelstein, S.E.; Theoret, M.R.; Rosenberg, S.A.; Restifo, N.P.; et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J. Clin. Investig. 2005, 15, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Gattinoni, L.; Restifo, N.P. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol. Rev. 2006, 211, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Gattinoni, L.; Palmer, D.C.; Muranski, P.; Ji, Y.; Hinrichs, C.S.; Borman, Z.A.; Kerkar, S.P.; Scott, C.D.; Finkelstein, S.E.; et al. Determinants of Successful CD8+ T-Cell Adoptive Immunotherapy for Large Established Tumors in Mice. Clin. Cancer Res. 2011, 17, 5343–5352. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, L.; Gurnani, K.; Dicaire, C.J.; van Faassen, H.; Zafer, A.; Kirschning, C.J.; Sad, S.; Sprott, G.D. Rapid clonal expansion and prolonged maintenance of memory CD8+ T cells of the effector (CD44highCD62Llow) and central (CD44highCD62Lhigh) phenotype by an archaeosome adjuvant independent of TLR2. J. Immunol. 2007, 178, 2396–2406. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.S.; Iskandar, M.; Mykytczuk, O.L.; Nash, J.H.E.; Krishnan, L.; Sad, S. A reduced antigen load in vivo, rather than weak inflammation, causes a substantial delay in CD8+ T cell priming against Mycobacterium bovis (bacillus Calmette-Guérin). J. Immunol. 2007, 179, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.M.; Fisher, T.L.; Wei, C.; Frelinger, J.G.; Lord, E.M. Tumours can act as adjuvants for humoral immunity. Immunology 2001, 102, 486–497. [Google Scholar] [CrossRef] [PubMed]
- Dudani, R.; Chapdelaine, Y.; Faassen, H.V.; Smith, D.K.; Shen, H.; Krishnan, L.; Sad, S. Multiple mechanisms compensate to enhance tumor-protective CD8(+) T cell response in the long-term despite poor CD8(+) T cell priming initially: Comparison between an acute versus a chronic intracellular bacterium expressing a model antigen. J. Immunol. 2002, 168, 5737–5745. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.L.; Wherry, E.J.; Ahmed, R. Cutting edge: Rapid in vivo killing by memory CD8 T cells. J. Immunol. 2003, 171, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Cheever, M.A.; Higano, C. PROVENGE (Sipuleucel-T) in Prostate Cancer: The First FDA Approved Therapeutic Cancer Vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Rosenberg, S.A. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat. Rev. Cancer 2003, 3, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Saito, K.; Shiiba, K.; Ohuchi, A.; Saigenji, K.; Nagura, H.; Ohtani, H. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998, 58, 3491–3494. [Google Scholar] [PubMed]
- Krishnan, L.; Sprott, G.D. Archaeosome adjuvants: Immunological capabilities and mechanism(s) of action. Vaccine 2008, 26, 2043–2055. [Google Scholar] [CrossRef] [PubMed]
- Hogquist, K.A.; Jameson, S.C.; Heath, W.R.; Howard, J.L.; Bevan, M.J.; Carbone, F.R. Pillars article: T cell receptor antagonist peptides induce positive selection. Cell. 1994. 76: 17–27. J. Immunol. Baltim. Md 1950 2012, 188, 2046–2056. [Google Scholar]
- Hogquist, K.A.; Jameson, S.C.; Heath, W.R.; Howard, J.L.; Bevan, M.J.; Carbone, F.R. T cell receptor antagonist peptides induce positive selection. Cell 1994, 76, 17–27. [Google Scholar] [CrossRef]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.; Gajewski, T.F.; Mackensen, A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: Implications for tumor immunotherapy. Cancer Immunol. Immunother. 2005, 54, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Dranoff, G. The biologic importance of tumor-infiltrating lymphocytes. J. Cutan. Pathol. 2010, 37 (Suppl. 1), 48–53. [Google Scholar] [CrossRef] [PubMed]
- Osada, T.; Patel, S.P.; Hammond, S.A.; Osada, K.; Morse, M.A.; Lyerly, H.K. CEA/CD3-bispecific T cell-engaging (BiTE) antibody-mediated T lymphocyte cytotoxicity maximized by inhibition of both PD1 and PD-L1. Cancer Immunol. Immunother. 2015, 64, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Peggs, K.S. Recent advances in antibody-based therapies for Hodgkin Lymphoma. Br. J. Haematol. 2015, 171, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Dudley, M.E. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr. Opin. Immunol. 2009, 21, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Lu, S. Heterologous prime-boost vaccination. Curr. Opin. Immunol. 2009, 21, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, H.; Song, G.-Y.; Srivastava, T.; Carroll, K.D.; Shahabi, V.; Manuel, E.R.; Diamond, D.J.; Ellenhorn, J.D.I. Heterologous prime/boost immunization with p53-based vaccines combined with Toll-like receptor stimulation enhances tumor regression. J. Immunother. 2010, 33, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Chapon, M.; Randriamampita, C.; Maubec, E.; Badoual, C.; Fouquet, S.; Wang, S.-F.; Marinho, E.; Farhi, D.; Garcette, M.; Jacobelli, S.; et al. Progressive Upregulation of PD-1 in Primary and Metastatic Melanomas Associated with Blunted TCR Signaling in Infiltrating T Lymphocytes. J. Investig. Dermatol. 2011, 131, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Inozume, T.; Hanada, K.-I.; Wang, Q.J.; Ahmadzadeh, M.; Wunderlich, J.R.; Rosenberg, S.A.; Yang, J.C. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J Immunother 2010, 33, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Stark, F.C.; Sad, S.; Krishnan, L. Intracellular bacterial vectors that induce CD8(+) T cells with similar cytolytic abilities but disparate memory phenotypes provide contrasting tumor protection. Cancer Res. 2009, 69, 4327–4334. [Google Scholar] [CrossRef] [PubMed]
- Sckisel, G.D.; Mirsoian, A.; Minnar, C.M.; Crittenden, M.; Curti, B.; Chen, J.Q.; Blazar, B.R.; Borowsky, A.D.; Monjazeb, A.M.; Murphy, W.J. Differential phenotypes of memory CD4 and CD8 T cells in the spleen and peripheral tissues following immunostimulatory therapy. J. Immunother. Cancer 2017, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, J.; Sun, Z.; Pagliano, O.; Chauvin, J.-M.; Sander, C.; Janjic, B.; Tarhini, A.A.; Tawbi, H.A.; Kirkwood, J.M.; Moschos, S.; et al. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8+ T cells induced by melanoma vaccines. Cancer Res. 2014, 74, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Badoual, C.; Hans, S.; Merillon, N.; Van Ryswick, C.; Ravel, P.; Benhamouda, N.; Levionnois, E.; Nizard, M.; Si-Mohamed, A.; Besnier, N.; et al. PD-1-expressing tumor-infiltrating T cells are a favorable prognostic biomarker in HPV-associated head and neck cancer. Cancer Res. 2013, 73, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Weber, J. Immune checkpoint proteins: A new therapeutic paradigm for cancer—preclinical background: CTLA-4 and PD-1 blockade. Semin. Oncol. 2010, 37, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.M.; Scotland, R.R.; Lau, R.L.; Wang, C.; Korman, A.J.; Kast, W.M.; Weber, J.S. Programmed death-1 blockade enhances expansion and functional capacity of human melanoma antigen-specific CTLs. Int. Immunol. 2007, 19, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Borkner, L.; Kaiser, A.; van de Kasteele, W.; Andreesen, R.; Mackensen, A.; Haanen, J.B.; Schumacher, T.N.; Blank, C. RNA interference targeting programmed death receptor-1 improves immune functions of tumor-specific T cells. Cancer Immunol. Immunother. 2010, 59, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.; Kuball, J.; Voelkl, S.; Wiendl, H.; Becker, B.; Walter, B.; Majdic, O.; Gajewski, T.F.; Theobald, M.; Andreesen, R.; et al. Blockade of PD-L1 (B7-H1) augments human tumor-specific T cell responses in vitro. Int. J. Cancer 2006, 119, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Berman, D.M.; Aznar, M.A.; Korman, A.J.; Pérez Gracia, J.L.; Haanen, J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat. Rev. Cancer 2015, 15, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Karaki, S.; Anson, M.; Tran, T.; Giusti, D.; Blanc, C.; Oudard, S.; Tartour, E. Is There Still Room for Cancer Vaccines at the Era of Checkpoint Inhibitors. Vaccines 2016, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Ock, C.-Y.; Keam, B.; Kim, S.; Lee, J.-S.; Kim, M.; Kim, T.M.; Jeon, Y.K.; Kim, D.-W.; Chung, D.H.; Heo, D.S. Pan-Cancer Immunogenomic Perspective on the Tumor Microenvironment Based on PD-L1 and CD8 T-Cell Infiltration. Clin. Cancer Res. 2016, 22, 2261–2270. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.C.; Engels, B.; Arina, A.; Yu, P.; Slauch, J.M.; Fu, Y.-X.; Karrison, T.; Burnette, B.; Idel, C.; Zhao, M.; et al. Antigen-specific bacterial vaccine combined with anti-PD-L1 rescues dysfunctional endogenous T cells to reject long-established cancer. Cancer Immunol. Res. 2013, 1, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [PubMed]
- Duraiswamy, J.; Kaluza, K.M.; Freeman, G.J.; Coukos, G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013, 73, 3591–3603. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stark, F.C.; Weeratna, R.D.; Deschatelets, L.; Gurnani, K.; Dudani, R.; McCluskie, M.J.; Krishnan, L. An Archaeosome-Adjuvanted Vaccine and Checkpoint Inhibitor Therapy Combination Significantly Enhances Protection from Murine Melanoma. Vaccines 2017, 5, 38. https://doi.org/10.3390/vaccines5040038
Stark FC, Weeratna RD, Deschatelets L, Gurnani K, Dudani R, McCluskie MJ, Krishnan L. An Archaeosome-Adjuvanted Vaccine and Checkpoint Inhibitor Therapy Combination Significantly Enhances Protection from Murine Melanoma. Vaccines. 2017; 5(4):38. https://doi.org/10.3390/vaccines5040038
Chicago/Turabian StyleStark, Felicity C., Risini D. Weeratna, Lise Deschatelets, Komal Gurnani, Renu Dudani, Michael J. McCluskie, and Lakshmi Krishnan. 2017. "An Archaeosome-Adjuvanted Vaccine and Checkpoint Inhibitor Therapy Combination Significantly Enhances Protection from Murine Melanoma" Vaccines 5, no. 4: 38. https://doi.org/10.3390/vaccines5040038