
Mesenchymal Stromal Cells Can Regulate the Immune Response in the Tumor Microenvironment


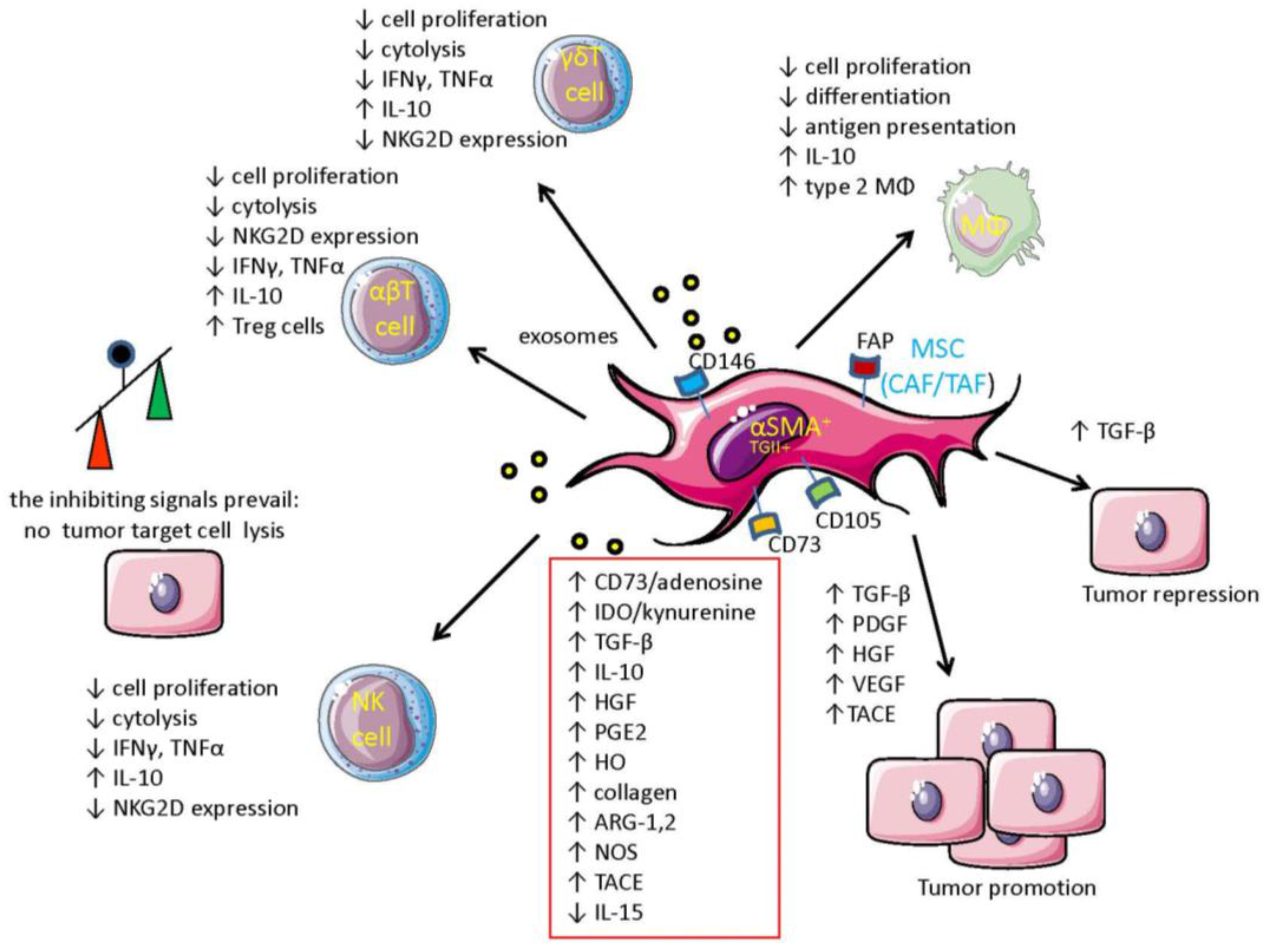
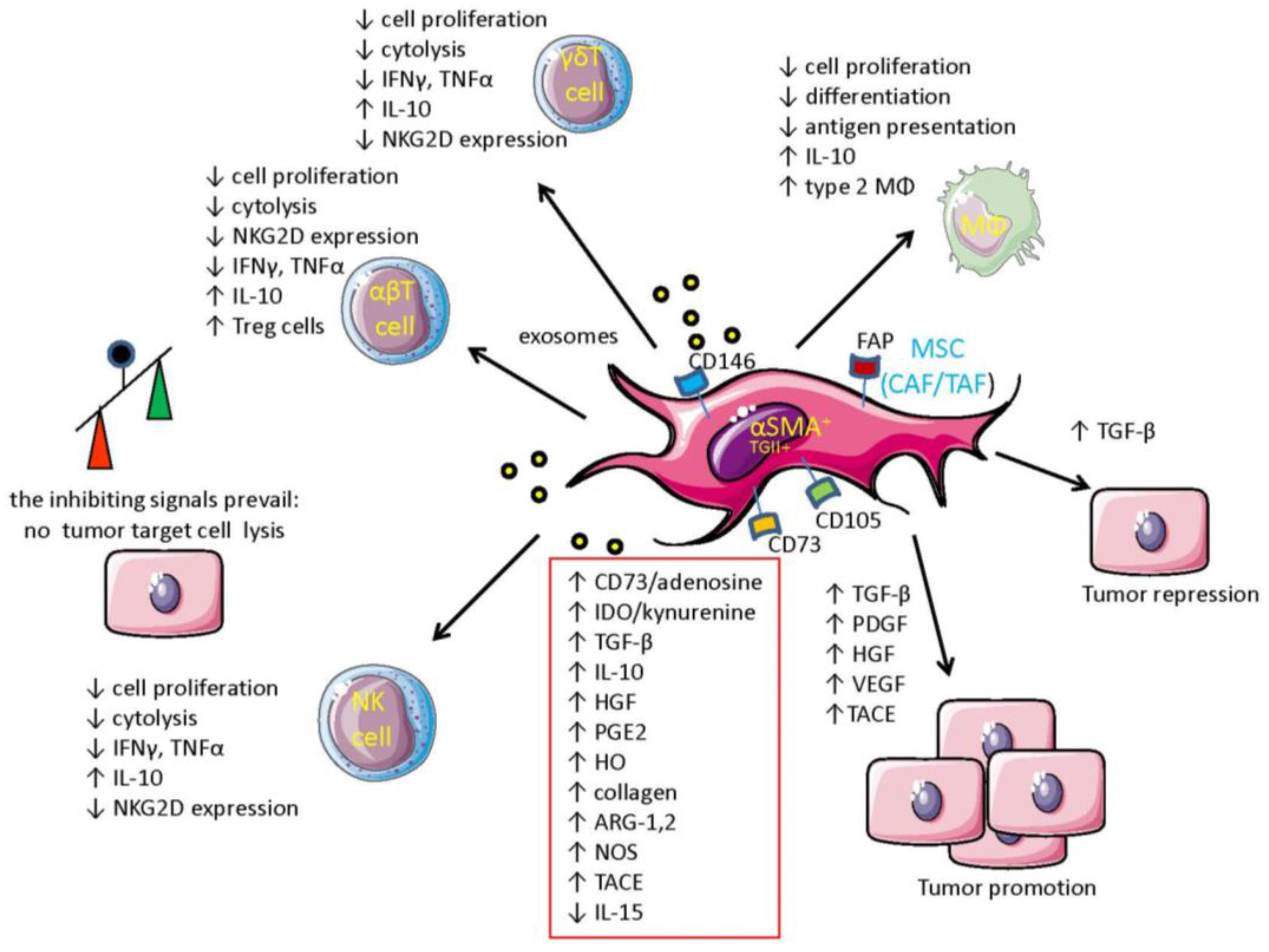{kind=link}
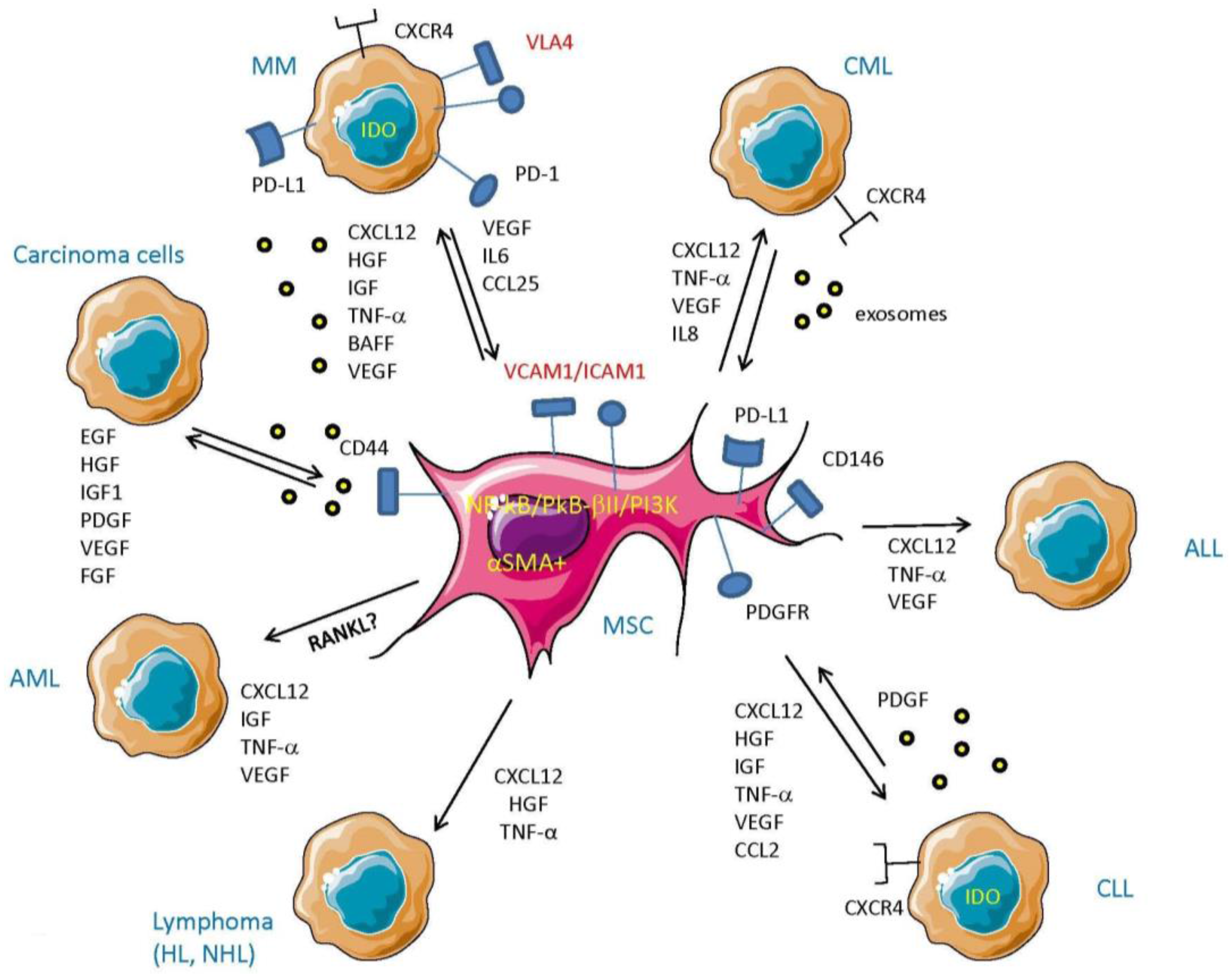{kind=link}
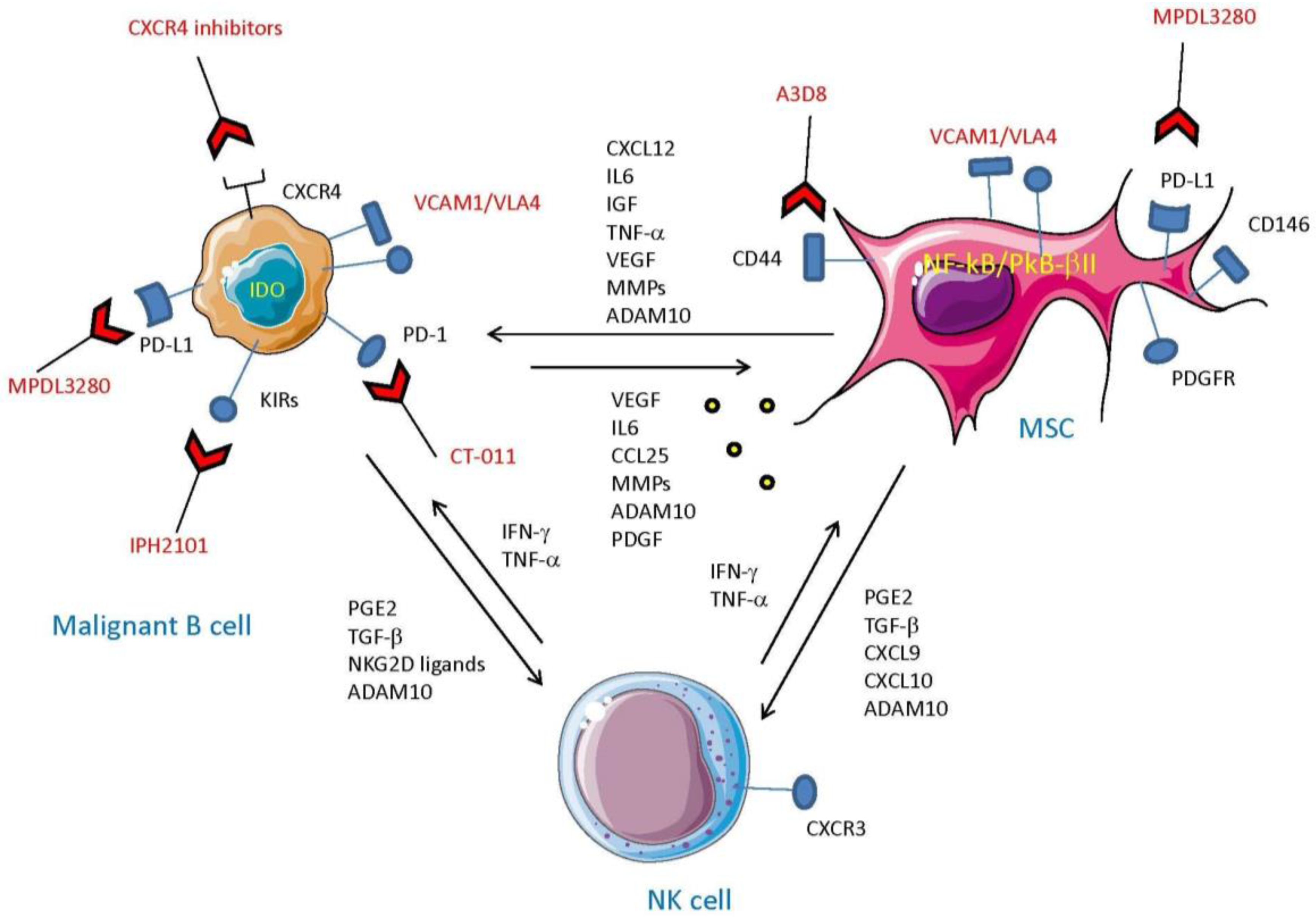{kind=link}
{kind=link}
Abstract
:1. Introduction
2. MSC Phenotypic and Functional Features Relevant in the Interaction with Tumor and Immune Cells
2.1. MSC Phenotypic Characterization and Function: Cell Membrane-Expressed Enzymes
2.2. Surface Molecules Expressed on MSC Involved in the Interaction with the Immune System
3. Molecular Mechanisms Responsible for the Immunosuppressive Effect Mediated by MSC in TME
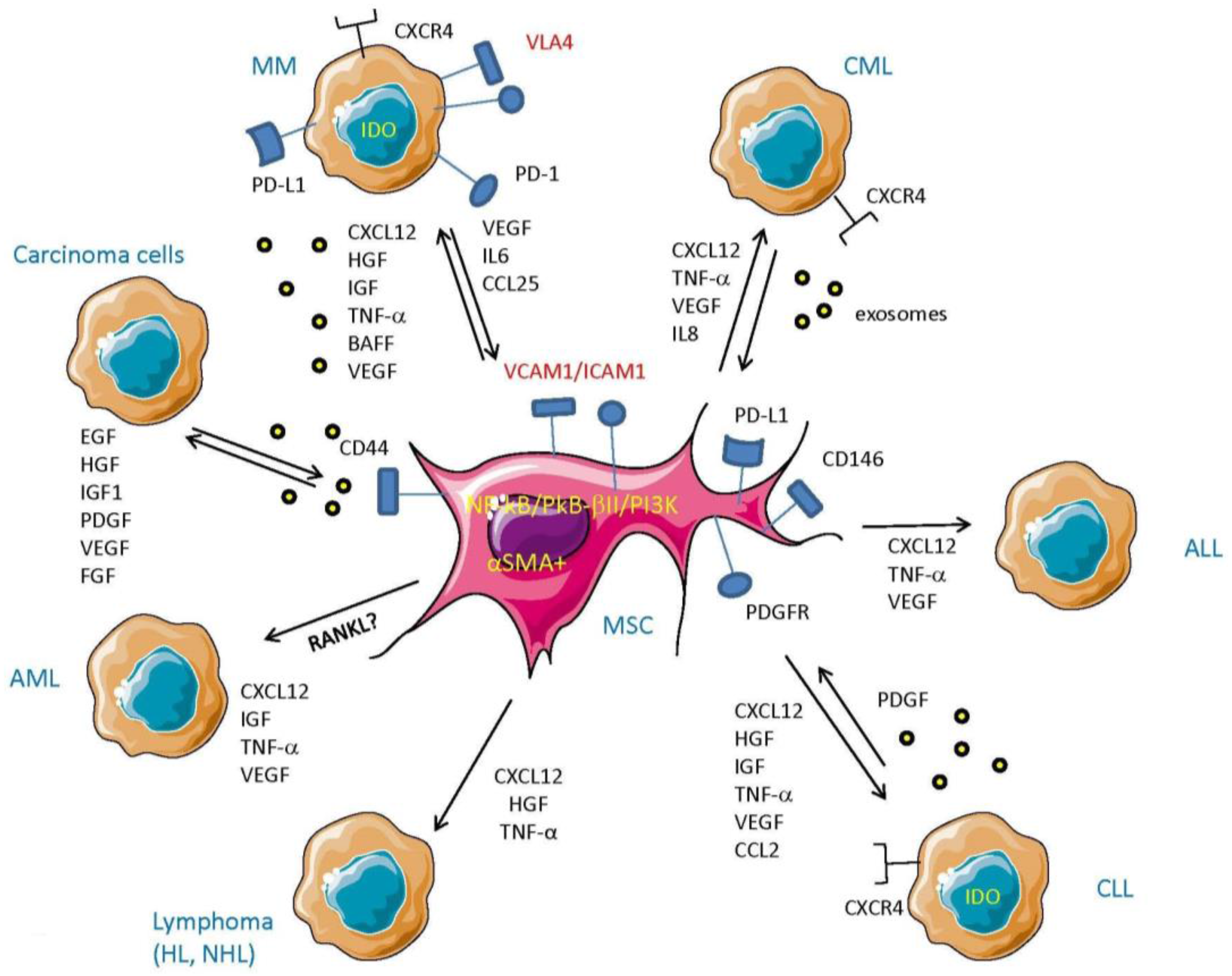
4. MSC Cross-Talk with Tumor Cells
4.1. MSC–Tumor Cell Cross-Talk in Hematological Malignancies
4.1.1. MSC in Chronic Lymphocytic Leukemia (CLL) and Chronic Myeloid Leukemia (CML)
4.1.2. MSC in Acute Myeloid Leukemia (AML)
4.1.3. MSC in Multiple Myeloma (MM)
4.2. MSC and Cross-Talk with Tumor Cells in Carcinomas
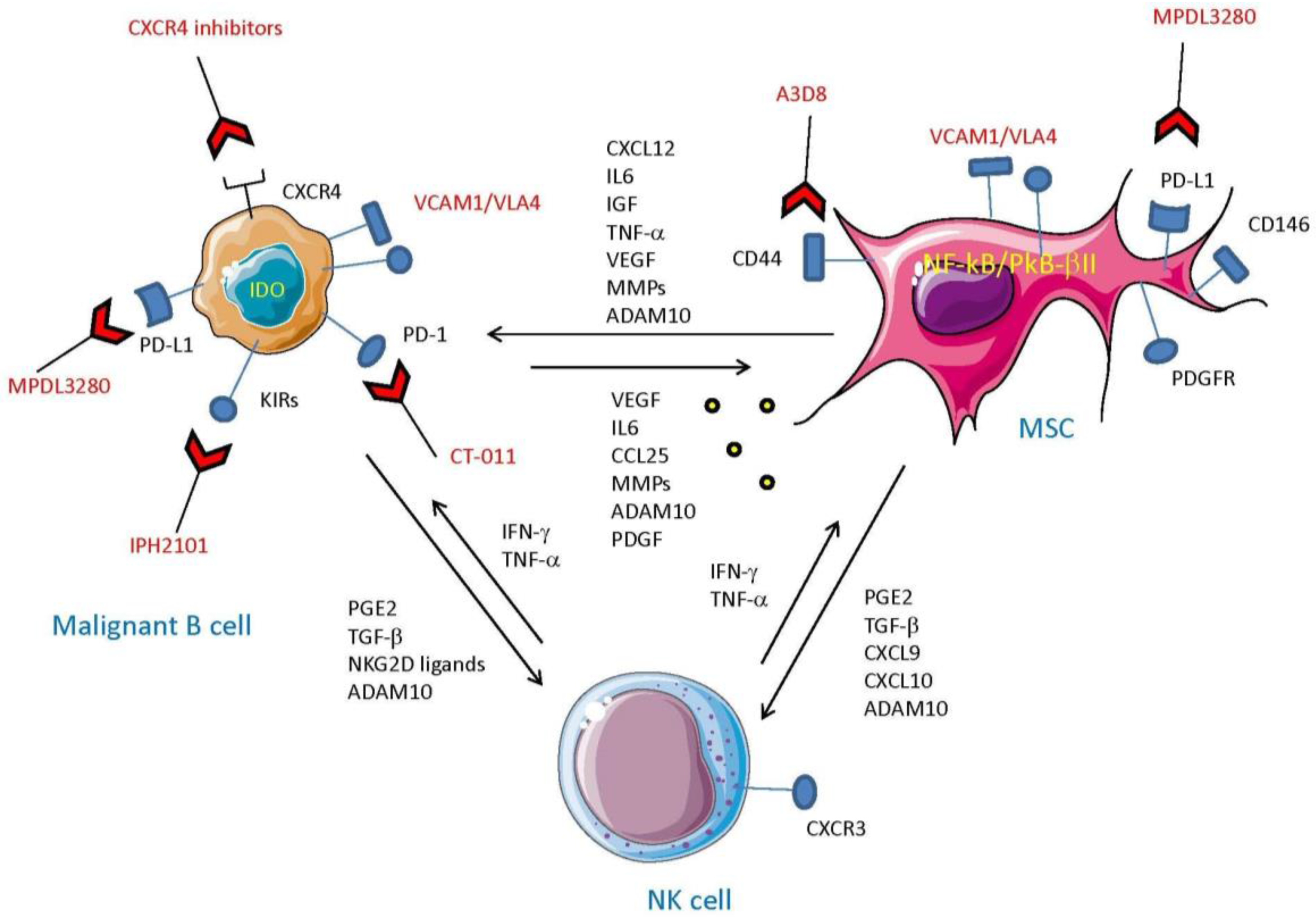
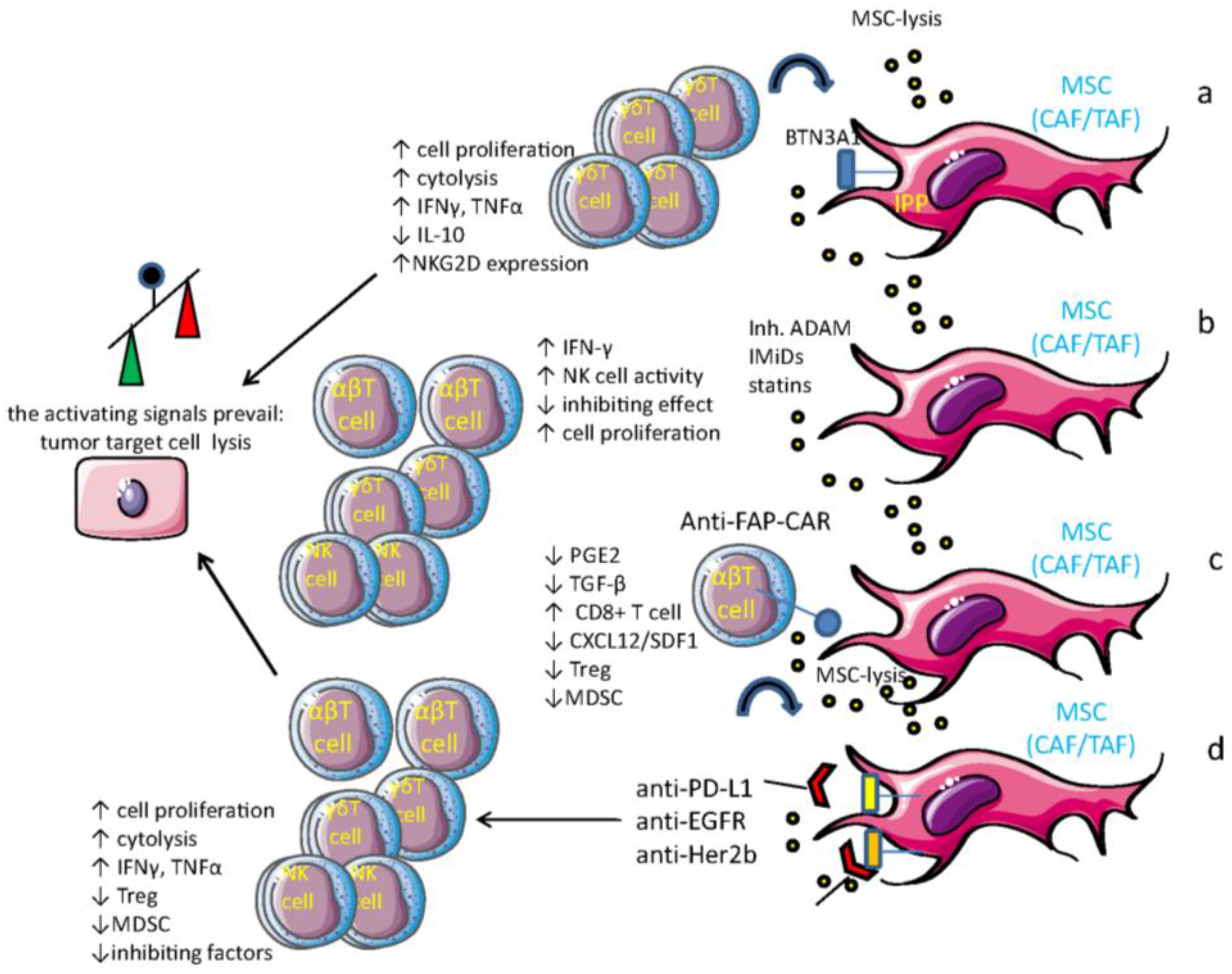
5. MSC and Tumor Cells: The Cross-Talk which Impairs Innate Cell-Mediated Surveillance
6. Drugs that Can Influence MSC-Mediated Immune Regulation
7. MSC as Target Cells for Anti-Tumor Vaccines
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ADAM | a disintegrin and metalloprotease |
| ADCC | antibody-dependent cellular cytotoxicity |
| ALL | acute lymphoblastic leukemia |
| AML | acute myeloid leukemia |
| AMP | adenosine monophosphate |
| BM | bone marrow |
| BMSC | bone marrow stromal cells |
| CAFs | carcinoma-associated fibroblasts |
| CAR | chimeric antigen receptor |
| CDC | complement-dependent cytotoxicity |
| CLIR | C-lectin type inhibitory receptors |
| CLL | chronic lymphocytic leukemia |
| CML | chronic myelogenous leukemia |
| CTL | cytolytic T lymphocytes |
| CTLA-4 | cytotoxic T lymphocyte antigen 4 |
| DC | dendritic cell |
| DNAM-1 | DNAX accessory molecule 1 |
| ECM | extracellular matrix component |
| EMT | epithelial–mesenchymal transition |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| FAP | fibroblast activation protein |
| PGE2 | prostaglandin E2 |
| HSC | hematopoietic stem cells |
| Her2b | epidermal growth receptor 2b |
| HGF | hepatocyte growth factor |
| HL | Hodgkin lymphoma |
| HLA | human class I leukocyte antigen |
| KIR | killer Ig-like inhibitory receptor |
| IDO | indoleamine 2,3, deoxigenase |
| IFNγ | interferon γ |
| IL | inteleukin-2, 6, 10, 12, 15, 18 |
| LILRB | leukocyte immunoglobulin-like receptors |
| LNMSC | lymph node MSC |
| MDSC | myeloid-derived suppressor cells |
| MICA/B | MHC class I polypeptide related sequence A/B |
| MM | multiple myeloma |
| MMP | membrane metalloprotease |
| MSC | mesenchymal stromal cells |
| NCR | natural cytotoxicity receptor |
| NHL | non-Hodgkin lymphoma |
| NK cell | natural killer cell |
| NKG2D | natural-killer group 2 member D |
| NKG2DL | NKG2D ligand |
| NKT | natural killer-like T cells |
| N-BPs | aminobisphosphonates |
| PD1 | programmed death-1 |
| PDL1 | programmed death ligand-1 |
| P4H | prolyl-4-idroxilase |
| SCID | severe combined immunodeficiency |
| SDF-1 | stromal-derived factor 1 |
| SMA | smooth muscle actin |
| TAA | tumor-associated antigen |
| TACE | tumor necrosis factor-α-converting enzyme |
| TAF | tumor-associated fibroblasts |
| TIGIT | T cell immunoglobulin and ITIM domain |
| TGFβ | transforming growth factor β |
| TNFα | tumor necrosis factor α |
| Treg | regulatory T cells |
| ULBP1-6 | UL16 binding protein 1–6 |
References
- Sato, R.; Semba, T.; Saya, H.; Arima, Y. Stem Cells and Epithelial-Mesenchymal Transition (EMT) in Cancer: Biological Implications and Therapeutic Targets. Stem Cells 2016, 34, 1997–2007. [Google Scholar] [CrossRef]
- Poggi, A.; Musso, A.; Dapino, I.; Zocchi, M.R. Mechanisms of tumor escape from immune system: Role of mesenchymal stromal cells. Immunol. Lett. 2014, 159, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015, 15, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Nickel, A.; Stadler, S.C. Epithelial-to mesenchymal transition of breast cancer cells Role of epigenetic mechanisms. Transl. Res. 2015, 165, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Nauta, A.J.; Fibbe, W.E. Immunomodulatory properties of mesenchymal stromal cells. Blood 2007, 110, 3499–3506. [Google Scholar] [CrossRef] [PubMed]
- Krampera, M. mesenchymal stromal cell “licensing”: A multistep process. Leukemia 2011, 25, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.; Adams, S.; Miller, G.T.; Jesson, M.I.; Watanabe, T.; Wallner, B.P. Hematopoietic stimulation by a dipeptidyl peptidase inhibitor reveals a novel regulatory mechanism and therapeutic treatment for blood cell deficiencies. Blood 2003, 102, 1641–1648. [Google Scholar] [CrossRef] [PubMed]
- Baird, S.K.; Rigopoulos, A.; Cao, D.; Allan, L.; Renner, C.; Scott, F.E.; Scott, A.M. Integral membrane protease fibroblast activation protein sensitizes fibrosarcoma to chemotherapy and alters cell death mechanisms. Apoptosis 2015, 20, 1483–1498. [Google Scholar] [CrossRef] [PubMed]
- Wolf, B.B.; Quan, C.; Tran, T.; Wiesmann, C.; Sutherlin, D. On the edge of validation–Cancer protease fibroblast activation protein. Mini Rev. Med. Chem. 2008, 8, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Puré, E. The road to integrative cancer therapies: Emergence of a tumor-associated fibroblast protease as a potential therapeutic targeting cancer. Expert Opin. Ther. Targets 2009, 13, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Tilan, J.U.; Everhart, L.; Czarnecka, M.; Soldin, S.J.; Mendu, D.R.; Jeha, D.; Hanafy, J.; Lee, C.K.; Sun, J.; et al. Dipeptidyl peptidases as survival factors in Ewing sarcoma family of tumors: Implications for tumor biology and therapy. J. Biol. Chem. 2011, 286, 27494–27505. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, H.; Liu, L.; Yu, J.; Ren, X. Fibroblast activation protein: A potential therapeutic targeting cancer. Cancer Biol. Ther. 2012, 13, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Allard, D.; Allard, B.; Gaudreau, P.O.; Chrobak, P.; Stagg, J. CD73-adenosine: A next-generation target in immuno-oncology. Immunotherapy 2016, 8, 145–163. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Mittal, D.; Stagg, J.; Smyth, M.J. Targeting cancer-derived adenosine: New therapeutic approaches. Cancer Discov. 2014, 4, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.A. Metabolic Immune Checkpoint: Adenosine in Tumor Microenvironment. Front. Immunol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Chang, W.; Lim, S.; Seo, H.S.; Shim, C.Y.; Park, S.; Yoo, K.J.; Kim, B.S.; Min, B.H.; Lee, H.; et al. Tissue transglutaminase is essential for integrin-mediated survival of bone marrow-derived mesenchymal stem cells. Stem Cells 2007, 25, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Shao, M.; Schilder, J.; Guise, T.; Mohammad, K.S.; Matei, D. Tissue transglutaminase links TGF-β, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene 2012, 31, 2521–2534. [Google Scholar] [CrossRef] [PubMed]
- Assi, J.; Srivastava, G.; Matta, A.; Chang, M.C.; Walfish, P.G.; Ralhan, R. Transglutaminase 2 overexpression in tumor stroma identifies invasive ductal carcinomas of breast at high risk of recurrence. PLoS ONE 2013, 8, e74437. [Google Scholar] [CrossRef] [PubMed]
- Kanchan, K.; Fuxreiter, M.; Fésüs, L. Physiological, pathological, and structural implications of non-enzymatic protein-protein interactions of the multifunctional human transglutaminase 2. Cell. Mol. Life Sci. 2015, 72, 3009–3035. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, A.M.; Liu, W. Transglutaminase 2 in cancer. Am. J. Cancer Res. 2015, 5, 2756–2776. [Google Scholar] [PubMed]
- Tatsukawa, H.; Furutani, Y.; Hitomi, K.; Kojima, S. Transglutaminase 2 has opposing roles in the regulation of cellular functions as well as cell growth and death. Cell Death Dis. 2016, 7, e2244. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.P.; Goh, B.C. Exosome-Mediated Metastasis: From Epithelial-Mesenchymal Transition to Escape from Immunosurveillance. Trends Pharmacol. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Roulis, M.; Flavell, R.A. Fibroblasts and myofibroblasts of the intestinal lamina propria in physiology and disease. Differentiation 2016. [Google Scholar] [CrossRef] [PubMed]
- Poggi, A.; Zocchi, M.R. How to exploit stress-related immunity against Hodgkin’s lymphoma: Targeting ERp5 and ADAM sheddases. Oncoimmunology 2013, 2, e27089. [Google Scholar] [CrossRef] [PubMed]
- Zocchi, M.R.; Catellani, S.; Canevali, P.; Tavella, S.; Garuti, A.; Villaggio, B.; Zunino, A.; Gobbi, M.; Fraternali-Orcioni, G.; Kunkl, A.; et al. High ERp5/ADAM10 expression in lymph node microenvironment and impaired NKG2D ligands recognition in Hodgkin lymphomas. Blood 2012, 119, 1479–1489. [Google Scholar] [CrossRef] [PubMed]
- Musso, A.; Catellani, S.; Canevali, P.; Tavella, S.; Venè, R.; Boero, S.; Pierri, I.; Gobbi, M.; Kunkl, A.; Ravetti, J.L.; et al. Aminobisphosphonates prevent the inhibitory effects exerted by lymph node stromal cells on anti-tumor Vδ 2 T lymphocytes in non-Hodgkin lymphomas. Haematologica 2014, 99, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Rocks, N.; Paulissen, G.; Quesada-Calvo, F.; Munaut, C.; Gonzalez, M.L.; Gueders, M.; Hacha, J.; Gilles, C.; Foidart, J.M.; Noel, A.; et al. ADAMTS-1 metalloproteinase promotes tumor development through the induction of a stromal reaction in vivo. Cancer Res. 2008, 68, 9541–9550. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Reiss, K. The “A Disintegrin And Metalloproteases” ADAM10 and ADAM17: Novel drug targets with therapeutic potential? Eur. J. Cell Biol. 2011, 90, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Mullooly, M.; O’Donovan, N.; Sukor, S.; Crown, J.; Pierce, A.; McGowan, P.M. The ADAMs family of proteases: New biomarkers and therapeutic targets for cancer? Clin. Proteom. 2011, 8, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Blobel, C.P. ADAMs: Key components in EGFR signalling and development. Nat. Rev. Cancer 2005, 6, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; McKiernan, E.; O’Donovan, N.; McGowan, P. Role of ADAMs in cancer formation and progression. Clin. Cancer Res. 2007, 13, 2335–2343. [Google Scholar] [CrossRef] [PubMed]
- Nausch, N.; Cerwenka, A. NKG2D ligands in tumor immunity. Oncogene 2008, 27, 5944–5958. [Google Scholar] [CrossRef] [PubMed]
- Champsaur, M.; Lanier, L.L. Effect of NKG2D ligand expression on host immune responses. Immunol. Rev. 2010, 235, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Salih, H.R.; Antropius, H.; Gieseke, F.; Lutz, S.Z.; Kanz, L.; Rammensee, H.G.; Steinle, A. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood 2003, 102, 1389–1396. [Google Scholar] [CrossRef] [PubMed]
- Zocchi, M.R.; Camodeca, C.; Nuti, E.; Rossello, A.; Venè, R.; Tosetti, F.; Dapino, I.; Costa, D.; Musso, A.; Poggi, A. ADAM10 new selective inhibitors reduce NKG2D ligand release sensitizing Hodgking-lymphoma cells to NKG2D-mediated killing. Oncoimmunology 2016, 5, e1123367. [Google Scholar] [CrossRef] [PubMed]
- Bottino, C.; Castriconi, R.; Pende, D.; Rivera, P.; Nanni, M.; Carnemolla, B.; et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J. Exp. Med. 2003, 198, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Zingoni, A.; Ardolino, M.; Santoni, A.; Cerboni, C. NKG2D and DNAM-1 activating receptors and their lignads in NK-T interactions: Role in NK cell-mediated negative regulation of T cell responses. Front. Immunol. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.J.; Dougall, W.C.; Miles, J.J.; Teng, M.W.; Smyth, M.J. Molecular Pathways: Targeting CD96 and TIGIT for Cancer Immunotherapy. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Correia, D.V.; Fogli, M.; Hudspeth, K.; da Silva, M.G.; Mavilio, D.; Silva-Santos, B. Differentiation of human peripheral blood Vδ1+ T cells expressing the natural cytotoxicity receptor NKp30 for recognition of lymphoid leukemia cells. Blood 2011, 118, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. NK cell recognition. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Kim, J.; Deng, M.; John, S.; Chen, H.; Wu, G.; Phan, H.; Zhang, C.C. Inhibitory leukocyte immunoglobulin-like receptors: Immune checkpoint proteins and tumor sustaining factors. Cell Cycle 2016, 15, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Mougiakakos, D.; Jitschin, R.; Johansson, C.C.; Okita, R.; Kiessling, R.; Le Blanc, K. The impact of inflammatory licensing on heme oxygenase-1-mediated induction of regulatory T cells by human mesenchymal stem cells. Blood 2011, 117, 4826–4835. [Google Scholar] [CrossRef] [PubMed]
- Uyttenhove, C.; Pilotte, L.; Theate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoural immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Vig, M.; Srivastava, S.; Kandpal, U.; Sade, H.; Lewis, V.; Sarin, A.; George, A.; Bal, V.; Durdik, J.M.; Rath, S. Inducible oxid nitric synthase in T cells regulates T cell death and immune memory. J. Clin. Invest. 2004, 113, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Ino, Y.; Yamazaki-Itoh, R.; Oguro, S.; Shimada, K.; Kosuge, T.; Zavada, J.; Kanai, Y.; Hiraoka, N. Arginase II expressed in cancer-associated fibroblasts indicates tissue hypoxia and predicts poor outcome in patients with pancreatic cancer. PLoS ONE 2013, 8, e55146. [Google Scholar] [CrossRef] [PubMed]
- Barnas, J.L.; Simpson-Abelson, M.R.; Brooks, S.P.; Kelleher, R.J., Jr.; Bankert, R.B. Reciprocal functional modulation of the activation of T lymphocytes and fibroblasts derived from human solid tumors. J. Immunol. 2010, 185, 2681–2692. [Google Scholar] [CrossRef] [PubMed]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, S.; Gianni, A.M. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting from immunosurveillance to tumour escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Von Boehmer, H. Mechanisms of suppression by suppressor T cells. Nat. Immunol. 2005, 6, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Chang, M.Y.; Mandik-Nayak, L.; Metz, R.; Muller, A.J. Indoleamine 2,3-dioxygenase as a modifier of pathogenic inflammation in cancer and other inflammation-associated diseases. Curr. Med. Chem. 2011, 18, 2257–2262. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Lal, G. Regulatory and effector functions of gamma-delta (γδ) T cells and their therapeutic potential in adoptive cellular therapy for cancer. Int. J. Cancer 2016. [Google Scholar] [CrossRef] [PubMed]
- Wistuba-Hamprecht, K.; Di Benedetto, S.; Schilling, B.; Sucker, A.; Schadendorf, D.; Garbe, C.; Weide, B.; Pawelec, G. Phenotypic characterization and prognostic impact of circulating γδ and αβ T-cells in metastatic malignant melanoma. Int. J. Cancer 2016, 138, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Balsamo, M.; Scordamaglia, F.; Pietra, G.; Manzini, C.; Cantoni, C.; Boitano, M.; Queirolo, P.; Vermi, W.; Facchetti, F.; Moretta, A.; Moretta, L.; Mingari, M.C.; Vitale, M. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 20847–20852. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Le Blanc, K.; Mougiakakos, D. Multipotent mesenchymal stromal cells and the innate immune system. Nat. Rev. Immunol. 2012, 12, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Baginska, J.; Viry, E.; Paggetti, J.; Medves, S.; Berchem, G.; Moussay, E.; Janji, B. The critical role of the tumor microenvironment in shaping natural killer cell-mediated anti-tumor immunity. Front. Immunol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Momin, E.N.; Vela, G.; Zaidi, H.A.; Quinones-Hinojosa, A. The oncogenic potential of mesenchymal stem cells in the treatment of cancer: Directions for future research. Curr. Immunol. Rev. 2010, 6, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; Diprete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Reagan, M.R.; Ghobrial, I.M. Multiple myeloma mesenchymal stem cells: Characterization, origin, and tumor-promoting effects. Clin. Cancer Res. 2012, 18, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Cirri, P.; Chiarugi, P. Cancer-associated-fibroblasts and tumour cells: A diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012, 31, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef] [PubMed]
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE 2009, 4, e4992. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-de-Souza, P.; Gori, V.; Bambi, F.; Chiarugi, P. Tumor microenvironment: Bone marrow-mesenchymal stem cells as key players. Biochim. Biophys. Acta 2013, 1836, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Jotzu, C.; Alt, E.; Welte, G.; Li, J.; Hennessy, B.T.; Devarajan, E.; Krishnappa, S.; Pinilla, S.; Droll, L.; Song, Y.H. Adipose tissue-derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor-derived factors. Anal.Cell. Pathol. (Amst.) 2010, 33, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Paggetti, J.; Haderk, F.; Seiffert, M.; Janji, B.; Distler, U.; Ammerlaan, W.; Kim, Y.J.; Adam, J.; Lichter, P.; Solary, E.; et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015, 126, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.C.; Carrasco, R.D. Pathogenesis of myeloma. Annu. Rev. Pathol. 2011, 6, 249–274. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, N.S.; Apollonio, B.; Ramsay, A.G. Tumor microenvironment (tme)-driven immune suppression in B cell malignancy. Biochim. Biophys. Acta 2016, 1863, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Raffaghello, L.; Vacca, A.; Pistoia, V.; Ribatti, D. Cancer associated fibroblasts in hematological malignancies. Oncotarget 2015, 6, 2589–2603. [Google Scholar] [CrossRef] [PubMed]
- Shain, K.H.; Dalton, W.S.; Tao, J. The tumor microenvironment shapes hallmarks of mature B-cell malignancies. Oncogene 2015, 34, 4673–4682. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Gribben, J.G. The microenvironment in chronic lymphocytic leukemia (CLL) and other b cell malignancies: Insight into disease biology and new targeted therapies. Semin. Cancer Biol. 2014, 24, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Lutzny, G.; Kocher, T.; Schmidt-Supprian, M.; Rudelius, M.; Klein-Hitpass, L.; Finch, A.J.; Durig, J.; Wagner, M.; Haferlach, C.; Kohlmann, A.; et al. Protein kinase c-beta-dependent activation of NF-kappab in stromal cells is indispensable for the survival of chronic lymphocytic leukemia b cells in vivo. Cancer Cell 2013, 23, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Seke Etet, P.F.; Vecchio, L.; Nwabo Kamdje, A.H. Signaling pathways in chronic myeloid leukemia and leukemic stem cell maintenance: Key role of stromal microenvironment. Cell. Signal. 2012, 24, 1883–1888. [Google Scholar] [CrossRef] [PubMed]
- Corrado, C.; Raimondo, S.; Saieva, L.; Flugy, A.M.; de Leo, G.; Alessandro, R. Exosome-mediated crosstalk between chronic myelogenous leukemia cells and human bone marrow stromal cells triggers an interleukin 8-dependent survival of leukemia cells. Cancer Lett. 2014, 348, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Le, Y.; Fraineau, S.; Chandran, P.; Sabloff, M.; Brand, M.; Lavoie, J.R.; Gagne, R.; Rosu-Myles, M.; Yauk, C.L.; Richardson, R.B.; et al. Adipogenic mesenchymal stromal cells from bone marrow and their hematopoietic supportive role: Towards understanding the permissive marrow microenvironment in acute myeloid leukemia. Stem Cell Rev. 2016, 12, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.C.; Basu, S.K.; Zhao, X.; Chien, S.; Fang, M.; Oehler, V.G.; Appelbaum, F.R.; Becker, P.S. Mesenchymal stromal cells derived from acute myeloid leukemia bone marrow exhibit aberrant cytogenetics and cytokine elaboration. Blood Cancer J. 2015, 5, e302. [Google Scholar] [CrossRef] [PubMed]
- Chandran, P.; Le, Y.; Li, Y.; Sabloff, M.; Mehic, J.; Rosu-Myles, M.; Allan, D.S. Mesenchymal stromal cells from patients with acute myeloid leukemia have altered capacity to expand differentiated hematopoietic progenitors. Leuk. Res. 2015, 39, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Geyh, S.; Rodriguez-Paredes, M.; Jager, P.; Khandanpour, C.; Cadeddu, R.P.; Gutekunst, J.; Wilk, C.M.; Fenk, R.; Zilkens, C.; Hermsen, D.; et al. Functional inhibition of mesenchymal stromal cells in acute myeloid leukemia. Leukemia 2016, 30, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Jacamo, R.; Chen, Y.; Wang, Z.; Ma, W.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal leukemia-stroma vcam-1/vla-4-dependent activation of NF-kappab mediates chemoresistance. Blood 2014, 123, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Fulzele, K.; Catic, A.; Sun, C.C.; Dombkowski, D.; Hurley, M.P.; Lezeau, S.; Attar, E.; Wu, J.Y.; Lin, H.Y.; et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat. Med. 2013, 19, 1513–1517. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Konopleva, M. Role of microenvironment in resistance to therapy in AML. Curr. Hematol. Malig. Rep. 2015, 10, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Vasu, S.; Caligiuri, M.A. Targeted immunotherapy for acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2011, 24, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Shi, Y.X.; Samudio, I.J.; Wang, R.Y.; Ling, X.; Frolova, O.; Levis, M.; Rubin, J.B.; Negrin, R.R.; Estey, E.H.; et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood 2009, 113, 6215–6224. [Google Scholar] [CrossRef] [PubMed]
- Rashidi, A.; DiPersio, J.F. Targeting the leukemia-stroma interaction in acute myeloid leukemia: Rationale and latest evidence. Ther. Adv. Hematol. 2016, 7, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Hilpert, J.; Grosse-Hovest, L.; Grunebach, F.; Buechele, C.; Nuebling, T.; Raum, T.; Steinle, A.; Salih, H.R. Comprehensive analysis of NKG2D ligand expression and release in leukemia: Implications for NKG2D-mediated NK cell responses. J. Immunol. 2012, 189, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Moschetta, M.; Manier, S.; Glavey, S.; Gorgun, G.T.; Roccaro, A.M.; Anderson, K.C.; Ghobrial, I.M. Targeting the bone marrow microenvironment in multiple myeloma. Immunol. Rev. 2015, 263, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Garayoa, M.; Garcia, J.L.; Santamaria, C.; Garcia-Gomez, A.; Blanco, J.F.; Pandiella, A.; Hernandez, J.M.; Sanchez-Guijo, F.M.; del Canizo, M.C.; Gutierrez, N.C.; et al. Mesenchymal stem cells from multiple myeloma patients display distinct genomic profile as compared with those from normal donors. Leukemia 2009, 23, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Garderet, L.; Mazurier, C.; Chapel, A.; Ernou, I.; Boutin, L.; Holy, X.; Gorin, N.C.; Lopez, M.; Doucet, C.; Lataillade, J.J. Mesenchymal stem cell abnormalities in patients with multiple myeloma. Leuk. Lymphoma 2007, 48, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Jurczyszyn, A.; Czepiel, J.; Gdula-Argasinska, J.; Perucki, W.; Skotnicki, A.B.; Majka, M. The analysis of the relationship between multiple myeloma cells and their microenvironment. J. Cancer 2015, 6, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, G.; Sciume, G.; Bosisio, D.; Morrone, S.; Sozzani, S.; Santoni, A. CCl3 and cxcl12 regulate trafficking of mouse bone marrow nk cell subsets. Blood 2008, 111, 3626–3634. [Google Scholar] [CrossRef] [PubMed]
- Corre, J.; Mahtouk, K.; Attal, M.; Gadelorge, M.; Huynh, A.; Fleury-Cappellesso, S.; Danho, C.; Laharrague, P.; Klein, B.; Reme, T.; et al. Bone marrow mesenchymal stem cells are abnormal in multiple myeloma. Leukemia 2007, 21, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gomez, A.; De Las Rivas, J.; Ocio, E.M.; Diaz-Rodriguez, E.; Montero, J.C.; Martin, M.; Blanco, J.F.; Sanchez-Guijo, F.M.; Pandiella, A.; San Miguel, J.F.; et al. Transcriptomic profile induced in bone marrow mesenchymal stromal cells after interaction with multiple myeloma cells: Implications in myeloma progression and myeloma bone disease. Oncotarget 2014, 5, 8284–8305. [Google Scholar] [CrossRef] [PubMed]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.M., Jr.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Andre, T.; Najar, M.; Stamatopoulos, B.; Pieters, K.; Pradier, O.; Bron, D.; Meuleman, N.; Lagneaux, L. Immune impairments in multiple myeloma bone marrow mesenchymal stromal cells. Cancer Immunol. Immunother. 2015, 64, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Ponzetta, A.; Benigni, G.; Antonangeli, F.; Sciume, G.; Sanseviero, E.; Zingoni, A.; Ricciardi, M.R.; Petrucci, M.T.; Santoni, A.; Bernardini, G. Multiple myeloma impairs bone marrow localization of effector natural killer cells by altering the chemokine microenvironment. Cancer Res. 2015, 75, 4766–4777. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Fu, J.; Chen, P.; Zhuang, W. Impairment in immunomodulatory function of mesenchymal stem cells from multiple myeloma patients. Arch. Med. Res. 2010, 41, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Menu, E.; De Becker, A.; Van Camp, B.; Vanderkerken, K.; Van Riet, I. Bone marrow-derived mesenchymal stromal cells are attracted by multiple myeloma cell-produced chemokine CCL25 and favor myeloma cell growth in vitro and in vivo. Stem Cells (Dayton, OH) 2012, 30, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Roccaro, A.M.; Sacco, A.; Maiso, P.; Azab, A.K.; Tai, Y.T.; Reagan, M.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E.; et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J. Clin. Investig. 2013, 123, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Abdi, J.; Qiu, L.; Chang, H. Micro-rnas, new performers in multiple myeloma bone marrow microenvironment. Biomark. Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Alsayed, Y.; Ngo, H.; Runnels, J.; Leleu, X.; Singha, U.K.; Pitsillides, C.M.; Spencer, J.A.; Kimlinger, T.; Ghobrial, J.M.; Jia, X.; et al. Mechanisms of regulation of CXCR4/SDF-1 (CXCL12)-dependent migration and homing in multiple myeloma. Blood 2007, 109, 2708–2717. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Ishibashi, M.; Yamashita, T.; Tanosaki, S.; Okuyama, N.; Kondo, A.; Hyodo, H.; Shinya, E.; Takahashi, H.; Dong, H.; et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia 2013, 27, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Yousef, S.; Marvin, J.; Steinbach, M.; Langemo, A.; Kovacsovics, T.; Binder, M.; Kroger, N.; Luetkens, T.; Atanackovic, D. Immunomodulatory molecule PD-L1 is expressed on malignant plasma cells and myeloma-propagating pre-plasma cells in the bone marrow of multiple myeloma patients. Blood Cancer J. 2015, 5, e285. [Google Scholar] [CrossRef] [PubMed]
- Yang, L. TGFbeta, a potent regulator of tumor microenvironment and host immune response, implication for therapy. Curr. Mol. Med. 2010, 10, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Rhee, K.J.; Lee, J.I.; Eom, Y.W. Mesenchymal Stem Cell-Mediated Effects of Tumor Support or Suppression. Int. J. Mol. Sci. 2015, 16, 30015–30033. [Google Scholar] [CrossRef] [PubMed]
- Shaked, Y. Balancing efficacy of and host immune responses to cancer therapy: The yin and yang effects. Nat. Rev. Clin. Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Adjei, A.A. Targeting Angiogenesis in Cancer Therapy: Moving Beyond Vascular Endothelial Growth Factor. Oncologist 2015, 20, 660–673. [Google Scholar] [CrossRef] [PubMed]
- Martin-Broto, J.; Hindi, N. Targeted treatments of sarcomas and connective tumors beside gastrointestinal stromal tumor. Curr. Opin. Oncol. 2016, 28, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Abarbanell, A.M.; Coffey, A.C.; Fehrenbacher, J.W.; Beckman, D.J.; Herrmann, J.L.; Weil, B.; et al. Proinflammatory cytokine effects on mesenchymal stem cell therapy for the ischemic heart. Ann. Thorac. Surg. 2009, 88, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Norozi, F.; Ahmadzadeh, A.; Shahrabi, S.; Vosoughi, T.; Saki, N. Mesenchymal stem cells as a double-edged sword in suppression or progression of solidtumor cells. Tumor Biol. 2016, 36, 11679–11689. [Google Scholar] [CrossRef] [PubMed]
- Lazennec, G.; Jorgensen, C. Concise review: Adult multipotent stromal cells and cancer: Risk or benefit? Stem Cells 2008, 26, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bucan, V.; Baehre, H.; von der Ohe, J.; Otte, A.; Hass, R. Acquisition of new tumor cell properties by MSC-derived exosomes. Int. J. Oncol. 2015, 47, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Otte, A.; Hass, R. Human mesenchymal stroma/stem cells exchange membrane proteins and alter functionality during interaction with different tumor cell lines. Stem Cells Dev. 2015, 24, 1205–1222. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Zhang, Q.; Xia, Y.; You, B.; Shan, Y.; Bao, L.; Li, L.; You, Y.; Gu, Z. Mesenchymal stem cell-derived exosomes facilitate nasopharyngeal carcinoma progression. Am. J. Cancer Res. 2016, 6, 459–472. [Google Scholar] [PubMed]
- Melzer, C.; Yang, Y.; Hass, R. Interaction of MSC with tumor cells. Cell Commun. Signal. 2016. [Google Scholar] [CrossRef] [PubMed]
- Vasold, J.; Wagner, M.; Drolle, H.; Deniffel, C.; Kutt, A.; Oostendorp, R.; Sironi, S.; Rieger, C.; Fiegl, M. The bone marrow microenvironment is a critical player in the NK cell response against acute myeloid leukaemia in vitro. Leuk. Res. 2015, 39, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, G.; Sciume, G.; Santoni, A. Differential chemotactic receptor requirements for NK cell subset trafficking into bone marrow. Front. Immunol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Entrena, A.; Varas, A.; Vazquez, M.; Melen, G.J.; Fernandez-Sevilla, L.M.; Garcia-Castro, J.; Ramirez, M.; Zapata, A.G.; Vicente, A. Mesenchymal stem cells derived from low risk acute lymphoblastic leukemia patients promote NK cell antitumor activity. Cancer Lett. 2015, 363, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Tauriello, D.V.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massagué, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Trapani, J.A. The dual adverse effects of TGF-beta secretion on tumor progression. Cancer Cell 2005, 8, 349–350. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Janji, B.; Berchem, G.; Chouaib, S. miR-210 and hypoxic microvesicles: Two critical components of hypoxia involved in the regulation of killercells function. Cancer Lett. 2016, 380, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Colio, L.M.; Muñoz-García, B.; Martín-Ventura, J.L.; Lorz, C.; Díaz, C.; Hernández, G.; Egido, J. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors decrease Fas ligand expression and cytotoxicity in activated human T lymphocytes. Circulation 2003, 108, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Katznelson, S.; Wang, X.M.; Chia, D.; Ozawa, M.; Zhong, H.P.; Hirata, M.; Terasaki, P.I.; Kobashigawa, J.A. The inhibitory effects of pravastatin on natural killer cell activity in vivo and on cytotoxic T lymphocyte activity in vitro. J. Heart Lung Trans. 1998, 17, 335–340. [Google Scholar]
- Poggi, A.; Boero, S.; Musso, A.; Zocchi, M.R. Selective role of mevalonate pathway in regulating perforin but not FasL and TNFalpha release in human Natural Killer cells. PLoS ONE 2013, 8, e62932. [Google Scholar] [CrossRef] [PubMed]
- Musso, A.; Zocchi, M.R.; Poggi, A. Relevance of the mevalonate biosynthetic pathway in the regulation of bone marrow mesenchymal stromal cell-mediated effects on T-cell proliferation and B-cell survival. Haematologica 2011, 96, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Poggi, A.; Zocchi, M.R. Modulating mesenchymal stromal cell function with cholesterol synthesis inhibitors. Curr. Med. Chem. 2011, 18, 5196–5205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ertl, H.C. Depletion of FAP+ cells reduces immunosuppressive cells and improves metabolism and functions CD8+T cells within tumors. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.; Wang, W.; Yan, J.; Tan, J.; Liao, L.; Shi, J.; Wei, C.; Xie, Y.; Jin, X.; Yang, L.; et al. Immunization of stromal cell targeting fibroblast activation protein providing immunotherapy to breast cancer mouse model. Tumour Biol. 2016, 22, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.; Zhao, Y.; Yang, H.; Tong, A.; Zhao, C.; Shi, H.; Li, Y.; Wang, Z.; Wei, Y. Immunotherapy of tumor with vaccine based on basic fibroblast growth factor-activated fibroblasts. J. Cancer Res. Clin. Oncol. 2014, 140, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Fassnacht, M.; Nair, S.; Boczkowski, D.; Gilboa, E. Tumor immunotherapy targeting fibroblast activation protein, a product expressed in tumor-associated fibroblasts. Cancer Res. 2005, 65, 11156–11163. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Lee, J.; Milazzo, C.; Boczkowski, D.; Su, Z.; Nair, S.; Gilboa, E. Induction of CD4(+) and CD8(+) T-cell responses to the human stromal antigen, fibroblast activation protein: Implication for cancer immunotherapy. Clin. Cancer Res. 2005, 11, 5566–5571. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Xiao, L.; Joo, K.I.; Liu, Y.; Zhang, C.; Liu, S.; Conti, P.S.; Li, Z.; Wang, P. A potent immunotoxin targeting fibroblast activation protein for treatment of breast cancer in mice. Int. J. Cancer 2016, 138, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, S.; Yu, F.; Ji, M.; Kakarla, S.; Song, X.T. A vaccine that co-targets tumor cells and cancer associated fibroblasts results in enhanced antitumor activity by inducing antigen spreading. PLoS ONE 2013, 8, e82658. [Google Scholar] [CrossRef] [PubMed]
- Klampfer, L. Cytokines, inflammation and colon cancer. Curr. Cancer Drug Targets 2011, 11, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Miller, G.T.; Jesson, M.I.; Watanabe, T.; Jones, B.; Wallner, B.P. PT-100, a small molecule dipeptidyl peptidase inhibitor, has potent antitumor effects and augments antibody-mediated cytotoxicity via a novel immune mechanism. Cancer Res. 2004, 64, 5471–5480. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Wang, C.T.; Ma, T.T.; Li, Z.Y.; Zhou, L.N.; Mu, B.; Leng, F.; Shi, H.S.; Li, Y.O.; Wei, Y.Q. Immunotherapy targeting fibroblast activation protein inhibits tumor growth and increases survival in a murine colon cancer model. Cancer Sci. 2010, 101, 2325–2332. [Google Scholar] [CrossRef] [PubMed]
- Petrausch, U.; Schuberth, P.C.; Hagedorn, C.; Soltermann, A.; Tomaszek, S.; Stahel, R.; Weder, W.; Renner, C. Re-directed T cells for the treatment of fibroblast activation protein (FAP)-positive malignant pleural mesothelioma (FAPME-1). BMC Cancer 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamson, E.J.; Keane, F.M.; Tholen, S.; Schilling, O.; Gorrell, M.D. Understanding fibroblast activation protein (FAP): Substrates, activities, expression and targeting for cancer therapy. Proteom. Clin. Appl. 2014. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.W.; Christiansen, V.J.; Yadav, V.R.; Silasi-Mansat, R.; Lupu, F.; Awasthi, V.; Zhang, R.R.; McKee, P.A. Suppression of tumor growth in mice by rationally designed pseudopeptide inhibitors of fibroblast activation protein and prolyloligopeptidase. Neoplasia 2015, 17, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Teichgräber, V.; Monasterio, C.; Chaitanya, K.; Boger, R.; Gordon, K.; Dieterle, T.; Jäger, D.; Bauer, S. Specific inhibition of fibroblast activation protein (FAP)-alpha prevents tumor progression in vitro. Adv. Med. Sci. 2015, 60, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Ohshio, Y.; Teramoto, K.; Hanaoka, J.; Tezuka, N.; Itoh, Y.; Asai, T.; Daigo, Y.; Ogasawara, K. Cancer-associated fibroblast-targeted strategy enhances antitumor immune responses in dendritic cell-based vaccine. Cancer Sci. 2015, 106, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, M.; Kr€uger, J.A.; Niethammer, A.G.; Reisfeld, R.A. Targeting tumor associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J. Clin. Invest. 2006, 116, 1955–1962. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Xiang, R.; Wen, Y.; Xu, G.; Wang, C.; Luo, S.; Yin, T.; Wei, X.; Shao, B.; Liu, N.; et al. A whole-cell tumor vaccine modified to express fibroblast activation protein induces antitumor immunity against both tumor cells and cancer-associated fibroblasts. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Luo, Y.; Markowitz, D.; Xiang, R.; Reisfeld, R.A. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS ONE 2009, 4, e7965. [Google Scholar] [CrossRef] [PubMed]
- Narunsky, L.; Oren, R.; Bochner, F.; Neeman, M. Imaging aspects of the tumor stroma with therapeutic implications. Pharmacol. Ther. 2014, 141, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Boise, L.H.; Kaufman, J.L.; Bahli, N.J.; Lonial, S.; Lee, K.P. The Tao of myeloma. Blood 2014, 124, 1873–1879. [Google Scholar] [CrossRef] [PubMed]
- Zeidner, J.F.; Foster, M.C. Immunomodulatory Drugs: IMiDs in Acute Myeloid Leukemia (AML). Curr. Drug Targets 2015, 16, 397–404. [Google Scholar] [CrossRef]
- Camicia, R.; Winkler, H.C.; Hassa, P.O. Novel drug targets for personalized precision medicine in relapsed/refractory diffuse large B-cell lymphoma: A comprehensive review. Mol. Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Henricks, L.M.; Schellens, J.H.; Huitema, A.D.; Beijnen, J.H. The use of combinations of monoclonal antibodies in clinical oncology. Cancer Treat. Rev. 2015, 41, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M. Targeting immune checkpoints in lymphoma. Curr. Opin. Hematol. 2015, 22, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Chitadze, G.; Lettau, M.; Luecke, S.; Wang, T.; Janssen, O.; Fürst, D.; Mytilineos, J.; Wesch, D.; Oberg, H.H.; Held-Feindt, J.; et al. NKG2D- and T-cell receptor-dependent lysis of malignant glioma cell lines by human γδ T cells: Modulation by temozolomide and A disintegrin and metalloproteases 10 and 17 inhibitors. Oncoimmunology 2015, 5, e1093276. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, L.H. Cancer vaccines. Br. Med. J. 2015. [Google Scholar] [CrossRef] [PubMed]
- Madorsky Rowdo, F.P.; Baron, A.; Urrutia, M.; Mordoh, J. Immunotherapy in Cancer: A Combat between Tumors and the Immune System; You Win Some, You Lose Some. Front. Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Dillner, J. Prevention of human papillomavirus-associated cancers. Semin. Oncol. 2015, 42, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Pizzurro, G.A.; Barrio, M.M. Dendritic cell-based vaccine efficacy: Aiming for hot spots. Front. Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Westdorp, H.; Fennemann, F.L.; Weren, R.D.; Bisseling, T.M.; Ligtenberg, M.J.; Figdor, C.G.; Schreibelt, G.; Hoogerbrugge, N.; Wimmers, F.; de Vries, I.J. Opportunities for immunotherapy in microsatellite instable colorectal cancer. Cancer Immunol. Immunother. 2016, 65, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Pampena, M.B.; Levy, E.M. Natural killer cells as helper cells in dendritic cell cancer vaccines. Front. Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Türeci, Ö.; Vormehr, M.; Diken, M.; Kreiter, S.; Huber, C.; Sahin, U. Targeting the Heterogeneity of Cancer with Individualized Neoepitope Vaccines. Clin. Cancer Res. 2016, 22, 1885–1896. [Google Scholar] [CrossRef] [PubMed]
- Desrichard, A.; Snyder, A.; Chan, T.A. Cancer Neoantigens and Applications for Immunotherapy. Clin. Cancer Res. 2016, 22, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.C.; Ichim, T.E.; Ma, H.; Szymanski, J.; Perez, J.A.; Lopez, J.; Bogin, V.; Patel, A.N.; Marincola, F.M.; Kesari, S. Cancer anti-angiogenesis vaccines: Is the tumor vasculature antigenically unique? J. Transl. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Schuberth, P.C.; Hagedorn, C.; Jensen, S.M.; Gulati, P.; van den Broek, M.; Mischo, A.; Soltermann, A.; Jüngel, A.; Marroquin Belaunzaran, O.; Stahel, R.; et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J. Transl. Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Kakarla, S.; Chow, K.K.; Mata, M.; Shaffer, D.R.; Song, X.T.; Wu, M.F.; Liu, H.; Wang, L.L.; Rowley, D.R.; Pfizenmaier, K.; et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol. Ther. 2013, 21, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Reisfeld, R.A. The tumor microenvironment: Atargetfor combination therapy of breast cancer. Crit. Rev. Oncog. 2013, 18, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Brennen, W.N.; Rosen, D.M.; Wang, H.; Isaacs, J.T.; Denmeade, S.R. Targeting carcinoma-associated fibroblasts within the tumor stroma with a fibroblast activation protein-activated prodrug. J. Natl. Cancer Inst. 2012, 104, 1320–1334. [Google Scholar] [CrossRef] [PubMed]
- Frey, A.B. Suppression of T cell responses in the tumor microenvironment. Vaccine 2015, 33, 7393–7400. [Google Scholar] [CrossRef] [PubMed]
- Hornig, N.; Kermer, V.; Frey, K.; Diebolder, P.; Kontermann, R.E.; Müller, D. Combination of a bispecific antibody and costimulatory antibody-ligand fusion proteins for targeted cancer immunotherapy. J. Immunother. 2012, 35, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Tsirigotis, P.; Savani, B.N.; Nagler, A. Programmed death-1 immune checkpoint blockade in the treatment of hematological malignancies. Ann. Med. 2016, 48, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.E.; Caenepeel, S.; Wu, L.C. Targeted Therapy and Checkpoint Immunotherapy Combinations for the Treatment of Cancer. Trends Immunol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Rudd, C.E. Diverse mechanisms regulate the surface expression of immunotherapeutic target CTLA-4. Front. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Philips, G.K.; Atkins, M. Therapeutic uses of anti-PD-1 and anti-PD-L1 antibodies. Int. Immunol. 2015, 27, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Ohashi, P.S. Clinical blockade of PD1 and LAG3—Potential mechanisms of action. Nat. Rev. Immunol. 2014, 15, 45–56. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poggi, A.; Giuliani, M. Mesenchymal Stromal Cells Can Regulate the Immune Response in the Tumor Microenvironment. Vaccines 2016, 4, 41. https://doi.org/10.3390/vaccines4040041
Poggi A, Giuliani M. Mesenchymal Stromal Cells Can Regulate the Immune Response in the Tumor Microenvironment. Vaccines. 2016; 4(4):41. https://doi.org/10.3390/vaccines4040041
Chicago/Turabian StylePoggi, Alessandro, and Massimo Giuliani. 2016. "Mesenchymal Stromal Cells Can Regulate the Immune Response in the Tumor Microenvironment" Vaccines 4, no. 4: 41. https://doi.org/10.3390/vaccines4040041