Enhanced Delivery and Potency of Self-Amplifying mRNA Vaccines by Electroporation in Situ

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
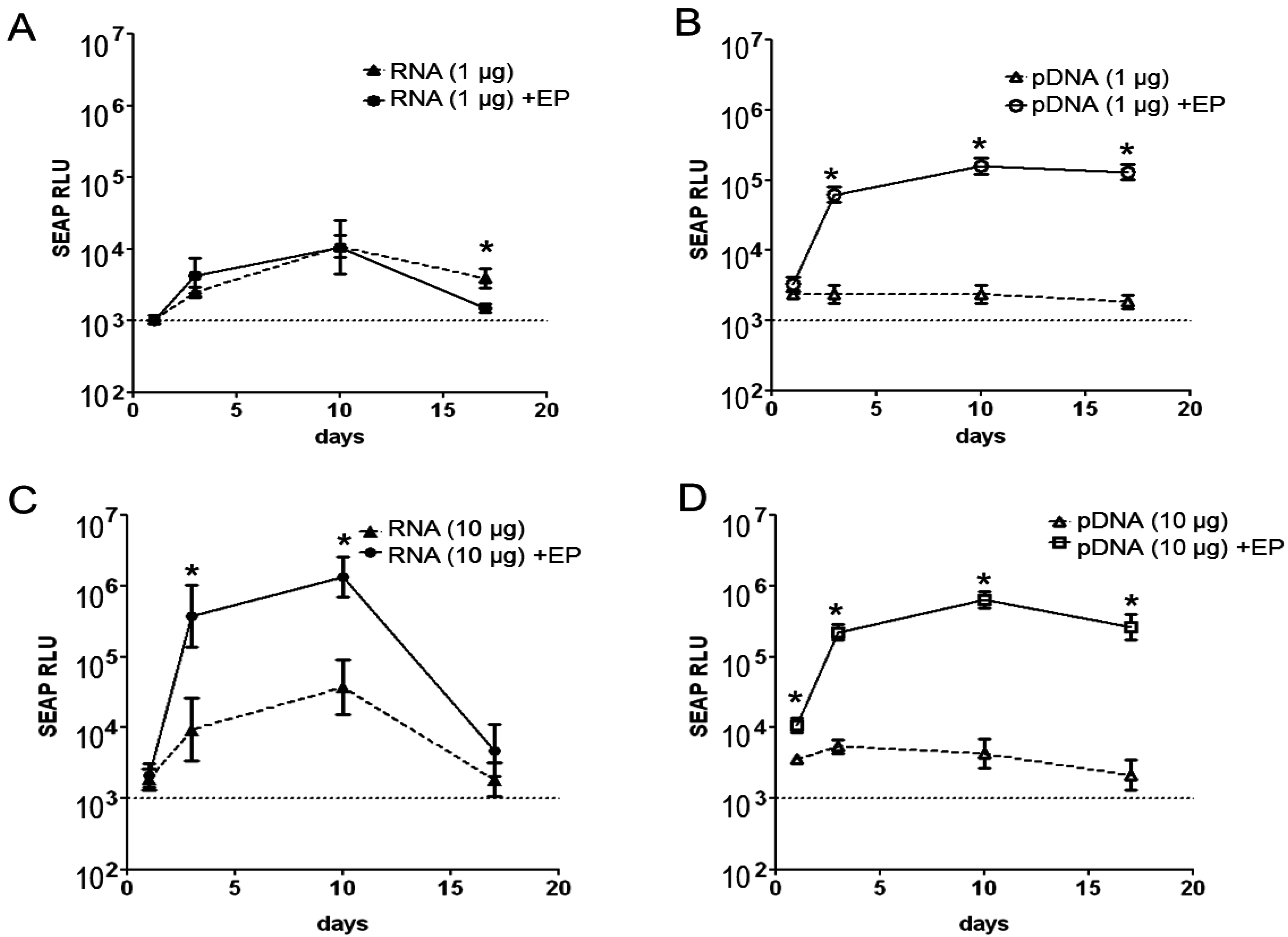
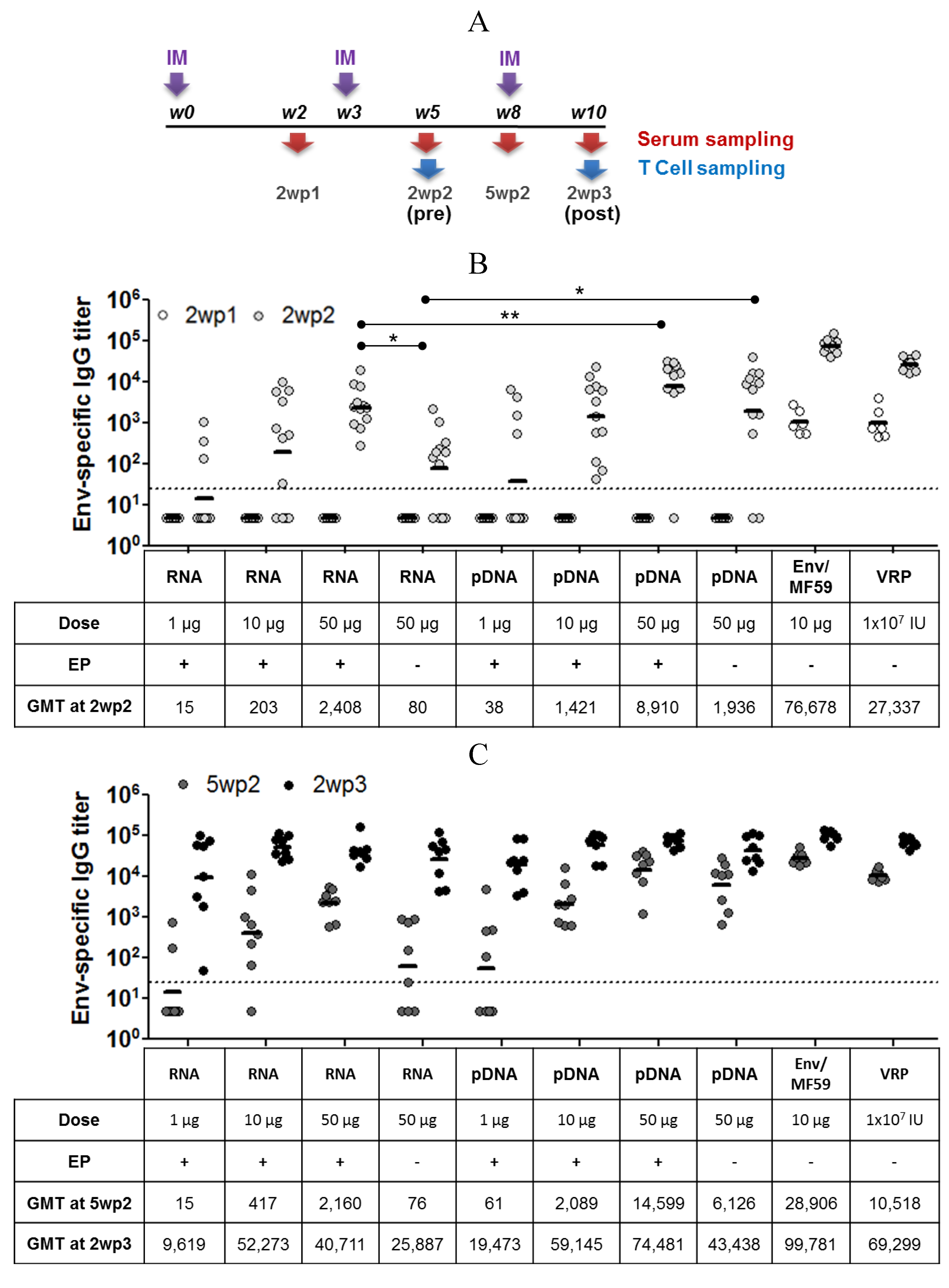
2.1. Magnitude and Kinetics of Gene Expression in Vivo

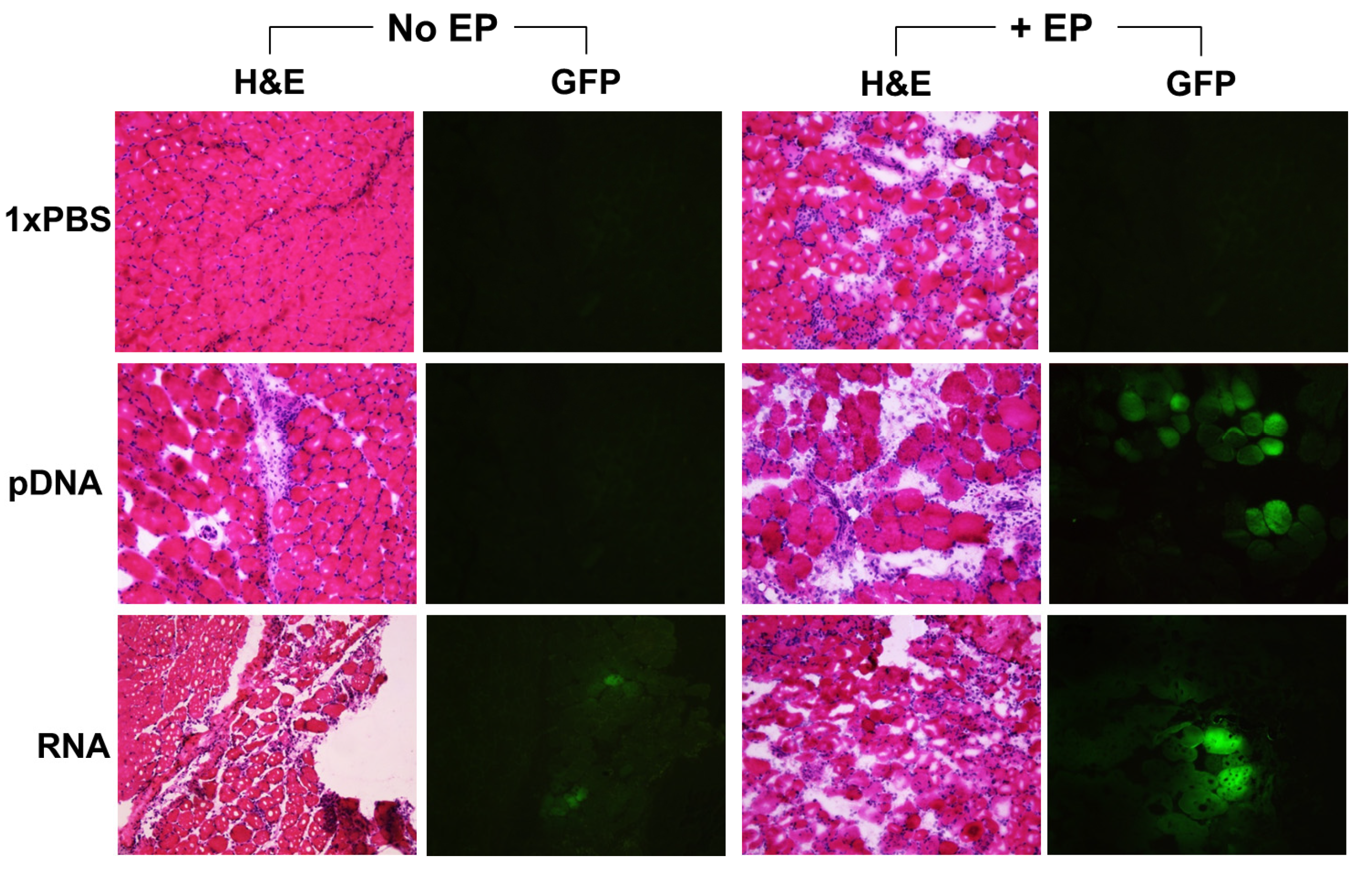
2.2. Reporter Gene Expression in Situ

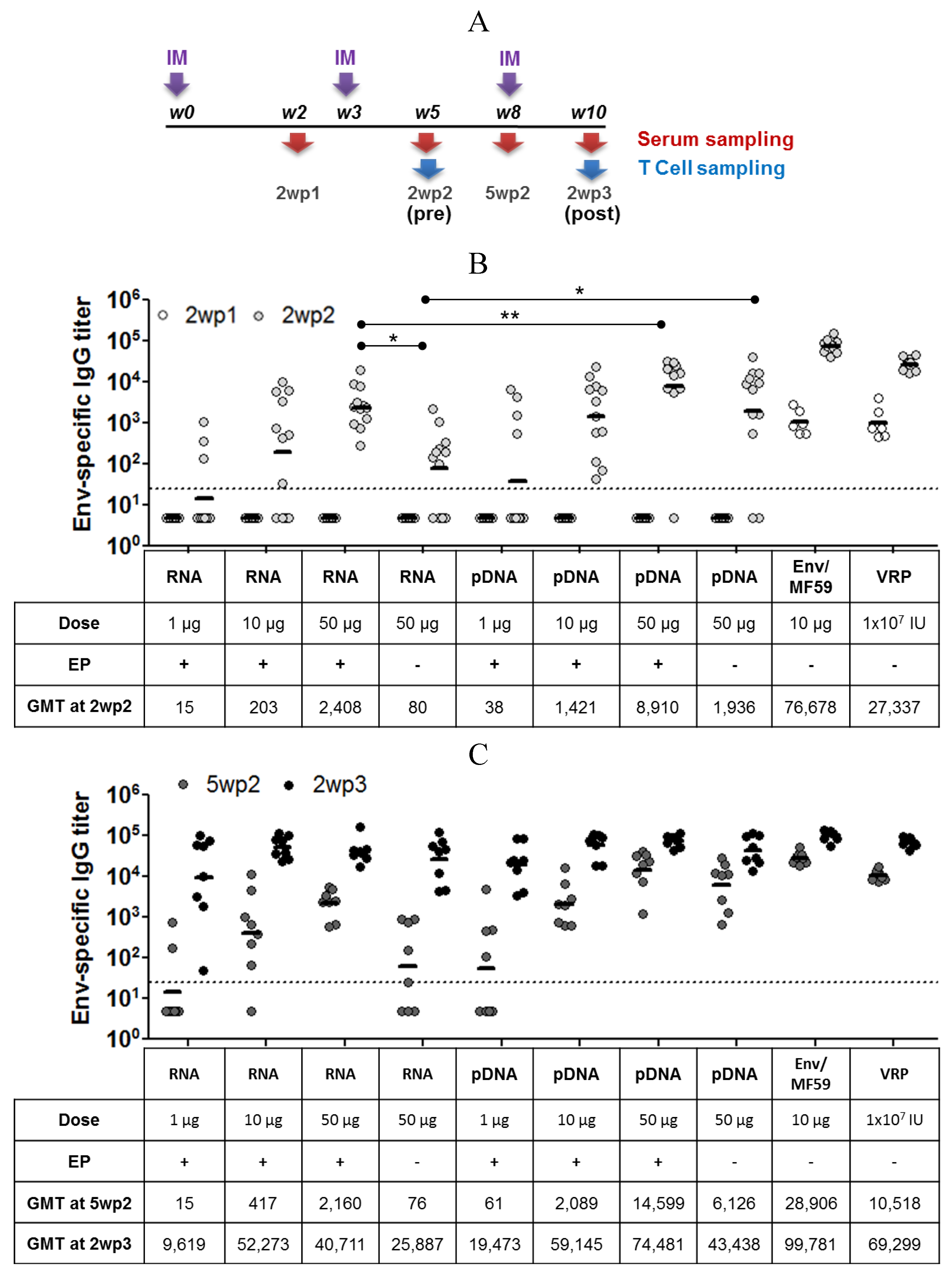
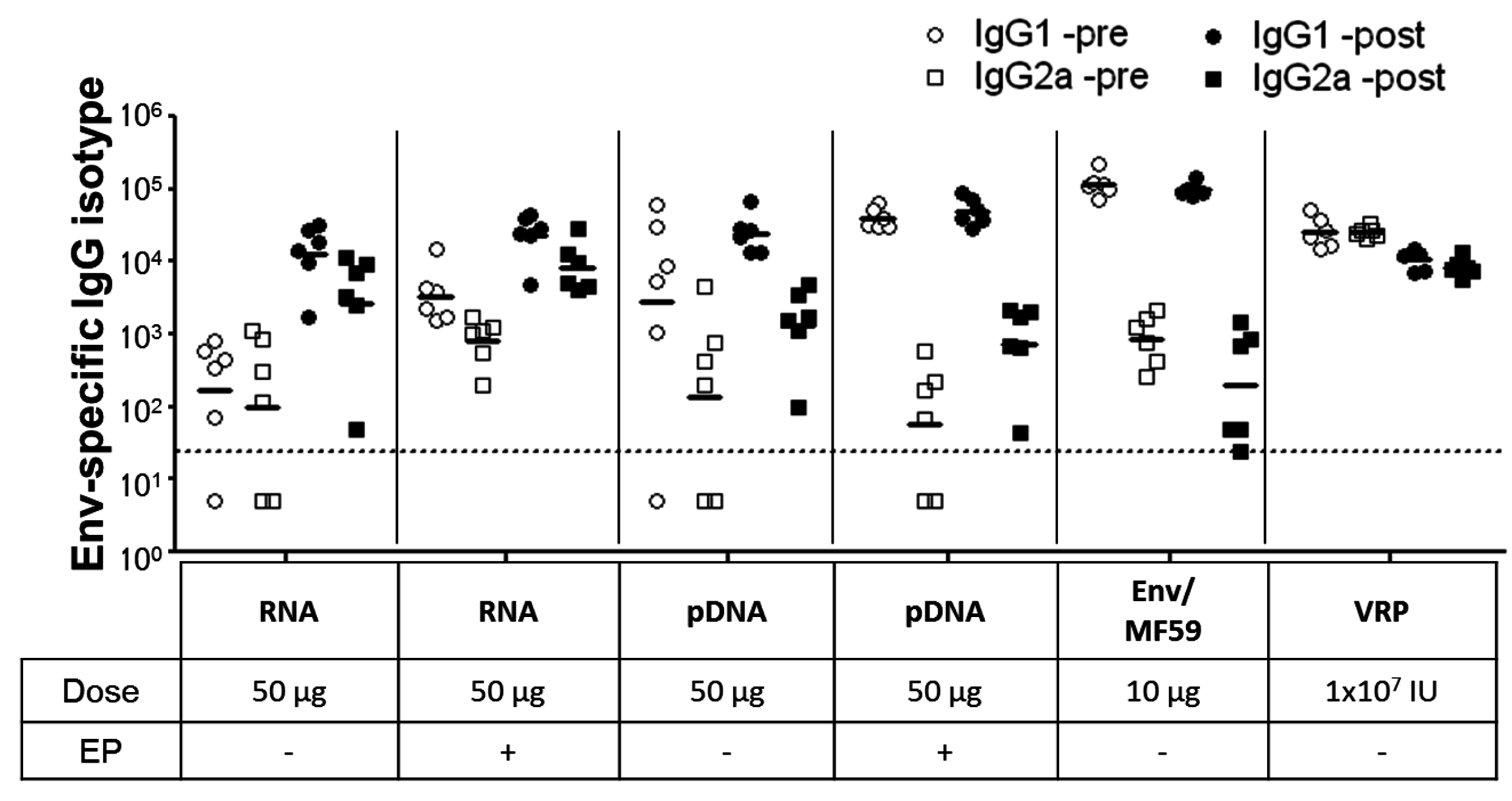
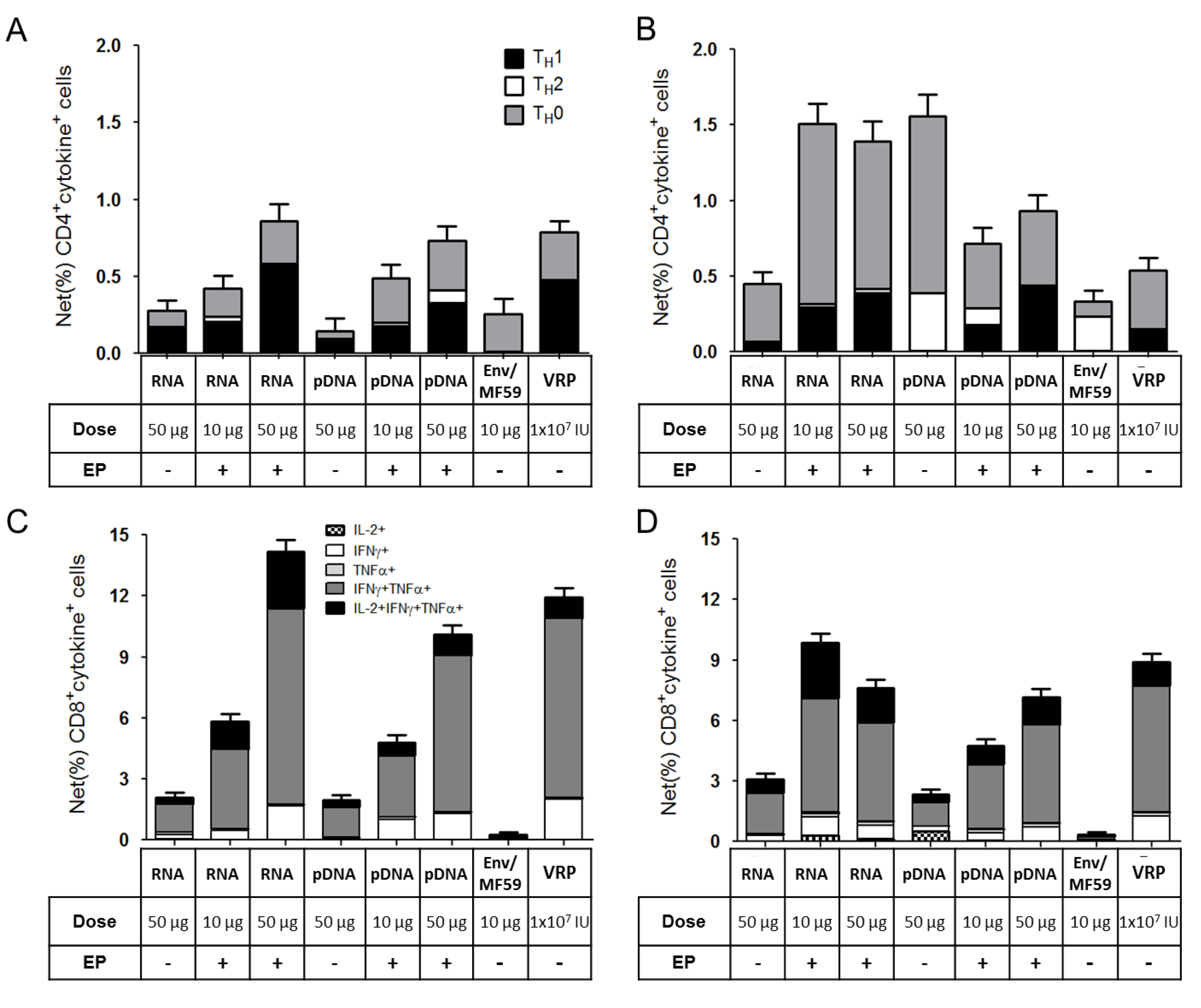
2.3. Immunogenicity of pDNA and Self-Amplifying mRNA Vaccines
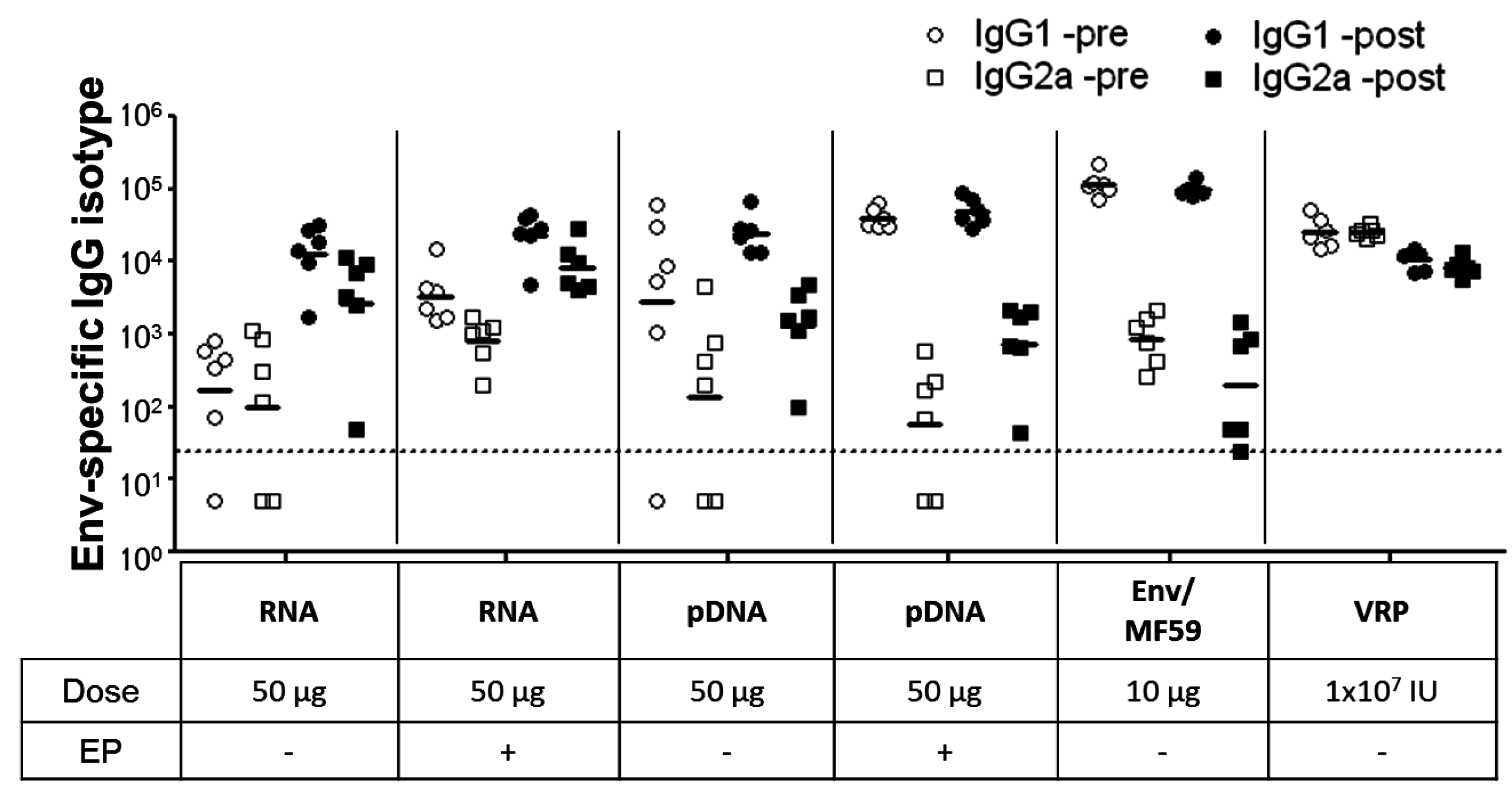
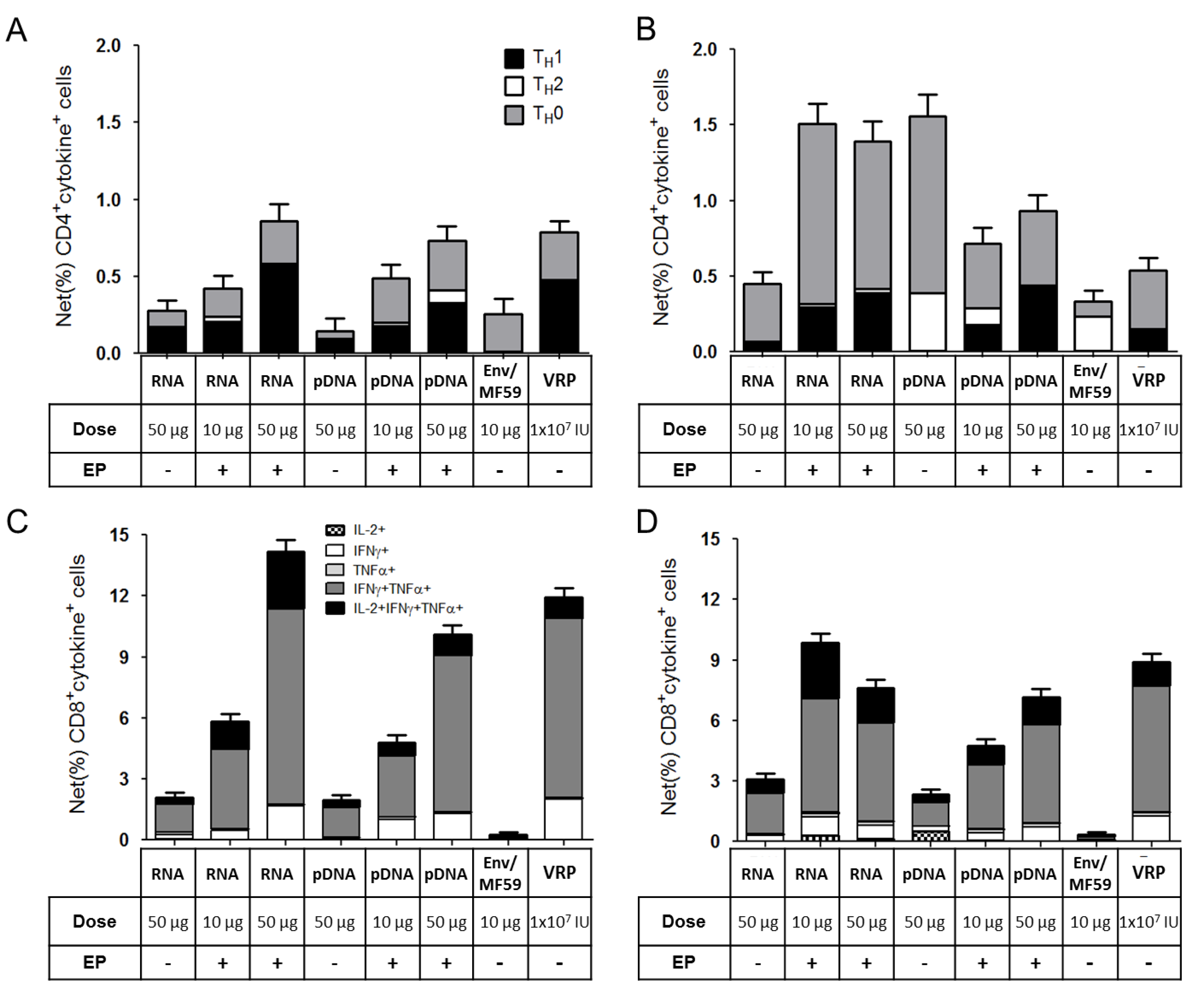
3. Discussion
4. Materials and Methods
4.1. RNA Synthesis
4.2. Plasmid DNA (pDNA) Preparation
4.3. Production of Viral Replicon Particles (VRPs)
4.4. MF59-Adjuvanted gp140.SF162 HIV Envelope Protein Subunit Vaccine Candidate
4.5. In Vivo Immunization
4.6. Histology
4.7. Quantitation of SEAP Expression
4.8. ELISA for Antigen-Specific Antibodies
4.9. Intracellular Cytokines Immunofluorescence Assay
4.10. Statistical Analysis and Graphing
5. Conclusions
Acknowledgements
Conflicts of Interest
References
- Wolff, J.A.; Malone, RW.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar]
- Davis, B.S.; Chang, G.J.; Cropp, B.; Roehrig, J.T.; Martin, D.A.; Mitchell, C.J.; Bowen, R.; Bunning, M.L. West Nile virus recombinant DNA vaccine protects mouse and horse from virus challenge and expresses in vitro a noninfectious recombinant antigen that can be used in enzyme-linked immunosorbent assays. J. Virol. 2001, 75, 4040–4047. [Google Scholar] [CrossRef]
- Garver, K.A.; LaPatra, S.E.; Kurath, G. Efficacy of an infectious hematopoietic necrosis (IHN) virus DNA vaccine in Chinook Oncorhynchus tshawytscha and sockeye O. nerka salmon. Dis. Aquat. Org. 2005, 64, 13–22. [Google Scholar] [CrossRef]
- Grosenbaugh, D.A.; Leard, A.T.; Bergman, P.J.; Klein, M.K.; Meleo, K.; Susaneck, S.; Hess, P.R.; Jankowski, M.K.; Jones, P.D.; Leibman, N.F.; et al. Safety and efficacy of a xenogeneic DNA vaccine encoding for human tyrosinase as adjunctive treatment for oral malignant melanoma in dogs following surgical excision of the primary tumor. Am. J. Vet. Res. 2011, 72, 1631–1638. [Google Scholar] [CrossRef]
- Draghia-Akli, R.; Li, X.; Schwartz, R.J. Enhanced growth by ectopic expression of growth hormone releasing hormone using an injectable myogenic vector. Nat. Biotechnol. 1997, 15, 1285–1289. [Google Scholar] [CrossRef]
- Kopycinski, J.; Cheeseman, H.; Ashraf, A.; Gill, D.; Hayes, P.; Hannaman, D.; Gilmour, J.; Cox, J.H.; Vasan, S. A DNA-based candidate HIV vaccine delivered via in vivo electroporation induces CD4 responses toward the alpha4beta7-binding V2 loop of HIV gp120 in healthy volunteers. Clin. Vaccine Immunol. 2012, 19, 1557–1559. [Google Scholar] [CrossRef]
- Vasan, S.; Hurley, A.; Schlesinger, S.J.; Hannaman, D.; Gardiner, D.F.; Dugin, D.P.; Boente-Carrera, M.; Vittorino, R.; Caskey, M.; Andersen, J.; et al. In vivo electroporation enhances the immunogenicity of an HIV-1 DNA vaccine candidate in healthy volunteers. PLoS One 2011, 6, e19252. [Google Scholar] [CrossRef] [Green Version]
- Daud, A.I.; DeConti, R.C.; Andrews, S.; Urbas, P.; Riker, A.I.; Sondak, V.K.; Munster, P.N.; Sullivan, D.M.; Ugen, K.E.; Messina, J.L.; et al. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J. Clin. Oncol. 2008, 26, 5896–5903. [Google Scholar]
- Sardesai, N.Y.; Weiner, D.B. Electroporation delivery of DNA vaccines: Prospects for success. Curr. Opin. Immunol. 2011, 23, 421–429. [Google Scholar] [CrossRef]
- Ramanathan, M.P.; Kutzler, M.A.; Kuo, Y.C.; Yan, J.; Liu, H.; Shah, V.; Bawa, A.; Selling, B.; Sardesai, N.Y.; Kim, J.J.; et al. Coimmunization with an optimized IL15 plasmid adjuvant enhances humoral immunity via stimulating B cells induced by genetically engineered DNA vaccines expressing consensus JEV and WNV E DIII. Vaccine 2009, 27, 4370–4380. [Google Scholar] [CrossRef]
- Hirao, L.A.; Wu, L.; Khan, A.S.; Hokey, D.A.; Yan, J.; Dai, A.; Betts, M.R.; Draghia-Akli, R.; Weiner, D.B. Combined effects of IL-12 and electroporation enhances the potency of DNA vaccination in macaques. Vaccine 2008, 26, 3112–3120. [Google Scholar] [CrossRef]
- Hutnick, N.A.; Myles, D.J.; Bian, C.B.; Muthumani, K.; Weiner, D.B. Selected approaches for increasing HIV DNA vaccine immunogenicity in vivo. Curr. Opin. Virol. 2011, 1, 233–240. [Google Scholar] [CrossRef]
- Kutzler, M.A.; Kraynyak, K.A.; Nagle, S.J.; Parkinson, R.M.; Zharikova, D.; Chattergoon, M.; Maguire, H.; Muthumani, K.; Ugen, K.; Weiner, D.B. Plasmids encoding the mucosal chemokines CCL27 and CCL28 are effective adjuvants in eliciting antigen-specific immunity in vivo. Gene Ther. 2010, 17, 72–82. [Google Scholar] [CrossRef]
- Geall, A.J.; Mandl, C.W.; Ulmer, J.B. RNA: The new revolution in nucleic acid vaccines. Semin. Immunol. 2013, 25, 152–159. [Google Scholar] [CrossRef]
- Pascolo, S. Messenger RNA-based vaccines. Expert Opin. Biol. Ther. 2004, 4, 1285–1294. [Google Scholar] [CrossRef]
- Fleeton, M.N.; Chen, M.; Berglund, P.; Rhodes, G.; Parker, S.E.; Murphy, M.; Atkins, G.J.; Liljestrom, P. Self-replicative RNA vaccines elicit protection against influenza A virus, respiratory syncytial virus, and a tickborne encephalitis virus. J. Infect. Dis. 2001, 183, 1395–1398. [Google Scholar] [CrossRef]
- Johanning, F.W.; Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.A.; Pike, M.J.; Curiel, D.T. A Sindbis virus mRNA polynucleotide vector achieves prolonged and high level heterologous gene expression in vivo. Nucleic Acids Res. 1995, 23, 1495–1501. [Google Scholar] [CrossRef]
- Rayner, J.O.; Dryga, S.A.; Kamrud, K.I. Alphavirus vectors and vaccination. Rev. Med. Virol. 2002, 12, 279–296. [Google Scholar] [CrossRef]
- Smerdou, C.; Liljestrom, P. Non-viral amplification systems for gene transfer: Vectors based on alphaviruses. Curr. Opin. Mol. Ther. 1999, 1, 244–251. [Google Scholar]
- Johansson, D.X.; Ljungberg, K.; Kakoulidou, M.; Liljestrom, P. Intradermal electroporation of naked replicon RNA elicits strong immune responses. PLoS One 2012, 7, e29732. [Google Scholar]
- Carralot, J.P.; Probst, J.; Hoerr, I.; Scheel, B.; Teufel, R.; Jung, G.; Rammensee, H.G.; Pascolo, S. Polarization of immunity induced by direct injection of naked sequence-stabilized mRNA vaccines. Cell Mol. Life Sci. 2004, 61, 2418–2424. [Google Scholar]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magne, R.; Gomard, E.; Guillet, J.G.; Levy, J.P.; Meulien, P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef]
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T.; et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA 2012, 109, 14604–14609. [Google Scholar] [CrossRef]
- Petsch, B.; Schnee, M.; Vogel, A.B.; Lange, E.; Hoffmann, B.; Voss, D.; Schlake, T.; Thess, A.; Kallen, K.J.; Stitz, L.; et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 2012, 30, 1210–1216. [Google Scholar] [CrossRef]
- Mandl, C.W.; Aberle, J.H.; Aberle, S.W.; Holzmann, H.; Allison, S.L.; Heinz, F.X. In vitro-synthesized infectious RNA as an attenuated live vaccine in a flavivirus model. Nat. Med. 1998, 4, 1438–1340. [Google Scholar] [CrossRef]
- Aberle, J.H.; Aberle, S.W.; Kofler, R.M.; Mandl, C.W. Humoral and cellular immune response to RNA immunization with flavivirus replicons derived from tick-borne encephalitis virus. J. Virol. 2005, 79, 15107–15113. [Google Scholar]
- Kofler, R.M.; Aberle, J.H.; Aberle, S.W.; Allison, S.L.; Heinz, F.X.; Mandl, C.W. Mimicking live flavivirus immunization with a noninfectious RNA vaccine. Proc. Natl. Acad. Sci. USA 2004, 101, 1951–1956. [Google Scholar]
- Piggott, J.M.; Sheahan, B.J.; Soden, D.M.; O’Sullivan, G.C.; Atkins, G.J. Electroporation of RNA stimulates immunity to an encoded reporter gene in mice. Mol. Med. Report 2009, 2, 753–756. [Google Scholar]
- Perri, S.; Greer, C.E.; Thudium, K.; Doe, B.; Legg, H.; Liu, H.; Romero, R.E.; Tang, Z.; Bin, Q.; Dubensky, T.W.; et al. An alphavirus replicon particle chimera derived from venezuelan equine encephalitis and sindbis viruses is a potent gene-based vaccine delivery vector. J. Virol. 2003, 77, 10394–10403. [Google Scholar] [CrossRef]
- Atkins, G.J.; Fleeton, M.N.; Sheahan, B.J. Therapeutic and prophylactic applications of alphavirus vectors. Expert Rev. Mol. Med. 2008, 10, e33. [Google Scholar]
- Barnett, S.W.; Burke, B.; Sun, Y.; Kan, E.; Legg, H.; Lian, Y.; Bost, K.; Zhou, F.; Goodsell, A.; Megede, J.Z.; et al. Antibody-mediated protection against mucosal simian-human immunodeficiency virus challenge of macaques immunized with alphavirus replicon particles and boosted with trimeric envelope glycoprotein in MF59 adjuvant. J. Virol. 2010, 84, 5975–5985. [Google Scholar] [CrossRef]
- Robert-Guroff, M. Replicating and non-replicating viral vectors for vaccine development. Curr. Opin. Biotechnol. 2007, 18, 546–556. [Google Scholar] [CrossRef]
- Zimmer, G. RNA replicons—A new approach for influenza virus immunoprophylaxis. Viruses 2010, 2, 413–434. [Google Scholar] [CrossRef]
- Gronevik, E.; von Steyern, F.V.; Kalhovde, J.M.; Tjelle, T.E.; Mathiesen, I. Gene expression and immune response kinetics using electroporation-mediated DNA delivery to muscle. J. Gene Med. 2005, 7, 218–227. [Google Scholar]
- Lin, F.; Shen, X.; McCoy, J.R.; Mendoza, J.M.; Yan, J.; Kemmerrer, S.V.; Khan, A.S.; Weiner, D.B.; Broderick, K.E.; Sardesai, N.Y. A novel prototype device for electroporation-enhanced DNA vaccine delivery simultaneously to both skin and muscle. Vaccine 2011, 29, 6771–6780. [Google Scholar] [CrossRef]
- Bagarazzi, M.L.; Yan, J.; Morrow, M.P.; Shen, X.; Parker, R.L.; Lee, J.C.; Giffear, M.; Pankhong, P.; Khan, A.S.; Broderick, K.E. Immunotherapy against HPV16/18 generates potent TH1 and cytotoxic cellular immune responses. Sci. Transl. Med. 2012. [Google Scholar] [CrossRef]
- Chiarella, P.; Massi, E.; de Robertis, M.; Sibilio, A.; Parrella, P.; Fazio, V.M.; Signori, E. Electroporation of skeletal muscle induces danger signal release and antigen-presenting cell recruitment independently of DNA vaccine administration. Expert Opin. Biol. Ther. 2008, 8, 1645–1657. [Google Scholar] [CrossRef]
- Liu, J.; Kjeken, R.; Mathiesen, I.; Barouch, D.H. Recruitment of antigen-presenting cells to the site of inoculation and augmentation of human immunodeficiency virus type 1 DNA vaccine immunogenicity by in vivo electroporation. J. Virol. 2008, 82, 5643–5649. [Google Scholar] [CrossRef]
- Dupuis, M.; Denis-Mize, K.; Woo, C.; Goldbeck, C.; Selby, M.J.; Chen, M.; Otten, G.R.; Ulmer, J.B.; Donnelly, J.J.; Ott, G. Distribution of DNA vaccines determines their immunogenicity after intramuscular injection in mice. J. Immunol. 2000, 165, 2850–2858. [Google Scholar]
- Mir, L.M.; Bureau, M.F.; Rangara, R.; Schwartz, B.; Scherman, D. Long-term, high level in vivo gene expression after electric pulse-mediated gene transfer into skeletal muscle. Comptes Rendus de l'Académie des Sciences—Series III—Sciences de la Vie 1998, 321, 893–899. [Google Scholar] [CrossRef]
- Barber, G.N. Cytoplasmic DNA innate immune pathways. Immunol. Rev. 2011, 243, 99–108. [Google Scholar] [CrossRef]
- Hornung, V.; Barchet, W.; Schlee, M.; Hartmann, G. RNA recognition via TLR7 and TLR8. Handb. Exp. Pharmacol. 2008, 183, 71–86. [Google Scholar] [CrossRef]
- Jensen, S.; Thomsen, A.R. Sensing of RNA viruses: A review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 2012, 86, 2900–2910. [Google Scholar] [CrossRef]
- Broderick, K.E.; Shen, X.; Soderholm, J.; Lin, F.; McCoy, J.; Khan, A.S.; Yan, J.; Morrow, M.P.; Patel, A.; Kobinger, G.P. Prototype development and preclinical immunogenicity analysis of a novel minimally invasive electroporation device. Gene Ther. 2011, 18, 258–265. [Google Scholar] [CrossRef]
- Scheerlinck, J.P.; Kalrlis, J.; Tjelle, T.E.; Presidente, P.J.; Mathiesen, I.; Newton, S.E. Electroporation improves immune responses to DNA vaccination in sheep. Vaccine 2004, 22, 1820–1825. [Google Scholar] [CrossRef]
- Widera, G.; Austin, M.; Rabussay, D.; Goldbeck, C.; Barnett, S.W.; Chen, M.; Leung, L.; Otten, G.R.; Thudium, K.; Selby, M.J.; et al. Increased DNA vaccine delivery and immunogenicity by electroporation in vivo. J. Immunol. 2000, 164, 4635–4640. [Google Scholar]
- Broderick, K.E.; Chan, A.; Lin, F.; Shen, X.; Kichaev, G.; Khan, A.S.; Aubin, J.; Zimmermann, T.S.; Sardesai, N.Y. Optimized in vivo transfer of small interfering rna targeting dermal tissue using in vivo surface electroporation. Mol. Ther. Nucleic Acids 2012, 1, e11. [Google Scholar] [CrossRef]
- Barnett, S.W.; Lu, S.; Srivastava, I.; Cherpelis, S.; Gettie, A.; Blanchard, J.; Wang, S.; Mboudjeka, I.; Leung, L.; Lian, Y.; et al. The ability of an oligomeric human immunodeficiency virus type 1 (HIV-1) envelope antigen to elicit neutralizing antibodies against primary HIV-1 isolates is improved following partial deletion of the second hypervariable region. J. Virol. 2001, 75, 5526–5540. [Google Scholar] [CrossRef]
- Srivastava, I.K.; Stamatatos, L.; Kan, E.; Vajdy, M.; Lian, Y.; Hilt, S.; Martin, L.; Vita, C.; Zhu, P.; et al. Purification, characterization, and immunogenicity of a soluble trimeric envelope protein containing a partial deletion of the V2 loop derived from SF162, an R5-tropic human immunodeficiency virus type 1 isolate. J. Virol. 2003, 77, 11244–11259. [Google Scholar] [CrossRef]
- Snedecor, G.W.; Cochran, W.G. Statistical Methods, 7th ed.; The Iowa State University Press: Ames, Iowa, IA, USA, 1980. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cu, Y.; Broderick, K.E.; Banerjee, K.; Hickman, J.; Otten, G.; Barnett, S.; Kichaev, G.; Sardesai, N.Y.; Ulmer, J.B.; Geall, A. Enhanced Delivery and Potency of Self-Amplifying mRNA Vaccines by Electroporation in Situ . Vaccines 2013, 1, 367-383. https://doi.org/10.3390/vaccines1030367
Cu Y, Broderick KE, Banerjee K, Hickman J, Otten G, Barnett S, Kichaev G, Sardesai NY, Ulmer JB, Geall A. Enhanced Delivery and Potency of Self-Amplifying mRNA Vaccines by Electroporation in Situ . Vaccines. 2013; 1(3):367-383. https://doi.org/10.3390/vaccines1030367
Chicago/Turabian StyleCu, Yen, Kate E. Broderick, Kaustuv Banerjee, Julie Hickman, Gillis Otten, Susan Barnett, Gleb Kichaev, Niranjan Y. Sardesai, Jeffrey B. Ulmer, and Andrew Geall. 2013. "Enhanced Delivery and Potency of Self-Amplifying mRNA Vaccines by Electroporation in Situ " Vaccines 1, no. 3: 367-383. https://doi.org/10.3390/vaccines1030367