Vector Design for Improved DNA Vaccine Efficacy, Safety and Production

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
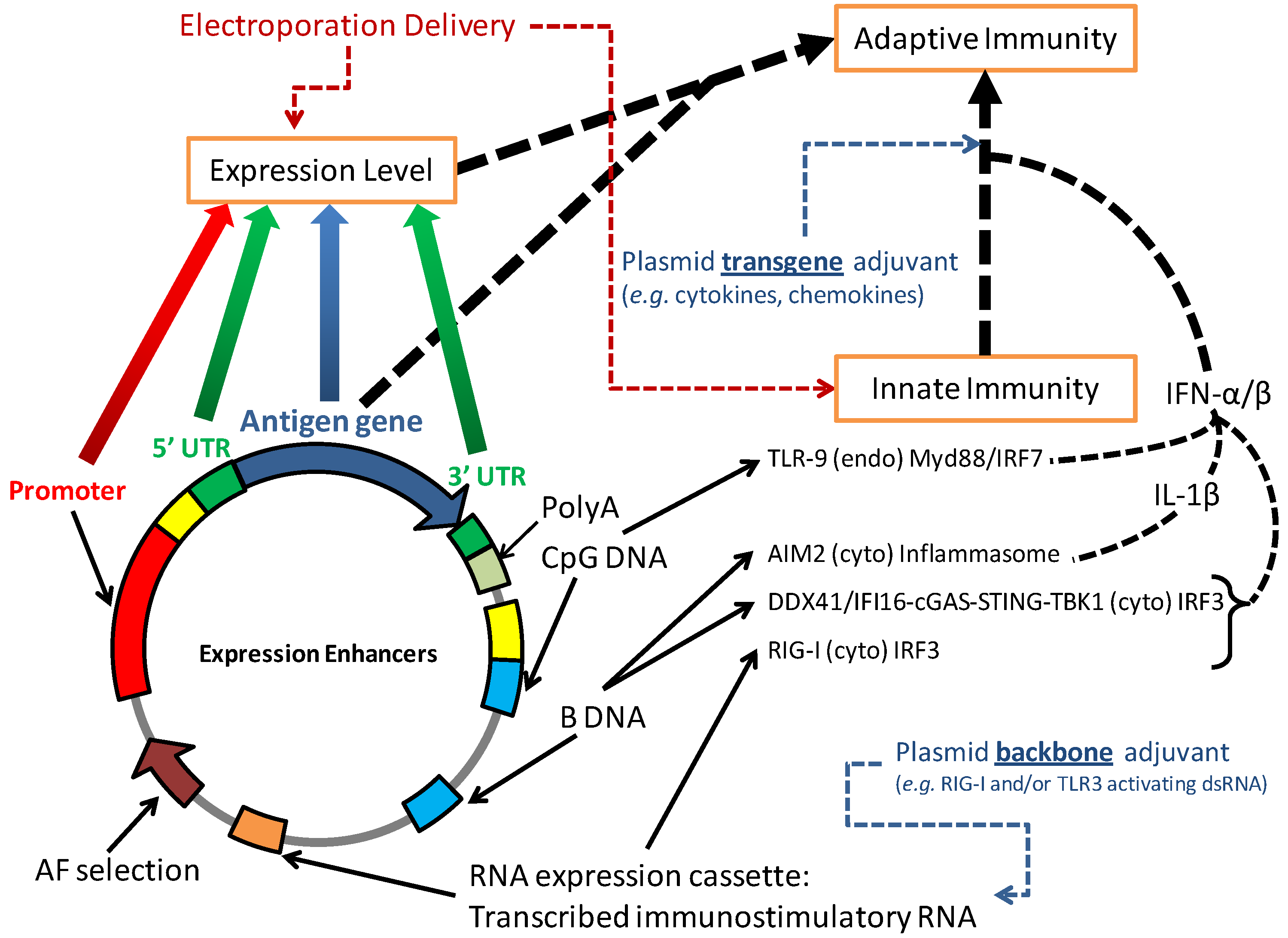{kind=link}
1. Introduction
2. Plasmid Design
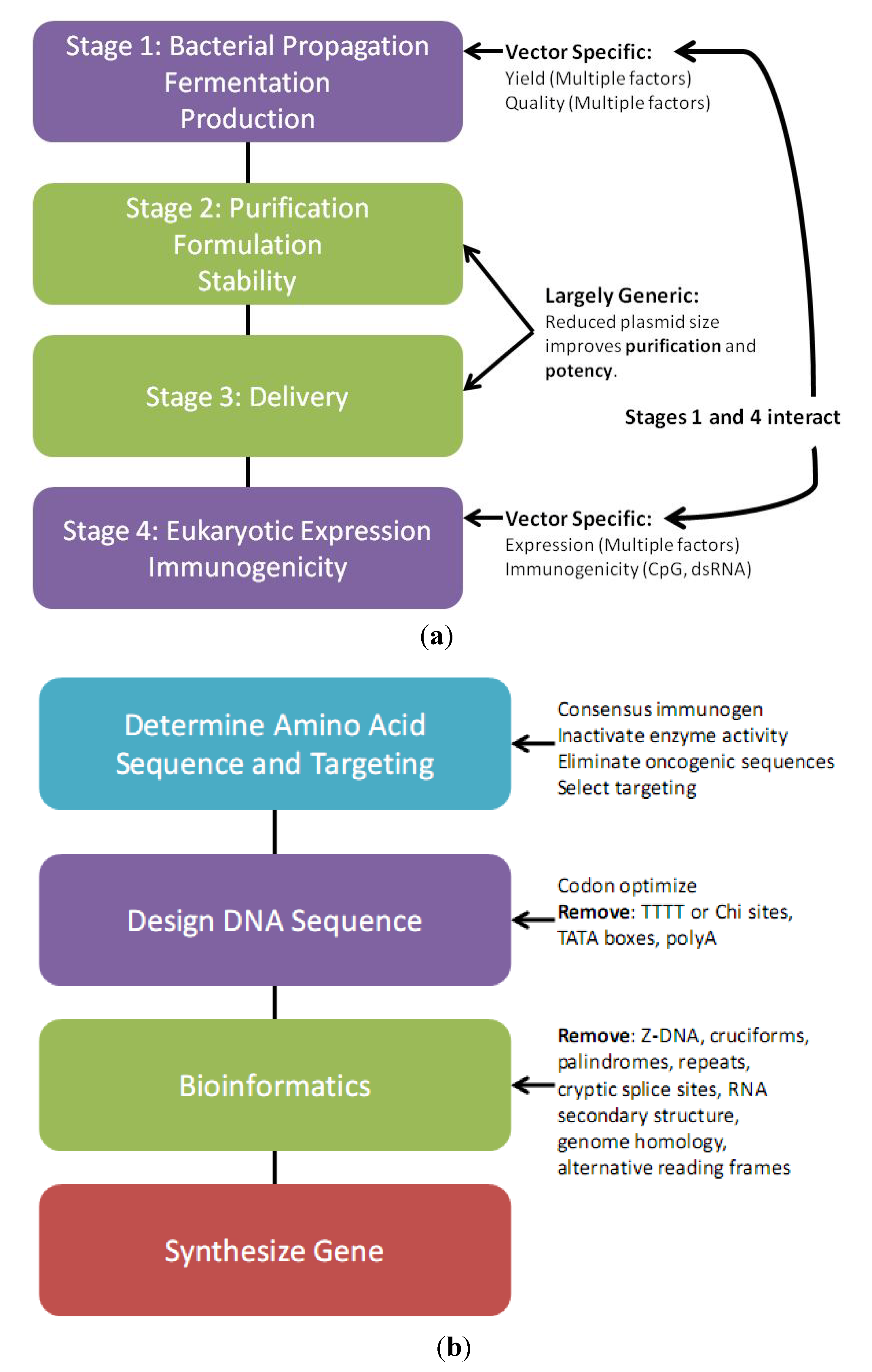
2.1. Vector Design Considerations
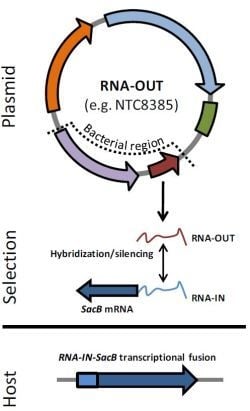
2.2. Antibiotic-Free Selection Using RNA Selection Markers

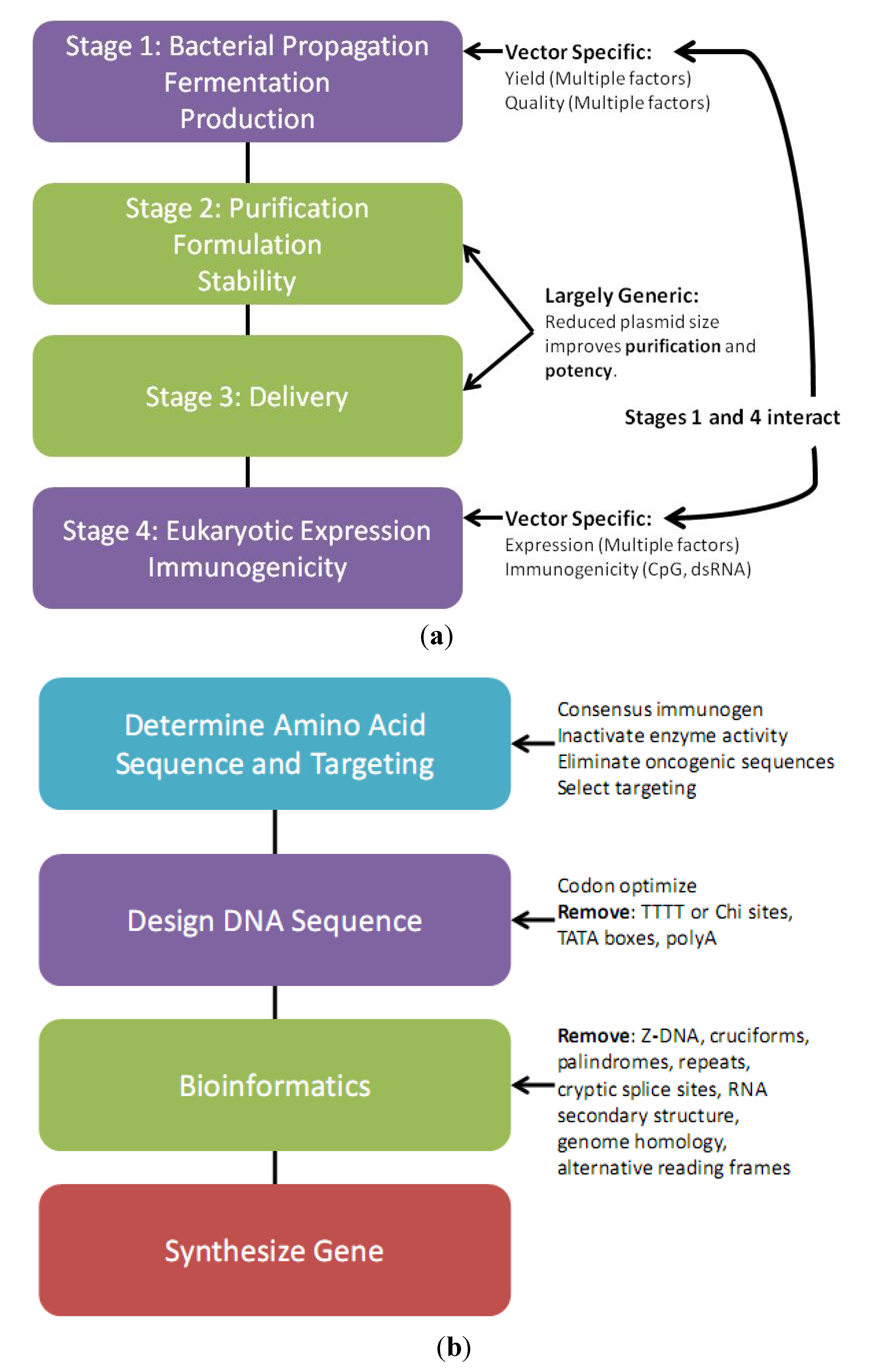
2.3. Transgene Design Considerations
3. Plasmid Manufacture
3.1. Plasmid Fermentation
3.2. Downstream Plasmid Purification
3.3. Plasmid Quality Control Considerations
3.4. Plasmid Host Strain and Growth Conditions Affect DNA Vaccine Performance
4. DNA Vaccine Immunology
4.1. DNA Vaccination Activates Innate Immunity
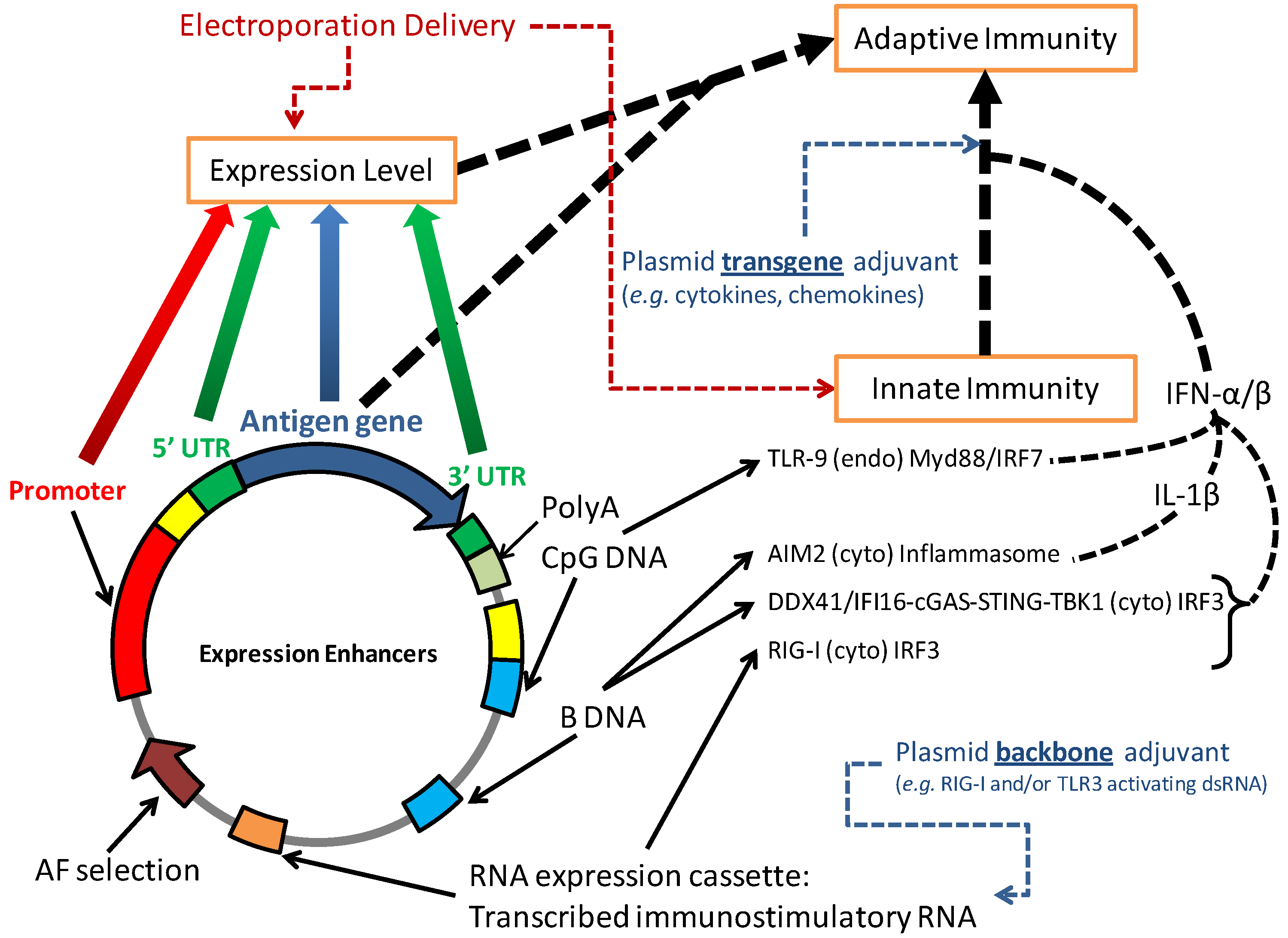
4.2. Vector Modifications to Increase Innate Immunity
5. New Developments
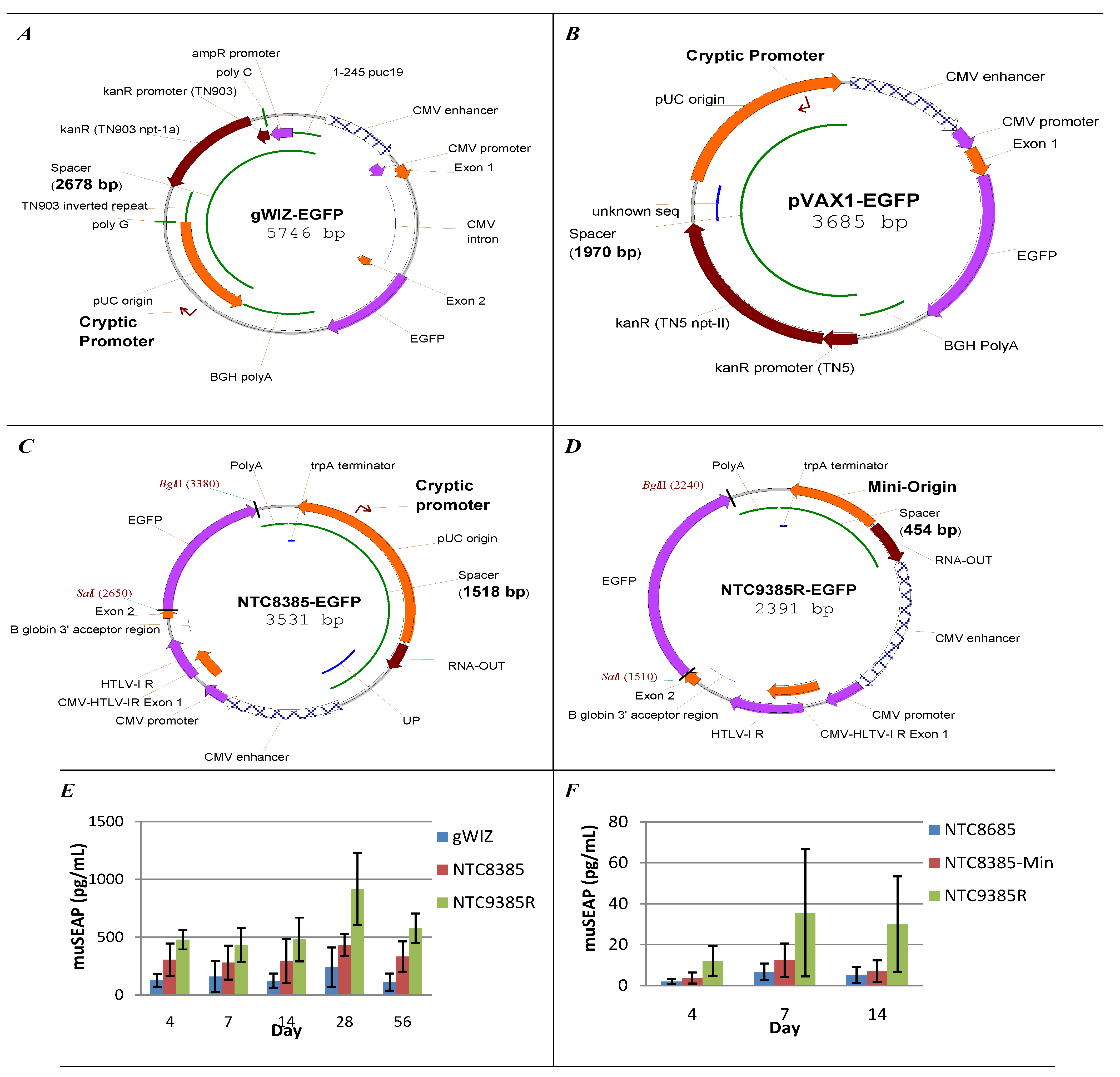
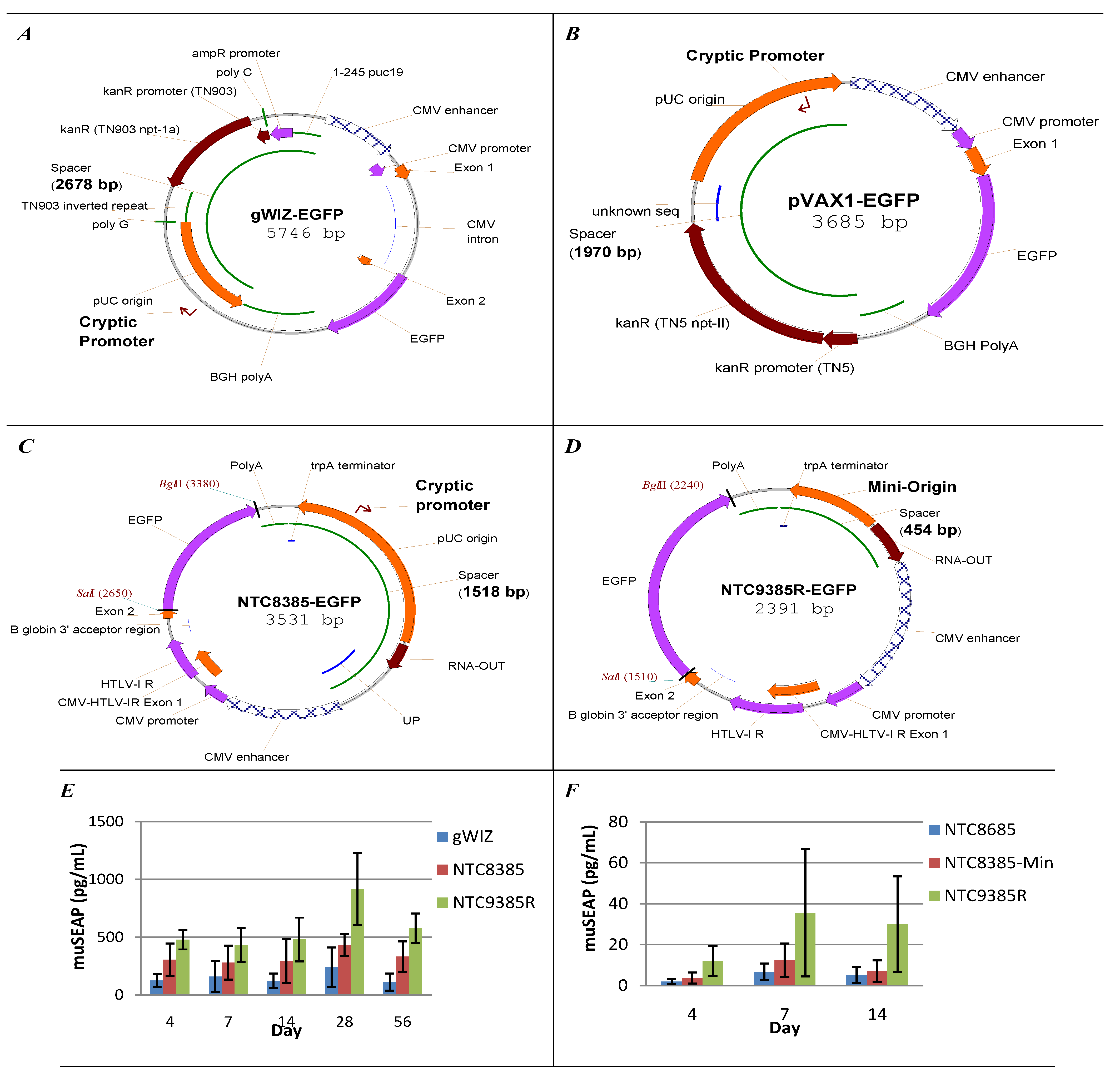
5.1. Vector Bacterial Region Inhibits Plasmid Expression
5.2. Minimal Backbone Vectors Dramatically Improve Plasmid Expression
6. Conclusions
Acknowledgments
Conflict of Interest
References
- Liu, M.A. DNA vaccines: An historical perspective and view to the future. Immunol. Rev. 2011, 239, 62–84. [Google Scholar] [CrossRef]
- Kutzler, M.A.; Weiner, D.B. DNA vaccines: Ready for prime time? Nat. Rev. Genet. 2008, 9, 776–788. [Google Scholar] [CrossRef]
- Lu, S.; Wang, S.; Grimes-Serrano, J.M. Current progress of DNA vaccine studies in humans. Expert Rev. Vaccines 2008, 7, 175–191. [Google Scholar] [CrossRef]
- Premenko-Lanier, M.; Rota, P.A.; Rhodes, G.H.; Bellini, W.J.; McChesney, M.B. Protection against challenge with measles virus (mv) in infant macaques by an mv DNA vaccine administered in the presence of neutralizing antibody. J. Infect. Dis. 2004, 189, 2064–2071. [Google Scholar] [CrossRef]
- Wang, S.; Parker, C.; Taaffe, J.; Solorzano, A.; Garcia-Sastre, A.; Lu, S. Heterologous HA DNA vaccine prime-inactivated influenza vaccine boost is more effective than using DNA or inactivated vaccine alone in eliciting antibody responses against H1 or H3 serotype influenza viruses. Vaccine 2008, 26, 3626–3633. [Google Scholar] [CrossRef]
- Wei, C.J.; Boyington, J.C.; McTamney, P.M.; Kong, W.P.; Pearce, M.B.; Xu, L.; Andersen, H.; Rao, S.; Tumpey, T.M.; Yang, Z.Y.; et al. Induction of broadly neutralizing H1N1 influenza antibodies by vaccination. Science 2010, 329, 1060–1064. [Google Scholar] [CrossRef]
- Ledgerwood, J.E.; Wei, C.J.; Hu, Z.; Gordon, I.J.; Enama, M.E.; Hendel, C.S.; McTamney, P.M.; Pearce, M.B.; Yassine, H.M.; Boyington, J.C.; et al. DNA priming and influenza vaccine immunogenicity: Two phase 1 open label randomised clinical trials. Lancet Infect. Dis. 2011, 11, 916–924. [Google Scholar] [CrossRef]
- Sardesai, N.Y.; Weiner, D.B. Electroporation delivery of DNA vaccines: Prospects for success. Curr. Opin. Immunol. 2011, 23, 421–429. [Google Scholar] [CrossRef]
- Ault, A.; Zajac, A.M.; Kong, W.P.; Gorres, J.P.; Royals, M.; Wei, C.J.; Bao, S.; Yang, Z.Y.; Reedy, S.E.; Sturgill, T.L.; et al. Immunogenicity and clinical protection against equine influenza by DNA vaccination of ponies. Vaccine 2012, 30, 3965–3974. [Google Scholar] [CrossRef]
- Gorres, J.P.; Lager, K.M.; Kong, W.P.; Royals, M.; Todd, J.P.; Vincent, A.L.; Wei, C.J.; Loving, C.L.; Zanella, E.L.; Janke, B.; et al. DNA vaccination elicits protective immune responses against pandemic and classic swine influenza viruses in pigs. Clin. Vaccine Immunol. 2011, 18, 1987–1995. [Google Scholar] [CrossRef]
- Sullivan, S.M.; Doukas, J.; Hartikka, J.; Smith, L.; Rolland, A. Vaxfectin: A versatile adjuvant for plasmid DNA- and protein-based vaccines. Expert Opin. Drug Deliv. 2010, 7, 1433–1446. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, Y.; Wang, X.; Huang, J.; Shang, J.; Sun, S. Human serum amyloid p functions as a negative regulator of the innate and adaptive immune responses to DNA vaccines. J. Immunol. 2011, 186, 2860–2870. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, Y.; Wang, X.; Huang, J.; Shang, J.; Sun, S. Serum amyloid p component facilitates DNA clearance and inhibits plasmid transfection: Implications for human DNA vaccine. Gene Ther. 2012, 19, 70–77. [Google Scholar] [CrossRef]
- Rosazza, C.; Escoffre, J.M.; Zumbusch, A.; Rols, M.P. The actin cytoskeleton has an active role in the electrotransfer of plasmid DNA in mammalian cells. Mol. Ther. 2011, 19, 913–921. [Google Scholar] [CrossRef]
- Lam, A.P.; Dean, D.A. Progress and prospects: Nuclear import of nonviral vectors. Gene Ther. 2010, 17, 439–447. [Google Scholar] [CrossRef]
- Li, C.; Goudy, K.; Hirsch, M.; Asokan, A.; Fan, Y.; Alexander, J.; Sun, J.; Monahan, P.; Seiber, D.; Sidney, J.; et al. Cellular immune response to cryptic epitopes during therapeutic gene transfer. Proc. Natl. Acad. Sci. USA 2009, 106, 10770–10774. [Google Scholar] [CrossRef]
- Hartikka, J.; Sawdey, M.; Cornefert-Jensen, F.; Margalith, M.; Barnhart, K.; Nolasco, M.; Vahlsing, H.L.; Meek, J.; Marquet, M.; Hobart, P.; et al. An improved plasmid DNA expression vector for direct injection into skeletal muscle. Hum. Gene Ther. 1996, 7, 1205–1217. [Google Scholar] [CrossRef]
- Williams, J.A.; Luke, J.; Johnson, L.; Hodgson, C. pDNAVACCultra vector family: High throughput intracellular targeting DNA vaccine plasmids. Vaccine 2006, 24, 4671–4676. [Google Scholar] [CrossRef]
- Lemp, N.A.; Hiraoka, K.; Kasahara, N.; Logg, C.R. Cryptic transcripts from a ubiquitous plasmid origin of replication confound tests for cis-regulatory function. Nucleic Acids Res. 2012, 40, 7280–7290. [Google Scholar] [CrossRef]
- Nejepinska, J.; Malik, R.; Moravec, M.; Svoboda, P. Deep sequencing reveals complex spurious transcription from transiently transfected plasmids. PLoS One 2012, 7, e43283. [Google Scholar] [CrossRef]
- Carnes, A.E.; Luke, J.M.; Vincent, J.M.; Anderson, S.; Schukar, A.; Hodgson, C.P.; Williams, J.A. Critical design criteria for minimal antibiotic-free plasmid vectors necessary to combine robust rna pol II and pol III-mediated eukaryotic expression with high bacterial production yields. J. Gene Med. 2010, 12, 818–831. [Google Scholar] [CrossRef]
- Williams, J.A.; Carnes, A.E.; Hodgson, C.P. Plasmid DNA vaccine vector design: Impact on efficacy, safety and upstream production. Biotechnol. Adv. 2009, 27, 353–370. [Google Scholar] [CrossRef]
- Mairhofer, J.; Grabherr, R. Rational vector design for efficient non-viral gene delivery: Challenges facing the use of plasmid DNA. Mol. Biotechnol. 2008, 39, 97–104. [Google Scholar] [CrossRef]
- Gill, D.R.; Pringle, I.A.; Hyde, S.C. Progress and prospects: The design and production of plasmid vectors. Gene Ther. 2009, 16, 165–171. [Google Scholar] [CrossRef]
- USA Food and Drug Administration. Guidance for Industry: Considerations for Plasmid DNA Vaccines for Infectious Disease Indications; Rockville, MD, USA, 2007. [Google Scholar]
- European Medicines Agency. Note for Guidance on the Quality, Preclinical and Clinical Aspects of Gene Transfer Medicinal Products; London, UK, 2001; CPMP/BWP/3088/99. [Google Scholar]
- European Medicines Agency. Presence of the Antibiotic Resistance Marker Gene nptII in GM Plants and Food and Feed Uses; London, UK, 2007; EMEA/CVMP/56937/2007. [Google Scholar]
- Luke, J.M.; Vincent, J.M.; Du, S.X.; Gerdemann, U.; Leen, A.M.; Whalen, R.G.; Hodgson, C.P.; Williams, J.A. Improved antibiotic-free plasmid vector design by incorporation of transient expression enhancers. Gene Ther. 2011, 18, 334–343. [Google Scholar] [CrossRef]
- Schirmbeck, R.; Riedl, P.; Fissolo, N.; Lemonnier, F.A.; Bertoletti, A.; Reimann, J. Translation from cryptic reading frames of DNA vaccines generates an extended repertoire of immunogenic, mhc class i-restricted epitopes. J. Immunol. 2005, 174, 4647–4656. [Google Scholar]
- Liu, H.X.; Zhang, M.; Krainer, A.R. Identification of functional exonic splicing enhancer motifs recognized by individual SR proteins. Genes Dev. 1998, 12, 1998–2012. [Google Scholar] [CrossRef]
- Fairbrother, W.G.; Yeh, R.F.; Sharp, P.A.; Burge, C.B. Predictive identification of exonic splicing enhancers in human genes. Science 2002, 297, 1007–1013. [Google Scholar]
- Wang, Y.; Ma, M.; Xiao, X.; Wang, Z. Intronic splicing enhancers, cognate splicing factors and context-dependent regulation rules. Nat. Struct. Mol. Biol. 2012, 19, 1044–1052. [Google Scholar] [CrossRef]
- Barouch, D.H.; Yang, Z.Y.; Kong, W.P.; Korioth-Schmitz, B.; Sumida, S.M.; Truitt, D.M.; Kishko, M.G.; Arthur, J.C.; Miura, A.; Mascola, J.R.; et al. A human T-cell leukemia virus type 1 regulatory element enhances the immunogenicity of human immunodeficiency virus type 1 DNA vaccines in mice and nonhuman primates. J. Virol. 2005, 79, 8828–8834. [Google Scholar] [CrossRef]
- Takahashi, Y.; Nishikawa, M.; Takiguchi, N.; Suehara, T.; Takakura, Y. Saturation of transgene protein synthesis from mRNA in cells producing a large number of transgene mRNA. Biotechnol. Bioeng. 2011, 108, 2380–2389. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, P.H.; Prather, K.J.; Prazeres, D.M.; Monteiro, G.A. Structural instability of plasmid biopharmaceuticals: Challenges and implications. Trends Biotechnol. 2009, 27, 503–511. [Google Scholar] [CrossRef]
- Luke, J.M.; Carnes, A.E.; Hodgson, C.P.; Williams, J.A. Vector insert-targeted integrative antisense expression system for plasmid stabilization. Mol. Biotechnol. 2011, 47, 43–49. [Google Scholar] [CrossRef]
- Oliveira, P.H.; Prather, K.L.; Prazeres, D.M.; Monteiro, G.A. Mutation detection in plasmid-based biopharmaceuticals. Biotechnol. J. 2011, 6, 378–391. [Google Scholar] [CrossRef]
- Chen, J.; Jin, M.; Qiu, Z.G.; Guo, C.; Chen, Z.L.; Shen, Z.Q.; Wang, X.W.; Li, J.W. A survey of drug resistance bla genes originating from synthetic plasmid vectors in six chinese rivers. Environ. Sci. Technol. 2012, 46, 13448–13454. [Google Scholar] [CrossRef]
- European Directorate for the Quality of Medicines (EDQM), Gene Transfer Medical Products for Human Use. In European Pharmacopoeia, 7.0 ed.; Council of Europe: Strasbourg, France, 2011; p. 648.
- Vandermeulen, G.; Marie, C.; Scherman, D.; Preat, V. New generation of plasmid backbones devoid of antibiotic resistance marker for gene therapy trials. Mol. Ther. 2011, 19, 1942–1949. [Google Scholar] [CrossRef]
- Luke, J.; Carnes, A.E.; Hodgson, C.P.; Williams, J.A. Improved antibiotic-free DNA vaccine vectors utilizing a novel rna based plasmid selection system. Vaccine 2009, 27, 6454–6459. [Google Scholar] [CrossRef]
- Soubrier, F.; Cameron, B.; Manse, B.; Somarriba, S.; Dubertret, C.; Jaslin, G.; Jung, G.; Caer, C.L.; Dang, D.; Mouvault, J.M.; et al. Pcor: A new design of plasmid vectors for nonviral gene therapy. Gene Ther. 1999, 6, 1482–1488. [Google Scholar] [CrossRef]
- Marie, C.; Vandermeulen, G.; Quiviger, M.; Richard, M.; Preat, V.; Scherman, D. pFARs, plasmids free of antibiotic resistance markers, display high-level transgene expression in muscle, skin and tumour cells. J. Gene Med. 2010, 12, 323–332. [Google Scholar] [CrossRef]
- Mairhofer, J.; Pfaffenzeller, I.; Merz, D.; Grabherr, R. A novel antibiotic free plasmid selection system: Advances in safe and efficient DNA therapy. Biotechnol. J. 2008, 3, 83–89. [Google Scholar] [CrossRef]
- Cranenburgh, R.M. Plasmid Maintenance. US Patent 7611883, 3 November 2009. [Google Scholar]
- Nelson, C.A.; Cai, Y.; Rodriguez, S.; Finlayson, N.; Williams, J.; Carnes, A.E. Antibiotic-free production of a herpes simplex virus 2 DNA vaccine in a high yield cGMP process. Hum. Vaccin. Immunother. 2013, 9. [Google Scholar]
- Mairhofer, J.; Cserjan-Puschmann, M.; Striedner, G.; Nobauer, K.; Razzazi-Fazeli, E.; Grabherr, R. Marker-free plasmids for gene therapeutic applications-lack of antibiotic resistance gene substantially improves the manufacturing process. J. Biotechnol. 2010, 146, 130–137. [Google Scholar] [CrossRef]
- Oliveira, P.H.; Mairhofer, J. Marker-free plasmids for biotechnological applications—implications and perspectives. Trends Biotechnol. 2013. [Google Scholar] [CrossRef]
- Dormitzer, P.R.; Ulmer, J.B.; Rappuoli, R. Structure-based antigen design: A strategy for next generation vaccines. Trends Biotechnol. 2008, 26, 659–667. [Google Scholar] [CrossRef]
- Laddy, D.J.; Yan, J.; Khan, A.S.; Andersen, H.; Cohn, A.; Greenhouse, J.; Lewis, M.; Manischewitz, J.; King, L.R.; Golding, H.; et al. Electroporation of synthetic DNA antigens offers protection in nonhuman primates challenged with highly pathogenic avian influenza virus. J. Virol. 2009, 83, 4624–4630. [Google Scholar] [CrossRef]
- Dupuy, L.C.; Locher, C.P.; Paidhungat, M.; Richards, M.J.; Lind, C.M.; Bakken, R.; Parker, M.D.; Whalen, R.G.; Schmaljohn, C.S. Directed molecular evolution improves the immunogenicity and protective efficacy of a venezuelan equine encephalitis virus DNA vaccine. Vaccine 2009, 27, 4152–4160. [Google Scholar] [CrossRef]
- Li, Z.; Howard, A.; Kelley, C.; Delogu, G.; Collins, F.; Morris, S. Immunogenicity of DNA vaccines expressing tuberculosis proteins fused to tissue plasminogen activator signal sequences. Infect. Immun. 1999, 67, 4780–4786. [Google Scholar]
- Wang, S.; Farfan-Arribas, D.J.; Shen, S.; Chou, T.H.; Hirsch, A.; He, F.; Lu, S. Relative contributions of codon usage, promoter efficiency and leader sequence to the antigen expression and immunogenicity of HIV-1 env DNA vaccine. Vaccine 2006, 24, 4531–4540. [Google Scholar] [CrossRef]
- Wang, S.; Hackett, A.; Jia, N.; Zhang, C.; Zhang, L.; Parker, C.; Zhou, A.; Li, J.; Cao, W.C.; Huang, Z.; et al. Polyvalent DNA vaccines expressing HA antigens of H5N1 influenza viruses with an optimized leader sequence elicit cross-protective antibody responses. PLoS One 2011, 6, e28757. [Google Scholar] [CrossRef]
- Rodriguez, F.; An, L.L.; Harkins, S.; Zhang, J.; Yokoyama, M.; Widera, G.; Fuller, J.T.; Kincaid, C.; Campbell, I.L.; Whitton, J.L. DNA immunization with minigenes: Low frequency of memory cytotoxic T lymphocytes and inefficient antiviral protection are rectified by ubiquitination. J. Virol. 1998, 72, 5174–5181. [Google Scholar]
- Wu, T.C.; Guarnieri, F.G.; Staveley-O’Carroll, K.F.; Viscidi, R.P.; Levitsky, H.I.; Hedrick, L.; Cho, K.R.; August, J.T.; Pardoll, D.M. Engineering an intracellular pathway for major histocompatibility complex class II presentation of antigens. Proc. Natl. Acad. Sci. USA 1995, 92, 11671–11675. [Google Scholar] [CrossRef]
- Rice, J.; Ottensmeier, C.H.; Stevenson, F.K. DNA vaccines: Precision tools for activating effective immunity against cancer. Nat. Rev. Cancer 2008, 8, 108–120. [Google Scholar] [CrossRef]
- Fath, S.; Bauer, A.P.; Liss, M.; Spriestersbach, A.; Maertens, B.; Hahn, P.; Ludwig, C.; Schafer, F.; Graf, M.; Wagner, R. Multiparameter RNA and codon optimization: A standardized tool to assess and enhance autologous mammalian gene expression. PLoS One 2011, 6, e17596. [Google Scholar] [CrossRef] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Wang, Z.; Troilo, P.J.; Wang, X.; Griffiths, T.G.; Pacchione, S.J.; Barnum, A.B.; Harper, L.B.; Pauley, C.J.; Niu, Z.; Denisova, L.; et al. Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther. 2004, 11, 711–721. [Google Scholar] [CrossRef]
- Carnes, A.E.; Williams, J.A. Plasmid DNA manufacturing technology. Recent Pat. Biotechnol. 2007, 1, 151–166. [Google Scholar] [CrossRef]
- Carnes, A.E.; Williams, J.A. Process for plasmid DNA fermentation. US Patent 7943377, 17 May 2011. [Google Scholar]
- Carnes, A.E.; Luke, J.M.; Vincent, J.M.; Schukar, A.; Anderson, S.; Hodgson, C.P.; Williams, J.A. Plasmid DNA fermentation strain and process-specific effects on vector yield, quality, and transgene expression. Biotechnol. Bioeng. 2011, 108, 354–363. [Google Scholar] [CrossRef]
- Urthaler, J.; Schuchnigg, H.; Garidel, P.; Huber, H. Industrial Manufacturing of Plasmid-DNA Products for Gene Vaccination and Therapy. In Gene Vaccines; Thalhamer, J., Weiss, R., Scheiblhofer, S., Eds.; SpringerWienNewYork: New York, NY, USA, 2012; Volume 3, pp. 311–330. [Google Scholar]
- Hoare, M.; Levy, M.S.; Bracewell, D.G.; Doig, S.D.; Kong, S.; Titchener-Hooker, N.; Ward, J.M.; Dunnill, P. Bioprocess engineering issues that would be faced in producing a DNA vaccine at up to 100 M3 fermentation scale for an influenza pandemic. Biotechnol. Prog. 2005, 21, 1577–1592. [Google Scholar] [CrossRef]
- Cai, Y.; Rodriguez, S.; Hebel, H. DNA vaccine manufacture: Scale and quality. Expert Rev. Vaccines 2009, 8, 1277–1291. [Google Scholar] [CrossRef]
- Sousa, A.; Sousa, F.; Queiroz, J.A. Advances in chromatographic supports for pharmaceutical-grade plasmid DNA purification. J. Sep. Sci. 2012, 35, 3046–3058. [Google Scholar] [CrossRef]
- Ghanem, A.; Healey, R.; Adly, F.G. Current trends in separation of plasmid DNA vaccines: A review. Anal. Chim. Acta 2013, 760, 1–15. [Google Scholar] [CrossRef]
- Wooddell, C.I.; Subbotin, V.M.; Sebestyen, M.G.; Griffin, J.B.; Zhang, G.; Schleef, M.; Braun, S.; Huss, T.; Wolff, J.A. Muscle damage after delivery of naked plasmid DNA into skeletal muscles is batch dependent. Hum. Gene Ther. 2011, 22, 225–235. [Google Scholar] [CrossRef]
- Bazzani, R.P.; Cai, Y.; Hebel, H.L.; Hyde, S.C.; Gill, D.R. The significance of plasmid DNA preparations contaminated with bacterial genomic DNA on inflammatory responses following delivery of lipoplexes to the murine lung. Biomaterials 2011, 32, 9854–9865. [Google Scholar] [CrossRef]
- Firozi, P.; Zhang, W.; Chen, L.; Quiocho, F.A.; Worley, K.C.; Templeton, N.S. Identification and removal of colanic acid from plasmid DNA preparations: Implications for gene therapy. Gene Ther. 2010, 17, 1484–1499. [Google Scholar] [CrossRef]
- Hidmark, A.; von Saint Paul, A.; Dalpke, A.H. Cutting edge: TLR13 is a receptor for bacterial RNA. J. Immunol. 2012, 189, 2717–2721. [Google Scholar] [CrossRef]
- Badger, C.V.; Richardson, J.D.; Dasilva, R.L.; Richards, M.J.; Josleyn, M.D.; Dupuy, L.C.; Hooper, J.W.; Schmaljohn, C.S. Development and application of a flow cytometric potency assay for DNA vaccines. Vaccine 2011, 29, 6728–6735. [Google Scholar] [CrossRef]
- Mahajan, R.; Feher, B.; Jones, B.; Jones, D.; Marjerison, L.; Sam, M.; Hartikka, J.; Wloch, M.; Lalor, P.; Kaslow, D.; et al. A taqman reverse transcription polymerase chain reaction (RT-PCR) in vitro potency assay for plasmid-based vaccine products. Mol. Biotechnol. 2008, 40, 47–57. [Google Scholar] [CrossRef]
- Goncalves, G.A.; Prazeres, D.M.; Monteiro, G.A.; Prather, K.L. De novo creation of MG1655-derived E. coli strains specifically designed for plasmid DNA production. Appl. Microbiol. Biotechnol. 2013, 97, 611–620. [Google Scholar] [CrossRef]
- Goncalves, G.A.; Bower, D.M.; Prazeres, D.M.; Monteiro, G.A.; Prather, K.L. Rational engineering of Escherichia coli strains for plasmid biopharmaceutical manufacturing. Biotechnol. J. 2012, 7, 251–261. [Google Scholar] [CrossRef]
- Williams, J.A.; Luke, J.; Langtry, S.; Anderson, S.; Hodgson, C.P.; Carnes, A.E. Generic plasmid DNA production platform incorporating low metabolic burden seed-stock and fed-batch fermentation processes. Biotechnol. Bioeng. 2009, 103, 1129–1143. [Google Scholar] [CrossRef]
- Yau, S.Y.; Keshavarz-Moore, E.; Ward, J. Host strain influences on supercoiled plasmid DNA production in Escherichia coli: Implications for efficient design of large-scale processes. Biotechnol. Bioeng. 2008, 101, 529–544. [Google Scholar] [CrossRef]
- Schvartzman, J.B.; Martinez-Robles, M.L.; Hernandez, P.; Krimer, D.B. Plasmid DNA replication and topology as visualized by two-dimensional agarose gel electrophoresis. Plasmid 2010, 63, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zechiedrich, E.L.; Khodursky, A.B.; Bachellier, S.; Schneider, R.; Chen, D.; Lilley, D.M.; Cozzarelli, N.R. Roles of topoisomerases in maintaining steady-state DNA supercoiling in Escherichia coli. J. Biol. Chem. 2000, 275, 8103–8113. [Google Scholar] [CrossRef]
- Deibler, R.W.; Rahmati, S.; Zechiedrich, E.L. Topoisomerase IV, alone, unknots DNA in E. coli. Genes Dev. 2001, 15, 748–761. [Google Scholar] [CrossRef]
- Goldstein, E.; Drlica, K. Regulation of bacterial DNA supercoiling: Plasmid linking numbers vary with growth temperature. Proc. Natl. Acad. Sci. USA 1984, 81, 4046–4050. [Google Scholar] [CrossRef]
- Balke, V.L.; Gralla, J.D. Changes in the linking number of supercoiled DNA accompany growth transitions in Escherichia coli. J. Bacteriol. 1987, 169, 4499–4506. [Google Scholar]
- Reyes-Dominguez, Y.; Contreras-Ferrat, G.; Ramirez-Santos, J.; Membrillo-Hernandez, J.; Gomez-Eichelmann, M.C. Plasmid DNA supercoiling and gyrase activity in Escherichia coli wild-type and rpoS stationary-phase cells. J. Bacteriol. 2003, 185, 1097–1100. [Google Scholar] [CrossRef]
- Weigl, D.; Molloy, M.J.; Clayton, T.M.; Griffith, J.; Smith, C.R.; Steward, T.; Merrill, B.M.; Deprince, R.B.; Jone, C.S.; Persmark, M. Characterization of a topologically aberrant plasmid population from pilot-scale production of clinical-grade DNA. J. Biotechnol. 2006, 121, 1–12. [Google Scholar]
- Benham, C.J. Duplex destabilization in superhelical DNA is predicted to occur at specific transcriptional regulatory regions. J. Mol. Biol. 1996, 255, 425–434. [Google Scholar] [CrossRef]
- Lim, H.M.; Lewis, D.E.; Lee, H.J.; Liu, M.; Adhya, S. Effect of varying the supercoiling of DNA on transcription and its regulation. Biochemistry 2003, 42, 10718–10725. [Google Scholar] [CrossRef]
- Prather, K.L.; Edmonds, M.C.; Herod, J.W. Identification and characterization of IS1 transposition in plasmid amplification mutants of E. coli clones producing DNA vaccines. Appl. Microbiol. Biotechnol. 2006, 73, 815–826. [Google Scholar] [CrossRef]
- Van der Heijden, I.; Gomez-Eerland, R.; van den Berg, J.H.; Oosterhuis, K.; Schumacher, T.N.; Haanen, J.B.; Beijnen, J.H.; Nuijen, B. Transposon leads to contamination of clinical pDNA vaccine. Vaccine 2013. [Google Scholar] [CrossRef]
- Cavlar, T.; Ablasser, A.; Hornung, V. Induction of type I IFNS by intracellular DNA-sensing pathways. Immunol. Cell. Biol. 2012, 90, 474–482. [Google Scholar] [CrossRef]
- Coban, C.; Koyama, S.; Takeshita, F.; Akira, S.; Ishii, K.J. Molecular and cellular mechanisms of DNA vaccines. Hum. Vaccin. 2008, 4, 453–456. [Google Scholar] [CrossRef]
- Liu, M.A. Immunologic basis of vaccine vectors. Immunity 2010, 33, 504–515. [Google Scholar] [CrossRef]
- Coban, C.; Kobiyama, K.; Aoshi, T.; Takeshita, F.; Horii, T.; Akira, S.; Ishii, K.J. Novel strategies to improve DNA vaccine immunogenicity. Curr. Gene Ther. 2011, 11, 479–484. [Google Scholar] [CrossRef]
- Pavlenko, M.; Leder, C.; Moreno, S.; Levitsky, V.; Pisa, P. Priming of CD8+ T-cell responses after DNA immunization is impaired in TLR9- and myd88-deficient mice. Vaccine 2007, 25, 6341–6347. [Google Scholar] [CrossRef]
- Rottembourg, D.; Filippi, C.M.; Bresson, D.; Ehrhardt, K.; Estes, E.A.; Oldham, J.E.; von Herrath, M.G. Essential role for TLR9 in prime but not prime-boost plasmid DNA vaccination to activate dendritic cells and protect from lethal viral infection. J. Immunol. 2010, 184, 7100–7107. [Google Scholar] [CrossRef]
- Hyde, S.C.; Pringle, I.A.; Abdullah, S.; Lawton, A.E.; Davies, L.A.; Varathalingam, A.; Nunez-Alonso, G.; Green, A.M.; Bazzani, R.P.; Sumner-Jones, S.G.; et al. CpG-free plasmids confer reduced inflammation and sustained pulmonary gene expression. Nat. Biotechol. 2008, 26, 549–551. [Google Scholar] [CrossRef]
- Warren, S.E.; Armstrong, A.; Hamilton, M.K.; Mao, D.P.; Leaf, I.A.; Miao, E.A.; Aderem, A. Cutting edge: Cytosolic bacterial DNA activates the inflammasome via aim2. J. Immunol. 2010, 185, 818–821. [Google Scholar]
- O’Neill, L.A. Immunology. Sensing the dark side of DNA. Science 2013, 339, 763–764. [Google Scholar] [CrossRef]
- Brazda, V.; Coufal, J.; Liao, J.C.; Arrowsmith, C.H. Preferential binding of ifi16 protein to cruciform structure and superhelical DNA. Biochem. Biophys. Res. Commun. 2012, 422, 716–720. [Google Scholar]
- Wang, X.D.; Tang, J.G.; Xie, X.L.; Yang, J.C.; Li, S.; Ji, J.G.; Gu, J. A comprehensive study of optimal conditions for naked plasmid DNA transfer into skeletal muscle by electroporation. J. Gene Med. 2005, 7, 1235–1245. [Google Scholar] [CrossRef]
- Hartikka, J.; Sukhu, L.; Buchner, C.; Hazard, D.; Bozoukova, V.; Margalith, M.; Nishioka, W.K.; Wheeler, C.J.; Manthorp, M.; Sawdey, M. Electroporation-facilitated delivery of plasmid DNA in skeletal muscle: Plasmid dependence of muscle damage and effect of poloxamer 188. Mol. Ther. 2001, 4, 407–415. [Google Scholar] [CrossRef]
- Mann, C.J.; Anguela, X.M.; Montane, J.; Obach, M.; Roca, C.; Ruzo, A.; Otaegui, P.; Mir, L.M.; Bosch, F. Molecular signature of the immune and tissue response to non-coding plasmid DNA in skeletal muscle after electrotransfer. Gene Ther. 2012, 19, 1177–1186. [Google Scholar] [CrossRef]
- Donate, A.; Heller, R. Assessment of delivery parameters with the multi-electrode array for development of a DNA vaccine against bacillus anthracis. Bioelectrochemistry 2013, 94C, 1–6. [Google Scholar] [CrossRef]
- Saade, F.; Petrovsky, N. Technologies for enhanced efficacy of DNA vaccines. Expert Rev. Vaccines 2012, 11, 189–209. [Google Scholar] [CrossRef]
- Desmet, C.J.; Ishii, K.J. Nucleic acid sensing at the interface between innate and adaptive immunity in vaccination. Nat. Rev. Immunol. 2012, 12, 479–491. [Google Scholar] [CrossRef]
- Bauer, S.; Kirschning, C.J.; Hacker, H.; Redecke, V.; Hausmann, S.; Akira, S.; Wagner, H.; Lipford, G.B. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. USA 2001, 98, 9237–9242. [Google Scholar] [CrossRef]
- Ohlschlager, P.; Spies, E.; Alvarez, G.; Quetting, M.; Groettrup, M. The combination of TLR-9 adjuvantation and electroporation-mediated delivery enhances in vivo antitumor responses after vaccination with hpv-16 E7 encoding DNA. Int. J. Cancer 2011, 128, 473–481. [Google Scholar] [CrossRef]
- Coban, C.; Ishii, K.J.; Gursel, M.; Klinman, D.M.; Kumar, N. Effect of plasmid backbone modification by different human CpG motifs on the immunogenicity of DNA vaccine vectors. J. Leukoc. Biol. 2005, 78, 647–655. [Google Scholar] [CrossRef]
- Li, D.; Liu, Y.; Zhang, Y.; Xu, J.; Hong, K.; Sun, M.; Shao, Y. Adjuvant effects of plasmid-generated hairpin rna molecules on DNA vaccination. Vaccine 2007, 25, 6992–7000. [Google Scholar] [CrossRef]
- Luke, J.M.; Simon, G.G.; Soderholm, J.; Errett, J.S.; August, J.T.; Gale, M., Jr.; Hodgson, C.P.; Williams, J.A. Coexpressed RIG-I agonist enhances humoral immune response to influenza virus DNA vaccine. J. Virol. 2011, 85, 1370–1383. [Google Scholar] [CrossRef]
- Wu, J.; Ma, H.; Qu, Q.; Zhou, W.J.; Luo, Y.P.; Thangaraj, H.; Lowrie, D.B.; Fan, X.Y. Incorporation of immunostimulatory motifs in the transcribed region of a plasmid DNA vaccine enhances TH1 immune responses and therapeutic effect against mycobacterium tuberculosis in mice. Vaccine 2011, 29, 7624–7630. [Google Scholar] [CrossRef]
- Artelt, P.; Grannemann, R.; Stocking, C.; Friel, J.; Bartsch, J.; Hauser, H. The prokaryotic neomycin-resistance-encoding gene acts as a transcriptional silencer in eukaryotic cells. Gene 1991, 99, 249–254. [Google Scholar] [CrossRef]
- Ribeiro, S.; Mairhofer, J.; Madeira, C.; Diogo, M.M.; Lobato da Silva, C.; Monteiro, G.; Grabherr, R.; Cabral, J.M. Plasmid DNA size does affect nonviral gene delivery efficiency in stem cells. Cell. Reprogram. 2012, 14, 130–137. [Google Scholar]
- Lukacs, G.L.; Haggie, P.; Seksek, O.; Lechardeur, D.; Freedman, N.; Verkman, A.S. Size-dependent DNA mobility in cytoplasm and nucleus. J. Biol. Chem. 2000, 275, 1625–1629. [Google Scholar]
- Kreiss, P.; Cameron, B.; Rangara, R.; Mailhe, P.; Aguerre-Charriol, O.; Airiau, M.; Scherman, D.; Crouzet, J.; Pitard, B. Plasmid DNA size does not affect the physicochemical properties of lipoplexes but modulates gene transfer efficiency. Nucleic Acids Res. 1999, 27, 3792–3798. [Google Scholar] [CrossRef]
- Yin, W.; Xiang, P.; Li, Q. Investigations of the effect of DNA size in transient transfection assay using dual luciferase system. Anal. Biochem. 2005, 346, 289–294. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, F.; Xu, S.; Fire, A.Z.; Kay, M.A. The extragenic spacer length between the 5' and 3' ends of the transgene expression cassette affects transgene silencing from plasmid-based vectors. Mol. Ther. 2012, 20, 2111–2119. [Google Scholar] [CrossRef]
- Gracey Maniar, L.E.; Maniar, J.M.; Chen, Z.Y.; Lu, J.; Fire, A.Z.; Kay, M.A. Minicircle DNA vectors achieve sustained expression reflected by active chromatin and transcriptional level. Mol. Ther. 2013, 21, 131–138. [Google Scholar] [CrossRef]
- Padegimas, L.; Kowalczyk, T.H.; Adams, S.; Gedeon, C.R.; Oette, S.M.; Dines, K.; Hyatt, S.L.; Sesenoglu-Laird, O.; Tyr, O.; Moen, R.C.; et al. Optimization of hCFTR lung expression in mice using DNA nanoparticles. Mol. Ther. 2012, 20, 63–72. [Google Scholar] [CrossRef]
- Hovav, A.H.; Panas, M.W.; Rahman, S.; Sircar, P.; Gillard, G.; Cayabyab, M.J.; Letvin, N.L. Duration of antigen expression in vivo following DNA immunization modifies the magnitude, contraction, and secondary responses of CD8+ t lymphocytes. J. Immunol. 2007, 179, 6725–6733. [Google Scholar]
- Finn, J.D.; Bassett, J.; Millar, J.B.; Grinshtein, N.; Yang, T.C.; Parsons, R.; Evelegh, C.; Wan, Y.; Parks, R.J.; Bramson, J.L. Persistence of transgene expression influences CD8+ T-cell expansion and maintenance following immunization with recombinant adenovirus. J. Virol. 2009, 83, 12027–12036. [Google Scholar] [CrossRef]
- Dietz, W.M.; Skinner, N.E.; Hamilton, S.E.; Jund, M.D.; Heitfeld, S.M.; Litterman, A.J.; Hwu, P.; Chen, Z.Y.; Salazar, A.M.; Ohlfest, J.R.; et al. Minicircle DNA is superior to plasmid DNA in eliciting antigen-specific CD8 T-cell responses. Mol. Ther. 2013. [Google Scholar] [CrossRef]
- Kay, M.A.; He, C.Y.; Chen, Z.Y. A robust system for production of minicircle DNA vectors. Nat. Biotechnol. 2010, 28, 1287–1289. [Google Scholar]
- Williams, J.A. DNA Plasmids with Improved Expression. US Patent Application PCT/US 13/00068, 14 March 2013. [Google Scholar]
- Willliams, J.A. Replicative Minicircle Vectors with Improved Expression. US Patent Application PCT/US 13/00067, 14 March 2013. [Google Scholar]
- Lu, J.; Zhang, F.; Kay, M.A. A mini-intronic plasmid (MIP): A novel robust transgene expression vector in vivo and in vitro. Mol. Ther. 2013, 21, 954–963. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Williams, J.A. Vector Design for Improved DNA Vaccine Efficacy, Safety and Production. Vaccines 2013, 1, 225-249. https://doi.org/10.3390/vaccines1030225
Williams JA. Vector Design for Improved DNA Vaccine Efficacy, Safety and Production. Vaccines. 2013; 1(3):225-249. https://doi.org/10.3390/vaccines1030225
Chicago/Turabian StyleWilliams, James A. 2013. "Vector Design for Improved DNA Vaccine Efficacy, Safety and Production" Vaccines 1, no. 3: 225-249. https://doi.org/10.3390/vaccines1030225