
Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases


{kind=link}
{kind=link}
Abstract
:1. Introduction
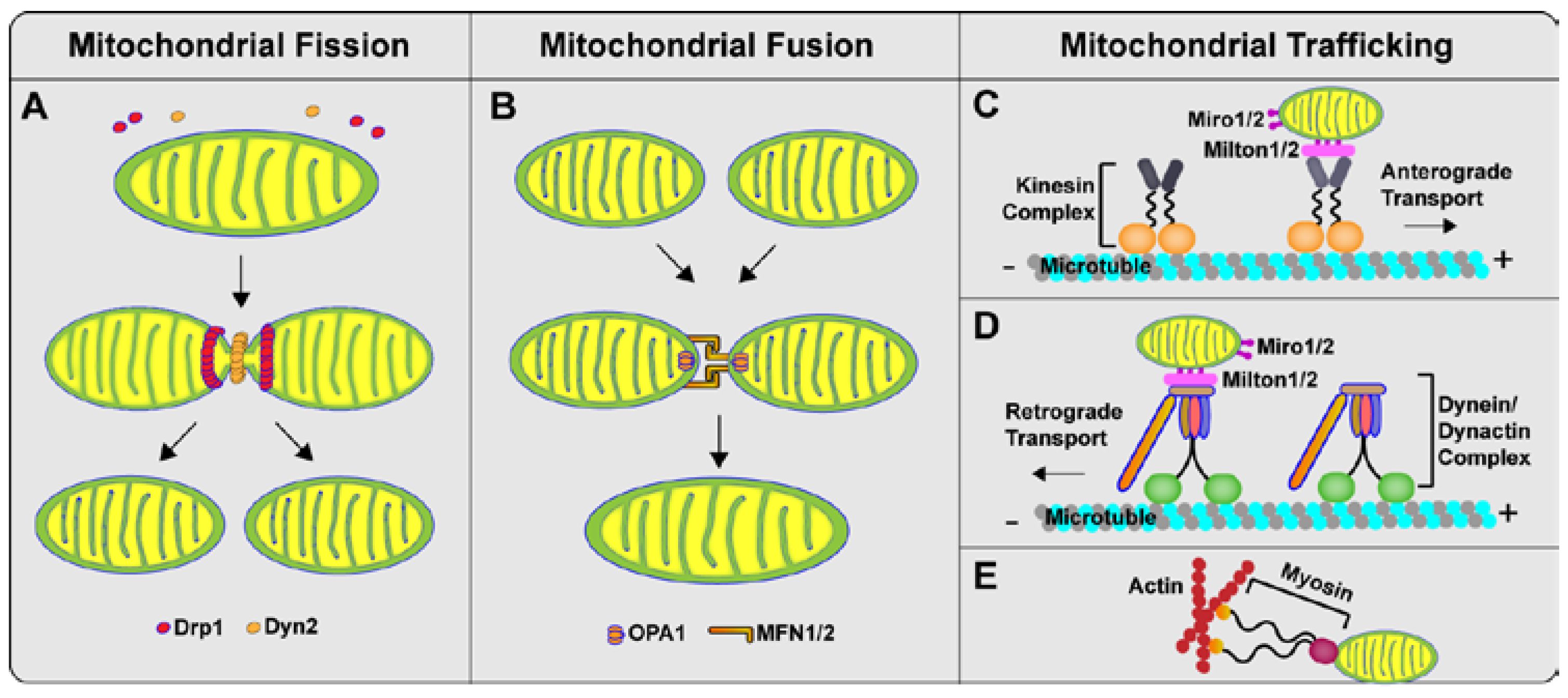
2. Mitochondrial Dynamics: Fission, Fusion and Trafficking
3. Mitochondrial Dynamics, Mitochondrial Function and Neuronal Function
3.1. Mitochondrial Dynamic Abnormalities in Alzheimer’s Disease (AD)
3.2. Mitochondrial Dynamic Abnormalities in Parkinson’s Disease (PD)
3.3. Mitochondrial Dynamic Abnormalities in Amyotrophic Lateral Sclerosis (ALS)
3.4. Mitochondrial Dynamic Abnormalities in Huntington’s Disease (HD)
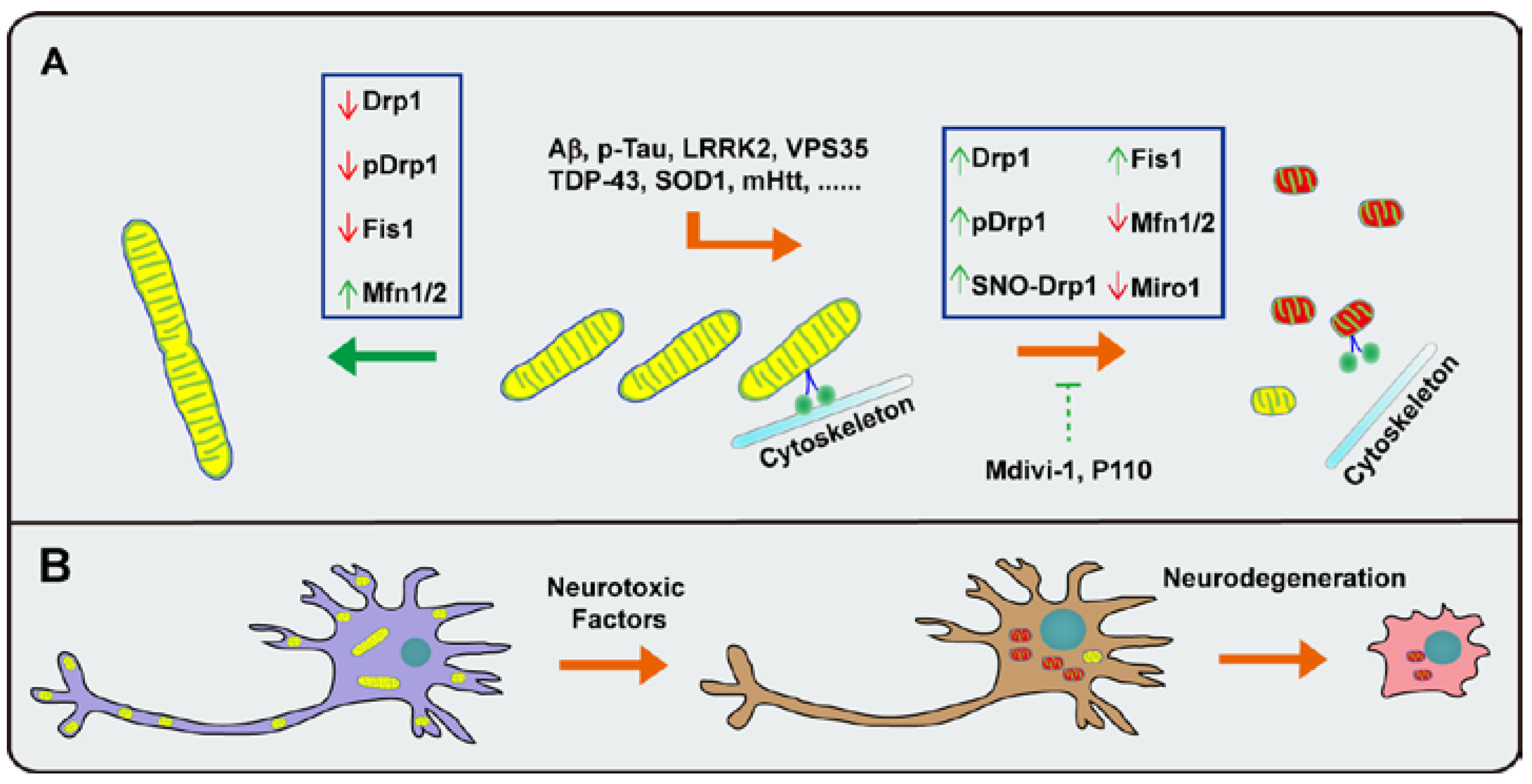
3.5. Perspective: Mitochondrial Dynamics as Common Therapeutic Targets for Neurodegeneration
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Suen, D.F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial dynamics—Fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2009, 109 (Suppl. 1), 153–159. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Kovacs, R. Mitochondria and neuronal activity. Am. J. Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Okamoto, K.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Blass, J.P.; Gibson, G.E. Cerebrometabolic aspects of delirium in relationship to dementia. Dement. Geriatr. Cogn. Disord. 1999, 10, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007, 120, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Frieden, M.; James, D.; Castelbou, C.; Danckaert, A.; Martinou, J.C.; Demaurex, N. Ca2+ homeostasis during mitochondrial fragmentation and perinuclear clustering induced by hFis1. J. Biol. Chem. 2004, 279, 22704–22714. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Simoni, A.M.; Chami, M.; Wieckowski, M.R.; Youle, R.J.; Rizzuto, R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol. Cell 2004, 16, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef]
- Sugioka, R.; Shimizu, S.; Tsujimoto, Y. Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J. Biol. Chem. 2004, 279, 52726–52734. [Google Scholar] [CrossRef] [PubMed]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Nakamura, K.; Iijima, M.; Sesaki, H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013, 23, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Burte, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. Embo Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- James, D.I.; Parone, P.A.; Mattenberger, Y.; Martinou, J.C. hFis1, a novel component of the mammalian mitochondrial fission machinery. J. Biol. Chem. 2003, 278, 36373–36379. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Westrate, L.M.; Wu, H.; Page, C.; Voeltz, G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef] [PubMed]
- Lackner, L.L.; Nunnari, J.M. The molecular mechanism and cellular functions of mitochondrial division. Biochim. Biophys. Acta 2009, 1792, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Zunino, R.; Braschi, E.; Xu, L.; McBride, H.M. Translocation of SenP5 from the nucleoli to the mitochondria modulates Drp1-dependent fission during mitosis. J. Biol. Chem. 2009, 284, 17783–17795. [Google Scholar] [CrossRef] [PubMed]
- Merrill, R.A.; Dagda, R.K.; Dickey, A.S.; Cribbs, J.T.; Green, S.H.; Usachev, Y.M.; Strack, S. Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP1. PLoS Biol. 2011, 9, e1000612. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, X.; Fujioka, H.; Hoppel, C.; Whone, A.L.; Caldwell, M.A.; Cullen, P.J.; Liu, J.; Zhu, X. Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med. 2016, 22, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [PubMed]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [PubMed]
- Zuchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Frohman, M.A. Role of mitochondrial lipids in guiding fission and fusion. J. Mol. Med. Berl. 2015, 93, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Huang, P.; Jenkins, G.M.; Chan, D.C.; Schiller, J.; Frohman, M.A. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat. Cell Biol. 2006, 8, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, X.; Bai, J.; Tian, X.; Zhao, X.; Liu, W.; Duan, X.; Shang, W.; Fan, H.Y.; Tong, C. Mitoguardin regulates mitochondrial fusion through mitopld and is required for neuronal homeostasis. Mol. Cell 2016, 61, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Montessuit, S.; Somasekharan, S.P.; Terrones, O.; Lucken-Ardjomande, S.; Herzig, S.; Schwarzenbacher, R.; Manstein, D.J.; Bossy-Wetzel, E.; Basanez, G.; Meda, P.; et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 2010, 142, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, P.J.; Stepanyants, N.; Mehrotra, N.; Mears, J.A.; Qi, X.; Sesaki, H.; Ramachandran, R. A dimeric equilibrium intermediate nucleates Drp1 reassembly on mitochondrial membranes for fission. Mol. Biol. Cell 2014, 25, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Frederick, R.L.; Shaw, J.M. Moving mitochondria: Establishing distribution of an essential organelle. Traffic 2007, 8, 1668–1675. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.H. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J. Cell Biol. 2014, 204, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Macleod, G.T.; Wellington, A.; Hu, F.; Panchumarthi, S.; Schoenfield, M.; Marin, L.; Charlton, M.P.; Atwood, H.L.; Zinsmaier, K.E. The GTPase dmiro is required for axonal transport of mitochondria to drosophila synapses. Neuron 2005, 47, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Glater, E.E.; Megeath, L.J.; Stowers, R.S.; Schwarz, T.L. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol. 2006, 173, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Hatch, A.L.; Gurel, P.S.; Higgs, H.N. Novel roles for actin in mitochondrial fission. J. Cell Sci. 2014, 127, 4549–4560. [Google Scholar] [CrossRef] [PubMed]
- Altmann, K.; Frank, M.; Neumann, D.; Jakobs, S.; Westermann, B. The class V myosin motor protein, myo2, plays a major role in mitochondrial motility in Saccharomyces cerevisiae. J. Cell Biol. 2008, 181, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I.R.; Yang, H.C.; Nowakowski, W.D.; Karmon, S.L.; Hays, L.G.; Yates, J.R., 3rd; Pon, L.A. Arp2/3 complex and actin dynamics are required for actin-based mitochondrial motility in yeast. Proc. Natl. Acad. Sci. USA 2001, 98, 3162–3167. [Google Scholar] [CrossRef] [PubMed]
- Griparic, L.; van der Wel, N.N.; Orozco, I.J.; Peters, P.J.; van der Bliek, A.M. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J. Biol. Chem. 2004, 279, 18792–18798. [Google Scholar] [CrossRef] [PubMed]
- Spinazzi, M.; Cazzola, S.; Bortolozzi, M.; Baracca, A.; Loro, E.; Casarin, A.; Solaini, G.; Sgarbi, G.; Casalena, G.; Cenacchi, G.; et al. A novel deletion in the GTPase domain of OPA1 causes defects in mitochondrial morphology and distribution, but not in function. Hum. Mol. Genet. 2008, 17, 3291–3302. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 2011, 17, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Fukumitsu, K.; Hatsukano, T.; Yoshimura, A.; Heuser, J.; Fujishima, K.; Kengaku, M. Mitochondrial fission protein Drp1 regulates mitochondrial transport and dendritic arborization in cerebellar Purkinje cells. Mol. Cell Neurosci. 2016, 71, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Misko, A.; Jiang, S.R.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.H.; Kim, K.Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [PubMed]
- Melentijevic, I.; Toth, M.L.; Arnold, M.L.; Guasp, R.J.; Harinath, G.; Nguyen, K.C.; Taub, D.; Parker, J.A.; Neri, C.; Gabel, C.V.; et al. Elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature 2017, 542, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Kanki, T.; Fukuoh, A.; Ohgaki, K.; Takeya, R.; Aoki, Y.; Hamasaki, N.; Kang, D. PDIP38 associates with proteins constituting the mitochondrial DNA nucleoid. J. Biochem. 2005, 138, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Arselin, G.; Vaillier, J.; Salin, B.; Schaeffer, J.; Giraud, M.F.; Dautant, A.; Brethes, D.; Velours, J. The modulation in subunits e and g amounts of yeast ATP synthase modifies mitochondrial cristae morphology. J. Biol. Chem. 2004, 279, 40392–40399. [Google Scholar] [CrossRef] [PubMed]
- Hackenbrock, C.R. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J. Cell Biol. 1966, 30, 269–297. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed]
- Scalettar, B.A.; Abney, J.R.; Hackenbrock, C.R. Dynamics, structure, and function are coupled in the mitochondrial matrix. Proc. Natl. Acad. Sci. USA 1991, 88, 8057–8061. [Google Scholar] [CrossRef] [PubMed]
- Benard, G.; Rossignol, R. Ultrastructure of the mitochondrion and its bearing on function and bioenergetics. Antioxid. Redox Signal. 2008, 10, 1313–1342. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Acin-Perez, R.; Geghman, K.D.; Manfredi, G.; Lu, B.; Li, C. Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc. Natl. Acad. Sci. USA 2011, 108, 12920–12924. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.I.; et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009, 11, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, J.; Zhang, Z.Y.; Wakabayashi, N.; Tamura, Y.; Fukaya, M.; Kensler, T.W.; Iijima, M.; Sesaki, H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 2009, 186, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Oh, S.S.; Weaver, D.; Lewandowska, A.; Maxfield, D.; Schuler, M.H.; Smith, N.K.; Macfarlane, J.; Saunders, G.; Palmer, C.A.; et al. Loss of Miro1-directed mitochondrial movement results in a novel murine model for neuron disease. Proc. Natl. Acad. Sci. USA 2014, 111, E3631–E3640. [Google Scholar] [CrossRef] [PubMed]
- Macaskill, A.F.; Rinholm, J.E.; Twelvetrees, A.E.; Arancibia-Carcamo, I.L.; Muir, J.; Fransson, A.; Aspenstrom, P.; Attwell, D.; Kittler, J.T. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 2009, 61, 541–555. [Google Scholar] [CrossRef] [PubMed]
- MacAskill, A.F.; Brickley, K.; Stephenson, F.A.; Kittler, J.T. GTPase dependent recruitment of Grif-1 by Miro1 regulates mitochondrial trafficking in hippocampal neurons. Mol. Cell Neurosci. 2009, 40, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Baloh, R.H. Mitochondrial dynamics and peripheral neuropathy. Neuroscientist 2008, 14, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Sleigh, J.N.; Grice, S.J.; Burgess, R.W.; Talbot, K.; Cader, M.Z. Neuromuscular junction maturation defects precede impaired lower motor neuron connectivity in Charcot-Marie-Tooth type 2D mice. Hum. Mol. Genet. 2014, 23, 2639–2650. [Google Scholar] [CrossRef] [PubMed]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A. Alzheimer disease. Int. Rev. Neurobiol. 1998, 42, 1–54. [Google Scholar] [CrossRef]
- Castellani, R.; Hirai, K.; Aliev, G.; Drew, K.L.; Nunomura, A.; Takeda, A.; Cash, A.D.; Obrenovich, M.E.; Perry, G.; Smith, M.A. Role of mitochondrial dysfunction in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [PubMed]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, R.A.; Matthews-Roberson, T.A.; Dolan, P.J.; Johnson, G.V. Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: Implications for the pathogenesis of Alzheimer disease. J. Biol. Chem. 2009, 284, 18754–18766. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimers disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef] [PubMed]
- DuBoff, B.; Gotz, J.; Feany, M.B. Tau promotes neurodegeneration via Drp1 mislocalization in vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Hu, Y.; Wang, Z.H.; Luo, Y.; Zhang, Y.; Liu, X.P.; Feng, Q.; Wang, Q.; Ye, K.; Liu, G.P.; et al. Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci. Rep. 2016, 6, 24756. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, D.M.; Fausett, H.J.; Page, K.J.; Kim, T.W.; Moir, R.D.; Merriam, D.E.; Hollister, R.D.; Hallmark, O.G.; Mancini, R.; Felsenstein, K.M.; et al. Alzheimer-associated presenilins 1 and 2: Neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat. Med. 1996, 2, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Begley, J.G.; Duan, W.; Chan, S.; Duff, K.; Mattson, M.P. Altered calcium homeostasis and mitochondrial dysfunction in cortical synaptic compartments of presenilin-1 mutant mice. J. Neurochem. 1999, 72, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Kilb, W.; Hartmann, D.; Saftig, P.; Luhmann, H.J. Altered morphological and electrophysiological properties of Cajal-Retzius cells in cerebral cortex of embryonic Presenilin-1 knockout mice. Eur. J. Neurosci. 2004, 20, 2749–2756. [Google Scholar] [CrossRef] [PubMed]
- Behbahani, H.; Shabalina, I.G.; Wiehager, B.; Concha, H.; Hultenby, K.; Petrovic, N.; Nedergaard, J.; Winblad, B.; Cowburn, R.F.; Ankarcrona, M. Differential role of presenilin-1 and -2 on mitochondrial membrane potential and oxygen consumption in mouse embryonic fibroblasts. J. Neurosci. Res. 2006, 84, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.M.; Burnett, P.; Thomas, A.P.; Tezapsidis, N. Calcium oscillations in type-1 astrocytes, the effect of a presenilin 1 (PS1) mutation. Neurosci. Lett. 2006, 395, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Velez-Pardo, C.; Arroyave, S.T.; Lopera, F.; Castano, A.D.; Jimenez Del Rio, M. Ultrastructure evidence of necrotic neural cell death in familial Alzheimer’s disease brains bearing presenilin-1 E280A mutation. J. Alzheimers Dis. 2001, 3, 409–415. [Google Scholar] [PubMed]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Manczak, M.; Fry, D.; Suneetha, Y.; Sesaki, H.; Reddy, P.H. Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 4881–4897. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Kandimalla, R.; Fry, D.; Sesaki, H.; Reddy, P.H. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 5148–5166. [Google Scholar] [CrossRef] [PubMed]
- Stokin, G.B.; Lillo, C.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davies, P.; Masliah, E.; Williams, D.S.; et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.; Tiwari, P.; Xie, Z.; Zheng, J.Q. Acute impairment of mitochondrial trafficking by beta-amyloid peptides in hippocampal neurons. J. Neurosci. 2006, 26, 10480–10487. [Google Scholar] [CrossRef] [PubMed]
- Shahpasand, K.; Uemura, I.; Saito, T.; Asano, T.; Hata, K.; Shibata, K.; Toyoshima, Y.; Hasegawa, M.; Hisanaga, S. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer’s disease. J. Neurosci. 2012, 32, 2430–2441. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Martin, T.; Pooler, A.M.; Lau, D.H.; Morotz, G.M.; De Vos, K.J.; Gilley, J.; Coleman, M.P.; Hanger, D.P. Reduced number of axonal mitochondria and tau hypophosphorylation in mouse p301l tau knockin neurons. Neurobiol. Dis. 2016, 85, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hajnoczky, G. Ca2+-dependent regulation of mitochondrial dynamics by the Miro-Milton complex. Int. J. Biochem. Cell Biol. 2009, 41, 1972–1976. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.A.H.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Ronnback, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Fasolato, C.; Pozzan, T.; Pizzo, P. Presenilin-2 modulation of ER-mitochondria interactions: FAD mutations, mechanisms and pathological consequences. Commun. Integr. Biol. 2011, 4, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Sun, X.; Starovoytov, V.; Cai, Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 2015, 24, 2938–2951. [Google Scholar] [CrossRef] [PubMed]
- Zare-shahabadi, A.; Masliah, E.; Johnson, G.V.W.; Rezaei, N. Autophagy in Alzheimer’s disease. Rev. Neurosci. 2015, 26, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G.E. Biology of Parkinson’s disease: Pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin. Neurosci. 2004, 6, 259–280. [Google Scholar] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.F.; David, S.N.; Newport, G.D.; Cadet, J.L.; Slikker, W., Jr. MPTP-induced oxidative stress and neurotoxicity are age-dependent: Evidence from measures of reactive oxygen species and striatal dopamine levels. Synapse 1994, 18, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Liu, W.; He, X.; Gao, Y.; Castellani, R.J.; Perry, G.; Smith, M.A.; Zhu, X. DLP1-dependent mitochondrial fragmentation mediates 1-methyl-4-phenylpyridinium toxicity in neurons: Implications for Parkinson’s disease. Aging Cell 2011, 10, 807–823. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.C.; Qi, X. Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum. Mol. Genet. 2013, 22, 4545–4561. [Google Scholar] [CrossRef] [PubMed]
- Mortiboys, H.; Johansen, K.K.; Aasly, J.O.; Bandmann, O. Mitochondrial impairment in patients with parkinson disease with the G2019S mutation in LRRK2. Neurology 2010, 75, 2017–2020. [Google Scholar] [CrossRef] [PubMed]
- Papkovskaia, T.D.; Chau, K.Y.; Inesta-Vaquera, F.; Papkovsky, D.B.; Healy, D.G.; Nishio, K.; Staddon, J.; Duchen, M.R.; Hardy, J.; Schapira, A.H.V.; et al. G2019S leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization. Hum. Mol. Genet. 2012, 21, 4201–4213. [Google Scholar] [CrossRef] [PubMed]
- Haylett, W.; Swart, C.; van der Westhuizen, F.; van Dyk, H.; van der Merwe, L.; van der Merwe, C.; Loos, B.; Carr, J.; Kinnear, C.; Bardien, S. Altered mitochondrial respiration and other features of mitochondrial function in Parkin-mutant fibroblasts from Parkinson’s disease patients. Parkinsons Dis. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.Y.; Giasson, B.I.; Bonini, N.M. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc. Natl. Acad. Sci. USA 2010, 107, 9747–9752. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.J.; McCoy, M.K.; Blackinton, J.; Beilina, A.; van der Brug, M.; Sandebring, A.; Miller, D.; Maric, D.; Cedazo-Minguez, A.; Cookson, M.R. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 2011, 20, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Sherer, T.B.; Betarbet, R.; Stout, A.K.; Lund, S.; Baptista, M.; Panov, A.V.; Cookson, M.R.; Greenamyre, J.T. An in vitro model of Parkinson’s disease: Linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J. Neurosci. 2002, 22, 7006–7015. [Google Scholar] [PubMed]
- Wang, X.L.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X.W. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef] [PubMed]
- Yue, M.; Hinkle, K.M.; Davies, P.; Trushina, E.; Fiesel, F.C.; Christenson, T.A.; Schroeder, A.S.; Zhang, L.; Bowles, E.; Behrouz, B.; et al. Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol. Dis. 2015, 78, 172–195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, L.; Jiang, X.; Zhai, S.; Xing, D. The essential role of Drp1 and its regulation by S-nitrosylation of Parkin in dopaminergic neurodegeneration: Implications for Parkinson’s disease. Antioxid. Redox Signal. 2016, 25, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Petrie, T.G.; Liu, Y.C.; Liu, J.; Fujioka, H.; Zhu, X.W. Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J. Neurochem. 2012, 121, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.E.; Dodson, M.W.; Jiang, C.G.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila PINK1 is required for mitochondrial function and interacts genetically with Parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rub, C.; Liu, Y.; Magrane, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. Alpha-synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Song, P.; Du, L.; Tian, W.; Yue, W.; Liu, M.; Li, D.; Wang, B.; Zhu, Y.; Cao, C.; et al. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: Implication of dysregulated mitochondrial dynamics in Parkinson disease. J. Biol. Chem. 2011, 286, 11649–11658. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Schapira, A.H.V. PINK1-Parkin-dependent mitophagy involves ubiquitination of mitofusins 1 and 2 implications for Parkinson disease pathogenesis. Autophagy 2011, 7, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Cooper, J.M.; Chau, K.Y.; Rojo, M.; Schapira, A.H.V.; Taanman, J.W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/Parkin-dependent manner upon induction of mitophagy (vol 19, pg 4861, 2010). Hum. Mol. Genet. 2013, 22, 1697. [Google Scholar] [CrossRef]
- Carelli, V.; Musumeci, O.; Caporali, L.; Zanna, C.; La Morgia, C.; Del Dotto, V.; Porcelli, A.M.; Rugolo, M.; Valentino, M.L.; Iommarini, L.; et al. Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann. Neurol. 2015, 78, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; Tocilescu, M.; Devries, R.L.; Alessi, D.M.; Jackson-Lewis, V.; Przedborski, S. Control of mitochondrial integrity in Parkinson’s disease. Prog. Brain Res. 2010, 183, 99–113. [Google Scholar] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Hirano, A.; Donnenfeld, H.; Sasaki, S.; Nakano, I. Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 1984, 43, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.E.; Gonzalez, D.M.; Monachelli, G.M.; Costa, J.J.; Nicola, A.F.; Sica, R.E. Morphological abnormalities in mitochondria of the skin of patients with sporadic amyotrophic lateral sclerosis. Arq. Neuropsiquiatr. 2012, 70, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Vande Velde, C.; McDonald, K.K.; Boukhedimi, Y.; McAlonis-Downes, M.; Lobsiger, C.S.; Bel Hadj, S.; Zandona, A.; Julien, J.P.; Shah, S.B.; Cleveland, D.W. Misfolded sod1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PLoS ONE 2011, 6, e22031. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Song, Y.; Kincaid, B.; Bossy, B.; Bossy-Wetzel, E. Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: Neuroprotection by SIRT3 and PGC-1alpha. Neurobiol. Dis. 2013, 51, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Vinsant, S.; Mansfield, C.; Jimenez-Moreno, R.; Del Gaizo Moore, V.; Yoshikawa, M.; Hampton, T.G.; Prevette, D.; Caress, J.; Oppenheim, R.W.; Milligan, C. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: Part II, results and discussion. Brain Behav. 2013, 3, 431–457. [Google Scholar] [CrossRef] [PubMed]
- Vinsant, S.; Mansfield, C.; Jimenez-Moreno, R.; Del Gaizo Moore, V.; Yoshikawa, M.; Hampton, T.G.; Prevette, D.; Caress, J.; Oppenheim, R.W.; Milligan, C. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: Part I, background and methods. Brain Behav. 2013, 3, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Magrane, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yamashita, T.; Tian, F.; Morimoto, N.; Ikeda, Y.; Deguchi, K.; Abe, K. Mitochondrial fusion and fission proteins expression dynamically change in a murine model of amyotrophic lateral sclerosis. Curr. Neurovasc. Res. 2013, 10, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.F.; Gendron, T.F.; Zhang, Y.J.; Lin, W.L.; D’Alton, S.; Sheng, H.; Casey, M.C.; Tong, J.; Knight, J.; Yu, X.; et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 2010, 30, 10851–10859. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.F.; Zhang, Y.J.; Lin, W.L.; Cao, X.; Stetler, C.; Dickson, D.W.; Lewis, J.; Petrucelli, L. Expression of mutant TDP-43 induces neuronal dysfunction in transgenic mice. Mol. Neurodegener. 2011, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Yang, M.; Chen, Y.; Chen, X.; Liu, J.; Sun, S.; Cheng, H.; Li, Y.; Bigio, E.H.; Mesulam, M.; et al. FUS interacts with HSP60 to promote mitochondrial damage. PLoS Genet. 2015, 11, e1005357. [Google Scholar] [CrossRef] [PubMed]
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.L.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells 2016, 34, 2063–2078. [Google Scholar] [PubMed]
- Altanbyek, V.; Cha, S.J.; Kang, G.U.; Im, D.S.; Lee, S.; Kim, H.J.; Kim, K. Imbalance of mitochondrial dynamics in drosophila models of amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2016, 481, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Iwata, M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Sotelo-Silveira, J.R.; Lepanto, P.; Elizondo, V.; Horjales, S.; Palacios, F.; Martinez-Palma, L.; Marin, M.; Beckman, J.S.; Barbeito, L. Axonal mitochondrial clusters containing mutant SOD1 in transgenic models of ALS. Antioxid. Redox Signal. 2009, 11, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; McLoughlin, D.M.; et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [CrossRef] [PubMed]
- Igaz, L.M.; Kwong, L.K.; Lee, E.B.; Chen-Plotkin, A.; Swanson, E.; Unger, T.; Malunda, J.; Xu, Y.; Winton, M.J.; Trojanowski, J.Q.; et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Investig. 2011, 121, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Janssens, J.; Wils, H.; Kleinberger, G.; Joris, G.; Cuijt, I.; Ceuterick-de Groote, C.; Van Broeckhoven, C.; Kumar-Singh, S. Overexpression of ALS-associated p.M337V human TDP-43 in mice worsens disease features compared to wild-type human TDP-43 mice. Mol. Neurobiol. 2013, 48, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Ligon, L.A.; LaMonte, B.H.; Wallace, K.E.; Weber, N.; Kalb, R.G.; Holzbaur, E.L. Mutant superoxide dismutase disrupts cytoplasmic dynein in motor neurons. Neuroreport 2005, 16, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.L.; Lin, Y.T.; Ting, C.H.; Lin-Chao, S.; Li, H.; Hsieh-Li, H.M. Stathmin, a microtubule-destabilizing protein, is dysregulated in spinal muscular atrophy. Hum. Mol. Genet. 2010, 19, 1766–1778. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, W.; Siedlak, S.L.; Liu, Y.; Liu, J.; Jiang, K.; Perry, G.; Zhu, X.; Wang, X. Miro1 deficiency in amyotrophic lateral sclerosis. Front. Aging Neurosci. 2015, 7, 100. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Vande Velde, C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H., Jr. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Vijayvergiya, C.; Beal, M.F.; Buck, J.; Manfredi, G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J. Neurosci. 2005, 25, 2463–2470. [Google Scholar] [CrossRef] [PubMed]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [PubMed]
- Sugars, K.L.; Rubinsztein, D.C. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003, 19, 233–238. [Google Scholar] [CrossRef]
- Waldvogel, H.J.; Kim, E.H.; Thu, D.C.; Tippett, L.J.; Faull, R.L. New perspectives on the neuropathology in Huntington’s disease in the human brain and its relation to symptom variation. J. Huntingt. Dis. 2012, 1, 143–153. [Google Scholar]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant Huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Moody, J.P.; Edgerly, C.K.; Bordiuk, O.L.; Cormier, K.; Smith, K.; Beal, M.F.; Ferrante, R.J. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum. Mol. Genet. 2010, 19, 3919–3935. [Google Scholar] [CrossRef] [PubMed]
- Guedes-Dias, P.; de Proenca, J.; Soares, T.R.; Leitao-Rocha, A.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M. HDAC6 inhibition induces mitochondrial fusion, autophagic flux and reduces diffuse mutant huntingtin in striatal neurons. Biochim. Biophys. Acta 2015, 1852, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Haun, F.; Nakamura, T.; Shiu, A.D.; Cho, D.H.; Tsunemi, T.; Holland, E.A.; La Spada, A.R.; Lipton, S.A. S-nitrosylation of dynamin-related protein 1 mediates mutant huntingtin-induced mitochondrial fragmentation and neuronal injury in Huntington’s disease. Antioxid. Redox Signal. 2013, 19, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Solesio, M.E.; Saez-Atienzar, S.; Jordan, J.; Galindo, M.F. 3-Nitropropionic acid induces autophagy by forming mitochondrial permeability transition pores rather than activatiing the mitochondrial fission pathway. Br. J. Pharmacol. 2013, 168, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Liot, G.; Bossy, B.; Lubitz, S.; Kushnareva, Y.; Sejbuk, N.; Bossy-Wetzel, E. Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Differ. 2009, 16, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Brouillet, E.; Jenkins, B.G.; Ferrante, R.J.; Kowall, N.W.; Miller, J.M.; Storey, E.; Srivastava, R.; Rosen, B.R.; Hyman, B.T. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J. Neurosci. 1993, 13, 4181–4192. [Google Scholar] [PubMed]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Dyer, R.B.; Badger, J.D., 2nd; Ure, D.; Eide, L.; Tran, D.D.; Vrieze, B.T.; Legendre-Guillemin, V.; McPherson, P.S.; Mandavilli, B.S.; et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell. Biol. 2004, 24, 8195–8209. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pages, M.; Dompierre, J.P.; Rangone, H.; Cordelieres, F.P.; De Mey, J.; MacDonald, M.E.; Lessmann, V.; Humbert, S.; et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.R.; Muyderman, H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim. Biophys. Acta 2010, 1802, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Bramlett, H.M.; Dietrich, W.D. Pathophysiology of cerebral ischemia and brain trauma: Similarities and differences. J. Cereb. Blood Flow Metab. 2004, 24, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Rappold, P.M.; Cui, M.; Grima, J.C.; Fan, R.Z.; de Mesy-Bentley, K.L.; Chen, L.; Zhuang, X.; Bowers, W.J.; Tieu, K. Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat. Commun. 2014, 5, 5244. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Disatnik, M.H.; Monbureau, M.; Shamloo, M.; Mochly-Rosen, D.; Qi, X. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J. Clin. Investig. 2013, 123, 5371–5388. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wang, X.; Bonda, D.; Perry, G.; Smith, M.; Zhu, X. Abnormal mitochondrial dynamics—A novel therapeutic target for Alzheimer’s disease? Mol. Neurobiol. 2010, 41, 87–96. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxidants 2017, 6, 25. https://doi.org/10.3390/antiox6020025
Gao J, Wang L, Liu J, Xie F, Su B, Wang X. Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxidants. 2017; 6(2):25. https://doi.org/10.3390/antiox6020025
Chicago/Turabian StyleGao, Ju, Luwen Wang, Jingyi Liu, Fei Xie, Bo Su, and Xinglong Wang. 2017. "Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases" Antioxidants 6, no. 2: 25. https://doi.org/10.3390/antiox6020025