
Genetic Inactivation of Peroxiredoxin-I Impairs the Growth of Human Pancreatic Cancer Cells



Abstract
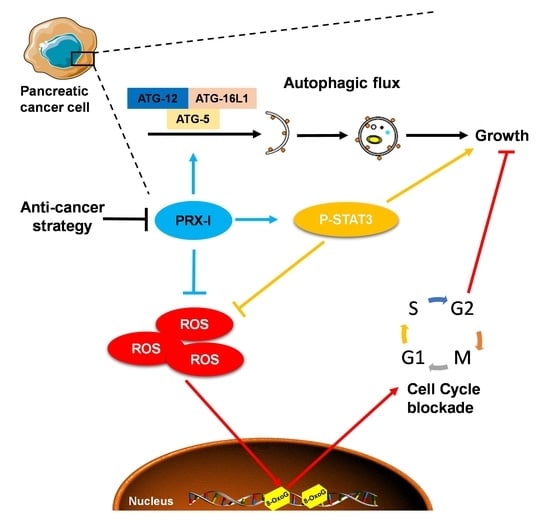
:
1. Introduction
2. Materials and Methods
2.1. Mice and Ethical Approval
2.2. Cell Lines, Small Interfering RNA (siRNA) Transfections and Pharmacological Treatments
2.3. Cell Counting and Crystal Violet Staining
2.4. Cell Cycle Analysis
2.5. Reactive Oxygen Species (ROS) Measurements
2.6. Western Blot
2.7. Immunofluorescence and Immunohistochemistry
2.8. Detection of 8-Oxoguanine (8-oxoG) Adducts
2.9. Statistical Analysis
3. Results and Discussion
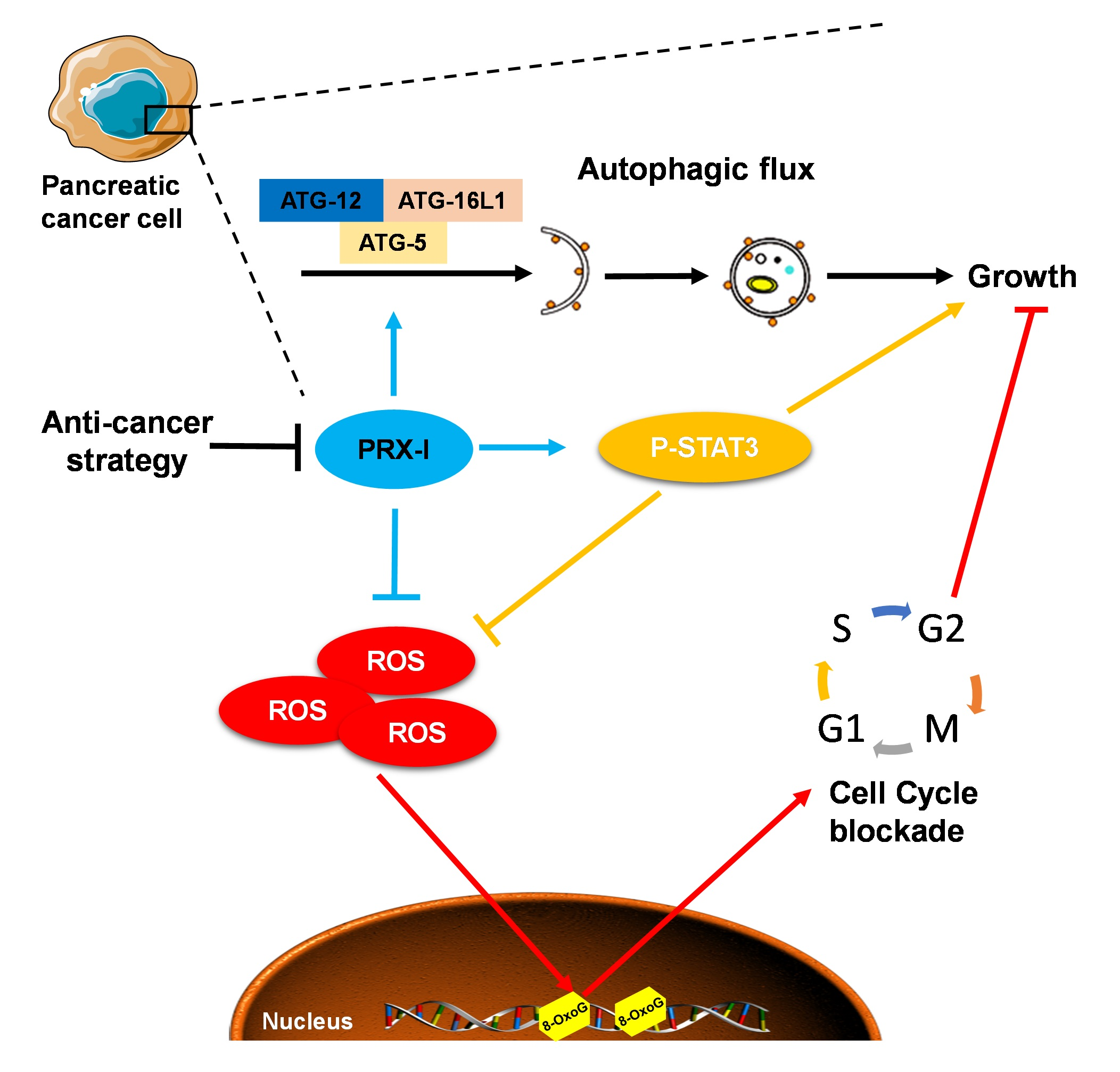
3.1. PRX-I Expression Is Induced during Pancreatic Carcinogenesis
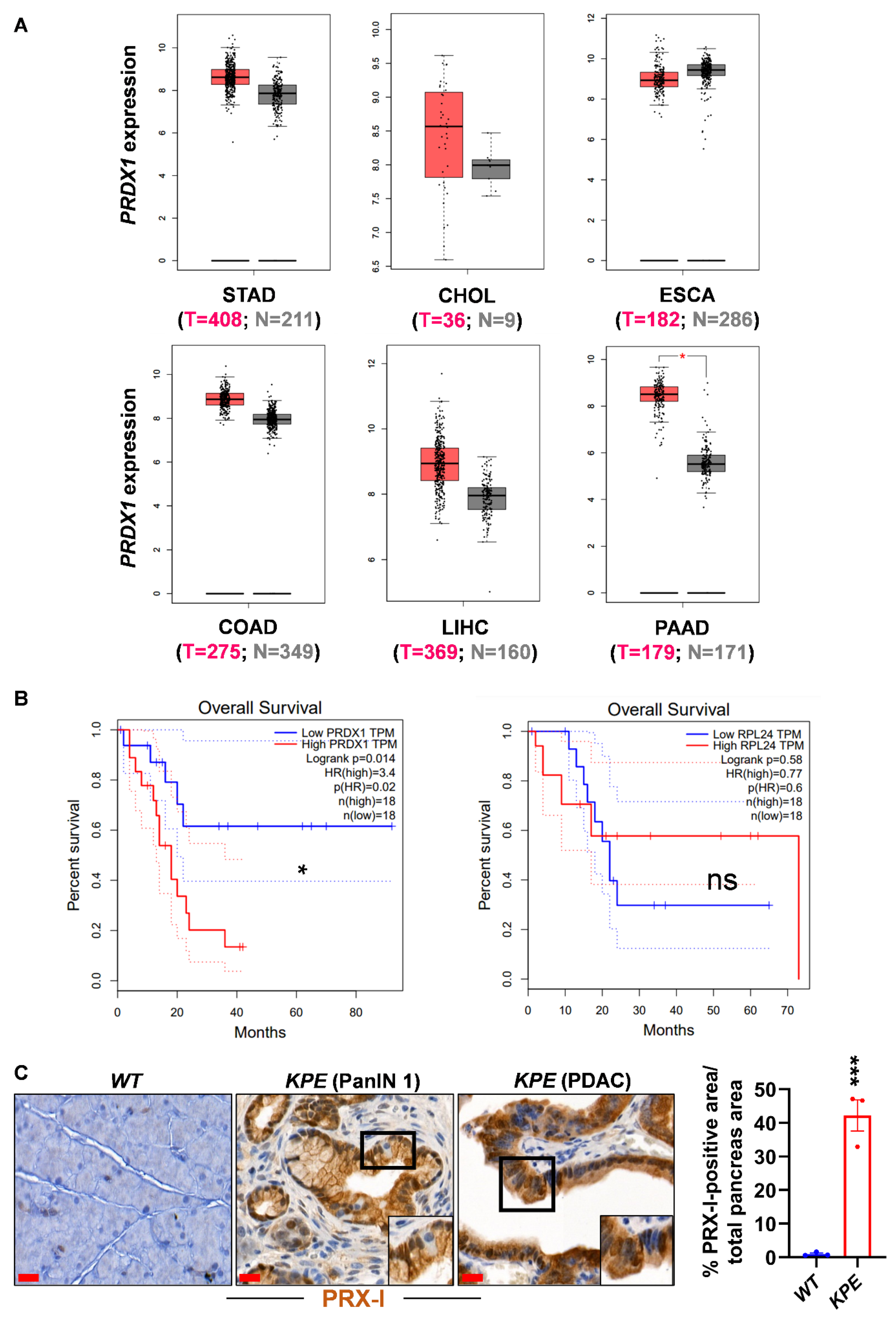
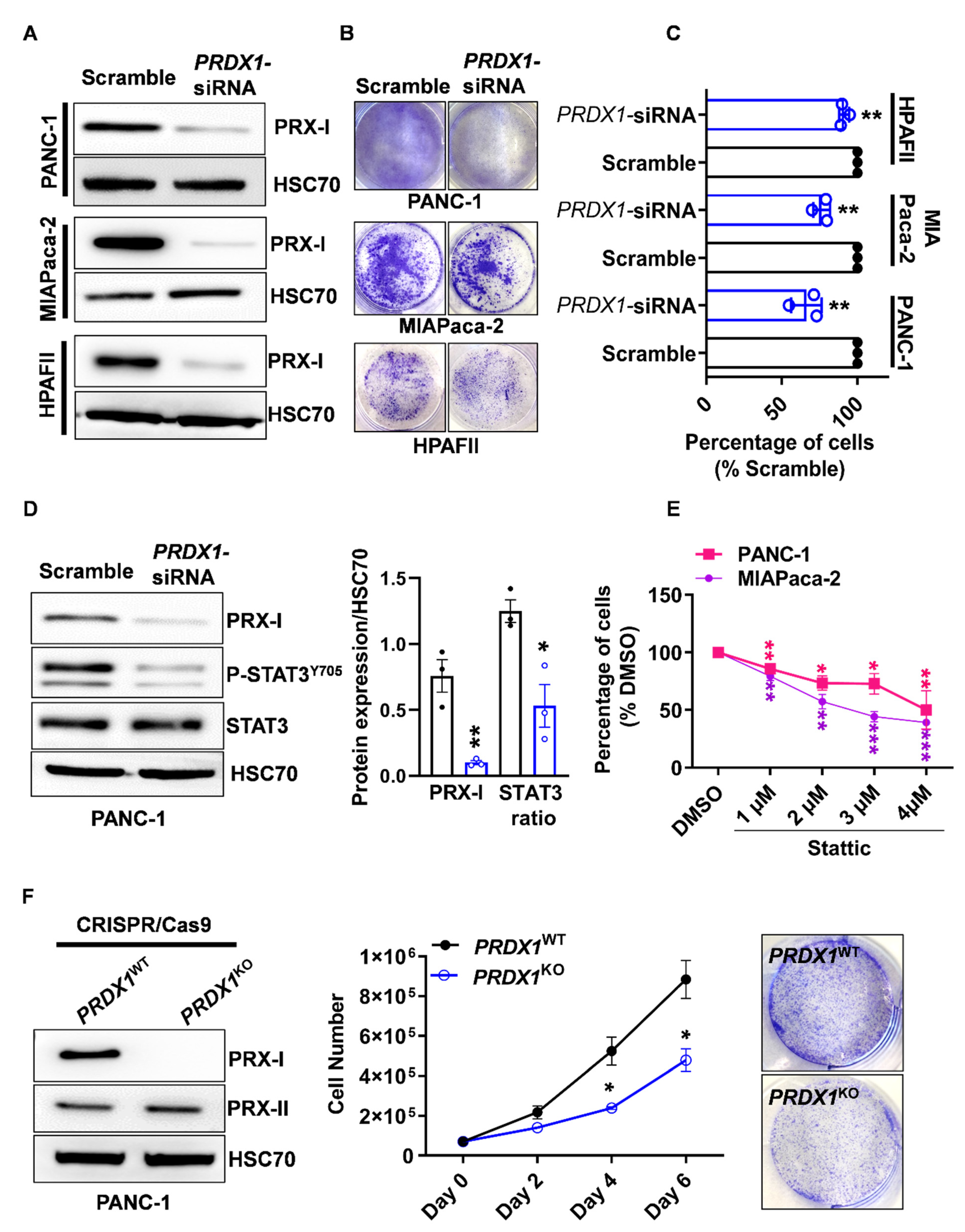
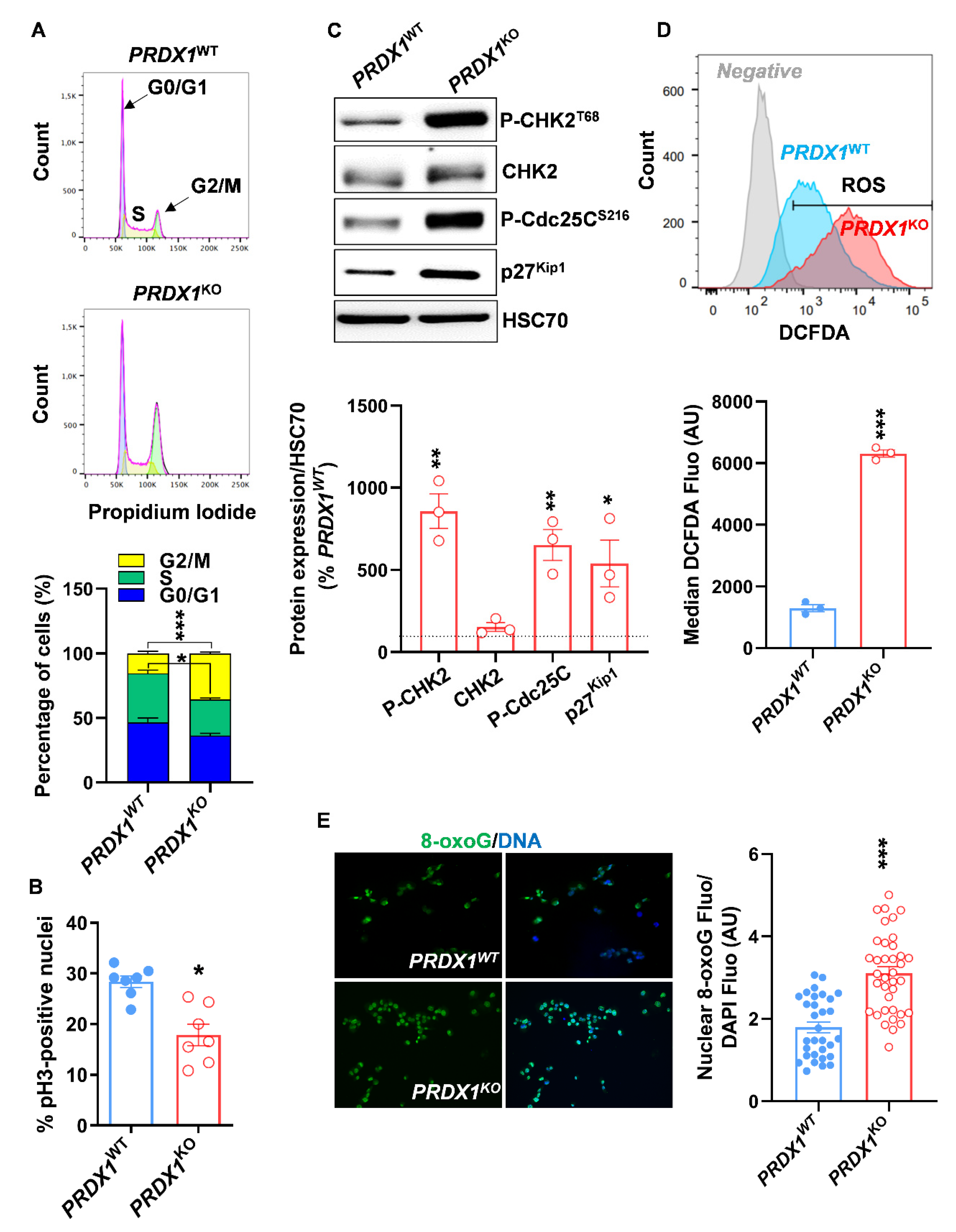
3.2. Genetic Ablation of PRX-I Induces a Prooxidant Response Leading to Cell Cycle Blockade and Reduced Growth of Human PDAC Cells
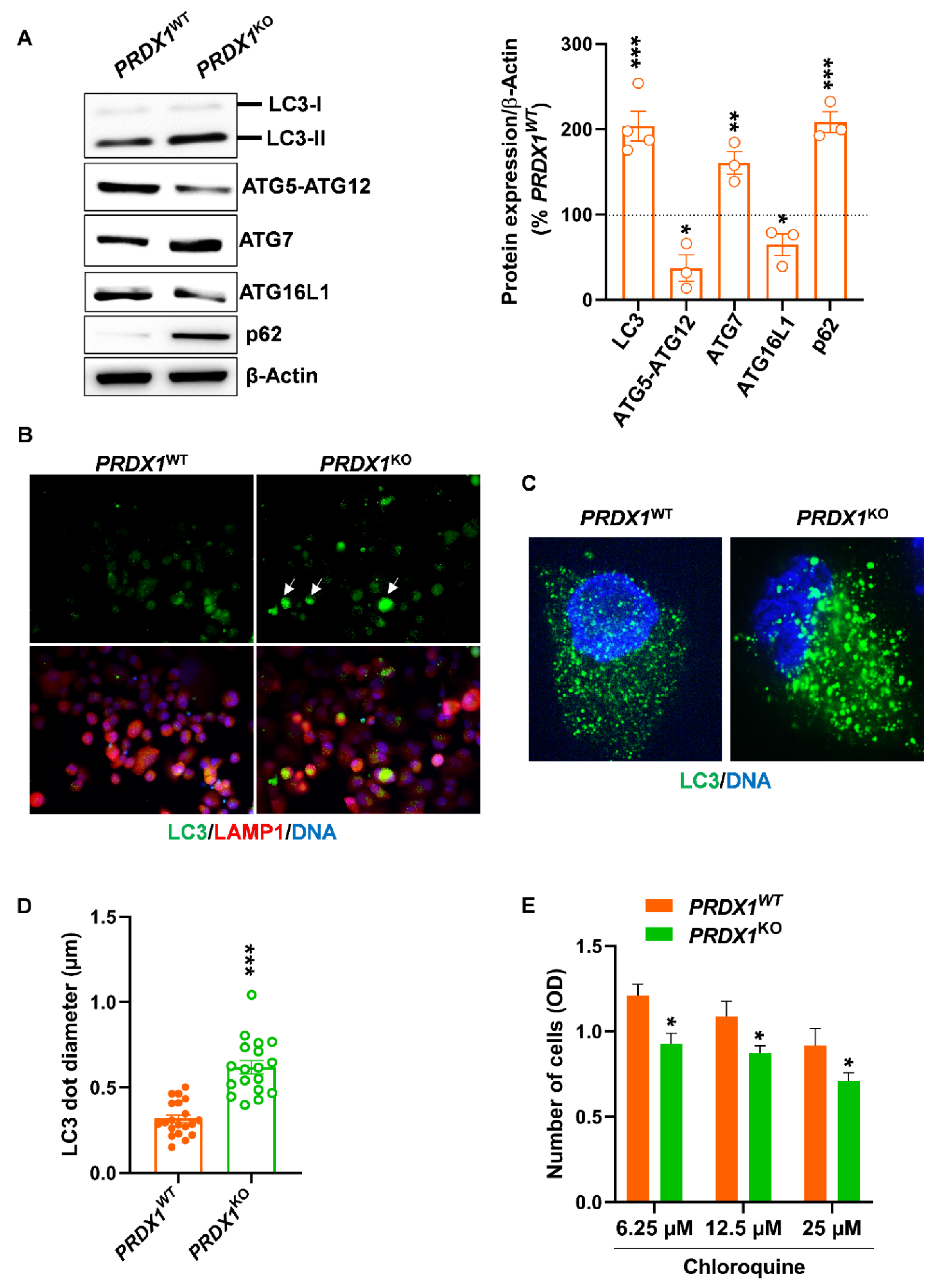
3.3. Genetic Ablation of PRX-I Alters the Autophagic Flux in PDAC Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 8-oxoG | 8-oxoguanine |
| ACC | Acetyl-CoA carboxylase |
| AMPK | AMP-activated protein kinase |
| ATG | Autophagy-related gene |
| Cdc25C | M-phase inducer phosphatase 3 |
| CHK2 | Checkpoint kinase 2 |
| ERK/MAPK | Extracellular signal-regulated kinases/mitogen-activated protein kinases |
| HRAS | Harvey rat sarcoma viral oncogene homolog protein |
| HSC70 | Heat-shock cognate 70 |
| JAK/STAT3 | Janus kinase/signal transducer and activator of transcription 3 |
| Kras | Kirsten rat sarcoma gene |
| KRAS | Kirsten rat sarcoma protein |
| LAMP1 | Lysosomal-associated membrane protein 1 |
| LC3 | Microtubule-associated protein 1A/1B-light chain |
| NRF2 | Nuclear factor erythroid 2-related factor 2 gene |
| NRF2 | Nuclear factor erythroid 2-related factor 2 protein |
| pH3 | Phospho-histone 3 |
| PRDX1 | Human Peroxiredoxin-1 gene |
| PRX-I | Peroxiredoxin-I protein |
| PRX-II | Peroxiredoxin-II protein |
| ROS | Reactive oxygen species |
References
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Tuveson, D.A.; Shaw, A.T.; Willis, N.A.; Silver, D.P.; Jackson, E.L.; Chang, S.; Mercer, K.L.; Grochow, R.; Hock, H.; Crowley, D.; et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 2004, 5, 375–387. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Guerra, C.; Schuhmacher, A.J.; Canamero, M.; Grippo, P.J.; Verdaguer, L.; Perez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assi, M.; Achouri, Y.; Loriot, A.; Dauguet, N.; Dahou, H.; Baldan, J.; Libert, M.; Fain, J.S.; Guerra, C.; Bouwens, L.; et al. Dynamic regulation of expression of KRAS and its effectors determines the ability to initiate tumorigenesis in pancreatic acinar cells. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Smith, J.P.; Solomon, T.E. Cholecystokinin and pancreatic cancer: The chicken or the egg? Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G91–G101. [Google Scholar] [CrossRef] [Green Version]
- Lamers, C.B.; Jansen, J.B.; Woutersen, R.A. Cholecystokinin and gastrointestinal cancer. J. Steroid Biochem. Mol. Biol. 1990, 37, 1069–1072. [Google Scholar] [CrossRef]
- Ceranowicz, P.; Warzecha, Z.; Dembinski, A. Peptidyl hormones of endocrine cells origin in the gut--their discovery and physiological relevance. J. Physiol. Pharmacol. 2015, 66, 11–27. [Google Scholar] [PubMed]
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Ohlund, D.; et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell 2016, 166, 963–976. [Google Scholar] [CrossRef] [Green Version]
- Cheung, E.C.; DeNicola, G.M.; Nixon, C.; Blyth, K.; Labuschagne, C.F.; Tuveson, D.A.; Vousden, K.H. Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 2020, 37, 168–182.e4. [Google Scholar] [CrossRef] [Green Version]
- Neumann, C.A.; Krause, D.S.; Carman, C.V.; Das, S.; Dubey, D.P.; Abraham, J.L.; Bronson, R.T.; Fujiwara, Y.; Orkin, S.H.; Van Etten, R.A. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature 2003, 424, 561–565. [Google Scholar] [CrossRef] [Green Version]
- Hampton, M.B.; Vick, K.A.; Skoko, J.J.; Neumann, C.A. Peroxiredoxin Involvement in the Initiation and Progression of Human Cancer. Antioxid. Redox Signal. 2018, 28, 591–608. [Google Scholar] [CrossRef] [PubMed]
- Ogusucu, R.; Rettori, D.; Munhoz, D.C.; Netto, L.E.; Augusto, O. Reactions of yeast thioredoxin peroxidases I and II with hydrogen peroxide and peroxynitrite: Rate constants by competitive kinetics. Free Radic. Biol. Med. 2007, 42, 326–334. [Google Scholar] [CrossRef]
- Park, Y.H.; Kim, S.U.; Lee, B.K.; Kim, H.S.; Song, I.S.; Shin, H.J.; Han, Y.H.; Chang, K.T.; Kim, J.M.; Lee, D.S.; et al. Prx I suppresses K-ras-driven lung tumorigenesis by opposing redox-sensitive ERK/cyclin D1 pathway. Antioxid. Redox Signal. 2013, 19, 482–496. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; Shin, H.J.; Bak, I.S.; Bak, Y.; Jeong, Y.L.; Kwon, T.; Park, Y.H.; Sun, H.N.; Kim, C.H.; Yu, D.Y. Peroxiredoxin I is important for cancer-cell survival in Ras-induced hepatic tumorigenesis. Oncotarget 2016, 7, 68044–68056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckens, H.; Pirenne, S.; Achouri, Y.; Baldan, J.; Dahou, H.; Bouwens, L.; Lemaigre, F.P.; Jacquemin, P.; Assi, M. Peroxiredoxin-I sustains inflammation during pancreatitis. Cell. Mol. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef]
- Taniuchi, K.; Furihata, M.; Hanazaki, K.; Iwasaki, S.; Tanaka, K.; Shimizu, T.; Saito, M.; Saibara, T. Peroxiredoxin 1 promotes pancreatic cancer cell invasion by modulating p38 MAPK activity. Pancreas 2015, 44, 331–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, B.M.; Oliver-Krasinski, J.; De Leon, D.D.; Farzad, C.; Hong, N.; Leach, S.D.; Stoffers, D.A. Preexisting pancreatic acinar cells contribute to acinar cell, but not islet beta cell, regeneration. J. Clin. Investig. 2007, 117, 971–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.Y.L.; Dubois, C.L.; Sarai, K.; Zarei, S.; Schaeffer, D.F.; Sander, M.; Kopp, J.L. Cell of origin affects tumour development and phenotype in pancreatic ductal adenocarcinoma. Gut 2019, 68, 487–498. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Assi, M.; Dauguet, N.; Jacquemin, P. DIE-RNA: A Reproducible Strategy for the Digestion of Normal and Injured Pancreas, Isolation of Pancreatic Cells from Genetically Engineered Mouse Models and Extraction of High Quality RNA. Front. Physiol. 2018, 9, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assi, M.; Pirlot, B.; Stroobant, V.; Thissen, J.P.; Jacquemin, P. A Novel KRAS Antibody Highlights a Regulation Mechanism of Post-Translational Modifications of KRAS during Tumorigenesis. Int. J. Mol. Sci. 2020, 21, 6361. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.Y.; Zhai, L.L.; Wu, Y.; Tang, Z.G. Expression and clinical value of peroxiredoxin-1 in patients with pancreatic cancer. Eur. J. Surg. Oncol. 2015, 41, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Froeling, F.E.M.; Swamynathan, M.M.; Deschenes, A.; Chio, I.I.C.; Brosnan, E.; Yao, M.A.; Alagesan, P.; Lucito, M.; Li, J.; Chang, A.Y.; et al. Bioactivation of Napabucasin Triggers Reactive Oxygen Species-Mediated Cancer Cell Death. Clin. Cancer Res. 2019, 25, 7162–7174. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Ahn, J.Y.; Liang, P.; Ip, C.; Zhang, Y.; Park, Y.M. Human prx1 gene is a target of Nrf2 and is up-regulated by hypoxia/reoxygenation: Implication to tumor biology. Cancer Res. 2007, 67, 546–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zhang, T.; Sun, W.; Wang, Z.; Zuo, D.; Zhou, Z.; Li, S.; Xu, J.; Yin, F.; Hua, Y.; et al. Erianin induces G2/M-phase arrest, apoptosis, and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells in vitro and in vivo. Cell Death Dis. 2016, 7, e2247. [Google Scholar] [CrossRef] [Green Version]
- Salehi, F.; Behboudi, H.; Kavoosi, G.; Ardestani, S.K. Oxidative DNA damage induced by ROS-modulating agents with the ability to target DNA: A comparison of the biological characteristics of citrus pectin and apple pectin. Sci. Rep. 2018, 8, 13902. [Google Scholar] [CrossRef]
- Lemoine, F.J.; Marriott, S.J. Accelerated G(1) phase progression induced by the human T cell leukemia virus type I (HTLV-I) Tax oncoprotein. J. Biol. Chem. 2001, 276, 31851–31857. [Google Scholar] [CrossRef] [Green Version]
- Arima, Y.; Nitta, M.; Kuninaka, S.; Zhang, D.; Fujiwara, T.; Taya, Y.; Nakao, M.; Saya, H. Transcriptional blockade induces p53-dependent apoptosis associated with translocation of p53 to mitochondria. J. Biol. Chem. 2005, 280, 19166–19176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runwal, G.; Stamatakou, E.; Siddiqi, F.H.; Puri, C.; Zhu, Y.; Rubinsztein, D.C. LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep. 2019, 9, 10147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G. AMPK and autophagy get connected. EMBO J. 2011, 30, 634–635. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Abudu, Y.P.; Claude-Taupin, A.; Gu, Y.; Kumar, S.; Choi, S.W.; Peters, R.; Mudd, M.H.; Allers, L.; Salemi, M.; et al. Galectins control MTOR and AMPK in response to lysosomal damage to induce autophagy. Autophagy 2019, 15, 169–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, A.T.; Hardie, D.G. The low activity of acetyl-CoA carboxylase in basal and glucagon-stimulated hepatocytes is due to phosphorylation by the AMP-activated protein kinase and not cyclic AMP-dependent protein kinase. FEBS Lett. 1988, 233, 294–298. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Ma, C.; Cao, Y.; Yang, L.; Huang, Z.; Qin, D.; Chen, Y.; Liu, C.; Xia, L.; Wang, T.; et al. Identification of H7 as a novel peroxiredoxin I inhibitor to induce differentiation of leukemia cells. Oncotarget 2016, 7, 3873–3883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Q.; Zhang, Y.; Cao, Y.; Wang, X.; Guo, Y.; Chen, J.; Horn, J.; Ponomareva, L.V.; Chaiswing, L.; Shaaban, K.A.; et al. Frenolicin B Targets Peroxiredoxin 1 and Glutaredoxin 3 to Trigger ROS/4E-BP1-Mediated Antitumor Effects. Cell Chem. Biol. 2019, 26, 366–377. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Reference | Dilution | Conditions |
|---|---|---|---|
| PRX-I | NBP1-82558, BioTechne Minneapolis, MN, USA | 1/1000 | 5% Milk, overnight, 4 °C |
| PRX-II | Sc-515428, SCB, Heidelberg, Germany | 1/500 | 5% Milk, overnight, 4 °C |
| P-STAT3Y705 | 9145S, CST, Leiden, The Netherlands | 1/1000 | 5% BSA, overnight, 4 °C |
| STAT3 | 12640S, CST, Leiden, The Netherlands | 1/1000 | 5% BSA, overnight, 4 °C |
| P-CHK2T68 | 2197S, CST, Leiden, The Netherlands | 1/1000 | 5% BSA, overnight, 4 °C |
| CHK2 | 2662S, CST, Leiden, The Netherlands | 1/1000 | 5% BSA, overnight, 4 °C |
| P-Cdc25CS216 | 4901S, CST, Leiden, The Netherlands | 1/1000 | 5% BSA, overnight, 4 °C |
| p27Kip1 | 3686S, CST, Leiden, The Netherlands | 1/1000 | 5% BSA, overnight, 4 °C |
| LC3 | 12741S, CST, Leiden, The Netherlands | 1/1000 | 5% BSA, overnight, 4 °C |
| ATG5-ATG12 | 4180T, CST, Leiden, The Netherlands | 1/1000 | 5% BSA, overnight, 4 °C |
| ATG7 | 8558T, CST, Leiden, The Netherlands | 1/1000 | 5% BSA, overnight, 4 °C |
| ATG16L1 | 8089T, CST, Leiden, The Netherlands | 1/1000 | 5% BSA, overnight, 4 °C |
| p62 | Sc-28359, SCB, Heidelberg, Germany | 1/500 | 5% Milk, overnight, 4 °C |
| HSC70 | Sc-7298, SCB, Heidelberg, Germany | 1/5000 | 5% Milk, overnight, 4 °C |
| β-Actin | A5441-100UL, SA, Overijse, Belgium | 1/5000 | 5% Milk, overnight, 4 °C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahou, H.; Minati, M.-A.; Jacquemin, P.; Assi, M. Genetic Inactivation of Peroxiredoxin-I Impairs the Growth of Human Pancreatic Cancer Cells. Antioxidants 2021, 10, 570. https://doi.org/10.3390/antiox10040570
Dahou H, Minati M-A, Jacquemin P, Assi M. Genetic Inactivation of Peroxiredoxin-I Impairs the Growth of Human Pancreatic Cancer Cells. Antioxidants. 2021; 10(4):570. https://doi.org/10.3390/antiox10040570
Chicago/Turabian StyleDahou, Hajar, Marie-Albane Minati, Patrick Jacquemin, and Mohamad Assi. 2021. "Genetic Inactivation of Peroxiredoxin-I Impairs the Growth of Human Pancreatic Cancer Cells" Antioxidants 10, no. 4: 570. https://doi.org/10.3390/antiox10040570
APA StyleDahou, H., Minati, M.-A., Jacquemin, P., & Assi, M. (2021). Genetic Inactivation of Peroxiredoxin-I Impairs the Growth of Human Pancreatic Cancer Cells. Antioxidants, 10(4), 570. https://doi.org/10.3390/antiox10040570