
Peroxiredoxins as Potential Targets for Cardiovascular Disease

1
Center for Cardiovascular Research, Washington University School of Medicine, St. Louis, MO 63110, USA
2
Biotherapeutics Translational Research Center, Korea Research Institute of Bioscience & Biotechnology (KRIBB), 125 Gwahak-ro, Yuseong-gu, Daejeon 34141, Korea
3
Department of Life Sciences, Heart-Immune-Brain Network Research Center, Ewha Womans University, 52 Ewhayeodae-gil, Seodaemun-gu, Seoul 03760, Korea
*
Authors to whom correspondence should be addressed.
Antioxidants 2021, 10(8), 1244; https://doi.org/10.3390/antiox10081244
Submission received: 22 July 2021
/
Accepted: 30 July 2021
/
Published: 3 August 2021
(This article belongs to the Special Issue Physiological and Pathological Significance of Peroxiredoxins)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Increased oxidative stress (OS) is considered a common etiology in the pathogenesis of cardiovascular disease (CVD). Therefore, the precise regulation of reactive oxygen species (ROS) in cardiovascular cells is essential to maintain normal physiological functions. Numerous regulators of cellular homeostasis are reportedly influenced by ROS. Hydrogen peroxide (H2O2), as an endogenous ROS in aerobic cells, is a toxic substance that can induce OS. However, many studies conducted over the past two decades have provided substantial evidence that H2O2 acts as a diffusible intracellular signaling messenger. Antioxidant enzymes, including superoxide dismutases, catalase, glutathione peroxidases, and peroxiredoxins (Prdxs), maintain the balance of ROS levels against augmentation of ROS production during the pathogenesis of CVD. Especially, Prdxs are regulatory sensors of transduced intracellular signals. The intracellular abundance of Prdxs that specifically react with H2O2 act as regulatory proteins. In this review, we focus on the role of Prdxs in the regulation of ROS-induced pathological changes in the development of CVD.
1. Introduction
1.1. Oxidative Stress (OS) and Cardiovascular Disease (CVD)
CVD is caused by various heterogeneous pathophysiological mechanisms, although increased OS is considered a potential common etiology [1,2,3]. Several factors control fluctuations in the concentrations of reactive oxygen species (ROS), including the sources of ROS production and antioxidant enzymes, as the precise regulation of ROS is essential to maintain normal physiological function of cardiovascular cells [4,5,6]. The regulation of cell differentiation and growth, as well as intracellular signaling molecules, such as phosphatases and kinases, are reportedly influenced by ROS to control cellular homeostasis [7,8,9,10,11,12]. However, an overabundance of ROS due to an imbalance between the production and removal of ROS damages intracellular molecules, including DNA, lipids, and proteins, leading to cellular dysfunctions and cell death via necrosis or apoptosis [13,14]. In cells, mitochondria and enzyme systems involving nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs) and xanthine oxidases are the main drivers of ROS production [15,16,17,18]. ROS constitute both nonradicals, including hydrogen peroxide (H2O2), hypochlorous acid, and ozone, and radicals, including superoxide, hydroxyl radicals, and peroxyl radicals [19]. Therefore, the actions of ROS differ according to the cell type, source, location, and concentration, which leads to a variety of physiological and pathophysiological functions [19].
Atherosclerosis is a chronic vascular inflammatory disease associated with the development of CVD, which is a leading cause of mortality and morbidity worldwide [20]. Atherogenic stimuli, including dyslipidemia, hypertension, and smoking, lead to endothelial dysfunction and structural alterations to the arterial walls and trigger the accumulation of circulating apolipoprotein B (apoB)-containing lipoproteins, mainly low-density lipoproteins (LDLs), in the intima region of the arterial wall [21,22]. Structural alterations to the aorta expose subendothelial proteoglycans, which contribute to the binding of apoB to LDLs under the endothelial layer [22]. ROS modify LDL particles, such as oxidized LDLs (oxLDLs), in the intima, which induces the expression of adhesion molecules, the release of chemokines by endothelial cells (ECs), and the recruitment of immune cells into the intima [3,23]. Monocytes recruited into the intima are differentiated into macrophages, which engulf oxLDLs through scavenger receptors and create inflammatory conditions in lesions by expressing various proinflammatory cytokines and the production of ROS and costimulatory molecules that activate other immune cells [23,24]. The excess engulfment of oxLDLs triggers the transformation of macrophages into foam cells, resulting in apoptotic cell death. The apoptosis of foam cells leads to the accumulation of cellular debris and cholesterol crystals that contribute to the development of the necrotic core, thereby inducing plaque destabilization [25]. Plaque rupture or erosion causes occlusive thrombosis, leading to acute cardiovascular events, which underlie mortality due to atherosclerotic CVD [24,26]. A schematic overview of ROS-mediated pathophysiology in the development of atherosclerosis is presented in Figure 1.
Numerous studies have demonstrated an important role of ROS in the development of atherosclerosis. NOX subunit gp91phox in macrophages and NOX4 in nonphagocytic vascular cells are upregulated in human atherosclerosis, and p47phox-deficient vascular smooth muscle cells (VSMCs) produce relatively lower levels of superoxide and undergo growth factor-induced proliferation [27,28]. NOX2-mediated superoxide production reduces the bioavailability of nitric oxide (NO) and augments the development of early atherosclerotic plaque formation in apolipoprotein E knockout (ApoE‒/‒) mice [29]. The deficiency of glutathione peroxidase (Gpx)-1 exacerbates atherosclerosis and loss of heme oxygenase-1 (HO-1) accelerates the development of atherosclerosis and vascular remodeling in ApoE‒/‒ mice [30,31].
The prevalence of abdominal aortic aneurysm (AAA) ranges from 2% to 8% in men older than 65 years and the mortality rate associated with ruptured AAA varies between 85% and 90% [20,32]. The development of AAA is associated with degraded plasticity of the tunica media with the activation of various proteases and inflammation caused by the accumulation of immune cells, angiogenesis, and necrosis [33]. ROS-induced OS is closely associated with inflammatory processes and the activation of matrix metalloproteinases (MMPs), which promote proteolytic degradation of structural proteins in the pathogenesis of AAA [34]. A schematic overview of ROS-mediated pathogenesis of AAA is presented in Figure 2.
The superoxide levels and lipid peroxidation are increased and the expression of the NOX subunits p47phox and p22phox are upregulated in the aneurysmal aorta as compared with the normal aorta of human patients [35]. Inducible NO synthase in human patients with AAA, but not normal human abdominal aortas, is identified by in situ hybridization and immunohistochemical analyses [36]. The deficiency of p47phox in ApoE‒/‒ mice attenuates angiotensin II (Ang II)-induced AAA formation with decreased OS, inflammation, and MMP-2 activity [37]. NOX1 deficiency reportedly prevents Ang II-induced aortic dissection with an increased expression of the tissue inhibitor of metalloproteinase-1 in mice as compared with the wild-type controls [38].
1.2. ROS-Mediated Pathophysiological Changes in Vascular Cells
OS and inflammation mainly induce dysfunction of ECs, resulting in the augmentation of vasoconstrictor and prothrombotic factors [39]. ECs modulate the vascular tone, cellular adhesion, thrombosis, smooth muscle cell proliferation, and vessel inflammation through the production of NO, prostaglandins, hyperpolarization factors, and contracting factors [40,41,42]. Therefore, the dysfunction of ECs is closely associated with an increased risk for the development of CVD and other diseases [39,43]. Endothelial nitric oxide synthase (NOS)-derived NO controls the contraction of vascular smooth muscle cells through the regulation of soluble guanylyl cyclase and cyclic guanosine monophosphate and inhibits leukocyte adhesion and platelet aggregation [42]. Cardiovascular risk factors, such as smoking, hypercholesterolemia, hypertension, and hyperglycemia, activate oxidative enzyme systems, including NOXs and uncoupled NOS, leading to the inactivation of NO by elevating superoxide levels [39,44]. The inactivation of NO induces an imbalance between EC-derived vasodilators and vasoconstrictors, resulting in endothelial dysfunction [42]. Inflammation also contributes to OS-mediated endothelial dysfunction in several human diseases, including CVD [45,46]. Proinflammatory stimuli, including oxLDL, tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1β), IL-6, and monocyte chemoattractant protein-1 (MCP-1), increase the production of ROS and induce endothelial dysfunction through a variety of signaling pathways involved in activation of the nuclear factor-kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) pathways [45,47,48,49]. The TNF-α-induced expression of vascular adhesion molecule-1 and intracellular adhesion molecule-1 in aortic ECs is inhibited by antioxidants and inhibitors of NF-κB and MAPK [50,51,52]. Inhibition of the oxidation of LDLs increases the expression of MCP-1 and adhesion molecules in ECs through the ROS- and NF-κB-dependent activation of lectin-like oxLDL receptor-1 [53,54]. In addition, OS and proinflammatory stimuli induce the apoptosis of ECs, resulting in atherogenesis and thrombosis, which are inhibited by superoxide dismutase (SOD), catalase, N-acetylcysteine (NAC), and vitamins [55,56]. Angiogenesis is also involved in the pathology of atherogenesis [57]. ROS regulates the migration, proliferation, and tubulogenesis of ECs by controlling intracellular signaling and angiogenic growth factor expression [43,57]. Taken together, these observations suggest that ROS is a critical factor for dysfunction and pathological signaling pathways in ECs in CVD (Figure 3).
ROS is closely involved in the regulation of the growth, migration, extracellular matrix production, and inflammatory gene expression in VSMCs [6]. The switching of VSMCs from the “contractile phenotype” to the “synthetic phenotype” is common in hypertension, atherosclerosis, aortic aneurysm, and restenosis after balloon angioplasty [58,59]. Phenotypic switching is a prerequisite for the proliferation and migration of VSMCs in the pathogenesis of CVD. Various factors, such as Ang II, platelet-derived growth factor (PDGF), and thrombin, induce ROS-dependent proliferation and the migration of VSMCs. Ang II induces the hypertrophy of VSMCs through the generation of ROS, which is inhibited by catalase and antisense p22phox [60,61]. PDGF and thrombin also induce the H2O2-dependent proliferation of VSMCs, which is inhibited by catalase, NAC, or a treatment with diphenyleneiodonium chloride [62,63]. The overexpression of catalase and peroxiredoxin (Prdx) 2 and inhibition of antioxidant production inhibit the PDGF-induced migration of VSMCs, implying that the PDGF-induced migration of VSMCs is dependent on ROS generation [11,62,64]. In addition, the production of the extracellular matrix and activation of inflammatory genes in VSMCs are regulated in a ROS-dependent manner. MMPs secreted from VSMCs are activated by ROS, and the NF-κB-mediated expression of inflammatory genes in VSMC is ROS-sensitive. The Ang II-induced expression of MCP-1 and IL-6 in VSMCs is also regulated in a ROS-dependent manner [65,66]. In addition, the TNF-α-induced MCP-1 expression in VSMCs is considered a redox-sensitive pathway [67]. Therefore, ROS-mediated alterations of the functions of VSMCs contribute to the progression of CVD (Figure 3).
Several antioxidant enzymes, including catalase, Gpxs, Prdxs, and SODs, are responsible for maintaining ROS levels against augmentation of ROS production during the pathogenesis of CVD. In this review, we focus on the role of Prdxs on the regulation of ROS-induced pathological changes in the development of CVD.
2. Prdxs in CVD
2.1. Overview of Prdxs
Prdxs, which consist of six Prdx isoforms (Prdx1–6) in mammalian cells, are conserved abundant cysteine (Cys)-based peroxidases involved in various biological functions [68,69]. Prdx1, 2, and 6 are localized in the cytosol and nucleus, while Prdx3 is restricted to the mitochondria, and Prdx5 is localized in the cytosol, mitochondria, and peroxisomes [70]. Prdxs are classified into typical 2-Cys, atypical 2-Cys, and 1-Cys Prdx subfamilies that possess diverse amino acid sequences, although all Prdxs have one conserved cysteine residue as the peroxidatic cysteine (CP) [68,71,72]. Typical 2-Cys Prdxs (Prdx1–4) contain two conserved cysteine residues (CP and a resolving Cys, CR) and consist of a homodimer formed via a disulfide bond between CP and CR in the chain of another subunit [68,72]. Atypical 2-Cys Prdx (Prdx5) contains an intrasubunit disulfide bond between CP and CR in the identical subunit, which is distinct from typical 2-Cys Prdxs [68,72]. In the 1-Cys Prdx (Prdx6), CP forms a disulfide bond with the CR of another protein or small thiol molecules because of the lack of CR in the 1-Cys Prdx [68,72]. A schematic of the catalytic mechanisms of Prdx isoforms is presented in Figure 4. By surrounding residues in the active site of Prdxs, the catalytic reactivity (rate constant, 106–108 M−1s−1) of CP for H2O2 is markedly augmented by up to one million-fold greater than other thiol proteins [73]. H2O2, as an endogenous form of ROS in aerobic cells, is considered a toxic substance that induces OS. However, many studies conducted over the past two decades have provided substantial evidence that H2O2 acts as a diffusible intracellular signaling messenger when it is transiently produced through the activation of cell surface receptors [74,75]. Prdxs rapidly react with H2O2, peroxynitrite, and other hydroperoxides but not with other electrophiles, indicating the target-specific reactivity of Prdxs [73,76]. Intracellular Prdxs that specifically react with H2O2 act as regulatory sensors during the transduction of intracellular signals [68,72].
2.2. Prdx1
Prdx1 is a typical 2-Cys Prdx that is expressed in the nucleus and cytosol of several cell types. Recent studies have elucidated various functional roles of Prdx1 in CVD. One found that laminar shear stress upregulates Prdx1, which functionally decreases the shear stress-dependent H2O2 levels and maintains the redox balance in ECs [77]. It is well-known that endogenous H2O2 formation induces the release of cytokines by activated ECs and inflammation, leading to atherosclerosis [78]. By scavenging H2O2, Prdx1 suppresses the excessive activation of ECs and consequent vascular inflammation, while Prdx1 deficiency induces early atherosclerosis in ApoE‒/‒ mice [79]. Moreover, another report suggested that lipophagy, the selective autophagic degradation of lipid in macrophages, is another possible protective role of Prdx1 in atherosclerosis [80]. Among the various antioxidant enzymes in macrophages, Prdx1 is highly expressed and is a major contributor to the maintenance of lipophagic flux and cholesterol homeostasis by regulating excessive H2O2 in macrophages and reducing foam cell formation and atherosclerosis [80]. While the mechanism underlying the role of Prdx1 in AAA is unknown, human studies have shown that the serum levels of Prdx1, which are increased in AAA patients [81], are positively correlated with the biomarkers plasmin-antiplasmin and myeloperoxidase and the diameter of AAA, suggesting that Prdx1 may be used as biomarker of AAA [81]. The overexpression of Prdx1 in cardiomyocytes of mice prevents transverse aortic constriction (TAC)-induced cardiac hypertrophy and heart failure, with decreased pressure overload-induced cardiac inflammation and OS via activation of the nuclear factor-erythroid 2-related factor 2/HO-1 signaling pathway [82]. In addition, the cardiomyocyte-specific expression of Prdx1 prevents doxorubicin-induced cardiotoxicity by reducing the OS and apoptosis [83]. Collectively, these results suggest that Prdx1 would be a useful target for the treatment of CVD.
2.3. Prdx2
Prdx2, a 2-Cys Prdx, is abundantly expressed in most cells and tissues. Although Prdx2 shares most of the features of typical 2-Cys Prdx with Prdx1, the expression patterns of Prdx2 in vascular cells appear to be relevant to its role in CVD. Prdx2 can suppress PDGF signaling by inhibiting protein tyrosine phosphatase in VSMCs and protects against PDGF-dependent neointimal thickening, which is a key feature of vascular remodeling [11]. The migration and proliferation of VSMCs during atherogenesis contribute to fibrous plaque formation, and PDGF is a strong growth factor that stimulates these pathologic processes [84]. The current evidence suggests that Prdx2 has a beneficial effect in CVD. One report demonstrated that Prdx2 is highly expressed in both vascular and immune cells, while Prdx2 deficiency accelerates atherosclerotic plaque formation and immune cell infiltration due to plaque progression, as compared with other types of antioxidant enzymes, such as Gpx1 and catalase [52]. Prdx2 expression is increased in the aneurysmal aorta of both human AAA patients and a mouse model of AAA, but the expression is relatively low in healthy humans and normal control mice, suggesting that Prdx2 is a potential biomarker of the progression of AAA [85]. In addition, Prdx2 deficiency induces the progression of AAA and most of the features of AAA tissue, including the OS, immune cell infiltration, MMP activation, elastin degradation, and aortic dilation [85]. Furthermore, Prdx2 negatively regulates H2O2 generation and thrombosis formation by platelets and VSMCs [86,87]. These findings suggest that Prdx2 may serve as a potential target for the treatment of CVD.
2.4. Prdx3
Prdx3 is a typical 2-Cys Prdx isoform that is exclusively localized in the mitochondria and contributes to the elimination of intracellular ROS, which is generated as a byproduct during energy production and cellular respiration. Mitochondria play central roles in various metabolic processes, including the production of cellular ATP through oxidative phosphorylation and the regulation of the intrinsic apoptosis pathway [88]. The increased mitochondrial production of ROS leads to mitochondrial DNA damage and mitochondrial dysfunction. To date, several kinds of intracellular antioxidant enzymes have been identified, especially Prdx3, which is a major antioxidant scavenger that can remove almost 90% of mitochondrial H2O2 to maintain mitochondrial homeostasis [89,90].
Prdx3 has protective roles in various cell types and ameliorates inflammation, which is highly associated with the pathogenesis of CVD. A recent study reported that the overexpression of Prdx3 protects the heart against left ventricular remodeling and failure after myocardial infarction (MI) [91]. The hallmarks of MI, such as reduced left ventricular cavity dilatation and dysfunction, myocyte hypertrophy, interstitial fibrosis, and apoptosis of the non-infarcted myocardium, are reduced in mice overexpressing Prdx3 [91]. Further, the redox state of Prdx3 significantly changes during ischemia–reperfusion heart injury [92]. The beneficial effects of Prdx-3 include maintaining cardiac function by the attenuation of the mitochondrial OS [92]. Although several studies have reported the protective roles of Prdx isoforms against atherosclerosis, the role of Prdx3 in the pathogenesis of atherosclerosis remains unclear. However, a possible mechanism of Prdx3 in atherosclerosis may be associated with mitophagy [93,94]. Mitophagy is a critical mechanism of cells to maintain metabolic homeostasis by eliminating damaged or long-lived mitochondria via autophagic degradation and has been linked with the pathogenesis of numerous human diseases, including cancers, neurodegenerative disorders, and CVD [95,96]. Although alterations to the expression of Prdx3 in mitophagy appear to be relevant, current data are insufficient to identify potential mechanisms in atherosclerosis. Nonetheless, recent studies have implicated potential roles of Prdx3 in various cell types associated with atherosclerosis [97,98].
2.5. Prdx4
Prdx4 is localized in the endoplasmic reticulum (ER), which is an important metabolic organelle involved in lipid and protein synthesis and transport by regulating the correct folding of proteins. The ER redox environment dictates the fate of entering proteins and the level of redox signaling mediators modulates the level of the ROS. Impairment of the ER leads to the cellular accumulation of ROS, and increased ER stress by ROS can induce apoptosis. Therefore, Prdx4 catabolizes H2O2 within the ER to provide a cytoprotective effect against the OS, acceleration of proper protein folding, and prevention of ER stress [99,100,101,102]. Prdx4 is a secretable peroxidase and, thus, a potential candidate biomarker. Prdx4 has recently been identified in the circulation of both healthy individuals and patients with peripheral artery disease (PAD) [103,104]. The overexpression of human Prdx4 in ApoE‒/‒ mice highly attenuated the development of atherosclerosis by limiting the infiltration of T-lymphocytes, reducing the OS, and ameliorating necrosis [105]. In ApoE‒/‒ mice with transplanted bone marrow, hematopoietic Prdx4 overexpression was sufficient to suppress the progression of atherosclerosis [105]. Therefore, Prdx4 is a potential target for treatment of CVD in the context of ER stress-induced apoptosis in the progression of atherosclerotic plaque formation [106].
2.6. Prdx5
Among the six Prdx isoforms, Prdx5 is the most recently discovered. Prdx5 is an atypical 2-Cys Prdx in mammals and is widely localized to the mitochondria, peroxisomes, cytosol, and nucleus. Although Prdx5 has unique features, such as localization in peroxisomes and expression as a short form and long form, which has a mitochondria targeting sequence, it has been studied less than other Prdxs [107]. Recent studies have found that Prdx5 prevents various pathological conditions by regulating the OS [108]. Prdx5 deficiency induces a susceptibility to high-fat diet-induced obesity and associated metabolic disorders [109]. Moreover, the depletion of Prdx5 significantly augmented kidney inflammation after renal ischemia–reperfusion injury [110]. Notably, extracellular Prdx5 is the strongest contributor among all Prdx isoforms to induce the expression of inflammatory cytokines through Toll-like receptor (TLR) 2 and TLR4 after a postischemic brain injury via damage-associated molecular patterns [111]. Additionally, Prdx5 can specifically bind with TLR4, and this interaction can modulate the release of proinflammatory cytokines and cell stiffening, which could be highly relevant in the inflammatory response [112]. However, several studies have indicated that intracellular Prdx5 is involved in the inhibition of inflammatory responses [113,114,115,116]. Prdx5 is constitutively expressed in human cartilage and is upregulated in osteoarthritic cartilage, suggesting that Prdx5 may have a protective function against OS in human cartilage [113]. In primary macrophages, Prdx5 expression is induced by inflammatory stimuli, such as lipopolysaccharides and interferon-γ, suggesting that Prdx5 plays a defensive role in activated macrophages [114]. However, further studies are needed to clarify the role of Prdx5 in the pathogenesis of CVD.
2.7. Prdx6
Prdx6 has a unique 1-Cys structure but similar functional and structural properties with other Prdx isoforms. Prdx6 has peroxidase activity via a single conserved Cys residue, short-chain organic fatty acids, and phospholipid hydroperoxides by using glutathione instead of thioredoxin as a physiological reductant. Moreover, Prdx6 is a bifunctional enzyme with glutathione peroxidase and phospholipase A2 (PLA2) activities, which characteristically responds to various extracellular stimuli [117,118]. Several recent studies have provided evidence of the role of Prdx6 in CVD. As a possible link between Prdx6 and atherosclerosis, as mentioned above, Prdx6 possesses PLA2 activity. Previous studies have demonstrated that members of the PLA2 family generate various active lipid metabolites that promote OS and inflammatory cytokine production, leading to the acceleration of inflammatory metabolic diseases, such as atherosclerosis and hyperlipidemia [119]. Additionally, Prdx6 has an important role in heart recovery following ischemia–reperfusion injury and can protect against phospholipid peroxidation-mediated membrane damage [120,121]. Prdx6-deficient mice are more susceptible to ischemia–reperfusion injury, with a reduced recovery of left ventricular function, increased MI area, and augmented apoptotic cell death [120]. The overexpression of Prdx6 in ECs prevents AngII-induced inflammation, OS, and endothelial dysfunction via inactivation of the p38 MAPK and c-Jun N-terminal kinase pathways [122]. In addition, a significant increase in plasma levels of Prdx6 has been positively correlated with endothelial dysfunction in diabetic patients with PAD [104]. Although Prdx6 has been extensively studied, more detailed studies are necessary to elucidate the role of Prdx6 in the pathogenesis of atherosclerosis. Prdx6 is reported to play a minor athero-protective role in mice with a mixed B6; 129 background [123]. Prdx6 deficiency induces higher plasma levels of lipid hydroperoxides and the oxidation of LDLs, whereas Prdx6 in transgenic mice was insufficient to protect against atherogenesis [123,124].
3. Conclusions and Future Perspectives
OS, which is a common feature of CVD, is thought to occur by an imbalance in the production and removal of ROS in cardiovascular cells. Various experimental studies of dietary antioxidants and antioxidant enzymes to block the abundance of OS in CVD revealed an efficient inverse correlation. However, dietary antioxidant therapies for patients with CVD have failed in many randomized trials. To overcome the limitation of dietary antioxidants, the development of antioxidant enzymes, such as Prdxs, is a potential therapeutic strategy against CVD. Prdxs are a family of abundant thiol-dependent peroxidases, consisting of six isoforms in mammalian cells. As sensor proteins, Prdxs are appropriate antioxidant enzymes for the regulation of OS and H2O2-mediated intracellular signaling. Various experimental studies have confirmed the potential of Prdxs as a therapeutic strategy against CVD (Figure 5). Therefore, the development of derivatives or mimetics of the catalytic activity of Prdxs offers great promise for antioxidant therapy in CVD. The evolution of biotechnologies in the pharmaceutical industry could provide unique derivatives or mimetics for each Prdx isoform to develop precise medications matched to the pathophysiology of CVD.
Author Contributions
Conceptualization, planning and execution: S.-J.J., J.-G.P.; conceptualization, review, and editing: G.T.O. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Korea Research Institute of Bioscience and Biotechnology (KRIBB) Research Initiative Program (grant no. KGM5272151), by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (grant no. 2020R1A3B2079811), and the Basic Science Research Program of the National Research Foundation of Korea (NRF) funded by the Ministry of Education (grant no. NRF-2021R1I1A2056805).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| EC | Endothelial cell |
| OS | Oxidative stress |
| CVD | Cardiovascular disease |
| ROS | Reactive oxygen species |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NOX | NADPH oxidase |
| ApoB | Apolipoprotein B |
| LDL | Low-density lipoprotein |
| oxLDL | oxidized LDL |
| VSMCs | Vascular smooth muscle cells |
| NO | Nitric oxide |
| ApoE‒/‒ | Apolipoprotein E knockout |
| Gpx | Glutathione peroxidase |
| HO-1 | Heme oxygenase-1 |
| AAA | Abdominal aortic aneurysm |
| MMPs | Matrix metalloproteinases |
| Ang II | Angiotensin II |
| TNF-α | Tumor necrosis factor-alpha |
| IL | Interleukin |
| MCP-1 | Monocyte chemoattractant protein-1 |
| NF-κB | Nuclear factor-kappa B |
| MAPK | Mitogen-activated protein kinase |
| SOD | Superoxide dismutase |
| NAC | N-acetylcysteine |
| PDGF | Platelet-derived growth factor |
| Prdx | Peroxiredoxin |
| Cys | Cysteine |
| CP | Peroxidatic cysteine |
| CR | Resolving Cys |
| TAC | Transverse aortic constriction |
| MI | Myocardial infarction |
| ER | Endoplasmic reticulum |
| PAD | Peripheral artery disease |
| TLR | Toll-like receptor |
| PLA2 | Phospholipase A2 |
References
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [Green Version]
- Baradaran, A.; Nasri, H.; Rafieian-Kopaei, M. Oxidative stress and hypertension: Possibility of hypertension therapy with antioxidants. J. Res. Med. Sci. 2014, 19, 358–367. [Google Scholar] [PubMed]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative stress in atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef]
- Cervantes Gracia, K.; Llanas-Cornejo, D.; Husi, H. Cvd and oxidative stress. J. Clin. Med. 2017, 6, 22. [Google Scholar] [CrossRef] [Green Version]
- Taniyama, Y.; Griendling, K.K. Reactive oxygen species in the vasculature: Molecular and cellular mechanisms. Hypertension 2003, 42, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Mittal, C.K.; Murad, F. Activation of guanylate cyclase by superoxide dismutase and hydroxyl radical: A physiological regulator of guanosine 3′,5′-monophosphate formation. Proc. Natl. Acad. Sci. USA 1977, 74, 4360–4364. [Google Scholar] [CrossRef] [Green Version]
- Olguin-Albuerne, M.; Moran, J. Ros produced by nox2 control in vitro development of cerebellar granule neurons development. ASN Neuro. 2015, 7, 1759091415578712. [Google Scholar] [CrossRef] [PubMed]
- Mandal, D.; Fu, P.; Levine, A.D. Redox regulation of il-13 signaling in intestinal epithelial cells: Usage of alternate pathways mediates distinct gene expression patterns. Cell Signal. 2010, 22, 1485–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abimannan, T.; Peroumal, D.; Parida, J.R.; Barik, P.K.; Padhan, P.; Devadas, S. Oxidative stress modulates the cytokine response of differentiated th17 and th1 cells. Free Radic. Biol. Med. 2016, 99, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.H.; Lee, I.K.; Kim, G.W.; Kim, B.U.; Han, Y.H.; Yu, D.Y.; Park, H.S.; Kim, K.Y.; Lee, J.S.; Choi, C.; et al. Regulation of pdgf signalling and vascular remodelling by peroxiredoxin ii. Nature 2005, 435, 347–353. [Google Scholar]
- Fujino, G.; Noguchi, T.; Matsuzawa, A.; Yamauchi, S.; Saitoh, M.; Takeda, K.; Ichijo, H. Thioredoxin and traf family proteins regulate reactive oxygen species-dependent activation of ask1 through reciprocal modulation of the n-terminal homophilic interaction of ask1. Mol. Cell Biol. 2007, 27, 8152–8163. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ros and rns sources in physiological and pathological conditions. Oxid. Med. Cell Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. Nadph oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [Green Version]
- Amanso, A.M.; Griendling, K.K. Differential roles of nadph oxidases in vascular physiology and pathophysiology. Front. BioSci. 2012, 4, 1044–1064. [Google Scholar]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase—Mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008, 275, 3278–3289. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart disease and stroke statistics-2018 update: A report from the american heart association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef] [PubMed]
- Maiolino, G.; Rossitto, G.; Caielli, P.; Bisogni, V.; Rossi, G.P.; Calo, L.A. The role of oxidized low-density lipoproteins in atherosclerosis: The myths and the facts. Mediat. Inflamm. 2013, 2013, 714653. [Google Scholar] [CrossRef] [Green Version]
- Kwon, G.P.; Schroeder, J.L.; Amar, M.J.; Remaley, A.T.; Balaban, R.S. Contribution of macromolecular structure to the retention of low-density lipoprotein at arterial branch points. Circulation 2008, 117, 2919–2927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef]
- Sakakura, K.; Nakano, M.; Otsuka, F.; Ladich, E.; Kolodgie, F.D.; Virmani, R. Pathophysiology of atherosclerosis plaque progression. Heart Lung Circ. 2013, 22, 399–411. [Google Scholar] [CrossRef] [Green Version]
- Yla-Herttuala, S.; Bentzon, J.F.; Daemen, M.; Falk, E.; Garcia-Garcia, H.M.; Herrmann, J.; Hoefer, I.; Jukema, J.W.; Krams, R.; Kwak, B.R.; et al. Stabilisation of atherosclerotic plaques. Position paper of the european society of cardiology (esc) working group on atherosclerosis and vascular biology. Thromb. Haemost. 2011, 106, 1–19. [Google Scholar]
- Sugiyama, S.; Okada, Y.; Sukhova, G.K.; Virmani, R.; Heinecke, J.W.; Libby, P. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am. J. Pathol. 2001, 158, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Sorescu, D.; Weiss, D.; Lassegue, B.; Clempus, R.E.; Szocs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [Green Version]
- Barry-Lane, P.A.; Patterson, C.; van der Merwe, M.; Hu, Z.; Holland, S.M.; Yeh, E.T.; Runge, M.S. P47phox is required for atherosclerotic lesion progression in apoe(-/-) mice. J. Clin. Investig. 2001, 108, 1513–1522. [Google Scholar] [CrossRef]
- Judkins, C.P.; Diep, H.; Broughton, B.R.; Mast, A.E.; Hooker, E.U.; Miller, A.A.; Selemidis, S.; Dusting, G.J.; Sobey, C.G.; Drummond, G.R. Direct evidence of a role for nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in apoe-/- mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H24–H32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torzewski, M.; Ochsenhirt, V.; Kleschyov, A.L.; Oelze, M.; Daiber, A.; Li, H.; Rossmann, H.; Tsimikas, S.; Reifenberg, K.; Cheng, F.; et al. Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein e-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 850–857. [Google Scholar] [CrossRef] [Green Version]
- Yet, S.F.; Layne, M.D.; Liu, X.; Chen, Y.H.; Ith, B.; Sibinga, N.E.; Perrella, M.A. Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 2003, 17, 1759–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, K.C. Clinical practice. Abdominal aortic aneurysms. N. Engl. J. Med. 2014, 371, 2101–2108. [Google Scholar] [CrossRef]
- Sakalihasan, N.; Limet, R.; Defawe, O.D. Abdominal aortic aneurysm. Lancet 2005, 365, 1577–1589. [Google Scholar] [CrossRef]
- Freestone, T.; Turner, R.J.; Coady, A.; Higman, D.J.; Greenhalgh, R.M.; Powell, J.T. Inflammation and matrix metalloproteinases in the enlarging abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.J., Jr.; Sharp, W.J.; Fang, X.; Oberley, L.W.; Oberley, T.D.; Weintraub, N.L. Oxidative stress in human abdominal aortic aneurysms: A potential mediator of aneurysmal remodeling. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 560–565. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Schmidt, J.; Ryschich, E.; Mueller-Schilling, M.; Schumacher, H.; Allenberg, J.R. Inducible nitric oxide synthase is present in human abdominal aortic aneurysm and promotes oxidative vascular injury. J. Vasc. Surg. 2003, 38, 360–367. [Google Scholar] [CrossRef]
- Thomas, M.; Gavrila, D.; McCormick, M.L.; Miller, F.J., Jr.; Daugherty, A.; Cassis, L.A.; Dellsperger, K.C.; Weintraub, N.L. Deletion of p47phox attenuates angiotensin ii-induced abdominal aortic aneurysm formation in apolipoprotein e-deficient mice. Circulation 2006, 114, 404–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavazzi, G.; Deffert, C.; Trocme, C.; Schappi, M.; Herrmann, F.R.; Krause, K.H. Nox1 deficiency protects from aortic dissection in response to angiotensin ii. Hypertension 2007, 50, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Hadi, H.A.; Carr, C.S.; Al Suwaidi, J. Endothelial dysfunction: Cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag. 2005, 1, 183–198. [Google Scholar] [PubMed]
- Godo, S.; Shimokawa, H. Endothelial functions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef] [Green Version]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Maulik, N.; Das, D.K. Redox signaling in vascular angiogenesis. Free Radic. Biol. Med. 2002, 33, 1047–1060. [Google Scholar] [CrossRef]
- Favero, G.; Paganelli, C.; Buffoli, B.; Rodella, L.F.; Rezzani, R. Endothelium and its alterations in cardiovascular diseases: Life style intervention. Biomed. Res. Int. 2014, 2014, 801896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, G.L.; Lesniewski, L.A.; Lawson, B.R.; Beske, S.D.; Seals, D.R. Nuclear factor-{kappa}b activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans. Circulation 2009, 119, 1284–1292. [Google Scholar] [CrossRef] [Green Version]
- Glass, C.K.; Witztum, J.L. Atherosclerosis. The road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Vora, D.K.; Fang, Z.T.; Liva, S.M.; Tyner, T.R.; Parhami, F.; Watson, A.D.; Drake, T.A.; Territo, M.C.; Berliner, J.A. Induction of p-selectin by oxidized lipoproteins. Separate effects on synthesis and surface expression. Circ. Res. 1997, 80, 810–818. [Google Scholar] [CrossRef]
- Takei, A.; Huang, Y.; Lopes-Virella, M.F. Expression of adhesion molecules by human endothelial cells exposed to oxidized low density lipoprotein. Influences of degree of oxidation and location of oxidized ldl. Atherosclerosis 2001, 154, 79–86. [Google Scholar] [CrossRef]
- Khan, B.V.; Parthasarathy, S.S.; Alexander, R.W.; Medford, R.M. Modified low density lipoprotein and its constituents augment cytokine-activated vascular cell adhesion molecule-1 gene expression in human vascular endothelial cells. J. Clin. Investig. 1995, 95, 1262–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marui, N.; Offermann, M.K.; Swerlick, R.; Kunsch, C.; Rosen, C.A.; Ahmad, M.; Alexander, R.W.; Medford, R.M. Vascular cell adhesion molecule-1 (vcam-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J. Clin. Investig. 1993, 92, 1866–1874. [Google Scholar] [CrossRef]
- Chappell, D.C.; Varner, S.E.; Nerem, R.M.; Medford, R.M.; Alexander, R.W. Oscillatory shear stress stimulates adhesion molecule expression in cultured human endothelium. Circ. Res. 1998, 82, 532–539. [Google Scholar] [CrossRef] [Green Version]
- Park, J.G.; Yoo, J.Y.; Jeong, S.J.; Choi, J.H.; Lee, M.R.; Lee, M.N.; Hwa Lee, J.; Kim, H.C.; Jo, H.; Yu, D.Y.; et al. Peroxiredoxin 2 deficiency exacerbates atherosclerosis in apolipoprotein e-deficient mice. Circ. Res. 2011, 109, 739–749. [Google Scholar] [CrossRef]
- Cushing, S.D.; Berliner, J.A.; Valente, A.J.; Territo, M.C.; Navab, M.; Parhami, F.; Gerrity, R.; Schwartz, C.J.; Fogelman, A.M. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc. Natl. Acad. Sci. USA 1990, 87, 5134–5138. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Masaki, T.; Sawamura, T. Lox-1, the receptor for oxidized low-density lipoprotein identified from endothelial cells: Implications in endothelial dysfunction and atherosclerosis. Pharmacol. Ther. 2002, 95, 89–100. [Google Scholar] [CrossRef]
- Dimmeler, S.; Zeiher, A.M. Reactive oxygen species and vascular cell apoptosis in response to angiotensin ii and pro-atherosclerotic factors. Regul. Pept. 2000, 90, 19–25. [Google Scholar] [CrossRef]
- Li, A.E.; Ito, H.; Rovira, I.I.; Kim, K.S.; Takeda, K.; Yu, Z.Y.; Ferrans, V.J.; Finkel, T. A role for reactive oxygen species in endothelial cell anoikis. Circ. Res. 1999, 85, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Camare, C.; Pucelle, M.; Negre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular smooth muscle cells in atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Petsophonsakul, P.; Furmanik, M.; Forsythe, R.; Dweck, M.; Schurink, G.W.; Natour, E.; Reutelingsperger, C.; Jacobs, M.; Mees, B.; Schurgers, L. Role of vascular smooth muscle cell phenotypic switching and calcification in aortic aneurysm formation. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1351–1368. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Zafari, A.M.; Fukui, T.; Ishizaka, N.; Griendling, K.K. P22phox is a critical component of the superoxide-generating nadh/nadph oxidase system and regulates angiotensin ii-induced hypertrophy in vascular smooth muscle cells. J. Biol. Chem. 1996, 271, 23317–23321. [Google Scholar] [CrossRef] [Green Version]
- Zafari, A.M.; Ushio-Fukai, M.; Akers, M.; Yin, Q.; Shah, A.; Harrison, D.G.; Taylor, W.R.; Griendling, K.K. Role of nadh/nadph oxidase-derived h2o2 in angiotensin ii-induced vascular hypertrophy. Hypertension 1998, 32, 488–495. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of h2o2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, C.; Ruef, J.; Madamanchi, N.R.; Barry-Lane, P.; Hu, Z.; Horaist, C.; Ballinger, C.A.; Brasier, A.R.; Bode, C.; Runge, M.S. Stimulation of a vascular smooth muscle cell nad(p)h oxidase by thrombin. Evidence that p47(phox) may participate in forming this oxidase in vitro and in vivo. J. Biol. Chem. 1999, 274, 19814–19822. [Google Scholar] [CrossRef] [Green Version]
- San Martin, A.; Griendling, K.K. Redox control of vascular smooth muscle migration. Antioxid. Redox Signal. 2010, 12, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Tummala, P.E.; Olbrych, M.T.; Alexander, R.W.; Medford, R.M. Angiotensin ii induces monocyte chemoattractant protein-1 gene expression in rat vascular smooth muscle cells. Circ. Res. 1998, 83, 952–959. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Runge, M.S.; Brasier, A.R. Angiotensin ii induces interleukin-6 transcription in vascular smooth muscle cells through pleiotropic activation of nuclear factor-kappa b transcription factors. Circ. Res. 1999, 84, 695–703. [Google Scholar] [CrossRef]
- De Keulenaer, G.W.; Ushio-Fukai, M.; Yin, Q.; Chung, A.B.; Lyons, P.R.; Ishizaka, N.; Rengarajan, K.; Taylor, W.R.; Alexander, R.W.; Griendling, K.K. Convergence of redox-sensitive and mitogen-activated protein kinase signaling pathways in tumor necrosis factor-alpha-mediated monocyte chemoattractant protein-1 induction in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 385–391. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.G.; Kang, S.W.; Chang, T.S.; Jeong, W.; Kim, K. Peroxiredoxin, a novel family of peroxidases. IUBMB Life 2001, 52, 35–41. [Google Scholar] [CrossRef]
- Szeliga, M. Peroxiredoxins in neurodegenerative diseases. Antioxidants 2020, 9, 1203. [Google Scholar] [CrossRef]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef]
- Chae, H.Z.; Robison, K.; Poole, L.B.; Church, G.; Storz, G.; Rhee, S.G. Cloning and sequencing of thiol-specific antioxidant from mammalian brain: Alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc. Natl. Acad. Sci. USA 1994, 91, 7017–7021. [Google Scholar] [CrossRef] [Green Version]
- Wood, Z.A.; Schroder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Peskin, A.V.; Low, F.M.; Paton, L.N.; Maghzal, G.J.; Hampton, M.B.; Winterbourn, C.C. The high reactivity of peroxiredoxin 2 with h(2)o(2) is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 2007, 282, 11885–11892. [Google Scholar] [CrossRef] [Green Version]
- Ying, J.; Clavreul, N.; Sethuraman, M.; Adachi, T.; Cohen, R.A. Thiol oxidation in signaling and response to stress: Detection and quantification of physiological and pathophysiological thiol modifications. Free Radic. Biol. Med. 2007, 43, 1099–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.G.; Bae, Y.S.; Lee, S.R.; Kwon, J. Hydrogen peroxide: A key messenger that modulates protein phosphorylation through cysteine oxidation. Sci. STKE 2000, 2000, pe1. [Google Scholar] [CrossRef]
- Hall, A.; Nelson, K.; Poole, L.B.; Karplus, P.A. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid. Redox Signal. 2011, 15, 795–815. [Google Scholar] [CrossRef] [Green Version]
- Mowbray, A.L.; Kang, D.H.; Rhee, S.G.; Kang, S.W.; Jo, H. Laminar shear stress up-regulates peroxiredoxins (prx) in endothelial cells: Prx 1 as a mechanosensitive antioxidant. J. Biol. Chem. 2008, 283, 1622–1627. [Google Scholar] [CrossRef] [Green Version]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III27–III32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisucka, J.; Chauhan, A.K.; Patten, I.S.; Yesilaltay, A.; Neumann, C.; Van Etten, R.A.; Krieger, M.; Wagner, D.D. Peroxiredoxin1 prevents excessive endothelial activation and early atherosclerosis. Circ. Res. 2008, 103, 598–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.J.; Kim, S.; Park, J.G.; Jung, I.H.; Lee, M.N.; Jeon, S.; Kweon, H.Y.; Yu, D.Y.; Lee, S.H.; Jang, Y.; et al. Prdx1 (peroxiredoxin 1) deficiency reduces cholesterol efflux via impaired macrophage lipophagic flux. Autophagy 2018, 14, 120–133. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Pinna, R.; Ramos-Mozo, P.; Madrigal-Matute, J.; Blanco-Colio, L.M.; Lopez, J.A.; Calvo, E.; Camafeita, E.; Lindholt, J.S.; Meilhac, O.; Delbosc, S.; et al. Identification of peroxiredoxin-1 as a novel biomarker of abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 935–943. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Yin, G.; Huang, C.; Wang, H.; Gao, J.; Luo, J.; Zhang, Z.; Wang, J.; Hong, J.; Chai, X. Peroxiredoxin-1 ameliorates pressure overload-induced cardiac hypertrophy and fibrosis. Biomed. Pharmacother. 2020, 129, 110357. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Gong, Y.; Hu, Y.; You, Y.; Wang, J.; Zhang, Z.; Wei, Z.; Tang, C. Peroxiredoxin-1 overexpression attenuates doxorubicin-induced cardiotoxicity by inhibiting oxidative stress and cardiomyocyte apoptosis. Oxid Med. Cell Longev. 2020, 2020, 2405135. [Google Scholar] [CrossRef] [PubMed]
- Basatemur, G.L.; Jorgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [CrossRef]
- Jeong, S.J.; Cho, M.J.; Ko, N.Y.; Kim, S.; Jung, I.H.; Min, J.K.; Lee, S.H.; Park, J.G.; Oh, G.T. Deficiency of peroxiredoxin 2 exacerbates angiotensin ii-induced abdominal aortic aneurysm. Exp. Mol. Med. 2020, 52, 1587–1601. [Google Scholar] [CrossRef]
- Jang, J.Y.; Wang, S.B.; Min, J.H.; Chae, Y.H.; Baek, J.Y.; Yu, D.Y.; Chang, T.S. Peroxiredoxin ii is an antioxidant enzyme that negatively regulates collagen-stimulated platelet function. J. Biol. Chem. 2015, 290, 11432–11442. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Febbraio, M.; Reddy, S.P.; Yu, D.Y.; Yamamoto, M.; Silverstein, R.L. Cd36 participates in a signaling pathway that regulates ros formation in murine vsmcs. J. Clin. Investig. 2010, 120, 3996–4006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Maharjan, S.; Oku, M.; Tsuda, M.; Hoseki, J.; Sakai, Y. Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Sci. Rep. 2014, 4, 5896. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J. Mitochondrial antioxidants and the maintenance of cellular hydrogen peroxide levels. Oxid. Med. Cell Longev. 2018, 2018, 7857251. [Google Scholar] [CrossRef]
- Matsushima, S.; Ide, T.; Yamato, M.; Matsusaka, H.; Hattori, F.; Ikeuchi, M.; Kubota, T.; Sunagawa, K.; Hasegawa, Y.; Kurihara, T.; et al. Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2006, 113, 1779–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Kitaeff, N.; Hampton, M.B.; Cannell, M.B.; Winterbourn, C.C. Reversible oxidation of mitochondrial peroxiredoxin 3 in mouse heart subjected to ischemia and reperfusion. FEBS Lett. 2009, 583, 997–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and mitophagy in cardiovascular disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Nikiforov, N.G.; Wu, W.K.; Kirichenko, T.V.; Orekhov, A.N. Autophagy and mitophagy as essential components of atherosclerosis. Cells 2021, 10, 443. [Google Scholar] [CrossRef]
- Vasquez-Trincado, C.; Garcia-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef]
- Um, J.H.; Yun, J. Emerging role of mitophagy in human diseases and physiology. BMB Rep. 2017, 50, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.Y.; Han, Z.D.; Li, W.; Yue, F.; Ye, J.; Li, B.; Cai, Z.; Lu, J.M.; Dong, W.; Jiang, X.; et al. Mitochondrion-associated protein peroxiredoxin 3 promotes benign prostatic hyperplasia through autophagy suppression and pyroptosis activation. Oncotarget 2017, 8, 80295–80302. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Lv, W.; Lu, C.; Zhao, X.; Zhang, C.; Song, H. Lats2 promotes cardiomyocyte h9c2 cells apoptosis via the prx3-mfn2-mitophagy pathways. J. Recept Signal. Transduct Res. 2019, 39, 470–478. [Google Scholar] [CrossRef]
- Zito, E.; Melo, E.P.; Yang, Y.; Wahlander, A.; Neubert, T.A.; Ron, D. Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Mol. Cell 2010, 40, 787–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Kojima, R.; Okumura, M.; Hagiwara, M.; Masui, S.; Maegawa, K.; Saiki, M.; Horibe, T.; Suzuki, M.; Inaba, K. Synergistic cooperation of pdi family members in peroxiredoxin 4-driven oxidative protein folding. Sci. Rep. 2013, 3, 2456. [Google Scholar] [CrossRef]
- Zito, E. Prdx4, an endoplasmic reticulum-localized peroxiredoxin at the crossroads between enzymatic oxidative protein folding and nonenzymatic protein oxidation. Antioxid. Redox Signal. 2013, 18, 1666–1674. [Google Scholar] [CrossRef]
- Abbasi, A.; Corpeleijn, E.; Postmus, D.; Gansevoort, R.T.; de Jong, P.E.; Gans, R.O.; Struck, J.; Schulte, J.; Hillege, H.L.; van der Harst, P.; et al. Peroxiredoxin 4, a novel circulating biomarker for oxidative stress and the risk of incident cardiovascular disease and all-cause mortality. J. Am. Heart Assoc. 2012, 1, e002956. [Google Scholar] [CrossRef] [Green Version]
- El Eter, E.; Al Masri, A.; Habib, S.; Al Zamil, H.; Al Hersi, A.; Al Hussein, F.; Al Omran, M. Novel links among peroxiredoxins, endothelial dysfunction, and severity of atherosclerosis in type 2 diabetic patients with peripheral atherosclerotic disease. Cell Stress Chaperones 2014, 19, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Yamada, S.; Tanimoto, A.; Ding, Y.; Wang, K.Y.; Shimajiri, S.; Murata, Y.; Kimura, S.; Tasaki, T.; Nabeshima, A.; et al. Overexpression of peroxiredoxin 4 attenuates atherosclerosis in apolipoprotein e knockout mice. Antioxid. Redox Signal. 2012, 17, 1362–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabas, I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ. Res. 2010, 107, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Knoops, B.; Goemaere, J.; Van der Eecken, V.; Declercq, J.P. Peroxiredoxin 5: Structure, mechanism, and function of the mammalian atypical 2-cys peroxiredoxin. Antioxid. Redox Signal. 2011, 15, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Sim, J.; Park, J.; Woo, H.A.; Rhee, S.G. Maturation of mitochondrially targeted prx v involves a second cleavage by mitochondrial intermediate peptidase that is sensitive to inhibition by h2o2. Antioxidants 2021, 10, 346. [Google Scholar] [CrossRef]
- Kim, M.H.; Park, S.J.; Kim, J.H.; Seong, J.B.; Kim, K.M.; Woo, H.A.; Lee, D.S. Peroxiredoxin 5 regulates adipogenesis-attenuating oxidative stress in obese mouse models induced by a high-fat diet. Free Radic. Biol. Med. 2018, 123, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, E.G.; Yi, H.J.; Kim, N.H.; Rhee, S.G.; Woo, H.A. Ablation of peroxiredoxin v exacerbates ischemia/reperfusion-induced kidney injury in mice. Antioxidants 2020, 9, 769. [Google Scholar] [CrossRef]
- Shichita, T.; Hasegawa, E.; Kimura, A.; Morita, R.; Sakaguchi, R.; Takada, I.; Sekiya, T.; Ooboshi, H.; Kitazono, T.; Yanagawa, T.; et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat. Med. 2012, 18, 911–917. [Google Scholar] [CrossRef]
- Knoops, B.; Becker, S.; Poncin, M.A.; Glibert, J.; Derclaye, S.; Clippe, A.; Alsteens, D. Specific interactions measured by afm on living cells between peroxiredoxin-5 and tlr4: Relevance for mechanisms of innate immunity. Cell Chem. Biol. 2018, 25, 550–559.e3. [Google Scholar] [CrossRef]
- Wang, M.X.; Wei, A.; Yuan, J.; Trickett, A.; Knoops, B.; Murrell, G.A. Expression and regulation of peroxiredoxin 5 in human osteoarthritis. FEBS Lett. 2002, 531, 359–362. [Google Scholar] [CrossRef] [Green Version]
- Abbas, K.; Breton, J.; Picot, C.R.; Quesniaux, V.; Bouton, C.; Drapier, J.C. Signaling events leading to peroxiredoxin 5 up-regulation in immunostimulated macrophages. Free Radic. Biol. Med. 2009, 47, 794–802. [Google Scholar] [CrossRef]
- Diet, A.; Abbas, K.; Bouton, C.; Guillon, B.; Tomasello, F.; Fourquet, S.; Toledano, M.B.; Drapier, J.C. Regulation of peroxiredoxins by nitric oxide in immunostimulated macrophages. J. Biol. Chem. 2007, 282, 36199–36205. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.N.; Kim, S.U.; Huang, S.M.; Kim, J.M.; Park, Y.H.; Kim, S.H.; Yang, H.Y.; Chung, K.J.; Lee, T.H.; Choi, H.S.; et al. Microglial peroxiredoxin v acts as an inducible anti-inflammatory antioxidant through cooperation with redox signaling cascades. J. Neurochem. 2010, 114, 39–50. [Google Scholar]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase a(2) activities. Antioxid. Redox Signal. 2011, 15, 831–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, A.B.; Dodia, C.; Chatterjee, S. A peptide inhibitor of peroxiredoxin 6 phospholipase a2 activity significantly protects against lung injury in a mouse model of ventilator induced lung injury (vili). Antioxidants 2021, 10, 925. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.Y. Phospholipase a(2) enzymes in metabolic and cardiovascular diseases. Curr. Opin. Lipidol. 2012, 23, 235–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, N.; Malik, G.; Fisher, A.B.; Das, D.K. Targeted disruption of peroxiredoxin 6 gene renders the heart vulnerable to ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2636–H2640. [Google Scholar] [CrossRef] [PubMed]
- Manevich, Y.; Sweitzer, T.; Pak, J.H.; Feinstein, S.I.; Muzykantov, V.; Fisher, A.B. 1-cys peroxiredoxin overexpression protects cells against phospholipid peroxidation-mediated membrane damage. Proc. Natl. Acad. Sci. USA 2002, 99, 11599–11604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.X.; Chen, W.; Jiang, Y.L.; Ni, J.Q.; Lu, L. Antioxidant protein peroxiredoxin 6 suppresses the vascular inflammation, oxidative stress and endothelial dysfunction in angiotensin ii-induced endotheliocyte. Gen. Physiol. Biophys. 2020, 39, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Phelan, S.A.; Petros, C.; Taylor, E.F.; Ledinski, G.; Jurgens, G.; Forsman-Semb, K.; Paigen, B. Peroxiredoxin 6 deficiency and atherosclerosis susceptibility in mice: Significance of genetic background for assessing atherosclerosis. Atherosclerosis 2004, 177, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Phelan, S.A.; Wang, X.; Wallbrandt, P.; Forsman-Semb, K.; Paigen, B. Overexpression of prdx6 reduces h2o2 but does not prevent diet-induced atherosclerosis in the aortic root. Free Radic. Biol. Med. 2003, 35, 1110–1120. [Google Scholar] [CrossRef]
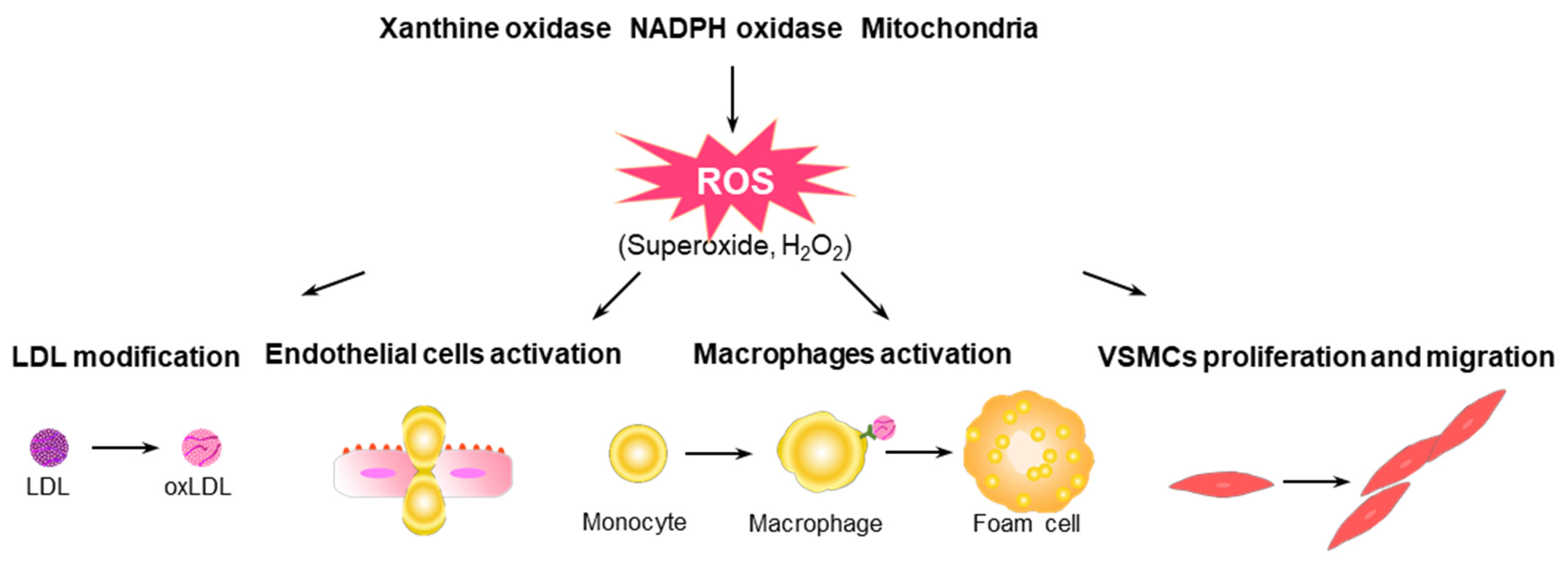
Figure 1.
Potential roles of ROS in the progression of atherosclerosis. ROS modify LDL particles, such as oxLDLs, in the intima, which induce the expression of adhesion molecules, the release of chemokines from ECs, and the recruitment of immune cells into the intima. Monocytes recruited into the intima differentiate into macrophages that engulf oxLDLs through scavenger receptors, and macrophages create inflammatory conditions in lesions by expressing various proinflammatory cytokines, ROS, and costimulatory molecules that activate other immune cells. Excess engulfment of oxLDLs triggers the transformation of macrophages into foam cells. ROS is closely involved in the regulation of the growth, migration, extracellular matrix production, and inflammatory gene expression in VSMCs.
Figure 1.
Potential roles of ROS in the progression of atherosclerosis. ROS modify LDL particles, such as oxLDLs, in the intima, which induce the expression of adhesion molecules, the release of chemokines from ECs, and the recruitment of immune cells into the intima. Monocytes recruited into the intima differentiate into macrophages that engulf oxLDLs through scavenger receptors, and macrophages create inflammatory conditions in lesions by expressing various proinflammatory cytokines, ROS, and costimulatory molecules that activate other immune cells. Excess engulfment of oxLDLs triggers the transformation of macrophages into foam cells. ROS is closely involved in the regulation of the growth, migration, extracellular matrix production, and inflammatory gene expression in VSMCs.

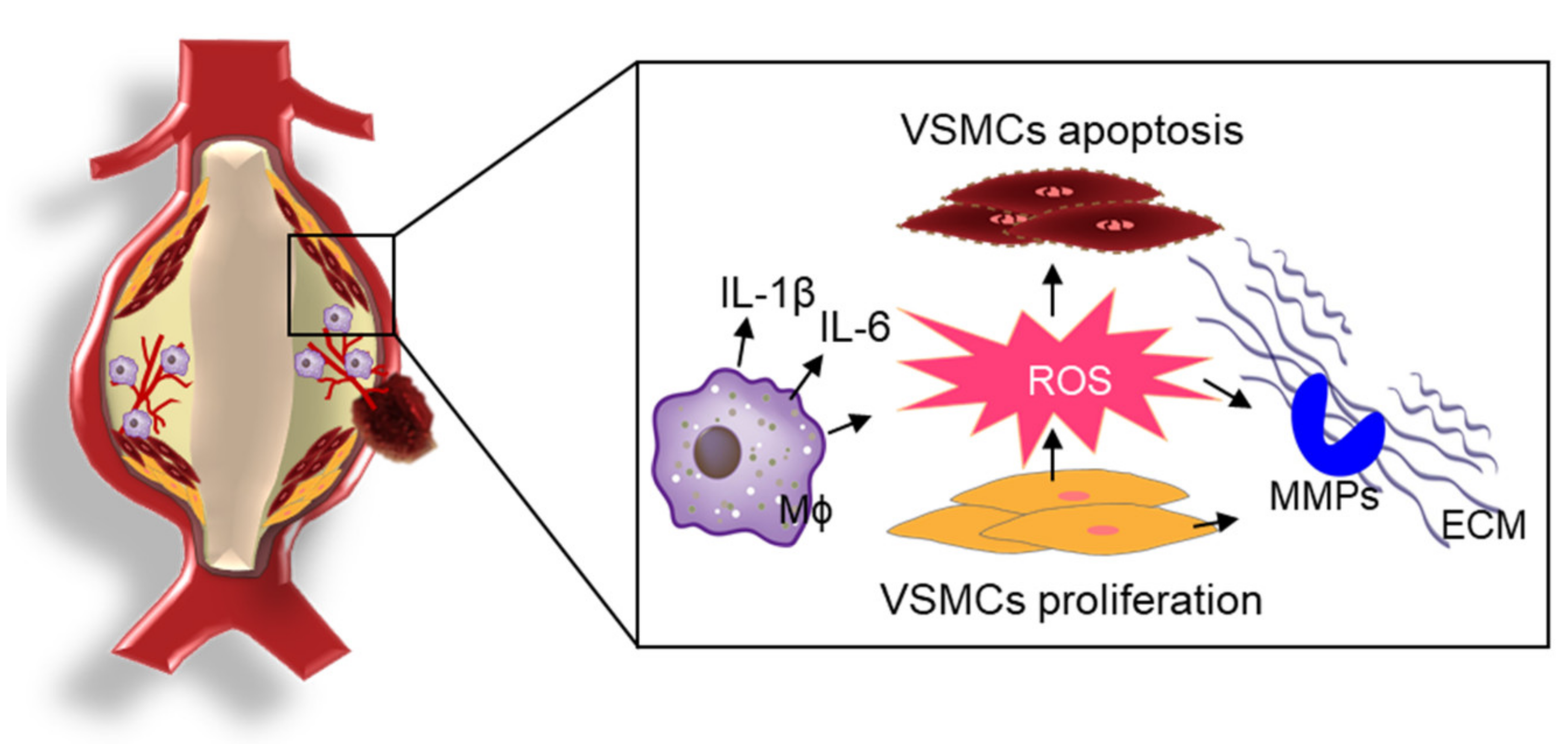
Figure 2.
ROS-mediated the pathophysiology of AAA. In the development of AAA, ROS-induced OS is closely associated with inflammatory processes, the proliferation/migration/apoptosis of VSMCs, and the activation of MMPs for proteolytic degradation of structural proteins in the pathogenesis of AAA.
Figure 2.
ROS-mediated the pathophysiology of AAA. In the development of AAA, ROS-induced OS is closely associated with inflammatory processes, the proliferation/migration/apoptosis of VSMCs, and the activation of MMPs for proteolytic degradation of structural proteins in the pathogenesis of AAA.

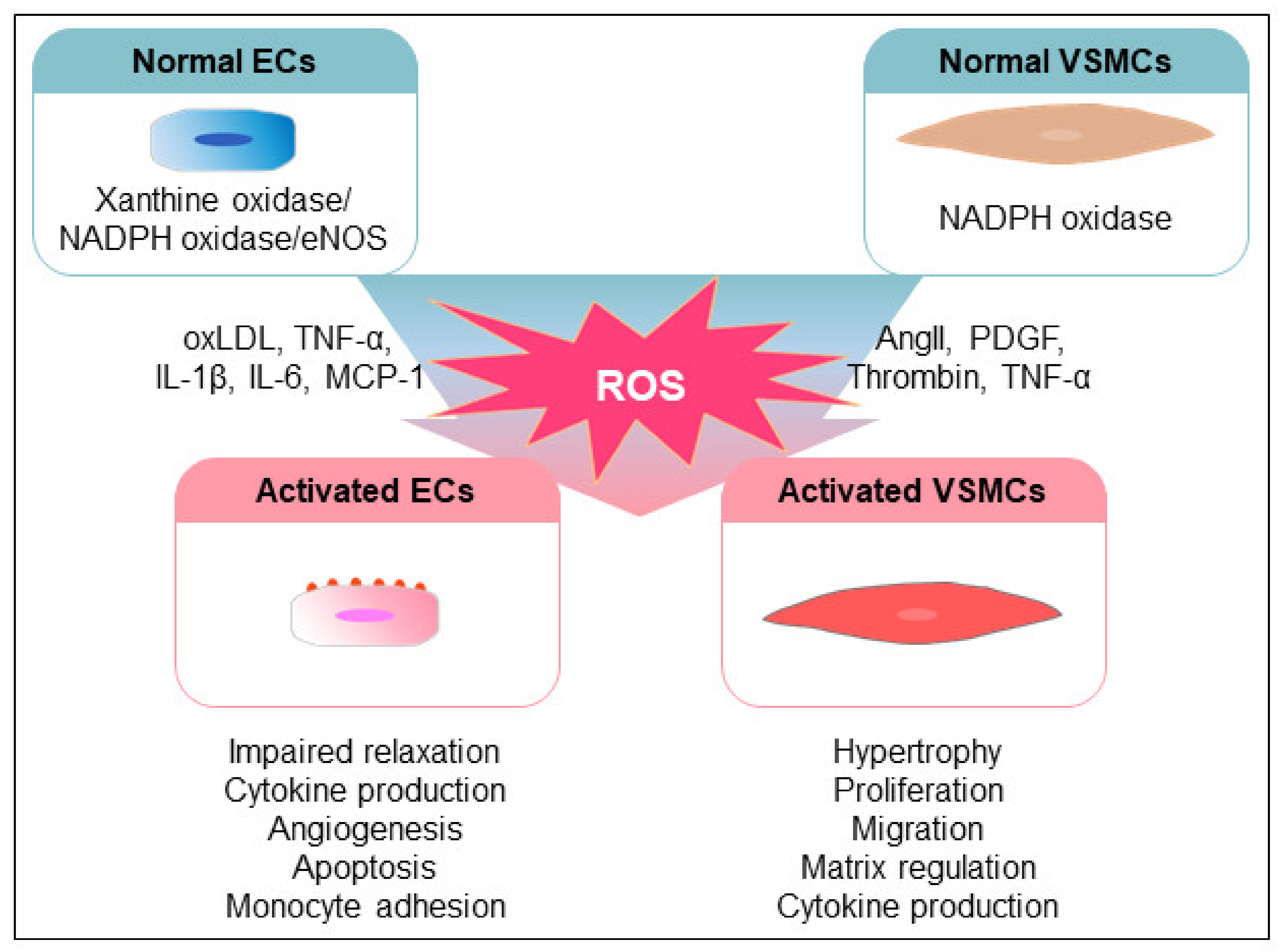
Figure 3.
Modulation of the vascular cells by ROS. Various extracellular stimuli increase the production of ROS through ROS-generated enzyme systems in vascular cells. oxLDLs and proinflammatory cytokines induce the augmentation of ROS, which leads to the dysfunction of ECs, including impaired vasorelaxation, cytokine production, angiogenesis, apoptosis, and immune cell adhesion. Additionally, VSMCs are activated by various stimuli, such as AngII, PDGF, thrombin, and TNF-α, leading to alterations of the cellular functions, including proliferation, migration, extracellular matrix formation, and cytokine production, through increased ROS production.
Figure 3.
Modulation of the vascular cells by ROS. Various extracellular stimuli increase the production of ROS through ROS-generated enzyme systems in vascular cells. oxLDLs and proinflammatory cytokines induce the augmentation of ROS, which leads to the dysfunction of ECs, including impaired vasorelaxation, cytokine production, angiogenesis, apoptosis, and immune cell adhesion. Additionally, VSMCs are activated by various stimuli, such as AngII, PDGF, thrombin, and TNF-α, leading to alterations of the cellular functions, including proliferation, migration, extracellular matrix formation, and cytokine production, through increased ROS production.

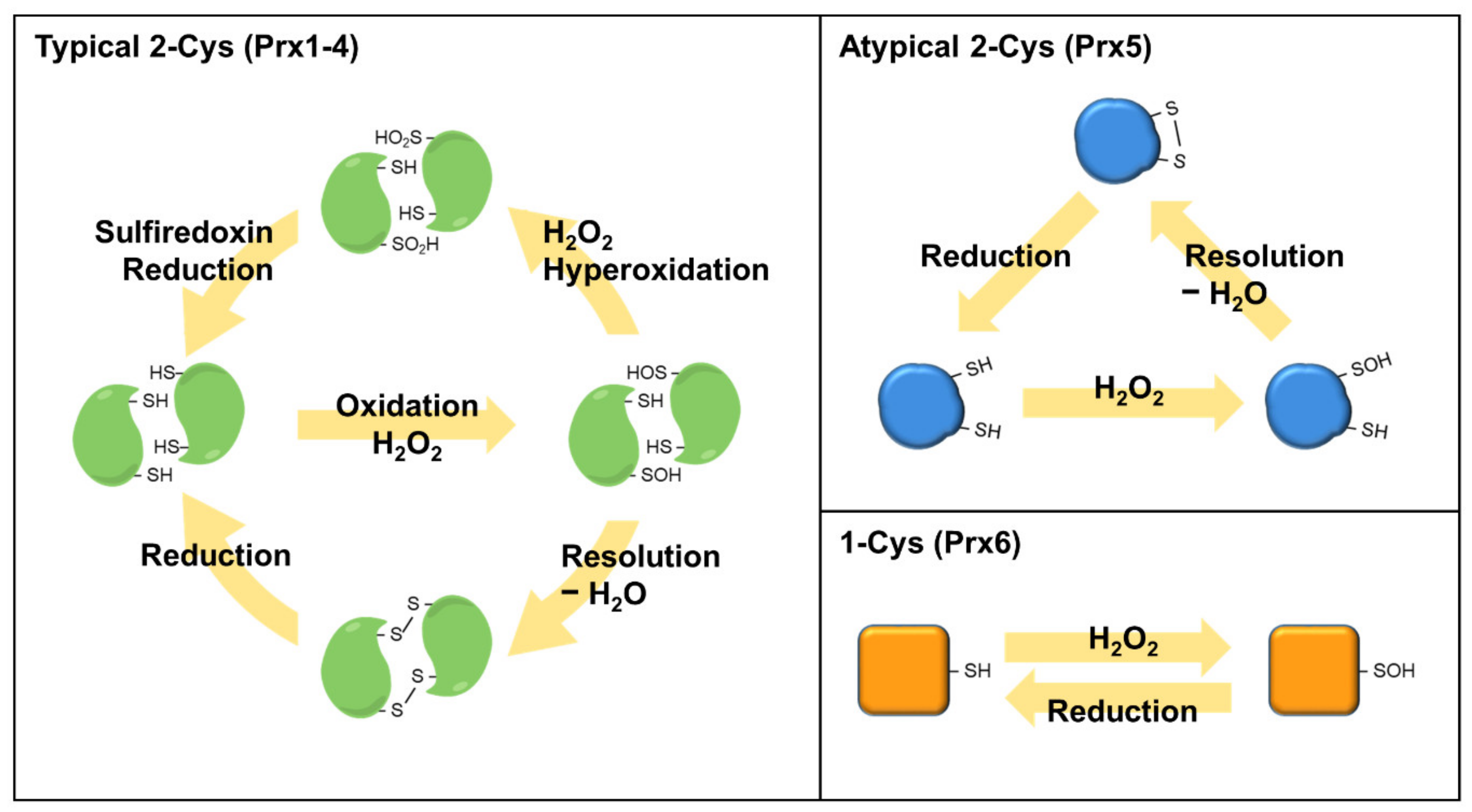
Figure 4.
Reaction mechanisms of Prdx isoforms. The peroxidatic cysteine residue of Prdxs reacts with H2O2 to release H2O and form a sulfenic acid (SOH) intermediate. SOH is resolved with a second monomer to form an intermolecular disulfide bond in typical 2-Cys Prdxs (Prdx1–4). Atypical 2-Cys Prdx (Prdx5) forms an intra-subunit disulfide in the identical subunit, which is distinct from typical 2-Cys Prdxs. The 1-Cys Prdx (Prdx6) react with H2O2 to form a SOH intermediate, which forms a disulfide bond with other proteins or small thiol molecules to reduce the peroxidatic cysteine.
Figure 4.
Reaction mechanisms of Prdx isoforms. The peroxidatic cysteine residue of Prdxs reacts with H2O2 to release H2O and form a sulfenic acid (SOH) intermediate. SOH is resolved with a second monomer to form an intermolecular disulfide bond in typical 2-Cys Prdxs (Prdx1–4). Atypical 2-Cys Prdx (Prdx5) forms an intra-subunit disulfide in the identical subunit, which is distinct from typical 2-Cys Prdxs. The 1-Cys Prdx (Prdx6) react with H2O2 to form a SOH intermediate, which forms a disulfide bond with other proteins or small thiol molecules to reduce the peroxidatic cysteine.

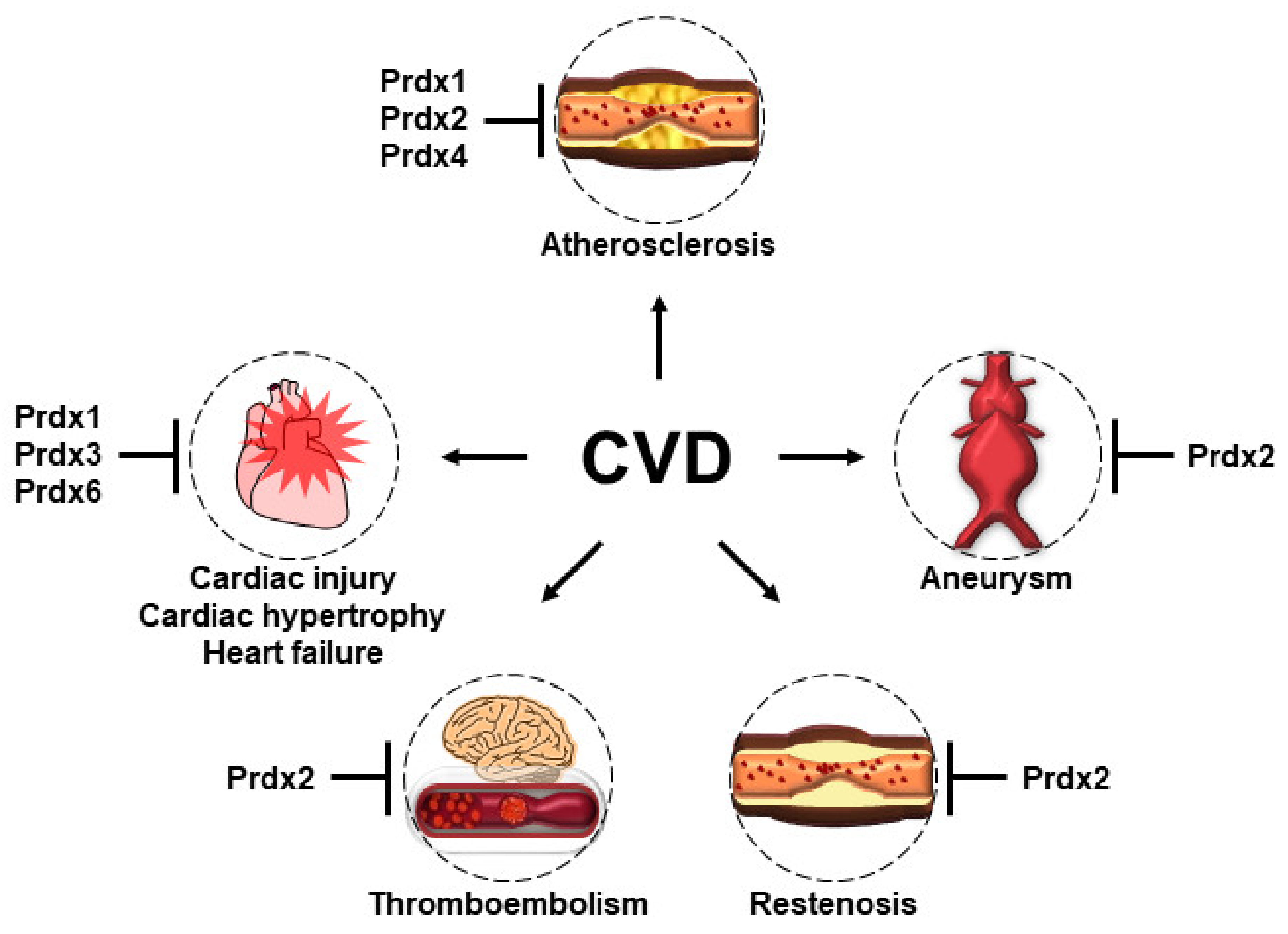
Figure 5.
The protective roles of Prdxs in the pathogenesis of CVD. Prdx 1, 2, and 4 play protective roles in the development of atherosclerosis, although through different mechanisms. Prdx1, which is highly expressed in macrophages, maintains lipophagic flux and cholesterol homeostasis and reduces the foam cell formation and atherosclerosis. Prdx2 deficiency accelerates atherosclerotic plaque formation and immune cell infiltration. The overexpression of human Prdx4 attenuates the development of atherosclerosis by limiting the infiltration of T-lymphocytes, reducing OS, and ameliorating necrosis. Prdx2 deficiency promotes the progression and most features of AAA. Prdx2 suppresses PDGF signaling by inhibiting the protein tyrosine phosphatase in VSMCs and protects against PDGF-dependent neointimal thickening. Prdx2 negatively regulates H2O2 generation and thrombosis formation by platelets and VSMCs. The overexpression of Prdx1 in the cardiomyocytes of mice prevents TAC-induced cardiac hypertrophy and heart failure. The overexpression of Prdx3 protects the heart against left ventricular remodeling and failure after MI. Prdx6-deficient mice have increased susceptibility to ischemia–reperfusion injury.
Figure 5.
The protective roles of Prdxs in the pathogenesis of CVD. Prdx 1, 2, and 4 play protective roles in the development of atherosclerosis, although through different mechanisms. Prdx1, which is highly expressed in macrophages, maintains lipophagic flux and cholesterol homeostasis and reduces the foam cell formation and atherosclerosis. Prdx2 deficiency accelerates atherosclerotic plaque formation and immune cell infiltration. The overexpression of human Prdx4 attenuates the development of atherosclerosis by limiting the infiltration of T-lymphocytes, reducing OS, and ameliorating necrosis. Prdx2 deficiency promotes the progression and most features of AAA. Prdx2 suppresses PDGF signaling by inhibiting the protein tyrosine phosphatase in VSMCs and protects against PDGF-dependent neointimal thickening. Prdx2 negatively regulates H2O2 generation and thrombosis formation by platelets and VSMCs. The overexpression of Prdx1 in the cardiomyocytes of mice prevents TAC-induced cardiac hypertrophy and heart failure. The overexpression of Prdx3 protects the heart against left ventricular remodeling and failure after MI. Prdx6-deficient mice have increased susceptibility to ischemia–reperfusion injury.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jeong, S.-J.; Park, J.-G.; Oh, G.T. Peroxiredoxins as Potential Targets for Cardiovascular Disease. Antioxidants 2021, 10, 1244. https://doi.org/10.3390/antiox10081244
AMA Style
Jeong S-J, Park J-G, Oh GT. Peroxiredoxins as Potential Targets for Cardiovascular Disease. Antioxidants. 2021; 10(8):1244. https://doi.org/10.3390/antiox10081244
Chicago/Turabian StyleJeong, Se-Jin, Jong-Gil Park, and Goo Taeg Oh. 2021. "Peroxiredoxins as Potential Targets for Cardiovascular Disease" Antioxidants 10, no. 8: 1244. https://doi.org/10.3390/antiox10081244
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.