1. Introduction
Nearly 14% of the United States population meets the diagnostic criteria for an alcohol use disorder (AUD) in any given year [
1]. Excessive alcohol consumption produces neurodegeneration in humans [
2,
3,
4], an effect that has been confirmed in various pre-clinical models [
5,
6,
7,
8]. Due to its preventable nature, alcoholism traditionally has not been defined as a neurodegenerative disorder, but chronic, excessive consumption may cause damage in the temporal lobe on par with diseases such as Alzheimer’s [
4]. Indeed, alcoholic-related dementia is the second leading cause of dementia in the United States only behind Alzheimer’s disease [
9,
10]. Even in the absence of dementia, cognitive deficits such as increased impulsivity and impaired executive decision-making are found in many with AUDs [
11,
12]. Alcohol-induced neurodegeneration and the associated cognitive deficits are thought to be critical factors in the development of AUDs [
13,
14,
15].
Despite the number of reports in human and preclinical models describing the neurotoxic effects of alcohol, the mechanism of how alcohol produces neurodegeneration is unclear [
16]. One such mechanism that has recently gained attention is the impact of excessive alcohol consumption on the neuroimmune system, and particularly, microglia [
17,
18]. Analysis of the brains of human alcoholics suggests that excessive alcohol consumption leads to microglial activation [
19,
20,
21], but whether this activation is the cause or consequence of alcohol-induced neurodegeneration is an active debate [
22]. This discussion is due, in part, to a lack of understanding of the effect of alcohol on microglia coupled with the recent appreciation of the role of microglia in both neurodegenerative and regenerative processes [
22,
23,
24,
25]. Although microglia have historically been discussed as the phagocytes of the central nervous system (CNS), these cells are far more complex, existing in a continuum of phenotypes or stages of activation [
26]. Microglia are constantly surveying the parenchyma in non-pathological conditions; where in response to even a subtle change in their environment, microglia alter their morphological and functional characteristics, a process termed microglial activation [
27]. The nomenclature for these stages or phenotypes vary. Terms like M1 and classical activation are applied when microglia have an amoeboid morphology and secrete pro-inflammatory cytokines, whereas M2 and alternative activation are used to describe microglia with bushier ramifications that secrete anti-inflammatory cytokines [
26,
28]. In neurodegenerative diseases where microglial activation drives neuronal loss, microglia are generally fully or classically activated (
i.e., M1 phenotype), secreting pro-inflammatory factors and undergoing uncontrolled phagocytosis [
25,
29]. How alcohol affects microglia is not well described and appears to vary depending on the model. Most reports of alcohol-induced microglia activation assume that all activated microglia are pro-inflammatory [
19,
23,
30]. However, in the one model with alcohol-induced neurodegeneration, the Majchrowicz four-day binge model, only a low level of activation or alternative (M2) phenotype has been observed [
22,
24,
31].
The variability of microglial phenotypes observed across different AUD models may be due to the pattern of alcohol exposure, specifically intermittent
versus sustained intoxication. Interestingly, the intermittent exposure models show stronger evidence of pro-inflammatory microglia even with lower doses of ethanol [
22,
30]. These disparate findings across models led us to question whether the initial hit of alcohol exposure “primes” microglia such that intermittent exposure leads to a potentiated response. Primed microglia have similar morphology and cytokine/growth factor profiles as the M2/alternative microglia, but primed microglial activation is potentiated when subsequent neuroimmunomodulators are applied [
28,
32,
33]. Ethanol’s ability to prime microglia and exacerbate the neuroimmune response to subsequent neuroimmune stimuli is suggested also by the enhanced microglia response to LPS following alcohol exposure [
23,
34,
35]. However, the ability of a second “hit” or insult of ethanol to potentiate the neuroimmune response (independent of peripheral immunomodulators) has not been examined. Therefore, the current study determines whether a second binge ethanol exposure can potentiate the microglia response to binge alcohol exposure. Investigating whether repeated ethanol exposure differentially affects microglia is important considering that the majority of individuals suffering from an AUD drink in a binge pattern that produces periods of high BECs interspersed with periods of withdrawal and abstinence [
36,
37,
38]. Specifically, this study examines both functional and morphological indices of microglial activation in the hippocampus and entorhinal cortex, regions consistently damaged in this model [
7,
8].
4. Discussion
These data collectively indicate that microglia previously activated by alcohol exposure can be further exacerbated by a second alcohol binge. This point was demonstrated by: (a) potentiated OX-42 immunoreactivity; (b) increased microglial number; and (c) increased TNF-α concentration in EtOH/EtOH (double binge) rats compared with Con/EtOH (single binge) rats. The alcohol model used produces a low-grade microglial activation state that is similar to an M2 phenotype [
22,
31]. However, as the subsequent binge produced more pro-inflammatory-like effects, these alcohol-activated microglia may also be primed. This enhanced response to a second binge aligns with the definition of microglial priming, which is where a stimulus changes microglia to be more susceptible to and over-respond to a second insult [
33,
63,
64]. Primed microglia and/or an exacerbated microglial response could lead to abnormally increased cell death and is a hypothesized etiology of neurodegenerative disorders [
28]. Furthermore, given that the majority of individuals with an AUD drink in an episodic binge pattern [
38,
65], the repeated cycles of binge drinking with periods of withdrawal, and therefore repeated microglial insult, may lead to even more dynamic microglial activation over time.
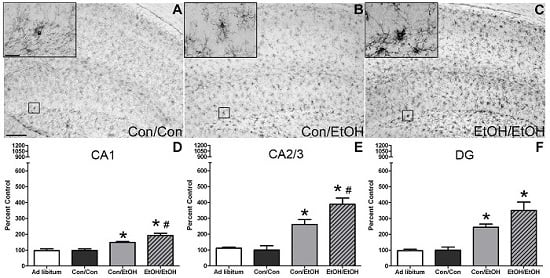
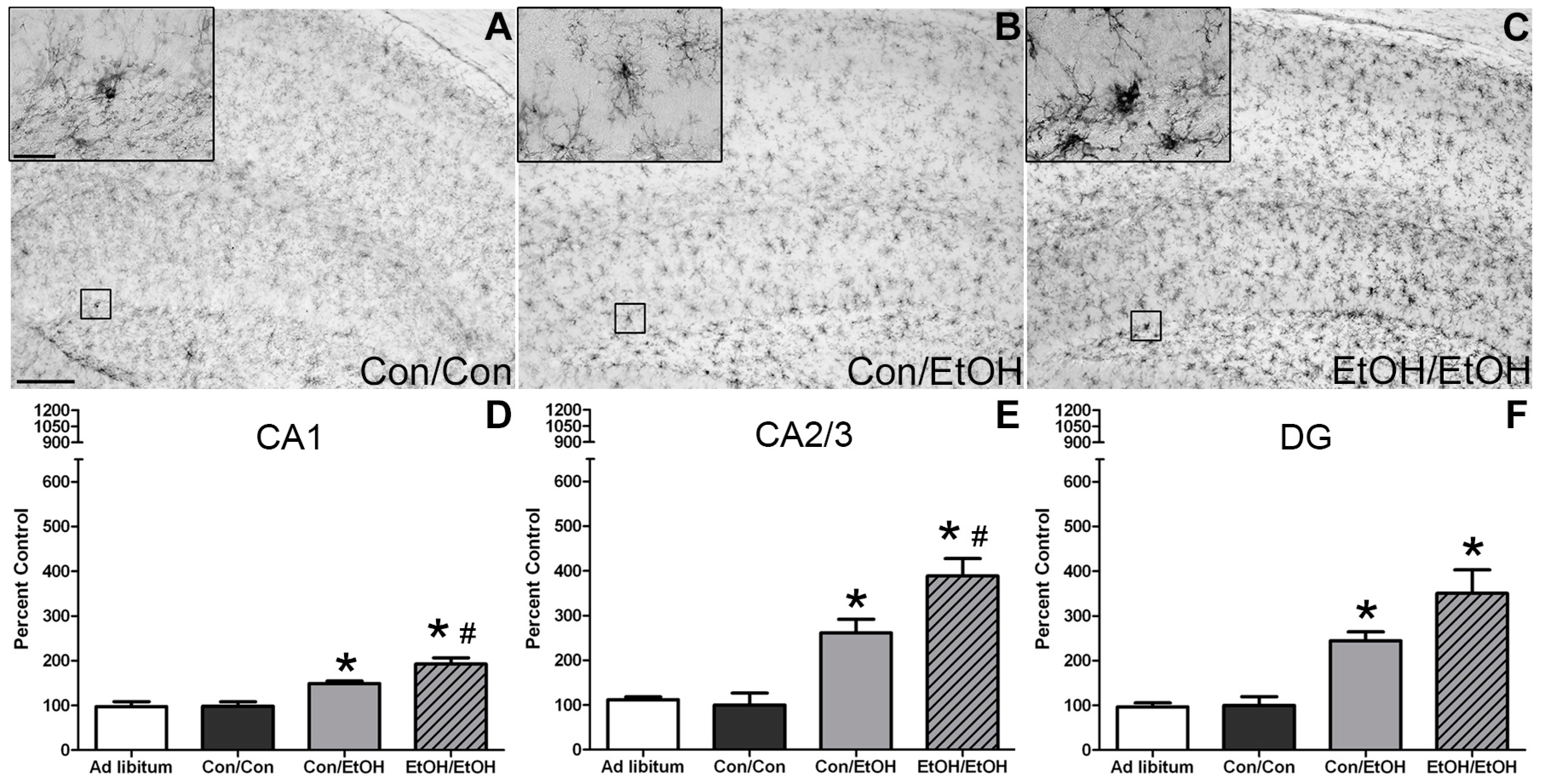
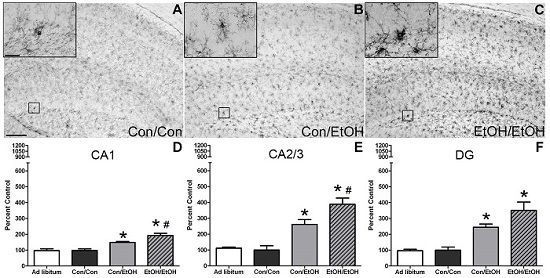
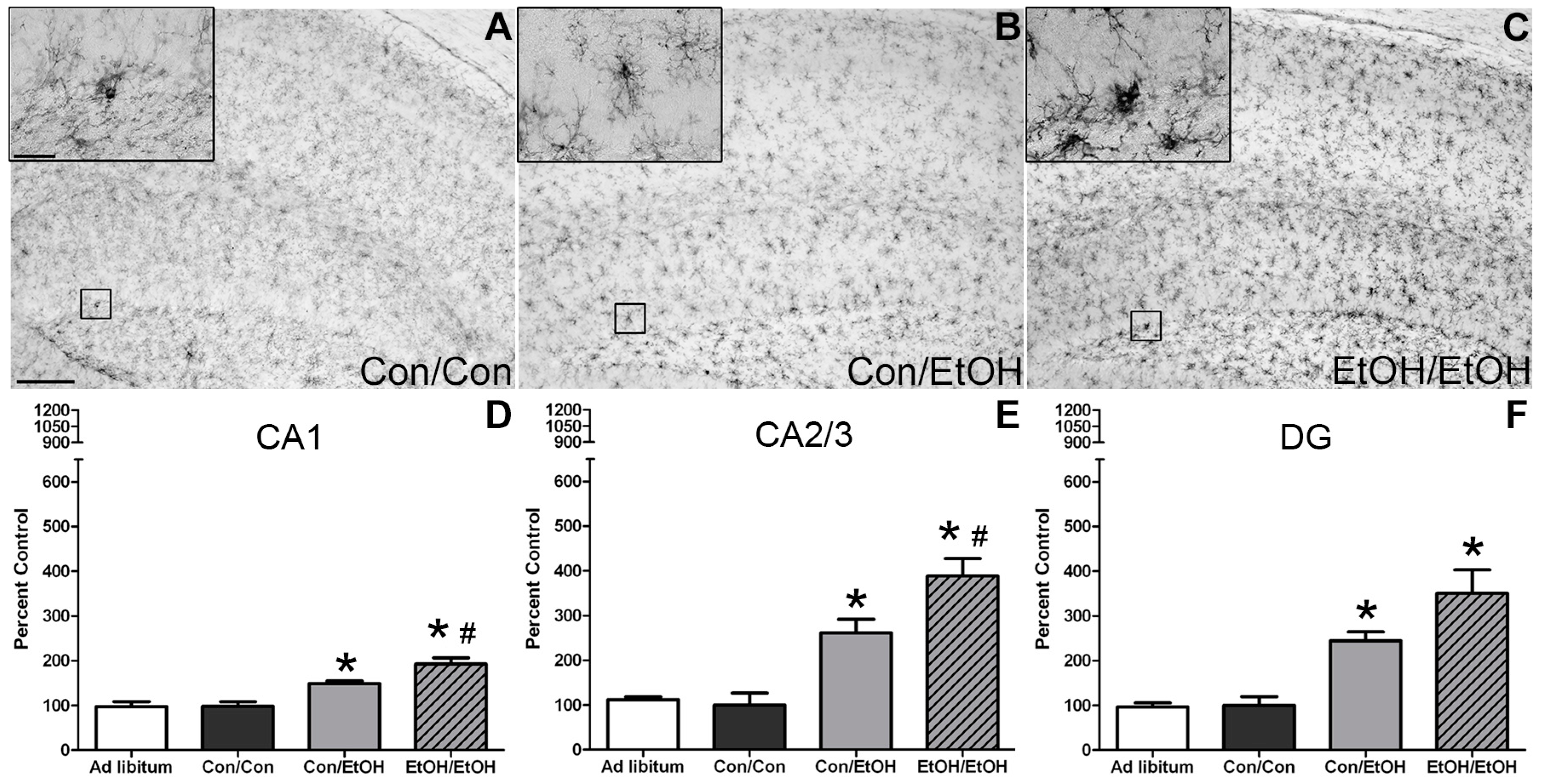
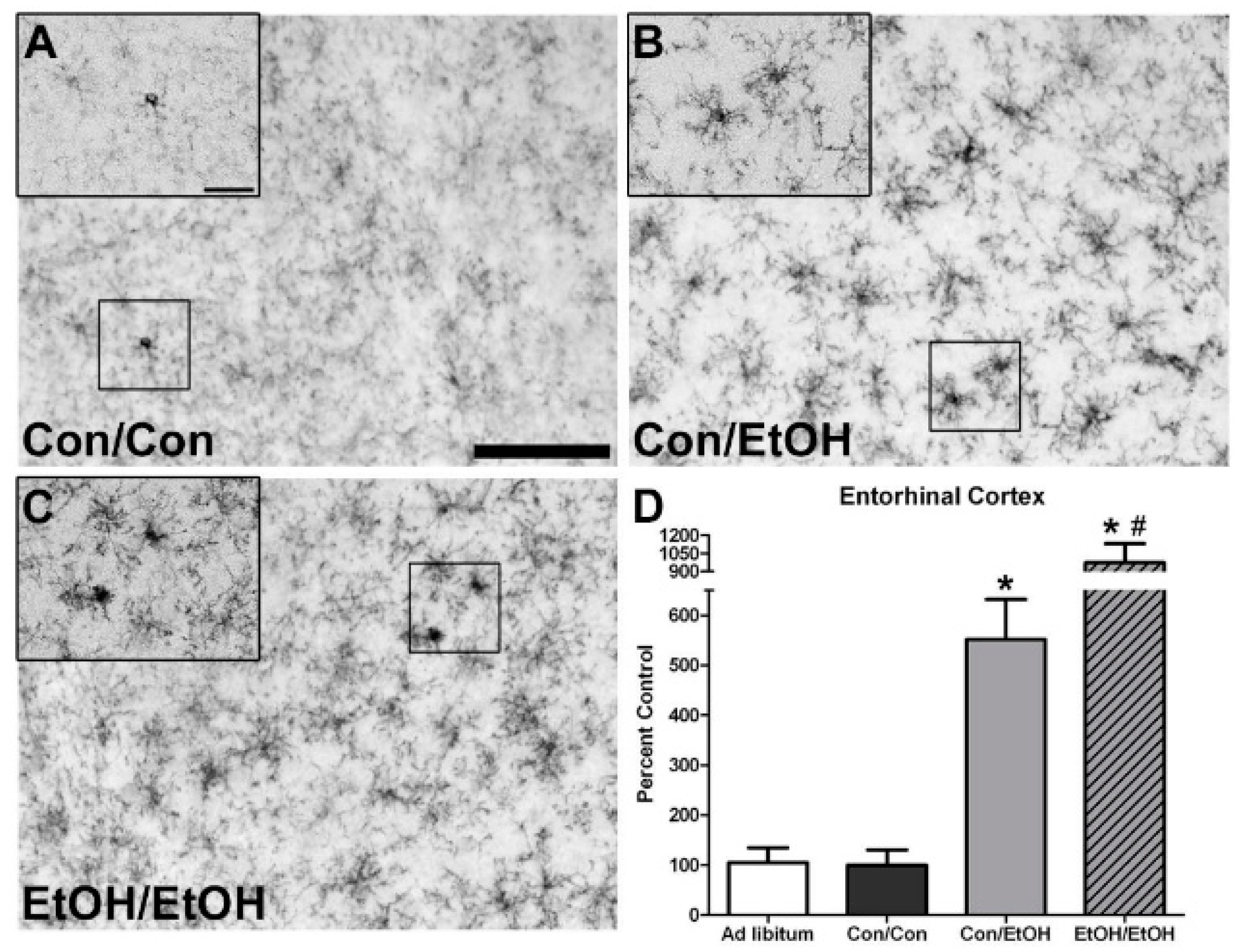
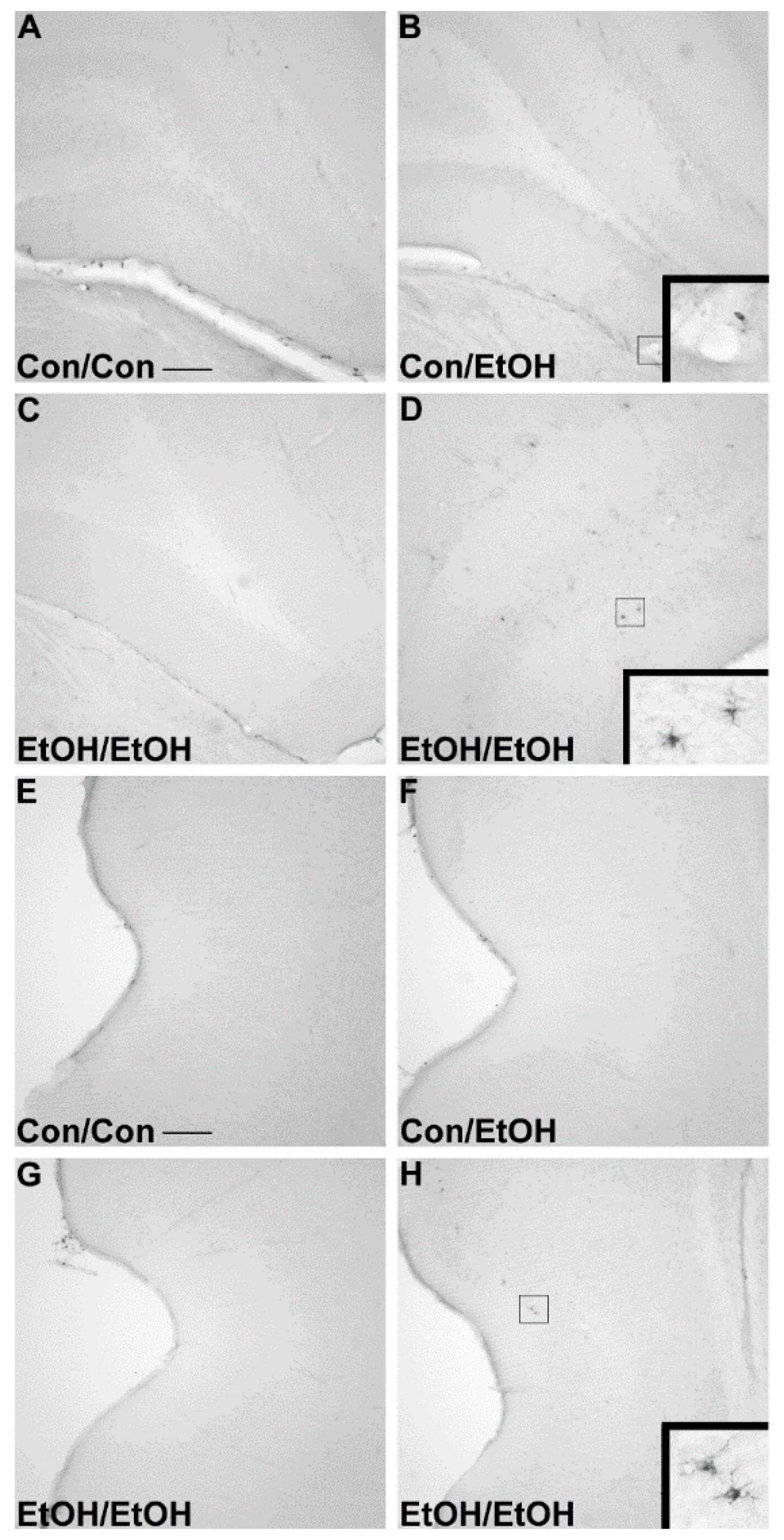
The first evidence of this potentiated microglia response in the double binge group was increased immunoreactivity to the OX-42 antibody. Increased OX-42 immunoreactivity, which labels CR3, is one of the earliest signs of microglial activation [
22,
46]. CR3 is associated with cell adhesion necessary for removing pathogens or damaged/dying neurons [
45,
66,
67]. Increased OX-42 staining has been reported in a number of animal models of ethanol exposure [
22,
68,
69,
70]. The current study confirms those findings; but furthers that work by showing that a second hit of binge ethanol exposure potentiates OX-42 immunoreactivity. A potentiated increase in OX-42 immunoreactivity, or CR3 density, by ethanol is particularly interesting because CR3 is intimately involved in microglial priming [
33]. The increased upregulation of CR3 in the EtOH/EtOH (double binge) rats compared with Con/EtOH (single binge) rats suggests that binge ethanol exposure acts as a priming stimulus to microglia. Morphology, though not specifically quantified, appeared consistent with a low grade/phenotype of activation as cells were ramified and not amoeboid (e.g.,
Figure 1) [
26]. A bushy, ramified microglial morphology is also consistent with that observed in other pathologies that report a primed microglia state [
33,
64,
71]. Furthermore, despite the potentiation of CR3 receptor density, no changes in ED-1 or OX-6 expression were seen following the second binge. The lack of visible ED-1+ or OX-6+ cells concurs with other reports in this model that do not show signs of classical microglial activation following ethanol exposure [
22,
31,
60].
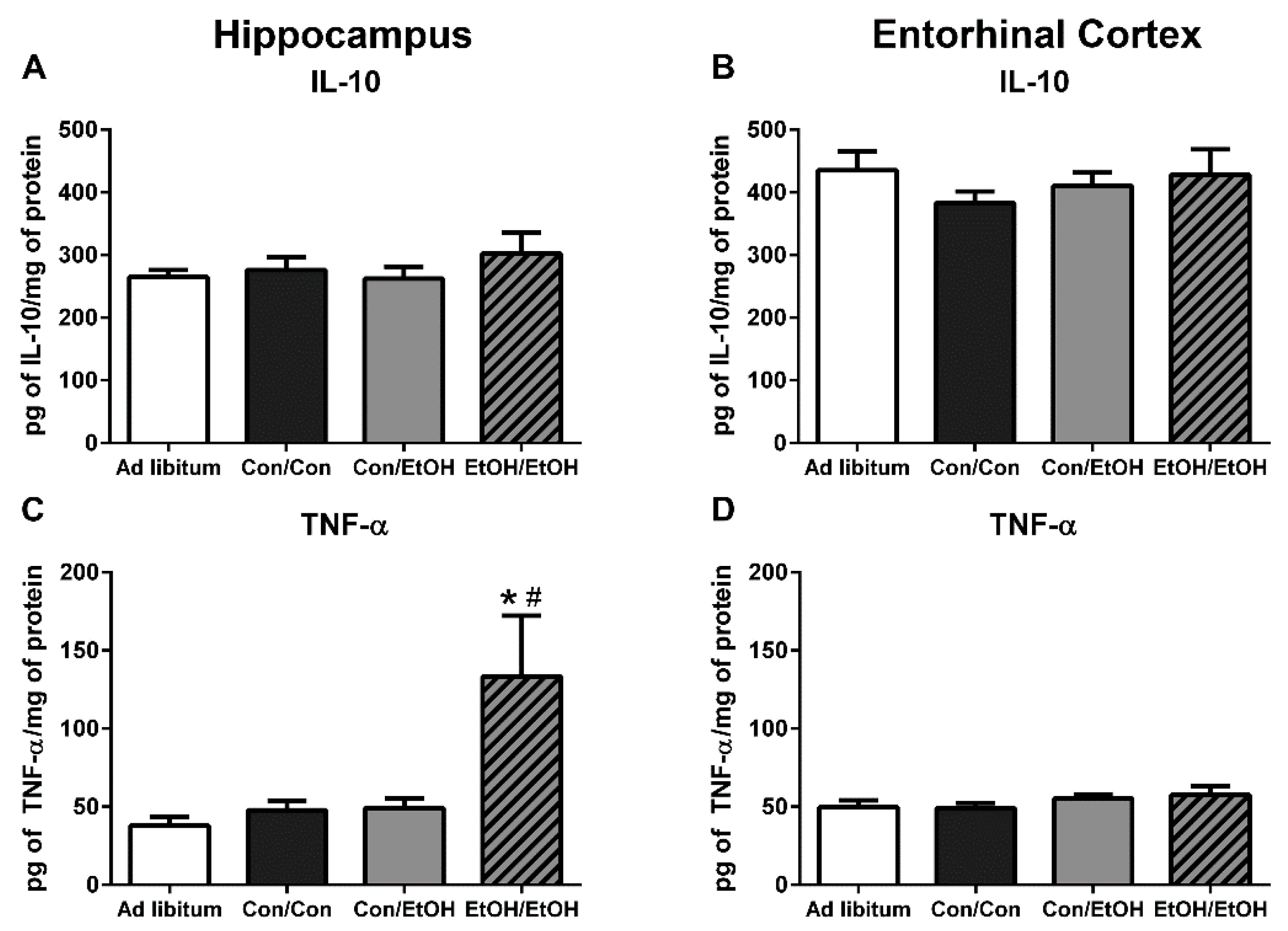
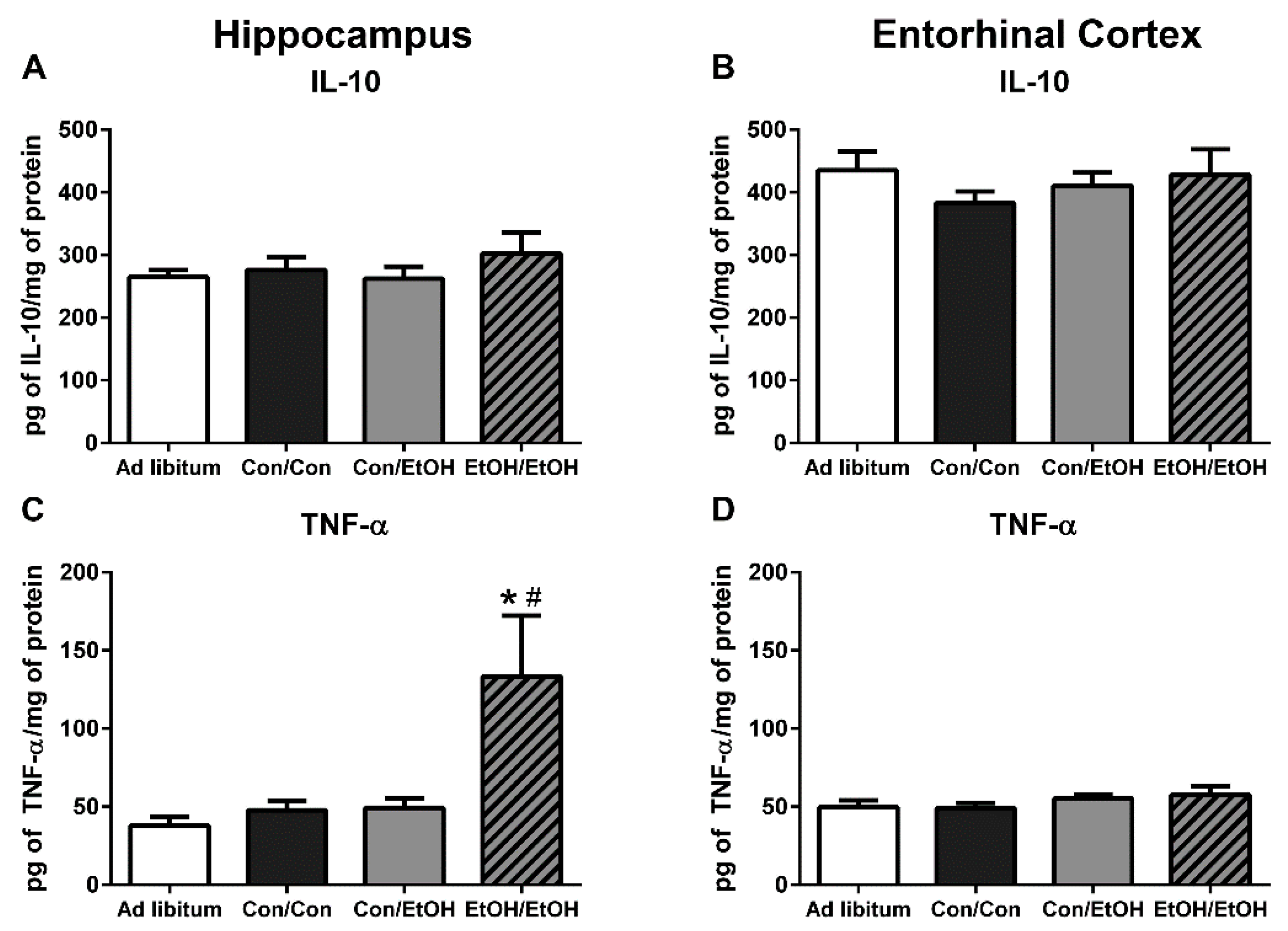
Because multiple endpoints should be measured to understand the phenotype of microglia after insult, functional outputs such as hallmark pro- and anti-inflammatory cytokines were measured to better understand the type of microglial activation associated with a second “hit” of ethanol exposure. No change in the concentration of the hallmark anti-inflammatory cytokine, IL-10, was observed in either ethanol exposure group in the hippocampus or entorhinal cortex. The lack of IL-10 response during intoxication confirms previous findings in this model, although IL-10 is decreased in a mouse AUD model [
22,
23]. However, upregulation of TNF-α in the hippocampus in the EtOH/EtOH group compared with all other groups suggests that the second binge promoted a pro-inflammatory state. This finding is highly distinct from multiple previous reports using Majchrowicz-like models where no effect of ethanol was observed on TNF-α concentrations [
22,
24,
31] and highlights the impact of repeated ethanol exposure on pro-inflammatory cytokine production and microglial activation. The potentiation of TNF-α expression by the second hit of ethanol, much like the morphological indices, is a common response for microglia that are primed and then hit with a secondary peripheral immune insult [
28,
64,
72]. In fact, alcohol and other drugs of abuse have been shown to prime the TNF-α response to other immune stimulators [
23,
63], but these finding specifically suggest that alcohol exposure can act as both the priming and secondary stimulus resulting in an increase in TNF-α.
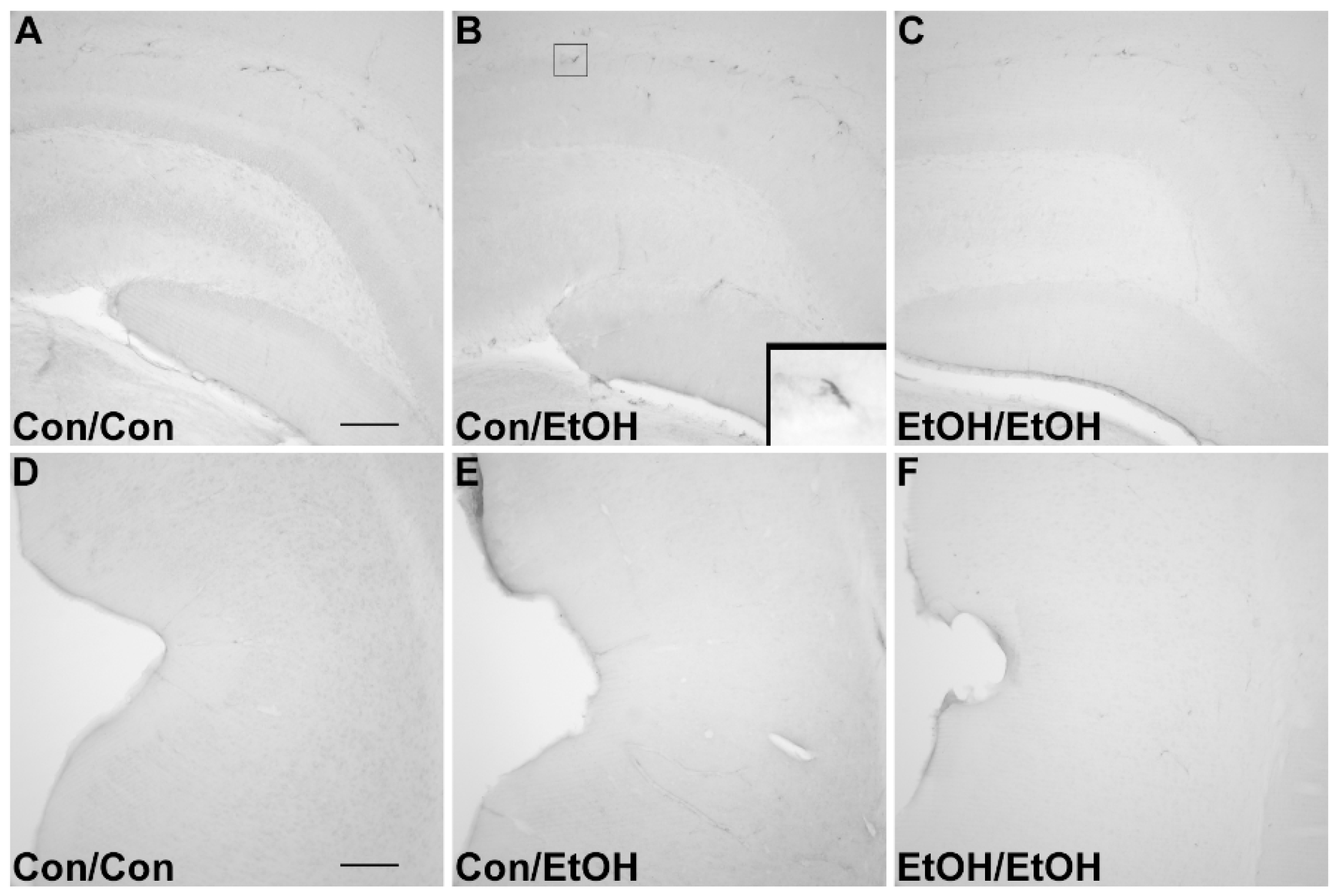
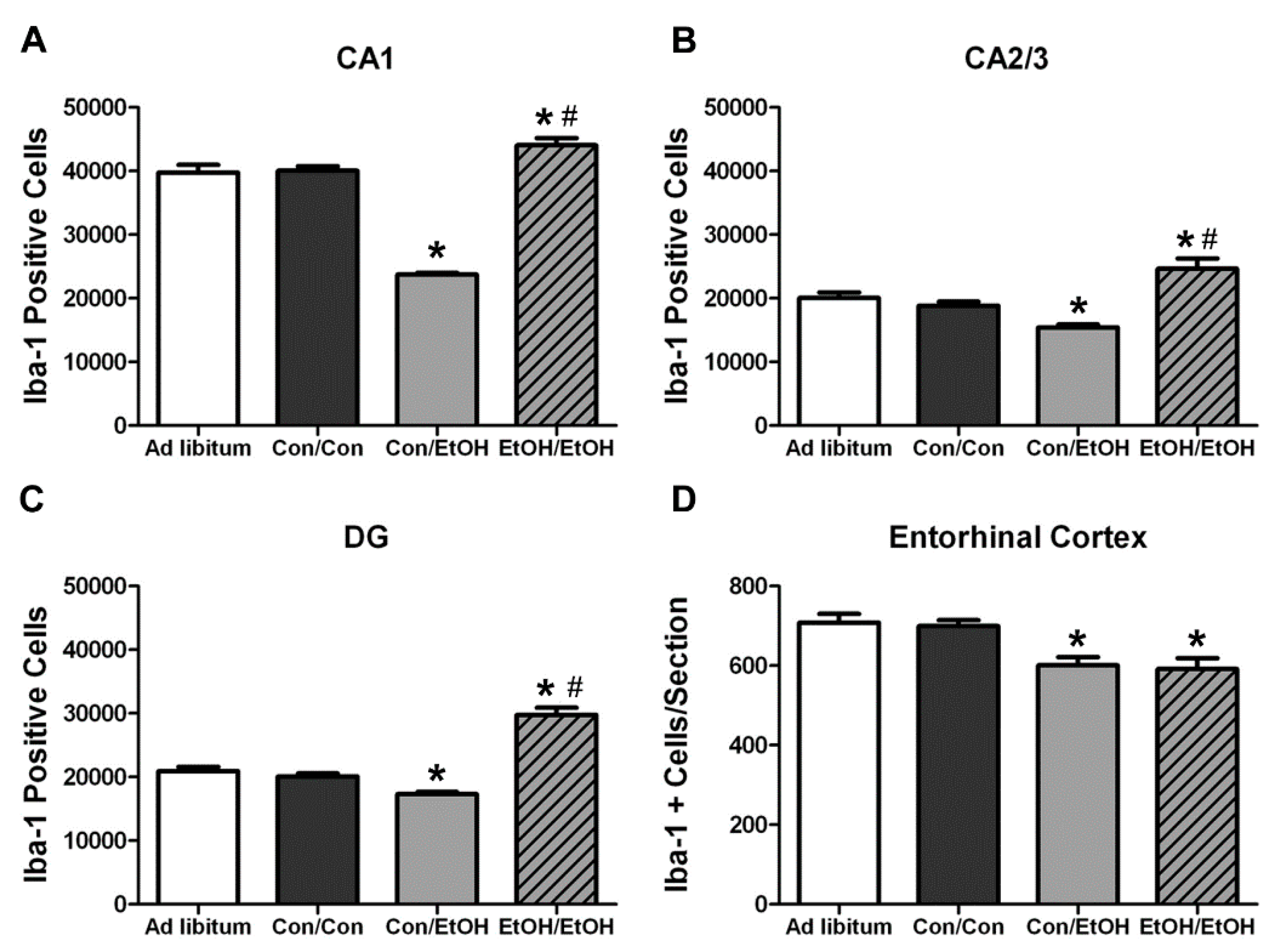
In the Majchrowicz model, microglia loss was observed during the last days of intoxication [
61], whereas microglia proliferation occurs after the cessation of alcohol exposure, on the second day of abstinence [
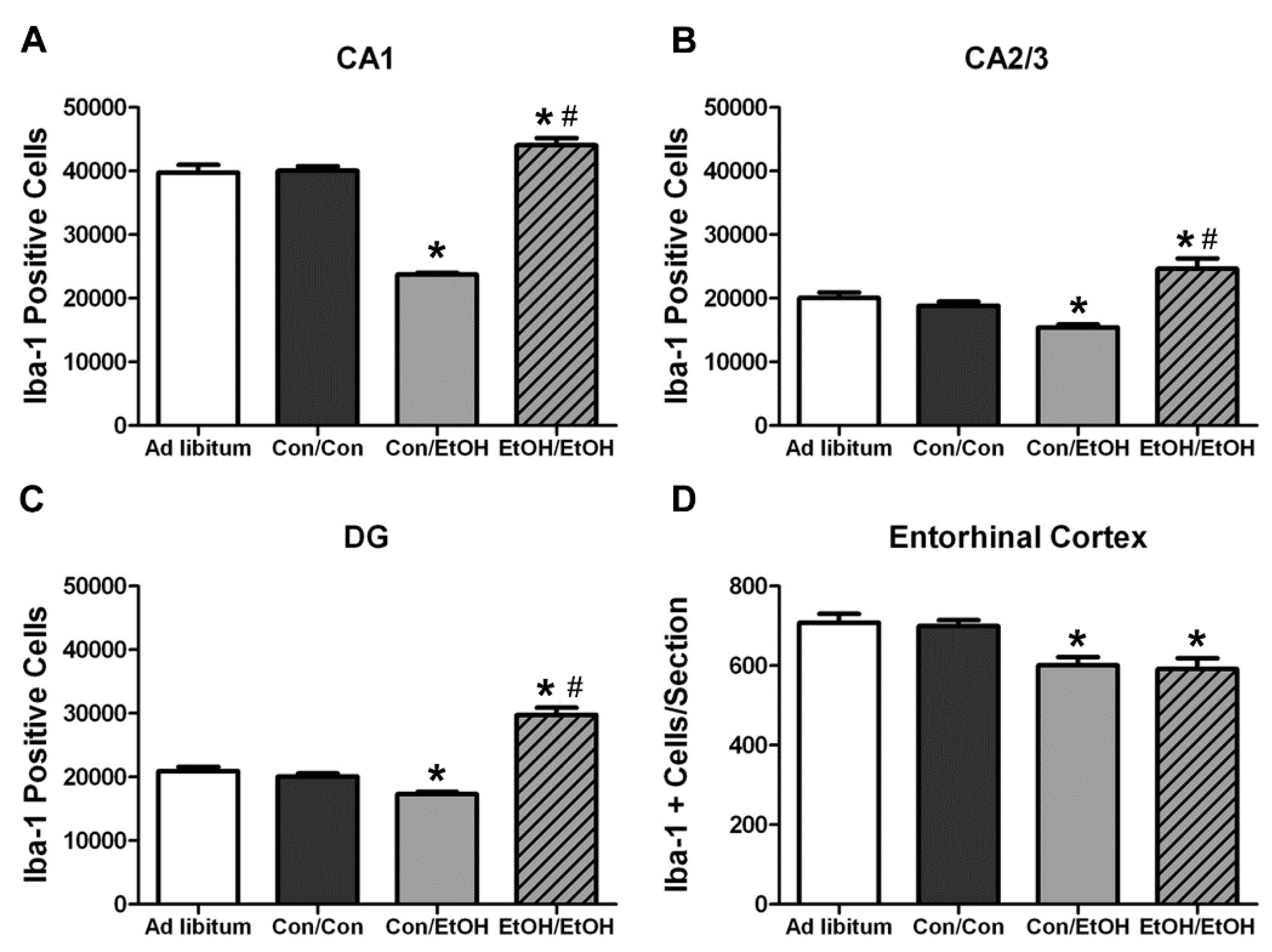
31,
60]. Therefore, Iba-1+ cell number was assessed to determine how multiple cycles of ethanol affects microglia number. The single ethanol binge (Con/EtOH group) reduced the number of Iba-1+ microglia, in both the hippocampus and entorhinal cortex. Our recent work supports that this reduction is likely due to degeneration of microglia following 4-day binge exposure [
61]. Interestingly, the second binge (EtOH/EtOH group) resulted in an increased number of Iba-1+ microglia in the hippocampus compared to either the control group or single binge (Con/EtOH) group. It is plausible that the increase in Iba-1+ cells in the EtOH/EtOH group observed in the hippocampus is due to microglial proliferation at two days following the first binge [
22,
31,
60]. This effect also suggests that ethanol does not significantly reduce these newly proliferated microglial cells. It is of note that the effect of ethanol on microglia number varied by region: in the entorhinal cortex, both the single and double binge resulted in a decrease in the number of microglia consistent with our recent report [
61]. The lack of increased Iba-1+ cells in the entorhinal cortex of EtOH/EtOH group is likely related to the finding that microglia neither proliferate dramatically at two days post-binge in the entorhinal cortex nor is there a significant increase in microglia number after seven days;, however, both proliferation and increased Iba-1+ cells have been observed in the hippocampus at this same time point [
22,
60]. Why microglia proliferate in the hippocampus but not entorhinal cortex after binge ethanol exposure is puzzling. Neurons in the entorhinal cortex degenerate more robustly, peaking at four days of exposure [
5,
7,
8], which is followed by other signs of reactive microgliosis [
22]. More studies are necessary to fully understand the dynamic effects of alcohol on microglia number, especially considering the recent discoveries that microglia contribute to synapse refinement and plasticity [
25]. These data support the hypothesis that a second binge alcohol exposure exacerbates the microglial response, since an increase in the number of activated microglia would likely result in a potentiated neuroimmune response during the second binge.
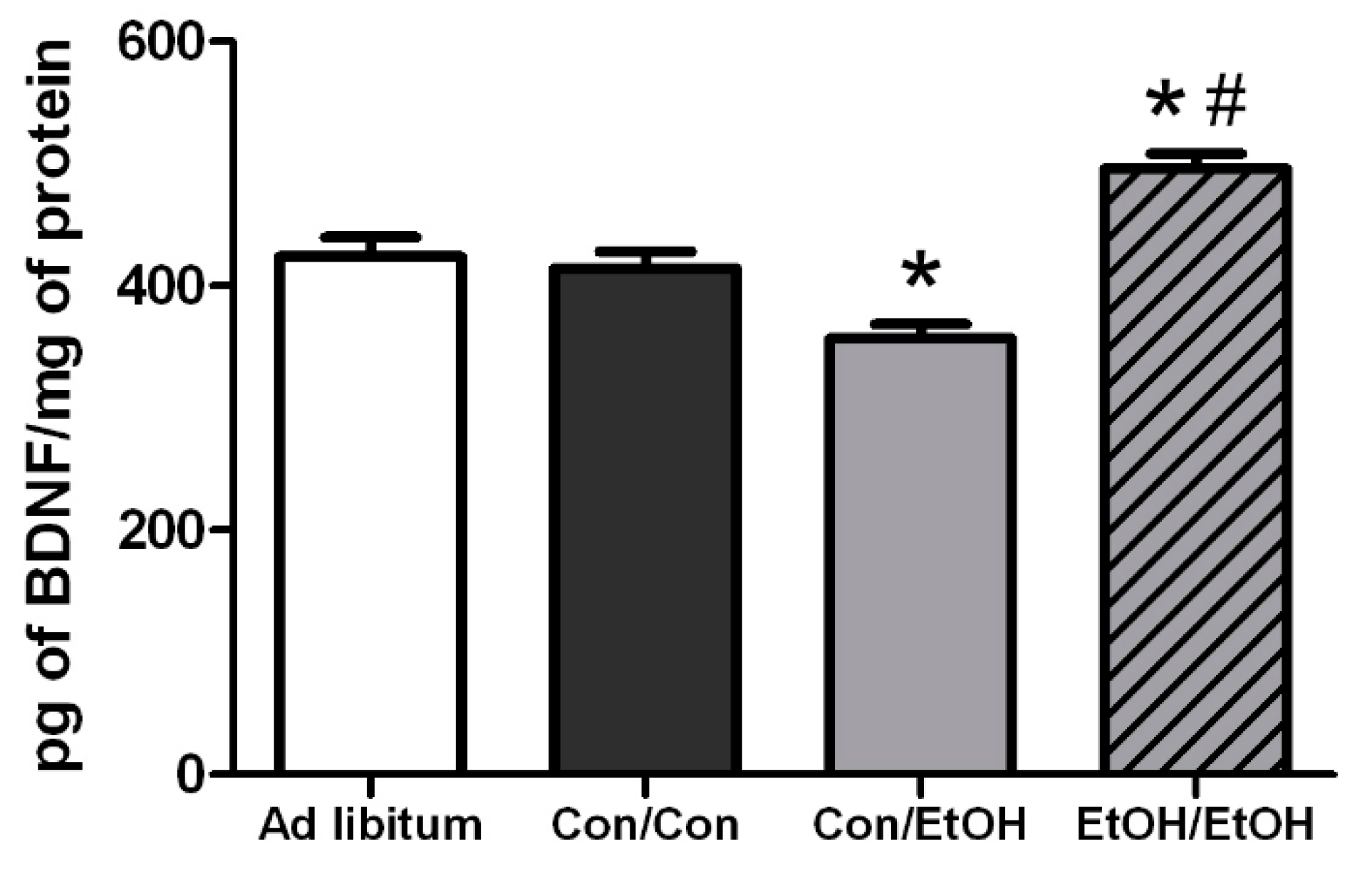
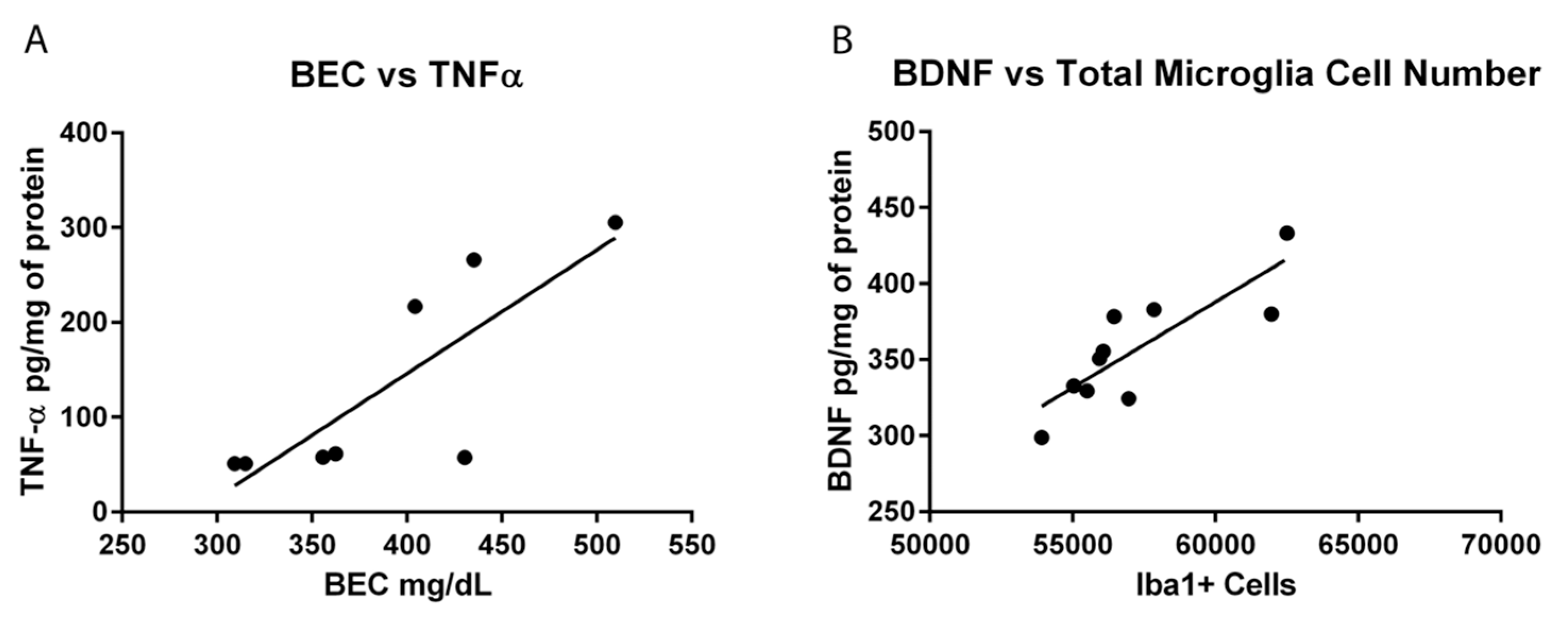
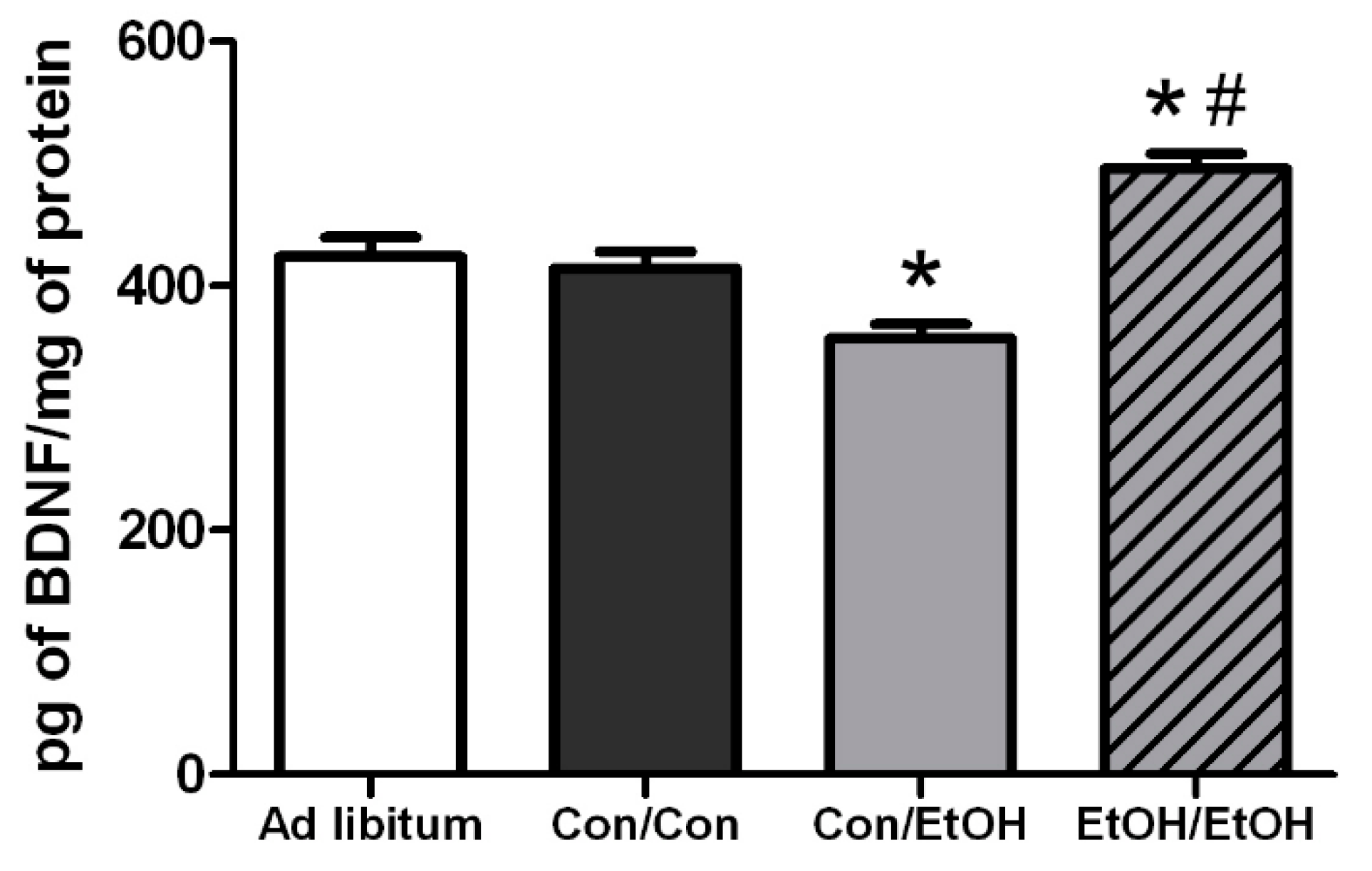
Hippocampal BDNF concentrations were determined in order to assess the impact of microglia reactivity and changes in microglia number on the surrounding environment. BDNF plays a pivotal role in neuronal integrity and its dysregulation is associated with neurodegeneration [
73]. In the Con/EtOH group, BDNF was decreased, the number of microglia were decreased and there was a significant correlation between the number of microglia and BDNF protein expression. However, in the EtOH/EtOH treated animals, where microglia were more activated and their numbers were increased, a significantly higher BDNF concentration was observed, though this value did not correlate significantly to microglia number. It is possible that the increase in BDNF concentrations is due to cells other than microglia, such as astrocytes, neurons, and other CNS cells secreting BDNF [
74]. In addition, the effect of ethanol on BDNF expression is quite complex [
75]. Nevertheless, the interplay between the increased cytokine and neurotrophin production observed in the EtOH/EtOH group requires further study to understand its functional implications.
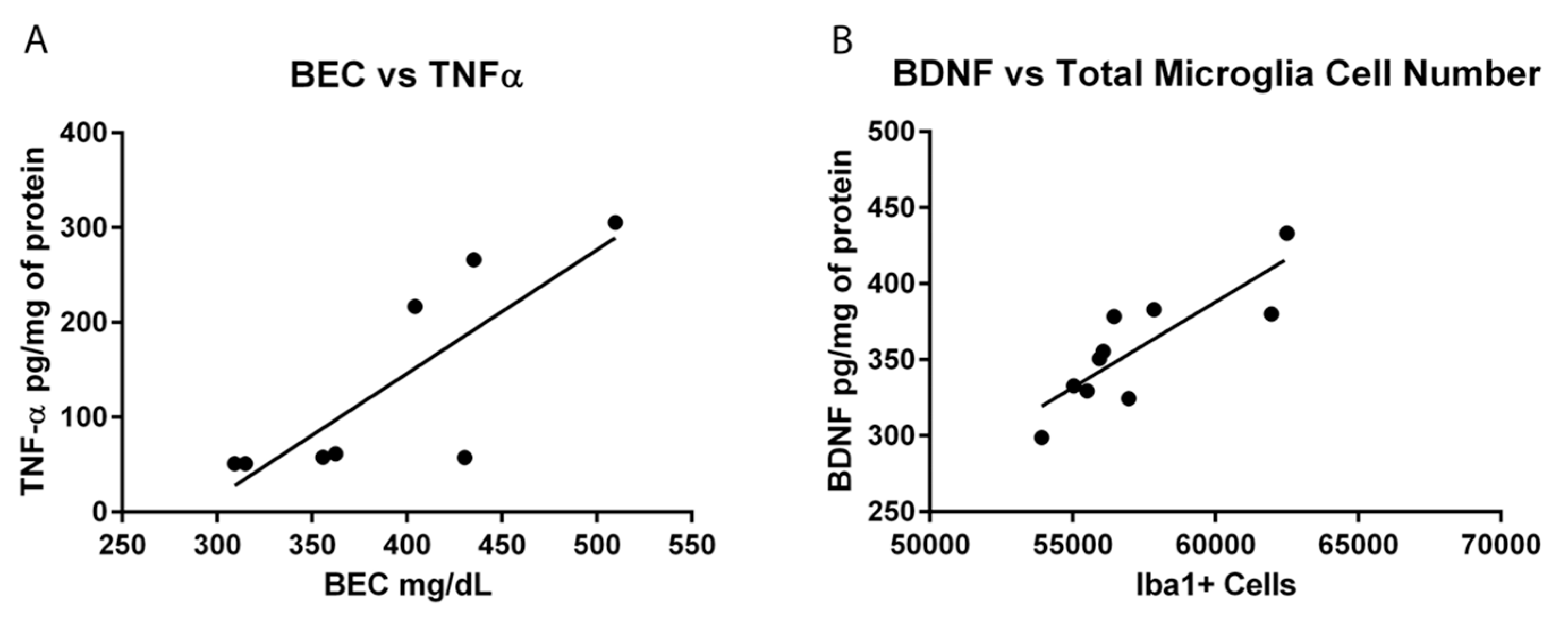
The experimental design to use the same animals for both immunohistochemical and ELISA experiments allowed for a series of correlations to help determine what aspect of ethanol exposure, in this AUD model, was associated with microglial reactivity. OX-42 immunoreactivity did not correlate to average dose per day or to the total dose of ethanol in either the Con/EtOH or EtOH/EtOH groups. This lack of correlation is important as immune modulators such as LPS have dose-dependent responses in microglia reactivity [
76]. The lack of correlation between OX-42 and total dose of ethanol suggests that ethanol potentiates the OX-42 response by acting as a secondary stimulus rather than an additive effect of the accumulative dose. Moreover, no relationship between the number of Iba-1+ cells and OX-42 immunoreactivity were observed, supporting that increased OX-42 immunoreactivity was a result of microglial activation and not an artifact of the change in cell number [
77]. Correlations were also used to examine the relationship between OX-42 immunoreactivity or Iba-1 cell number and functional indices (cytokine/neurotrophin production). However, neither CR3 receptor (OX-42) upregulation nor Iba-1+ cell number was significantly correlated with TNF-α expression. Interestingly, the bimodal distribution of TNF-α production observed in the EtOH/EtOH group did map on to BECs. Although the mechanism by which BECs are related to TNF-α were not measured, at minimum, this correlation suggests that as BECs increase with repeated exposure, a primed microglial state may cause increased pro-inflammatory cytokines. Finally, in relation to BDNF, only microglia cell number in the Con/EtOH group showed a significant correlation with BDNF concentrations supporting the idea that microglial dysfunction and subsequent loss of trophic factors may contribute to neurodegeneration, especially alcoholic brain damage [
61]. Correlations are not being interpreted as causation, but they do provide direction for what aspects of alcohol-exposure impact microglia reactivity leading to a primed microglial state.
Some evidence of classical activation has been observed in other AUD models, an effect that may be attributable to species differences and/or variations in the duration and pattern of exposure [
30,
69]. While previous reports suggested that the difference in microglia reactivity was due to these aforementioned variations in AUD models, the current data in this report more definitively indicates that it is the repeated insult that may drive the greater microglial response. For example, a model of alcohol exposure with lower total doses of alcohol dispersed over a longer period of time produced more OX-6 positive cells than the exposure used herein, where OX-6 expression may have been an anomaly in a single animal [
30]. The appearance of the OX-6+ cells, however, in both models still appeared to be the bushy, ramified morphology associated with a low-level or M2-like activation. Indeed, the only alcohol study, human or animal, where ED-1+ microglia have been observed, is from a study in which rats underwent four cycles of a Majchrowicz-like model with three days between binges. However, the high mortality rate and severe weight loss of rats in that report make interpretations difficult. One interpretation is that microglial activation may have occurred due to the stress of repetitive gavage and/or weight loss [
57,
78,
79,
80]. Thus, the current study specifically used a seven day abstinence period to allow rats to recover from four days of intoxication and the significant withdrawal sequelae that occurs in this model [
40]. Moreover, because some weight loss is observed in the Majchrowicz model [
40], repetitive gavage may be stressful [
81], and both of these aspects modulate microglial reactivity [
78,
82], a group with
ad libitum access to food and water was included. None of the measures of microglia activation were different between the
ad libitum group and the Con/Con group despite their slight weight loss and experience with gavage. Moreover, weight loss did not correlate with any measure of microglial activation in animals receiving ethanol.
The potentiated microglia activation seen in this double binge AUD model suggests that the microglial response can be altered by ethanol alone and supports the idea that chronic ethanol exposure can elicit a more pro-inflammatory state than a single bout of binge exposure. The lack of expression of ED-1 and morphology of OX-42+ and Iba-1+ cells support that even with the two binges, cells are not fully or classically activated. However, microglia are “further” down the spectrum towards classical activation than a single binge alone. These data coupled with the lack of evidence for classically activated microglia in human alcoholic brain—whether the markers are not expressed or no one has examined those particular markers—supports that initially microglia activation is likely a consequence of alcoholic neuropathology and not a cause. Whether this increased response causes microglia to over-respond to insult or if it makes the brain more susceptible to ongoing neuroinflammation should be considered in future experiments. In addition, how these effects relate to changes in neurodegeneration, specifically neuronal or volume loss, is an important area for future study. Because microglia have the capacity to maintain low grade activation or a primed state for extensive periods following insult, including alcohol exposure [
22,
83], the episodic nature of binge drinking would lead to a cycle of repeated priming and over-response in individuals suffering from an AUD [
18,
38,
65]. Understanding the mechanisms that underlie or contribute to alcohol-induced neurodegeneration may provide a novel therapeutic target to ameliorate damage and prevent the downward spiral into an AUD [
14,
84].





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}