Sleep, Plasticity and the Pathophysiology of Neurodevelopmental Disorders: The Potential Roles of Protein Synthesis and Other Cellular Processes

Abstract
:1. Introduction
2. Sleep-Dependent Memory during Normal Development
3. Disturbances of Sleep in Neurodevelopmental Disorders
3.1. Autism
3.2. Tuberous Sclerosis Complex
3.3. Fragile X Syndrome
3.4. Rett Syndrome
3.5. Prader–Willi Syndrome
3.6. Angelman Syndrome
3.7. Williams Syndrome
3.8. Down Syndrome
3.9. Attention-Deficit/Hyperactivity Disorder
4. The Cellular Consequences of Normal and Prolonged Wakefulness and Their Effects on Plasticity
4.1. Sleep Regulatory Substances
4.1.1. Adenosine
4.1.2. Prostaglandins
4.1.3. Nitric Oxide
4.1.4. Cytokines
4.2. Sleep Regulatory Processes
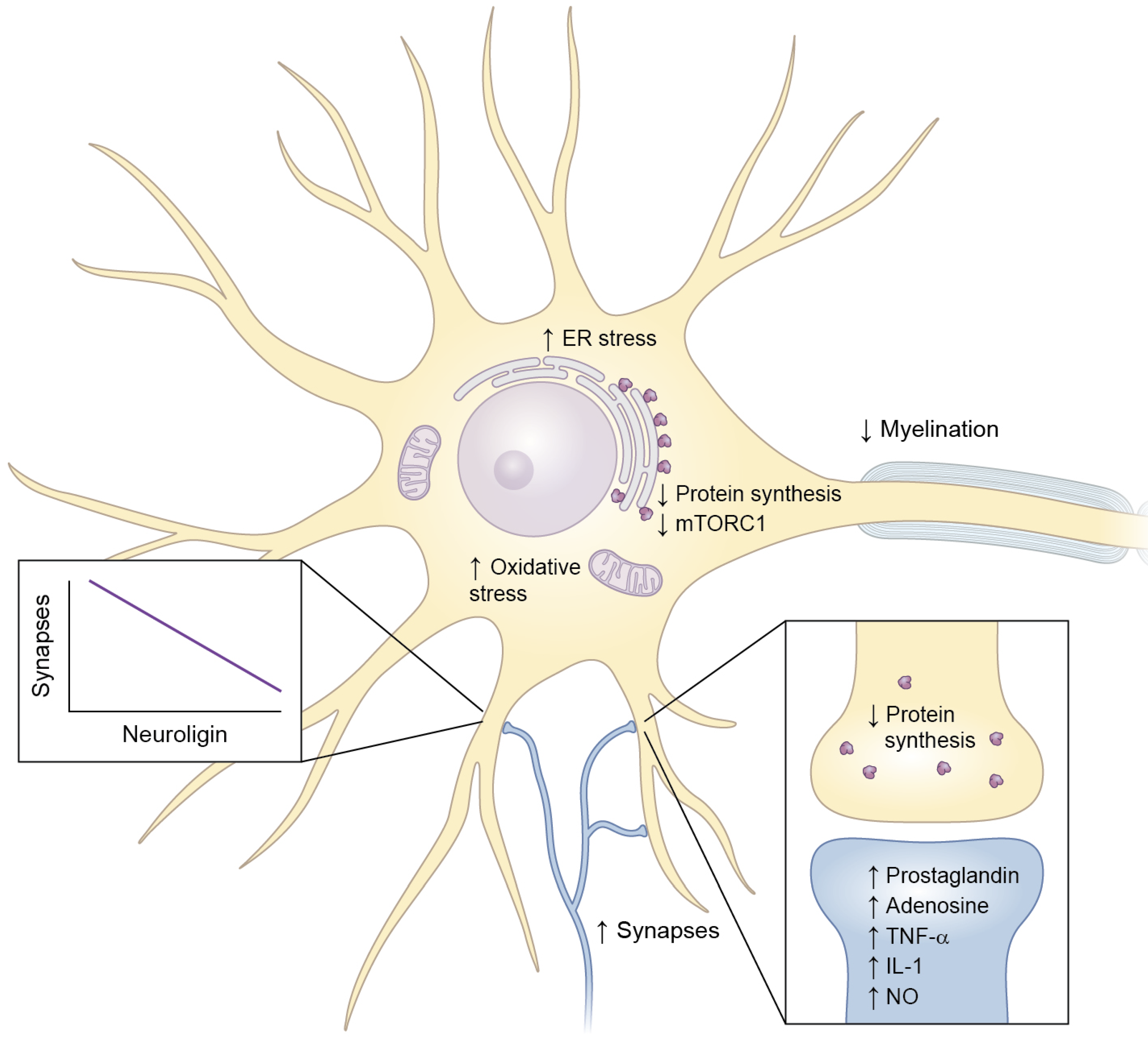
4.2.1. Synapse Formation and Synaptic Activity
4.2.2. Myelination
4.2.3. Cellular Toxicity
4.2.3.1. The Glymphatic System
4.2.3.2. Unfolded Protein Response (UPR)/Endoplasmic Reticulum (ER) Stress
4.2.3.3. Oxidative Stress
4.2.3.4. Neurodegeneration Following Cellular Stress
5. The Role of Protein Synthesis in Sleep-Dependent Plasticity
5.1. Transcription versus Translation
5.2. Manipulating Protein Synthesis

{kind=link}
| Year, First Author | Inhibitor | Administration Method/Location | NR | R |
|---|---|---|---|---|
| 1975, Drucker-Colin [284] | Anisomycin | Intraperitoneal | 0 | – |
| 1977, Rojas-Ramirez [285] | Anisomycin | Intraperitoneal | 0 | – |
| 1979, Drucker-Colin [286] | Anisomycin | Intraperitoneal | 0 | – |
| 1980, Gutwein [287] | Anisomycin | Subcutaneous | + | – |
| 2008, Methippara [288] | Anisomycin | Ventrolateral Preoptic Nucleus | + | 0 |
| 2008, Methippara [288] | Anisomycin | Lateral Hypothalamus | 0 | + |
| 2008, Methippara [288] | Anisomycin | Hippocampus | 0 | 0 |
| 1975, Kitahama [289] | Chloramphenicol | Oral | – | – |
| 1977, Rojas-Ramirez [285] | Chloramphenicol | Intraperitoneal | 0 | – |
| 1979, Drucker-Colin [286] | Chloramphenicol | Intraperitoneal | + | – |
| 1979, Petitjean [290] | Chloramphenicol | Oral | – | – |
| 1980, Drucker-Colin [291] | Chloramphenicol | Oral | 0 | – |
| 1982, Bowersox [292] | Chloramphenicol | Oral | 0 | – |
| 2005, Moulin-Sallanon [293] | Chloramphenicol | Intraperitoneal | – | – |
| 1972, Stern [294] | Cycloheximide | Intracerebroventricular | + | + |
| 1972, Stern [294] | Cycloheximide | Intraperitoneal | – | 0 |
| 1973, Pegram [295] | Cycloheximide | Subcutaneous | + | – |
| 1981, Uezu [296] | Cycloheximide | Intracerebroventricular | 0 | – |
| 1981, Uezu [296] | Cycloheximide | Intraperitoneal | 0 | – |
| 1979, Petitjean [290] | Erythromycin | Oral | 0 | – |
| 1983, Nonaka [297] | Minocycline | Oral | – | 0 |
| 1979, Petitjean [290] | Oxytetracycline | Oral | 0 | 0 |
| 1977, Rojas-Ramirez [285] | Penicillin G | Intraperitoneal | 0 | 0 |
| 1972, Stern [294] | Puromycin | Intracerebroventricular | 0 | 0 |
| 1972, Stern [294] | Puromycin | Intraperitoneal | 0 | 0 |
| 1981, Uezu [296] | Puromycin | Intracerebroventricular | 0 | – |
| 1981, Uezu [296] | Puromycin | Intraperitoneal | 0 | 0 |
| 2009, Methippara [249] | Salubrinal | Intracerebroventricular | + | 0 |
| 2012, Methippara [250] | Salubrinal | Intracerebroventricular | + | 0 |
| 1975, Kitahama [289] | Thiamphenicol | Oral | 0 | 0 |
| 1979, Petitjean [290] | Thiamphenicol | Oral | 0 | 0 |
| 1982, Bowersox [292] | Thiamphenicol | Oral | 0 | 0 |
5.3. Measuring Protein Synthesis: Sleep Deprivation
5.4. Measuring Protein Synthesis: Normal Sleep
5.5. Sleep, Protein Synthesis and Plasticity
6. The Potential Role of Disordered Sleep in the Pathophysiology of Neurodevelopmental Disorders
6.1. Sleepiness/Inattention
6.2. Accumulation of Sleep Regulatory Substances
6.3. Cellular Stress
6.4. Myelination
6.5. Synaptic Regulation
6.6. mTORC1
6.7. Protein Synthesis
7. The Potential Role of Disordered Sleep in the Pathophysiology of Neurodegenerative Diseases
8. Future Studies
9. Conclusion
Acknowledgments
Conflicts of Interest
References
- Diekelmann, S.; Wilhelm, I.; Born, J. The whats and whens of sleep-dependent memory consolidation. Sleep Med. Rev. 2009, 13, 309–321. [Google Scholar] [CrossRef]
- Stickgold, R. How do I remember? Let me count the ways. Sleep Med. Rev. 2009, 13, 305–308. [Google Scholar] [CrossRef]
- Wilhelm, I.; Prehn-Kristensen, A.; Born, J. Sleep-dependent memory consolidation—what can be learnt from children? Neurosci. Biobehav. Rev. 2012, 36, 1718–1728. [Google Scholar] [CrossRef]
- Deregnaucourt, S.; Mitra, P.P.; Feher, O.; Pytte, C.; Tchernichovski, O. How sleep affects the developmental learning of bird song. Nature 2005, 433, 710–716. [Google Scholar] [CrossRef]
- Gomez, R.L.; Bootzin, R.R.; Nadel, L. Naps promote abstraction in language-learning infants. Psychol. Sci. 2006, 17, 670–674. [Google Scholar] [CrossRef]
- Hupbach, A.; Gomez, R.L.; Bootzin, R.R.; Nadel, L. Nap-dependent learning in infants. Dev. Sci. 2009, 12, 1007–1012. [Google Scholar] [CrossRef]
- Backhaus, J.; Hoeckesfeld, R.; Born, J.; Hohagen, F.; Junghanns, K. Immediate as well as delayed post learning sleep but not wakefulness enhances declarative memory consolidation in children. Neurobiol. Learn. Mem. 2008, 89, 76–80. [Google Scholar] [CrossRef]
- Fischer, S.; Wilhelm, I.; Born, J. Developmental differences in sleep’s role for implicit off-line learning: Comparing children with adults. J. Cogn. Neurosci. 2007, 19, 214–227. [Google Scholar] [CrossRef]
- Prehn-Kristensen, A.; Goder, R.; Chirobeja, S.; Bressmann, I.; Ferstl, R.; Baving, L. Sleep in children enhances preferentially emotional declarative but not procedural memories. J. Exp. Child Psychol. 2009, 104, 132–139. [Google Scholar] [CrossRef]
- Wilhelm, I.; Diekelmann, S.; Born, J. Sleep in children improves memory performance on declarative but not procedural tasks. Learn. Mem. 2008, 15, 373–377. [Google Scholar] [CrossRef]
- Wilhelm, I.; Metzkow-Meszaros, M.; Knapp, S.; Born, J. Sleep-dependent consolidation of procedural motor memories in children and adults: The pre-sleep level of performance matters. Dev. Sci. 2012, 15, 506–515. [Google Scholar] [CrossRef]
- Wilhelm, I.; Rose, M.; Imhof, K.I.; Rasch, B.; Buchel, C.; Born, J. The sleeping child outplays the adult’s capacity to convert implicit into explicit knowledge. Nat. Neurosci. 2013, 16, 391–393. [Google Scholar] [CrossRef]
- Schreck, K.A.; Mulick, J.A.; Smith, A.F. Sleep problems as possible predictors of intensified symptoms of autism. Res. Dev. Disabil. 2004, 25, 57–66. [Google Scholar] [CrossRef]
- Sikora, D.M.; Johnson, K.; Clemons, T.; Katz, T. The relationship between sleep problems and daytime behavior in children of different ages with autism spectrum disorders. Pediatrics 2012, 130 (Suppl. 2), S83–S90. [Google Scholar]
- Taylor, M.A.; Schreck, K.A.; Mulick, J.A. Sleep disruption as a correlate to cognitive and adaptive behavior problems in autism spectrum disorders. Res. Dev. Disabil. 2012, 33, 1408–1417. [Google Scholar] [CrossRef]
- Van Eeghen, A.M.; Numis, A.I.; Staley, B.A.; Therrien, S.E.; Thibert, R.L.; Thiele, E.A. Characterizing sleep disorders of adults with tuberous sclerosis complex: A questionnaire-based study and review. Epilepsy Behav. 2011, 20, 68–74. [Google Scholar] [CrossRef]
- Kronk, R.; Bishop, E.E.; Raspa, M.; Bickel, J.O.; Mandel, D.A.; Bailey, D.B., Jr. Prevalence, nature, and correlates of sleep problems among children with fragile X syndrome based on a large scale parent survey. Sleep 2010, 33, 679–687. [Google Scholar]
- Gamble, K.L.; May, R.S.; Besing, R.C.; Tankersly, A.P.; Fargason, R.E. Delayed sleep timing and symptoms in adults with attention-deficit/hyperactivity disorder: A controlled actigraphy study. Chronobiol. Int. 2013, 30, 598–606. [Google Scholar] [CrossRef]
- Prevalence of Autism Spectrum Disorders—Autism and Developmental Disabilities Monitoring Network, 14 Sites, United States, 2008; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2012.
- Cappon, D. Clinical manifestations of autism and schizophrenia in childhood. Can. Med. Assoc. J. 1953, 69, 44–49. [Google Scholar]
- Le Couteur, A.; Rutter, M.; Lord, C.; Rios, P.; Robertson, S.; Holdgrafer, M.; McLennan, J. Autism diagnostic interview: A standardized investigator-based instrument. J. Autism Dev. Disord. 1989, 19, 363–387. [Google Scholar] [CrossRef]
- Richdale, A.L. Sleep problems in autism: Prevalence, cause, and intervention. Dev. Med. Child Neurol. 1999, 41, 60–66. [Google Scholar] [CrossRef]
- Oyane, N.M.; Bjorvatn, B. Sleep disturbances in adolescents and young adults with autism and Asperger syndrome. Autism 2005, 9, 83–94. [Google Scholar] [CrossRef]
- Tani, P.; Lindberg, N.; Nieminen-von Wendt, T.; von Wendt, L.; Alanko, L.; Appelberg, B.; Porkka-Heiskanen, T. Insomnia is a frequent finding in adults with Asperger syndrome. BMC Psychiatry 2003, 3, 12. [Google Scholar] [CrossRef]
- Cortesi, F.; Giannotti, F.; Ivanenko, A.; Johnson, K. Sleep in children with autistic spectrum disorder. Sleep Med. 2010, 11, 659–664. [Google Scholar] [CrossRef]
- Souders, M.C.; Mason, T.B.; Valladares, O.; Bucan, M.; Levy, S.E.; Mandell, D.S.; Weaver, T.E.; Pinto-Martin, J. Sleep behaviors and sleep quality in children with autism spectrum disorders. Sleep 2009, 32, 1566–1578. [Google Scholar]
- Kotagal, S.; Broomall, E. Sleep in children with autism spectrum disorder. Pediatric Neurol. 2012, 47, 242–251. [Google Scholar] [CrossRef]
- Xue, M.; Brimacombe, M.; Chaaban, J.; Zimmerman-Bier, B.; Wagner, G.C. Autism spectrum disorders: Concurrent clinical disorders. J. Child Neurol. 2008, 23, 6–13. [Google Scholar]
- Patzold, L.M.; Richdale, A.L.; Tonge, B.J. An investigation into sleep characteristics of children with autism and Asperger’s Disorder. J. Paediatr. Child Health 1998, 34, 528–533. [Google Scholar] [CrossRef]
- Anders, T.F.; Iosif, A.M.; Schwichtenberg, A.J.; Tang, K.; Goodlin-Jones, B.L. Six-month sleep-wake organization and stability in preschool-age children with autism, developmental delay, and typical development. Behav. Sleep Med. 2011, 9, 92–106. [Google Scholar] [CrossRef]
- Miano, S.; Bruni, O.; Elia, M.; Trovato, A.; Smerieri, A.; Verrillo, E.; Roccella, M.; Terzano, M.G.; Ferri, R. Sleep in children with autistic spectrum disorder: A questionnaire and polysomnographic study. Sleep Med. 2007, 9, 64–70. [Google Scholar] [CrossRef]
- Goodlin-Jones, B.L.; Tang, K.; Liu, J.; Anders, T.F. Sleep patterns in preschool-age children with autism, developmental delay, and typical development. J. Am. Acad. Child Adolesc. Psychiatry 2008, 47, 930–938. [Google Scholar] [CrossRef]
- Buckley, A.W.; Rodriguez, A.J.; Jennison, K.; Buckley, J.; Thurm, A.; Sato, S.; Swedo, S. Rapid eye movement sleep percentage in children with autism compared with children with developmental delay and typical development. Arch. Pediatr. Adolesc. Med. 2010, 164, 1032–1037. [Google Scholar]
- Buckley, A.; Wingert, K.; Swedo, S.; Thurm, A.; Sato, S.; Appel, S.; Rodriguez, A.J. First night effect analysis in a cohort of young children with autism spectrum disorder. J. Clin. Sleep Med. 2013, 9, 67–70. [Google Scholar]
- Elia, M.; Ferri, R.; Musumeci, S.A.; Del Gracco, S.; Bottitta, M.; Scuderi, C.; Miano, G.; Panerai, S.; Bertrand, T.; Grubar, J.C. Sleep in subjects with autistic disorder: A neurophysiological and psychological study. Brain Dev. 2000, 22, 88–92. [Google Scholar] [CrossRef]
- Honomichl, R.D.; Goodlin-Jones, B.L.; Burnham, M.; Gaylor, E.; Anders, T.F. Sleep patterns of children with pervasive developmental disorders. J. Autism Dev. Disord. 2002, 32, 553–561. [Google Scholar] [CrossRef]
- Allik, H.; Larsson, J.O.; Smedje, H. Insomnia in school-age children with Asperger syndrome or high-functioning autism. BMC Psychiatry 2006, 6, 18. [Google Scholar] [CrossRef]
- Limoges, E.; Mottron, L.; Bolduc, C.; Berthiaume, C.; Godbout, R. Atypical sleep architecture and the autism phenotype. Brain 2005, 128, 1049–1061. [Google Scholar] [CrossRef]
- Hare, D.J.; Jones, S.; Evershed, K. A comparative study of circadian rhythm functioning and sleep in people with Asperger syndrome. Autism 2006, 10, 565–575. [Google Scholar] [CrossRef]
- Limoges, E.; Bolduc, C.; Berthiaume, C.; Mottron, L.; Godbout, R. Relationship between poor sleep and daytime cognitive performance in young adults with autism. Res. Dev. Disabil. 2013, 34, 1322–1335. [Google Scholar] [CrossRef]
- Diomedi, M.; Curatolo, P.; Scalise, A.; Placidi, F.; Caretto, F.; Gigli, G.L. Sleep abnormalities in mentally retarded autistic subjects: Down’s syndrome with mental retardation and normal subjects. Brain Dev. 1999, 21, 548–553. [Google Scholar] [CrossRef]
- Baker, E.; Richdale, A.; Short, M.; Gradisar, M. An investigation of sleep patterns in adolescents with high-functioning autism spectrum disorder compared with typically developing adolescents. Dev. Neurorehabil. 2013, 16, 155–165. [Google Scholar] [CrossRef]
- Cipolli, C.; Mazzetti, M.; Plazzi, G. Sleep-dependent memory consolidation in patients with sleep disorders. Sleep Med. Rev. 2013, 17, 91–103. [Google Scholar] [CrossRef]
- Dresler, M.; Kluge, M.; Pawlowski, M.; Schussler, P.; Steiger, A.; Genzel, L. A double dissociation of memory impairments in major depression. J. Psychiatr. Res. 2011, 45, 1593–1599. [Google Scholar] [CrossRef]
- Tessier, S.; Lambert, A.; Rochette, A.; Mottron, L.; Godbout, R. REM sleep and face perception in children with autism. Sleep 2010, 33, A242–A243. [Google Scholar]
- Maski, K.; Holbrook, H.; Hanson, E.; Manoach, D.; Stickgold, R. Sleep dependent memory consolidation in children with autism spectrum disorders (ASD) using a probabalistic categorical learning task. Sleep 2012, 35, A369. [Google Scholar]
- Osborne, J.P.; Fryer, A.; Webb, D. Epidemiology of tuberous sclerosis. Ann. N. Y. Acad. Sci. 1991, 615, 125–127. [Google Scholar] [CrossRef]
- Curatolo, P.; Verdecchia, M.; Bombardieri, R. Tuberous sclerosis complex: A review of neurological aspects. Eur. J. Paediatr. Neurol. 2002, 6, 15–23. [Google Scholar] [CrossRef]
- Gillberg, I.C.; Gillberg, C.; Ahlsen, G. Autistic behaviour and attention deficits in tuberous sclerosis: A population-based study. Dev. Med. Child Neurol. 1994, 36, 50–56. [Google Scholar] [CrossRef]
- Numis, A.L.; Major, P.; Montenegro, M.A.; Muzykewicz, D.A.; Pulsifer, M.B.; Thiele, E.A. Identification of risk factors for autism spectrum disorders in tuberous sclerosis complex. Neurology 2011, 76, 981–987. [Google Scholar] [CrossRef]
- Bruni, O.; Cortesi, F.; Giannotti, F.; Curatolo, P. Sleep disorders in tuberous sclerosis: A polysomnographic study. Brain Dev. 1995, 17, 52–56. [Google Scholar] [CrossRef]
- Hunt, A.; Stores, G. Sleep disorder and epilepsy in children with tuberous sclerosis: A questionnaire-based study. Dev. Med. Child Neurol. 1994, 36, 108–115. [Google Scholar] [CrossRef]
- Hunt, A. Development, behaviour and seizures in 300 cases of tuberous sclerosis. J. Intell. Disabil. Res. 1993, 37, 41–51. [Google Scholar] [CrossRef]
- van Golde, E.G.; Gutter, T.; de Weerd, A.W. Sleep disturbances in people with epilepsy; prevalence, impact and treatment. Sleep Med. Rev. 2011, 15, 357–368. [Google Scholar] [CrossRef]
- Bailey, D.B., Jr.; Mesibov, G.B.; Hatton, D.D.; Clark, R.D.; Roberts, J.E.; Mayhew, L. Autistic behavior in young boys with fragile X syndrome. J. Autism Dev. Disord 1998, 28, 499–508. [Google Scholar] [CrossRef]
- Hagerman, R.; Hoem, G.; Hagerman, P. Fragile X and autism: Intertwined at the molecular level leading to targeted treatments. Mol. Autism 2010, 1, 12. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Jackson, A.W., 3rd; Levitas, A.; Rimland, B.; Braden, M. An analysis of autism in fifty males with the fragile X syndrome. Am. J. Med. Genet. 1986, 23, 359–374. [Google Scholar] [CrossRef]
- Schaefer, G.B.; Mendelsohn, N.J. Genetics evaluation for the etiologic diagnosis of autism spectrum disorders. Genet. Med. 2008, 10, 4–12. [Google Scholar] [CrossRef]
- Muhle, R.; Trentacoste, S.V.; Rapin, I. The genetics of autism. Pediatrics 2004, 113, e472–e486. [Google Scholar] [CrossRef]
- Turner, G.; Webb, T.; Wake, S.; Robinson, H. Prevalence of fragile X syndrome. Am. J. Med. Genet. 1996, 64, 196–197. [Google Scholar] [CrossRef]
- Penagarikano, O.; Mulle, J.G.; Warren, S.T. The pathophysiology of fragile X syndrome. Ann. Rev. Genomics Human Genet. 2007, 8, 109–129. [Google Scholar] [CrossRef]
- Gould, E.L.; Loesch, D.Z.; Martin, M.J.; Hagerman, R.J.; Armstrong, S.M.; Huggins, R.M. Melatonin profiles and sleep characteristics in boys with fragile X syndrome: A preliminary study. Am. J. Med. Genet. 2000, 95, 307–315. [Google Scholar] [CrossRef]
- Musumeci, S.A.; Ferri, R.; Elia, M.; DalGracco, S.; Scuderi, C.; Stefanini, M.C.; Castano, A.; Azan, G. Sleep neurophysiology in fragile X syndrome patients. Dev. Brain Dysf. 1995, 8, 218–222. [Google Scholar]
- Miano, S.; Bruni, O.; Elia, M.; Scifo, L.; Smerieri, A.; Trovato, A.; Verrillo, E.; Terzano, M.G.; Ferri, R. Sleep phenotypes of intellectual disability: A polysomnographic evaluation in subjects with Down syndrome and Fragile-X syndrome. Clin. Neurophysiol. 2008, 119, 1242–1247. [Google Scholar] [CrossRef]
- Bushey, D.; Tononi, G.; Cirelli, C. The Drosophila fragile X mental retardation gene regulates sleep need. J. Neurosci. 2009, 29, 1948–1961. [Google Scholar] [CrossRef]
- Zhang, J.; Fang, Z.; Jud, C.; Vansteensel, M.J.; Kaasik, K.; Lee, C.C.; Albrecht, U.; Tamanini, F.; Meijer, J.H.; Oostra, B.A.; Nelson, D.L. Fragile X-related proteins regulate mammalian circadian behavioral rhythms. Am. J. Human Genet. 2008, 83, 43–52. [Google Scholar] [CrossRef]
- Goncalves, J.T.; Anstey, J.E.; Golshani, P.; Portera-Cailliau, C. Circuit level defects in the developing neocortex of Fragile X mice. Nat. Neurosci. 2013, 16, 903–909. [Google Scholar] [CrossRef]
- Neul, J.L.; Zoghbi, H.Y. Rett syndrome: A prototypical neurodevelopmental disorder. Neuroscientist 2004, 10, 118–128. [Google Scholar] [CrossRef]
- Marcus, C.L.; Carroll, J.L.; McColley, S.A.; Loughlin, G.M.; Curtis, S.; Pyzik, P.; Naidu, S. Polysomnographic characteristics of patients with Rett syndrome. J. Pediatr. 1994, 125, 218–224. [Google Scholar] [CrossRef]
- Carotenuto, M.; Esposito, M.; D’Aniello, A.; Rippa, C.D.; Precenzano, F.; Pascotto, A.; Bravaccio, C.; Elia, M. Polysomnographic findings in Rett syndrome: A case-control study. Sleep Breath. 2013, 17, 93–98. [Google Scholar] [CrossRef]
- Piazza, C.C.; Fisher, W.; Kiesewetter, K.; Bowman, L.; Moser, H. Aberrant sleep patterns in children with the Rett syndrome. Brain Dev. 1990, 12, 488–493. [Google Scholar] [CrossRef]
- McArthur, A.J.; Budden, S.S. Sleep dysfunction in Rett syndrome: A trial of exogenous melatonin treatment. Dev. Med. Child Neurol. 1998, 40, 186–192. [Google Scholar] [CrossRef]
- Moretti, P.; Bouwknecht, J.A.; Teague, R.; Paylor, R.; Zoghbi, H.Y. Abnormalities of social interactions and home-cage behavior in a mouse model of Rett syndrome. Human Mol. Genet. 2005, 14, 205–220. [Google Scholar]
- De Filippis, B.; Ricceri, L.; Laviola, G. Early postnatal behavioral changes in the Mecp2-308 truncation mouse model of Rett syndrome. Genes Brain Behav. 2010, 9, 213–223. [Google Scholar]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef]
- Holm, V.A.; Cassidy, S.B.; Butler, M.G.; Hanchett, J.M.; Greenswag, L.R.; Whitman, B.Y.; Greenberg, F. Prader-Willi syndrome: Consensus diagnostic criteria. Pediatrics 1993, 91, 398–402. [Google Scholar]
- Cassidy, S.B. Prader-Willi syndrome. J. Med. Genet. 1997, 34, 917–923. [Google Scholar] [CrossRef]
- Kleppe, S.A.; Katayama, K.M.; Shipley, K.G.; Foushee, D.R. The speech and language characteristics of children with Prader-Willi syndrome. J. Speech Hear. Disord. 1990, 55, 300–309. [Google Scholar]
- Kim, J.W.; Yoo, H.J.; Cho, S.C.; Hong, K.E.; Kim, B.N. Behavioral characteristics of Prader-Willi syndrome in Korea: Comparison with children with mental retardation and normal controls. J. Child Neurol. 2005, 20, 134–138. [Google Scholar] [CrossRef]
- Stores, G.; Wiggs, L. Sleep Disturbance in Children and Adolescents with Disorders of Development: Its Significance and Management; Mac Keith Press: London, UK, 2001; p. 221. [Google Scholar]
- Joo, E.Y.; Hong, S.B.; Sohn, Y.B.; Kwak, M.J.; Kim, S.J.; Choi, Y.O.; Kim, S.W.; Paik, K.H.; Jin, D.K. Plasma adiponectin level and sleep structures in children with Prader-Willi syndrome. J. Sleep Res. 2010, 19, 248–254. [Google Scholar] [CrossRef]
- Gibbs, S.; Wiltshire, E.; Elder, D. Nocturnal sleep measured by actigraphy in children with Prader-Willi Syndrome. J. Pediatr. 2013, 162, 765–769. [Google Scholar] [CrossRef]
- Helbing-Zwanenburg, B.; Kamphuisen, H.A.; Mourtazaev, M.S. The origin of excessive daytime sleepiness in the Prader-Willi syndrome. J. Intell. Disabil. Res. 1993, 37, 533–541. [Google Scholar] [CrossRef]
- Vela-Bueno, A.; Kales, A.; Soldatos, C.R.; Dobladez-Blanco, B.; Campos-Castello, J.; Espino-Hurtado, P.; Olivan-Palacios, J. Sleep in the Prader-Willi syndrome. Clinical and polygraphic findings. Arch. Neurol. 1984, 41, 294–296. [Google Scholar] [CrossRef]
- Nevsimalova, S.; Vankova, J.; Stepanova, I.; Seemanova, E.; Mignot, E.; Nishino, S. Hypocretin deficiency in Prader-Willi syndrome. Eur. J. Neurol. 2005, 12, 70–72. [Google Scholar] [CrossRef]
- Richdale, A.L.; Cotton, S.; Hibbit, K. Sleep and behaviour disturbance in Prader-Willi syndrome: A questionnaire study. J. Intell. Disabil. Res. 1999, 43, 380–392. [Google Scholar] [CrossRef]
- Clarke, D.J.; Waters, J.; Corbett, J.A. Adults with Prader-Willi syndrome: Abnormalities of sleep and behaviour. J. R. Soc. Med. 1989, 82, 21–24. [Google Scholar]
- Manni, R.; Politini, L.; Nobili, L.; Ferrillo, F.; Livieri, C.; Veneselli, E.; Biancheri, R.; Martinetti, M.; Tartara, A. Hypersomnia in the Prader Willi syndrome: Clinical-electrophysiological features and underlying factors. Clin. Neurophysiol. 2001, 112, 800–805. [Google Scholar] [CrossRef]
- De Cock, V.C.; Diene, G.; Molinas, C.; Masson, V.D.; Kieffer, I.; Mimoun, E.; Tiberge, M.; Tauber, M. Efficacy of modafinil on excessive daytime sleepiness in Prader-Willi syndrome. Am. J. Med. Genet. A 2011, 155A, 1552–1557. [Google Scholar]
- Vgontzas, A.N.; Bixler, E.O.; Kales, A.; Centurione, A.; Rogan, P.K.; Mascari, M.; Vela-Bueno, A. Daytime sleepiness and REM abnormalities in Prader-Willi syndrome: Evidence of generalized hypoarousal. Int. J. Neurosci. 1996, 87, 127–139. [Google Scholar] [CrossRef]
- Steffenburg, S.; Gillberg, C.L.; Steffenburg, U.; Kyllerman, M. Autism in Angelman syndrome: A population-based study. Pediatr. Neurol. 1996, 14, 131–136. [Google Scholar] [CrossRef]
- Petersen, M.B.; Brondum-Nielsen, K.; Hansen, L.K.; Wulff, K. Clinical, cytogenetic, and molecular diagnosis of Angelman syndrome: Estimated prevalence rate in a Danish county. Am. J. Med. Genet. 1995, 60, 261–262. [Google Scholar] [CrossRef]
- Peters, S.U.; Beaudet, A.L.; Madduri, N.; Bacino, C.A. Autism in Angelman syndrome: Implications for autism research. Clin. Genet. 2004, 66, 530–536. [Google Scholar] [CrossRef]
- Williams, C.A.; Angelman, H.; Clayton-Smith, J.; Driscoll, D.J.; Hendrickson, J.E.; Knoll, J.H.; Magenis, R.E.; Schinzel, A.; Wagstaff, J.; Whidden, E.M.; et al. Angelman syndrome: Consensus for diagnostic criteria. Angelman Syndrome Foundation. Am. J. Med. Genet. 1995, 56, 237–238. [Google Scholar] [CrossRef]
- Barry, R.J.; Leitner, R.P.; Clarke, A.R.; Einfeld, S.L. Behavioral aspects of Angelman syndrome: A case control study. Am. J. Med. Genet. A 2005, 132A, 8–12. [Google Scholar] [CrossRef]
- Walz, N.C.; Beebe, D.; Byars, K. Sleep in individuals with Angelman syndrome: Parent perceptions of patterns and problems. Am. J. Mental Retard. 2005, 110, 243–252. [Google Scholar] [CrossRef]
- Clayton-Smith, J. Clinical research on Angelman syndrome in the United Kingdom: Observations on 82 affected individuals. Am. J. Med. Genet. 1993, 46, 12–15. [Google Scholar] [CrossRef]
- Smith, A.; Wiles, C.; Haan, E.; McGill, J.; Wallace, G.; Dixon, J.; Selby, R.; Colley, A.; Marks, R.; Trent, R.J. Clinical features in 27 patients with Angelman syndrome resulting from DNA deletion. J. Med. Genet. 1996, 33, 107–112. [Google Scholar] [CrossRef]
- Miano, S.; Bruni, O.; Leuzzi, V.; Elia, M.; Verrillo, E.; Ferri, R. Sleep polygraphy in Angelman syndrome. Clin. Neurophysiol. 2004, 115, 938–945. [Google Scholar] [CrossRef]
- Miano, S.; Bruni, O.; Elia, M.; Musumeci, S.A.; Verrillo, E.; Ferri, R. Sleep breathing and periodic leg movement pattern in Angelman Syndrome: A polysomnographic study. Clin. Neurophysiol. 2005, 116, 2685–2692. [Google Scholar]
- Moncla, A.; Malzac, P.; Livet, M.O.; Voelckel, M.A.; Mancini, J.; Delaroziere, J.C.; Philip, N.; Mattei, J.F. Angelman syndrome resulting from UBE3A mutations in 14 patients from eight families: Clinical manifestations and genetic counselling. J. Med. Genet. 1999, 36, 554–560. [Google Scholar]
- Huang, H.S.; Burns, A.J.; Nonneman, R.J.; Baker, L.K.; Riddick, N.V.; Nikolova, V.D.; Riday, T.T.; Yashiro, K.; Philpot, B.D.; Moy, S.S. Behavioral deficits in an Angelman syndrome model: Effects of genetic background and age. Behav. Brain Res. 2013, 243, 79–90. [Google Scholar] [CrossRef]
- Colas, D.; Wagstaff, J.; Fort, P.; Salvert, D.; Sarda, N. Sleep disturbances in Ube3a maternal-deficient mice modeling Angelman syndrome. Neurobiol. Dis. 2005, 20, 471–478. [Google Scholar] [CrossRef]
- Stromme, P.; Bjornstad, P.G.; Ramstad, K. Prevalence estimation of Williams syndrome. J. Child Neurol. 2002, 17, 269–271. [Google Scholar] [CrossRef]
- Riby, D.M.; Hancock, P.J. Viewing it differently: Social scene perception in Williams syndrome and autism. Neuropsychologia 2008, 46, 2855–2860. [Google Scholar] [CrossRef]
- Ypsilanti, A.; Grouios, G.; Alevriadou, A.; Tsapkini, K. Expressive and receptive vocabulary in children with Williams and Down syndromes. J. Intell. Disabil. Res. 2005, 49, 353–364. [Google Scholar] [CrossRef]
- Fishman, I.; Yam, A.; Bellugi, U.; Mills, D. Language and sociability: Insights from Williams syndrome. J. Neurodev. Disord. 2011, 3, 185–192. [Google Scholar] [CrossRef]
- Karmiloff-Smith, A. Perspectives on the dynamic development of cognitive capacities: Insights from Williams syndrome. Curr. Opin. Neurol. 2012, 25, 106–111. [Google Scholar] [CrossRef]
- Annaz, D.; Hill, C.M.; Ashworth, A.; Holley, S.; Karmiloff-Smith, A. Characterisation of sleep problems in children with Williams syndrome. Res. Dev. Disabil. 2011, 32, 164–169. [Google Scholar] [CrossRef]
- Mason, T.B.; Arens, R.; Sharman, J.; Bintliff-Janisak, B.; Schultz, B.; Walters, A.S.; Cater, J.R.; Kaplan, P.; Pack, A.I. Sleep in children with Williams Syndrome. Sleep Med. 2011, 12, 892–897. [Google Scholar] [CrossRef]
- Goldman, S.E.; Malow, B.A.; Newman, K.D.; Roof, E.; Dykens, E.M. Sleep patterns and daytime sleepiness in adolescents and young adults with Williams syndrome. J. Intell. Disabil. Res. 2009, 53, 182–188. [Google Scholar] [CrossRef]
- Gombos, F.; Bodizs, R.; Kovacs, I. Atypical sleep architecture and altered EEG spectra in Williams syndrome. J. Intell. Disabil. Res. 2011, 55, 255–262. [Google Scholar] [CrossRef]
- Ashworth, A.; Hill, C.M.; Karmiloff-Smith, A.; Dimitriou, D. Cross syndrome comparison of sleep problems in children with Down syndrome and Williams syndrome. Res. Dev. Disabil. 2013, 34, 1572–1580. [Google Scholar] [CrossRef]
- Arens, R.; Wright, B.; Elliott, J.; Zhao, H.; Wang, P.P.; Brown, L.W.; Namey, T.; Kaplan, P. Periodic limb movement in sleep in children with Williams syndrome. J. Pediatr. 1998, 133, 670–674. [Google Scholar] [CrossRef]
- Bodizs, R.; Gombos, F.; Kovacs, I. Sleep EEG fingerprints reveal accelerated thalamocortical oscillatory dynamics in Williams syndrome. Res. Dev. Disabil. 2012, 33, 153–164. [Google Scholar] [CrossRef]
- Roizen, N.J.; Patterson, D. Down’s syndrome. Lancet 2003, 361, 1281–1289. [Google Scholar] [CrossRef]
- Cotton, S.; Richdale, A. Brief report: Parental descriptions of sleep problems in children with autism, Down syndrome, and Prader-Willi syndrome. Res. Dev. Disabil. 2006, 27, 151–161. [Google Scholar] [CrossRef]
- Stores, R. Sleep and down syndrome. In Sleep Disturbances in Children and Adolescents with Disorders of Development: Its Significance and Management; Stores, G., Wiggs, L., Eds.; Mac Keith Press: London, UK, 2001; pp. 53–59. [Google Scholar]
- Marcus, C.L.; Keens, T.G.; Bautista, D.B.; von Pechmann, W.S.; Ward, S.L. Obstructive sleep apnea in children with Down syndrome. Pediatrics 1991, 88, 132–139. [Google Scholar]
- Clausen, J.; Sersen, E.A.; Lidsky, A. Sleep patterns in mental retardation: Down’s syndrome. Electroencephalogr. Clin. Neurophysiol. 1977, 43, 183–191. [Google Scholar] [CrossRef]
- Hamaguchi, H.; Hashimoto, T.; Mori, K.; Tayama, M. Sleep in the Down syndrome. Brain Dev. 1989, 11, 399–406. [Google Scholar] [CrossRef]
- Levanon, A.; Tarasiuk, A.; Tal, A. Sleep characteristics in children with Down syndrome. J. Pediatr. 1999, 134, 755–760. [Google Scholar] [CrossRef]
- Stores, R.; Stores, G.; Buckley, S. The Pattern of Sleep Problems in Children with Down’s Syndrome and Other Intellectual Disabilities. J. Appl. Res. Intellect. Disabil. 1996, 9, 145–159. [Google Scholar] [CrossRef]
- Carter, M.; McCaughey, E.; Annaz, D.; Hill, C.M. Sleep problems in a Down syndrome population. Arch. Dis. Childhood 2009, 94, 308–310. [Google Scholar] [CrossRef]
- Stores, G.; Stores, R. Sleep disorders and their clinical significance in children with Down syndrome. Dev. Med. Child Neurol. 2013, 55, 126–130. [Google Scholar] [CrossRef]
- de Miguel-Diez, J.; Villa-Asensi, J.R.; Alvarez-Sala, J.L. Prevalence of sleep-disordered breathing in children with Down syndrome: Polygraphic findings in 108 children. Sleep 2003, 26, 1006–1009. [Google Scholar]
- Ferri, R.; Curzi-Dascalova, L.; Del Gracco, S.; Elia, M.; Musumeci, S.A.; Stefanini, M.C. Respiratory patterns during sleep in Down’s syndrome: Importance of central apnoeas. J. Sleep Res. 1997, 6, 134–141. [Google Scholar] [CrossRef]
- Colas, D.; Valletta, J.S.; Takimoto-Kimura, R.; Nishino, S.; Fujiki, N.; Mobley, W.C.; Mignot, E. Sleep and EEG features in genetic models of Down syndrome. Neurobiol. Dis. 2008, 30, 1–7. [Google Scholar] [CrossRef]
- Colas, D.; Chuluun, B.; Hagiwara, G.; Mobley, W.; Garner, C.; Heller, H. Genetic determinant and cognitive consequences of sleep disturbances in mice models of Down syndrome [Abstract]. Sleep 2011, 34, A78. [Google Scholar]
- Prehn-Kristensen, A.; Goder, R.; Fischer, J.; Wilhelm, I.; Seeck-Hirschner, M.; Aldenhoff, J.; Baving, L. Reduced sleep-associated consolidation of declarative memory in attention-deficit/hyperactivity disorder. Sleep Med. 2011, 12, 672–679. [Google Scholar] [CrossRef]
- Yoon, S.Y.; Jain, U.; Shapiro, C. Sleep in attention-deficit/hyperactivity disorder in children and adults: Past, present, and future. Sleep Med. Rev. 2012, 16, 371–388. [Google Scholar] [CrossRef]
- Frankland, P.W.; Bontempi, B. The organization of recent and remote memories. Nat. Rev. Neurosci. 2005, 6, 119–130. [Google Scholar] [CrossRef]
- Dang-Vu, T.T.; Desseilles, M.; Peigneux, P.; Maquet, P. A role for sleep in brain plasticity. Pediatr. Rehabil. 2006, 9, 98–118. [Google Scholar]
- Frank, M.G. Sleep and developmental plasticity not just for kids. Prog. Brain Res. 2011, 193, 221–232. [Google Scholar] [CrossRef]
- Havekes, R.; Vecsey, C.G.; Abel, T. The impact of sleep deprivation on neuronal and glial signaling pathways important for memory and synaptic plasticity. Cell. Signal. 2012, 24, 1251–1260. [Google Scholar] [CrossRef]
- Poe, G.R.; Walsh, C.M.; Bjorness, T.E. Cognitive neuroscience of sleep. Prog. Brain Res. 2010, 185, 1–19. [Google Scholar] [CrossRef]
- Achermann, P.; Borbely, A.A. Sleep homeostasis and models of sleep regulation. In Principles and Practice of Sleep Medicine, 5th ed.; Kryger, M.H., Roth, T., Dement, W.C., Eds.; Elsevier: St. Louis, MI, USA, 2011; pp. 431–444. [Google Scholar]
- Borbely, A.A. A two process model of sleep regulation. Human Neurobiol. 1982, 1, 195–204. [Google Scholar]
- Wurts, S.W.; Edgar, D.M. Circadian and homeostatic control of rapid eye movement (REM) sleep: Promotion of REM tendency by the suprachiasmatic nucleus. J. Neurosci. 2000, 20, 4300–4310. [Google Scholar]
- Obal, F., Jr.; Krueger, J.M. Biochemical regulation of non-rapid-eye-movement sleep. Front. Biosci. 2003, 8, d520–d550. [Google Scholar] [CrossRef]
- Basheer, R.; Strecker, R.E.; Thakkar, M.M.; McCarley, R.W. Adenosine and sleep-wake regulation. Prog. Neurobiol. 2004, 73, 379–396. [Google Scholar] [CrossRef]
- Basheer, R.; Porkka-Heiskanen, T.; Stenberg, D.; McCarley, R.W. Adenosine and behavioral state control: Adenosine increases c-Fos protein and AP1 binding in basal forebrain of rats. Brain Res. Mol. Brain Res. 1999, 73, 1–10. [Google Scholar] [CrossRef]
- Porkka-Heiskanen, T.; Strecker, R.E.; McCarley, R.W. Brain site-specificity of extracellular adenosine concentration changes during sleep deprivation and spontaneous sleep: An in vivo microdialysis study. Neuroscience 2000, 99, 507–517. [Google Scholar] [CrossRef]
- Porkka-Heiskanen, T.; Strecker, R.E.; Thakkar, M.; Bjorkum, A.A.; Greene, R.W.; McCarley, R.W. Adenosine: A mediator of the sleep-inducing effects of prolonged wakefulness. Science 1997, 276, 1265–1268. [Google Scholar] [CrossRef]
- Portas, C.M.; Thakkar, M.; Rainnie, D.G.; Greene, R.W.; McCarley, R.W. Role of adenosine in behavioral state modulation: A microdialysis study in the freely moving cat. Neuroscience 1997, 79, 225–235. [Google Scholar] [CrossRef]
- Jones, B.E. Arousal systems. Frontiers in Bioscience 2003, 8, s438–s451. [Google Scholar] [CrossRef]
- Huston, J.P.; Haas, H.L.; Boix, F.; Pfister, M.; Decking, U.; Schrader, J.; Schwarting, R.K. Extracellular adenosine levels in neostriatum and hippocampus during rest and activity periods of rats. Neuroscience 1996, 73, 99–107. [Google Scholar] [CrossRef]
- Goodman, R.R.; Synder, S.H. Autoradiographic localization of adenosine receptors in rat brain using [3H]cyclohexyladenosine. J. Neurosci. 1982, 2, 1230–1241. [Google Scholar]
- De Mendonca, A.; Ribeiro, J.A. Adenosine and synaptic plasticity. Drug Dev. Res. 2001, 52, 283–290. [Google Scholar] [CrossRef]
- Hellman, K.M.; Abel, T. Molecular mechanisms of memory consolidation. In Sleep and Brain Plasticity; Maquet, P., Smith, C., Stickgold, R., Eds.; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Hayaishi, O. Molecular mechanisms of sleep-wake regulation: Roles of prostaglandins D2 and E2. FASEB J. 1991, 5, 2575–2581. [Google Scholar]
- Hayaishi, O. Molecular mechanisms of sleep-wake regulation: A role of prostaglandin D2. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000, 355, 275–280. [Google Scholar] [CrossRef]
- Toyomoto, M.; Ohta, M.; Okumura, K.; Yano, H.; Matsumoto, K.; Inoue, S.; Hayashi, K.; Ikeda, K. Prostaglandins are powerful inducers of NGF and BDNF production in mouse astrocyte cultures. FEBS Lett. 2004, 562, 211–215. [Google Scholar] [CrossRef]
- Althaus, H.H. Remyelination in multiple sclerosis: A new role for neurotrophins? Prog. Brain Res. 2004, 146, 415–432. [Google Scholar] [CrossRef]
- Gautier-Sauvigne, S.; Colas, D.; Parmantier, P.; Clement, P.; Gharib, A.; Sarda, N.; Cespuglio, R. Nitric oxide and sleep. Sleep Med. Rev. 2005, 9, 101–113. [Google Scholar] [CrossRef]
- Cespuglio, R.; Amrouni, D.; Meiller, A.; Buguet, A.; Gautier-Sauvigne, S. Nitric oxide in the regulation of the sleep-wake states. Sleep Med. Rev. 2012, 16, 265–279. [Google Scholar] [CrossRef]
- Leonard, T.O.; Lydic, R. Pontine nitric oxide modulates acetylcholine release, rapid eye movement sleep generation, and respiratory rate. J. Neurosci. 1997, 17, 774–785. [Google Scholar]
- Lowenstein, C.J.; Dinerman, J.L.; Snyder, S.H. Nitric oxide: A physiologic messenger. Ann. Intern. Med. 1994, 120, 227–237. [Google Scholar] [CrossRef]
- Wink, D.A.; Hanbauer, I.; Krishna, M.C.; DeGraff, W.; Gamson, J.; Mitchell, J.B. Nitric oxide protects against cellular damage and cytotoxicity from reactive oxygen species. Proc. Natl. Acad. Sci. USA 1993, 90, 9813–9817. [Google Scholar] [CrossRef]
- Robb, S.J.; Connor, J.R. Nitric oxide protects astrocytes from oxidative stress. Ann. N. Y. Acad. Sci. 2002, 962, 93–102. [Google Scholar] [CrossRef]
- Zhuo, M.; Small, S.A.; Kandel, E.R.; Hawkins, R.D. Nitric oxide and carbon monoxide produce activity-dependent long-term synaptic enhancement in hippocampus. Science 1993, 260, 1946–1950. [Google Scholar]
- Schuman, E.M.; Madison, D.V. A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science 1991, 254, 1503–1506. [Google Scholar]
- Krueger, J.M.; Obal, F.J.; Fang, J.; Kubota, T.; Taishi, P. The role of cytokines in physiological sleep regulation. Ann. N. Y. Acad. Sci. 2001, 933, 211–221. [Google Scholar]
- Bachwich, P.R.; Chensue, S.W.; Larrick, J.W.; Kunkel, S.L. Tumor necrosis factor stimulates interleukin-1 and prostaglandin E2 production in resting macrophages. Biochem. Biophys. Res. Commun. 1986, 136, 94–101. [Google Scholar] [CrossRef]
- Knofler, M.; Kiss, H.; Mosl, B.; Egarter, C.; Husslein, P. Interleukin-1 stimulates tumor necrosis factor-alpha (TNF-alpha) release from cytotrophoblastic BeWo cells independently of induction of the TNF-alpha mRNA. FEBS Lett. 1997, 405, 213–218. [Google Scholar] [CrossRef]
- Luk, W.P.; Zhang, Y.; White, T.D.; Lue, F.A.; Wu, C.; Jiang, C.G.; Zhang, L.; Moldofsky, H. Adenosine: A mediator of interleukin-1beta-induced hippocampal synaptic inhibition. J. Neurosci. 1999, 19, 4238–4244. [Google Scholar]
- Terao, A.; Matsumura, H.; Saito, M. Interleukin-1 induces slow-wave sleep at the prostaglandin D2-sensitive sleep-promoting zone in the rat brain. J. Neurosci. 1998, 18, 6599–6607. [Google Scholar]
- Ishizaka, M.; Ohe, Y.; Senbongi, T.; Wakabayashi, K.; Ishikawa, K. Inflammatory stimuli increase prostaglandin D synthase levels in cerebrospinal fluid of rats. Neuroreport 2001, 12, 1161–1165. [Google Scholar] [CrossRef]
- Southern, C.; Schulster, D.; Green, I.C. Inhibition of insulin secretion by interleukin-1 beta and tumour necrosis factor-alpha via an l-arginine-dependent nitric oxide generating mechanism. FEBS Lett. 1990, 276, 42–44. [Google Scholar] [CrossRef]
- Schneider, H.; Pitossi, F.; Balschun, D.; Wagner, A.; del Rey, A.; Besedovsky, H.O. A neuromodulatory role of interleukin-1beta in the hippocampus. Proc. Natl. Acad. Sci. USA 1998, 95, 7778–7783. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, L.S.; Gibboni, R.; Weiner, B.; Bao, S. Impaired development and competitive refinement of the cortical frequency map in tumor necrosis factor-alpha-deficient mice. Cereb. Cortex 2013. [Google Scholar] [CrossRef]
- Avital, A.; Goshen, I.; Kamsler, A.; Segal, M.; Iverfeldt, K.; Richter-Levin, G.; Yirmiya, R. Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus 2003, 13, 826–834. [Google Scholar] [CrossRef]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-alpha. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef]
- Tononi, G.; Cirelli, C. Sleep and synaptic homeostasis: A hypothesis. Brain Res. Bull. 2003, 62, 143–150. [Google Scholar] [CrossRef]
- Tononi, G.; Cirelli, C. Sleep function and synaptic homeostasis. Sleep Med. Rev. 2006, 10, 49–62. [Google Scholar] [CrossRef]
- Gilestro, G.F.; Tononi, G.; Cirelli, C. Widespread changes in synaptic markers as a function of sleep and wakefulness in Drosophila. Science 2009, 324, 109–112. [Google Scholar] [CrossRef]
- Vyazovskiy, V.V.; Cirelli, C.; Pfister-Genskow, M.; Faraguna, U.; Tononi, G. Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat. Neurosci. 2008, 11, 200–208. [Google Scholar] [CrossRef]
- Bushey, D.; Tononi, G.; Cirelli, C. Sleep and synaptic homeostasis: Structural evidence in Drosophila. Science 2011, 332, 1576–1581. [Google Scholar] [CrossRef]
- Donlea, J.M.; Ramanan, N.; Shaw, P.J. Use-dependent plasticity in clock neurons regulates sleep need in Drosophila. Science 2009, 324, 105–108. [Google Scholar] [CrossRef]
- Maret, S.; Faraguna, U.; Nelson, A.B.; Cirelli, C.; Tononi, G. Sleep and waking modulate spine turnover in the adolescent mouse cortex. Nat. Neurosci. 2011, 14, 1418–1420. [Google Scholar] [CrossRef]
- Yang, G.; Gan, W.B. Sleep contributes to dendritic spine formation and elimination in the developing mouse somatosensory cortex. Dev. Neurobiol. 2012, 72, 1391–1398. [Google Scholar] [CrossRef]
- Frank, M.G. Erasing synapses in sleep: Is it time to be SHY? Neural Plasticity 2012, 2012, 264–378. [Google Scholar] [CrossRef]
- Aton, S.J.; Seibt, J.; Dumoulin, M.; Jha, S.K.; Steinmetz, N.; Coleman, T.; Naidoo, N.; Frank, M.G. Mechanisms of sleep-dependent consolidation of cortical plasticity. Neuron 2009, 61, 454–466. [Google Scholar] [CrossRef]
- Scheiffele, P.; Fan, J.; Choih, J.; Fetter, R.; Serafini, T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 2000, 101, 657–669. [Google Scholar] [CrossRef]
- Graf, E.R.; Zhang, X.; Jin, S.X.; Linhoff, M.W.; Craig, A.M. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 2004, 119, 1013–1026. [Google Scholar] [CrossRef]
- Chih, B.; Engelman, H.; Scheiffele, P. Control of excitatory and inhibitory synapse formation by neuroligins. Science 2005, 307, 1324–1328. [Google Scholar] [CrossRef]
- Prange, O.; Wong, T.P.; Gerrow, K.; Wang, Y.T.; El-Husseini, A. A balance between excitatory and inhibitory synapses is controlled by PSD-95 and neuroligin. Proc. Natl. Acad. Sci. USA 2004, 101, 13915–13920. [Google Scholar] [CrossRef]
- Bourgeron, T. The possible interplay of synaptic and clock genes in autism spectrum disorders. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 645–654. [Google Scholar] [CrossRef]
- Varoqueaux, F.; Aramuni, G.; Rawson, R.L.; Mohrmann, R.; Missler, M.; Gottmann, K.; Zhang, W.; Sudhof, T.C.; Brose, N. Neuroligins determine synapse maturation and function. Neuron 2006, 51, 741–754. [Google Scholar] [CrossRef]
- Jamain, S.; Quach, H.; Betancur, C.; Rastam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; Bourgeron, T. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef]
- Ylisaukko-oja, T.; Rehnstrom, K.; Auranen, M.; Vanhala, R.; Alen, R.; Kempas, E.; Ellonen, P.; Turunen, J.A.; Makkonen, I.; Riikonen, R.; et al. Analysis of four neuroligin genes as candidates for autism. Eur. J. Human Genet. 2005, 13, 1285–1292. [Google Scholar] [CrossRef]
- Daoud, H.; Bonnet-Brilhault, F.; Vedrine, S.; Demattei, M.V.; Vourc’h, P.; Bayou, N.; Andres, C.R.; Barthelemy, C.; Laumonnier, F.; Briault, S. Autism and nonsyndromic mental retardation associated with a de novo mutation in the NLGN4X gene promoter causing an increased expression level. Biol. Psychiatry 2009, 66, 906–910. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Briesemann, M.; Yan, J. Computational analysis of gene regulation in animal sleep deprivation. Physiol. Genomics 2010, 42, 427–436. [Google Scholar] [CrossRef]
- Porter, N.M.; Bohannon, J.H.; Curran-Rauhut, M.; Buechel, H.M.; Dowling, A.L.; Brewer, L.D.; Popovic, J.; Thibault, V.; Kraner, S.D.; Chen, K.C.; Blalock, E.M. Hippocampal CA1 transcriptional profile of sleep deprivation: Relation to aging and stress. PLoS One 2012, 7, e40128. [Google Scholar] [CrossRef]
- El Helou, J.; Belanger-Nelson, E.; Freyburger, M.; Dorsaz, S.; Curie, T.; La Spada, F.; Gaudreault, P.O.; Beaumont, E.; Pouliot, P.; Lesage, F.; Frank, M.G.; Franken, P.; Mongrain, V. Neuroligin-1 links neuronal activity to sleep-wake regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 9974–9979. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, Z.; Zhang, X.; Tong, H.; Li, P.; Zhang, Z.C.; Jia, Z.; Xie, W.; Han, J. Drosophila neuroligin 4 regulates sleep through modulating GABA transmission. J. Neurosci. 2013, 33, 15545–15554. [Google Scholar] [CrossRef]
- Kim, J.; Jung, S.Y.; Lee, Y.K.; Park, S.; Choi, J.S.; Lee, C.J.; Kim, H.S.; Choi, Y.B.; Scheiffele, P.; Bailey, C.H.; et al. Neuroligin-1 is required for normal expression of LTP and associative fear memory in the amygdala of adult animals. Proc. Natl. Acad. Sci. USA 2008, 105, 9087–9092. [Google Scholar] [CrossRef]
- Blundell, J.; Blaiss, C.A.; Etherton, M.R.; Espinosa, F.; Tabuchi, K.; Walz, C.; Bolliger, M.F.; Sudhof, T.C.; Powell, C.M. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J. Neurosci. 2010, 30, 2115–2129. [Google Scholar] [CrossRef]
- Dahlhaus, R.; Hines, R.M.; Eadie, B.D.; Kannangara, T.S.; Hines, D.J.; Brown, C.E.; Christie, B.R.; El-Husseini, A. Overexpression of the cell adhesion protein neuroligin-1 induces learning deficits and impairs synaptic plasticity by altering the ratio of excitation to inhibition in the hippocampus. Hippocampus 2010, 20, 305–322. [Google Scholar]
- Cirelli, C.; Gutierrez, C.M.; Tononi, G. Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron 2004, 41, 35–43. [Google Scholar] [CrossRef]
- Cirelli, C. A molecular window on sleep: Changes in gene expression between sleep and wakefulness. Neuroscientist 2005, 11, 63–74. [Google Scholar] [CrossRef]
- Bellesi, M.; Pfister-Genskow, M.; Maret, S.; Keles, S.; Tononi, G.; Cirelli, C. Effects of sleep and wake on oligodendrocytes and their precursors. J. Neurosci. 2013, 33, 14288–14300. [Google Scholar] [CrossRef]
- Cirelli, C. Cellular consequences of sleep deprivation in the brain. Sleep Med. Rev. 2006, 10, 307–321. [Google Scholar] [CrossRef]
- Mongrain, V.; Hernandez, S.A.; Pradervand, S.; Dorsaz, S.; Curie, T.; Hagiwara, G.; Gip, P.; Heller, H.C.; Franken, P. Separating the contribution of glucocorticoids and wakefulness to the molecular and electrophysiological correlates of sleep homeostasis. Sleep 2010, 33, 1147–1157. [Google Scholar]
- Sternberger, N.H.; Itoyama, Y.; Kies, M.W.; Webster, H.D. Myelin basic protein demonstrated immunocytochemically in oligodendroglia prior to myelin sheath formation. Proc. Natl. Acad. Sci. USA 1978, 75, 2521–2524. [Google Scholar] [CrossRef]
- Goldstein, R. A rebound-like pattern of REM sleep induced by guinea pig myelin basic protein (MBP) in cats. Endocrinologie 1990, 28, 15–20. [Google Scholar]
- Stevens, B.; Porta, S.; Haak, L.L.; Gallo, V.; Fields, R.D. Adenosine: A neuron-glial transmitter promoting myelination in the CNS in response to action potentials. Neuron 2002, 36, 855–868. [Google Scholar] [CrossRef]
- Scherfler, C.; Frauscher, B.; Schocke, M.; Nocker, M.; Gschliesser, V.; Ehrmann, L.; Niederreiter, M.; Esterhammer, R.; Seppi, K.; Brandauer, E.; et al. White and gray matter abnormalities in narcolepsy with cataplexy. Sleep 2012, 35, 345–351. [Google Scholar]
- Hor, H.; Bartesaghi, L.; Kutalik, Z.; Vicario, J.L.; de Andres, C.; Pfister, C.; Lammers, G.J.; Guex, N.; Chrast, R.; Tafti, M.; Peraita-Adrados, R. A missense mutation in myelin oligodendrocyte glycoprotein as a cause of familial narcolepsy with cataplexy. Am. J. Human Genet. 2011, 89, 474–479. [Google Scholar] [CrossRef]
- Lunde, H.M.; Bjorvatn, B.; Myhr, K.M.; Bo, L. Clinical assessment and management of sleep disorders in multiple sclerosis: A literature review. Acta Neurol. Scand. Suppl. 2013, 24–30. [Google Scholar] [CrossRef]
- Gallup, A.C.; Gallup, G.G., Jr.; Feo, C. Yawning, sleep, and symptom relief in patients with multiple sclerosis. Sleep Med. 2010, 11, 329–330. [Google Scholar] [CrossRef]
- Anch, A.M.; Laposky, A.D. Rat sleep and eye movement density as biological markers of demyelinating disease. Physiol. Behav. 2000, 71, 269–275. [Google Scholar] [CrossRef]
- Cortes Mdel, C.; Gavito, B.; Ita, M.L.; Valencia, J.; Eguibar, J.R. Characterization of the spontaneous and gripping-induced immobility episodes on taiep rats. Synapse 2005, 58, 95–101. [Google Scholar]
- Fields, R.D. Myelination: An overlooked mechanism of synaptic plasticity? Neuroscientist 2005, 11, 528–531. [Google Scholar] [CrossRef]
- Markham, J.A.; Greenough, W.T. Experience-driven brain plasticity: Beyond the synapse. Neuron Glia Biol. 2004, 1, 351–363. [Google Scholar] [CrossRef]
- Gyllensten, L.; Malmfors, T. Myelinization of the optic nerve and its dependence on visual function—a quantitative investigation in mice. J. Embryol. Exp. Morphol. 1963, 11, 255–266. [Google Scholar]
- Tauber, H.; Waehneldt, T.V.; Neuhoff, V. Myelination in rabbit optic nerves is accelerated by artificial eye opening. Neurosci. Lett. 1980, 16, 235–238. [Google Scholar] [CrossRef]
- Artola, A.; von Frijtag, J.C.; Fermont, P.C.; Gispen, W.H.; Schrama, L.H.; Kamal, A.; Spruijt, B.M. Long-lasting modulation of the induction of LTD and LTP in rat hippocampal CA1 by behavioural stress and environmental enrichment. Eur. J. Neurosci. 2006, 23, 261–272. [Google Scholar] [CrossRef]
- Juraska, J.M.; Kopcik, J.R. Sex and environmental influences on the size and ultrastructure of the rat corpus callosum. Brain Res. 1988, 450, 1–8. [Google Scholar] [CrossRef]
- Giedd, J.N. Structural magnetic resonance imaging of the adolescent brain. Ann. N. Y. Acad. Sci. 2004, 1021, 77–85. [Google Scholar]
- Benes, F.M.; Turtle, M.; Khan, Y.; Farol, P. Myelination of a key relay zone in the hippocampal formation occurs in the human brain during childhood, adolescence, and adulthood. Arch. Gen. Psychiatry 1994, 51, 477–484. [Google Scholar] [CrossRef]
- Nunez, J.L.; Nelson, J.; Pych, J.C.; Kim, J.H.; Juraska, J.M. Myelination in the splenium of the corpus callosum in adult male and female rats. Brain Res. Dev. Brain Res. 2000, 120, 87–90. [Google Scholar] [CrossRef]
- Lebel, C.; Walker, L.; Leemans, A.; Phillips, L.; Beaulieu, C. Microstructural maturation of the human brain from childhood to adulthood. NeuroImage 2008, 40, 1044–1055. [Google Scholar] [CrossRef]
- Jernigan, T.L.; Tallal, P. Late childhood changes in brain morphology observable with MRI. Dev. Med. Child Neurol. 1990, 32, 379–385. [Google Scholar] [CrossRef]
- Rosenzweig, I.; Vukadinovic, Z.; Turner, A.J.; Catani, M. Neuroconnectivity and valproic acid: The myelin hypothesis. Neurosci. Biobehav. Rev. 2012, 36, 1848–1856. [Google Scholar] [CrossRef]
- Sowell, E.R.; Thompson, P.M.; Tessner, K.D.; Toga, A.W. Mapping continued brain growth and gray matter density reduction in dorsal frontal cortex: Inverse relationships during postadolescent brain maturation. J. Neurosci. 2001, 21, 8819–8829. [Google Scholar]
- Pujol, J.; Vendrell, P.; Junque, C.; Marti-Vilalta, J.L.; Capdevila, A. When does human brain development end? Evidence of corpus callosum growth up to adulthood. Ann. Neurol. 1993, 34, 71–75. [Google Scholar] [CrossRef]
- Bengtsson, S.L.; Nagy, Z.; Skare, S.; Forsman, L.; Forssberg, H.; Ullen, F. Extensive piano practicing has regionally specific effects on white matter development. Nat. Neurosci. 2005, 8, 1148–1150. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep drives metabolite clearance from the adult brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef]
- Inoue, S.; Honda, K.; Komoda, Y. Sleep as neuronal detoxification and restitution. Behavi. Brain Res. 1995, 69, 91–96. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Ron, D. Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 2006, 86, 1133–1149. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar]
- Mattson, M.P.; LaFerla, F.M.; Chan, S.L.; Leissring, M.A.; Shepel, P.N.; Geiger, J.D. Calcium signaling in the ER: Its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2000, 23, 222–229. [Google Scholar] [CrossRef]
- Naidoo, N.; Giang, W.; Galante, R.J.; Pack, A.I. Sleep deprivation induces the unfolded protein response in mouse cerebral cortex. J. Neurochem. 2005, 92, 1150–1157. [Google Scholar] [CrossRef]
- Methippara, M.M.; Bashir, T.; Kumar, S.; Alam, N.; Szymusiak, R.; McGinty, D. Sleep fragmentation in rats increases endoplasmic reticulum stress in basal forebrain neurons as shown by the expression of p-eIF2a. Sleep 2008, 31, A362. [Google Scholar]
- Vecsey, C.G.; Peixoto, L.; Choi, J.H.; Wimmer, M.; Jaganath, D.; Hernandez, P.J.; Blackwell, J.; Meda, K.; Park, A.J.; Hannenhalli, S.; Abel, T. Genomic analysis of sleep deprivation reveals translational regulation in the hippocampus. Physiol. Genomics 2012, 44, 981–991. [Google Scholar] [CrossRef]
- Poirrier, J.E.; Guillonneau, F.; Renaut, J.; Sergeant, K.; Luxen, A.; Maquet, P.; Leprince, P. Proteomic changes in rat hippocampus and adrenals following short-term sleep deprivation. Proteome Sci. 2008, 6, 14. [Google Scholar] [CrossRef]
- Pawlyk, A.C.; Ferber, M.; Shah, A.; Pack, A.I.; Naidoo, N. Proteomic analysis of the effects and interactions of sleep deprivation and aging in mouse cerebral cortex. J. Neurochem. 2007, 103, 2301–2313. [Google Scholar] [CrossRef]
- Mackiewicz, M.; Shockley, K.R.; Romer, M.A.; Galante, R.J.; Zimmerman, J.E.; Naidoo, N.; Baldwin, D.A.; Jensen, S.T.; Churchill, G.A.; Pack, A.I. Macromolecule biosynthesis: A key function of sleep. Physiol. Genomics 2007, 31, 441–457. [Google Scholar] [CrossRef]
- Jones, S.; Pfister-Genskow, M.; Benca, R.M.; Cirelli, C. Molecular correlates of sleep and wakefulness in the brain of the white-crowned sparrow. J. Neurochem. 2008, 105, 46–62. [Google Scholar] [CrossRef]
- Terao, A.; Steininger, T.L.; Hyder, K.; Apte-Deshpande, A.; Ding, J.; Rishipathak, D.; Davis, R.W.; Heller, H.C.; Kilduff, T.S. Differential increase in the expression of heat shock protein family members during sleep deprivation and during sleep. Neuroscience 2003, 116, 187–200. [Google Scholar] [CrossRef]
- Terao, A.; Wisor, J.P.; Peyron, C.; Apte-Deshpande, A.; Wurts, S.W.; Edgar, D.M.; Kilduff, T.S. Gene expression in the rat brain during sleep deprivation and recovery sleep: An Affymetrix GeneChip study. Neuroscience 2006, 137, 593–605. [Google Scholar] [CrossRef]
- Cirelli, C.; Faraguna, U.; Tononi, G. Changes in brain gene expression after long-term sleep deprivation. J. Neurochem. 2006, 98, 1632–1645. [Google Scholar] [CrossRef]
- Naidoo, N.; Casiano, V.; Cater, J.; Zimmerman, J.; Pack, A.I. A role for the molecular chaperone protein BiP/GRP78 in Drosophila sleep homeostasis. Sleep 2007, 30, 557–565. [Google Scholar]
- Cirelli, C.; Tononi, G. Gene expression in the brain across the sleep-waking cycle. Brain Res. 2000, 885, 303–321. [Google Scholar] [CrossRef]
- Zimmerman, J.E.; Rizzo, W.; Shockley, K.R.; Raizen, D.M.; Naidoo, N.; Mackiewicz, M.; Churchill, G.A.; Pack, A.I. Multiple mechanisms limit the duration of wakefulness in Drosophila brain. Physiol. Genomics 2006, 27, 337–350. [Google Scholar] [CrossRef]
- Cirelli, C.; Shaw, P.J.; Rechtschaffen, A.; Tononi, G. No evidence of brain cell degeneration after long-term sleep deprivation in rats. Brain Res. 1999, 840, 184–193. [Google Scholar] [CrossRef]
- Methippara, M.M.; Bashir, T.; Kumar, S.; Alam, N.; Szymusiak, R.; McGinty, D. Salubrinal, an inhibitor of protein synthesis, promotes deep slow wave sleep. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R178–R184. [Google Scholar]
- Methippara, M.; Mitrani, B.; Schrader, F.X.; Szymusiak, R.; McGinty, D. Salubrinal, an endoplasmic reticulum stress blocker, modulates sleep homeostasis and activation of sleep- and wake-regulatory neurons. Neuroscience 2012, 209, 108–118. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Koumenis, C.; Naczki, C.; Koritzinsky, M.; Rastani, S.; Diehl, A.; Sonenberg, N.; Koromilas, A.; Wouters, B.G. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol. Cell. Biol. 2002, 22, 7405–7416. [Google Scholar] [CrossRef]
- Estes, S.D.; Stoler, D.L.; Anderson, G.R. Normal fibroblasts induce the C/EBP beta and ATF-4 bZIP transcription factors in response to anoxia. Exp. Cell Res. 1995, 220, 47–54. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA 2008, 105, 18525–18530. [Google Scholar]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef]
- Song, B.; Scheuner, D.; Ron, D.; Pennathur, S.; Kaufman, R.J. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Investig. 2008, 118, 3378–3389. [Google Scholar] [CrossRef]
- Moller-Levet, C.S.; Archer, S.N.; Bucca, G.; Laing, E.E.; Slak, A.; Kabiljo, R.; Lo, J.C.; Santhi, N.; von Schantz, M.; Smith, C.P.; Dijk, D.J. Effects of insufficient sleep on circadian rhythmicity and expression amplitude of the human blood transcriptome. Proc. Natl. Acad. Sci. USA 2013, 110, E1132–E1141. [Google Scholar] [CrossRef]
- Lungato, L.; Marques, M.S.; Pereira, V.G.; Hix, S.; Gazarini, M.L.; Tufik, S.; D’Almeida, V. Sleep deprivation alters gene expression and antioxidant enzyme activity in mice splenocytes. Scand. J. Immunol. 2013, 77, 195–199. [Google Scholar] [CrossRef]
- Gulec, M.; Ozkol, H.; Selvi, Y.; Tuluce, Y.; Aydin, A.; Besiroglu, L.; Ozdemir, P.G. Oxidative stress in patients with primary insomnia. Progress Neuropsychopharmacol. Biol. Psychiatry 2012, 37, 247–251. [Google Scholar] [CrossRef]
- Vollert, C.; Zagaar, M.; Hovatta, I.; Taneja, M.; Vu, A.; Dao, A.; Levine, A.; Alkadhi, K.; Salim, S. Exercise prevents sleep deprivation-associated anxiety-like behavior in rats: Potential role of oxidative stress mechanisms. Behav. Brain Res. 2011, 224, 233–240. [Google Scholar] [CrossRef]
- Suer, C.; Dolu, N.; Artis, A.S.; Sahin, L.; Yilmaz, A.; Cetin, A. The effects of long-term sleep deprivation on the long-term potentiation in the dentate gyrus and brain oxidation status in rats. Neurosci. Res. 2011, 70, 71–77. [Google Scholar] [CrossRef]
- Khadrawy, Y.A.; Nour, N.A.; Aboul Ezz, H.S. Effect of oxidative stress induced by paradoxical sleep deprivation on the activities of Na+, K+-ATPase and acetylcholinesterase in the cortex and hippocampus of rat. Transl. Res. 2011, 157, 100–107. [Google Scholar] [CrossRef]
- Everson, C.A.; Laatsch, C.D.; Hogg, N. Antioxidant defense responses to sleep loss and sleep recovery. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R374–R383. [Google Scholar]
- Singh, R.; Kiloung, J.; Singh, S.; Sharma, D. Effect of paradoxical sleep deprivation on oxidative stress parameters in brain regions of adult and old rats. Biogerontology 2008, 9, 153–162. [Google Scholar] [CrossRef]
- Silva, R.H.; Abilio, V.C.; Takatsu, A.L.; Kameda, S.R.; Grassl, C.; Chehin, A.B.; Medrano, W.A.; Calzavara, M.B.; Registro, S.; Andersen, M.L.; et al. Role of hippocampal oxidative stress in memory deficits induced by sleep deprivation in mice. Neuropharmacology 2004, 46, 895–903. [Google Scholar] [CrossRef]
- Melgarejo-Gutierrez, M.; Acosta-Pena, E.; Venebra-Munoz, A.; Escobar, C.; Santiago-Garcia, J.; Garcia-Garcia, F. Sleep deprivation reduces neuroglobin immunoreactivity in the rat brain. Neuroreport 2013, 24, 120–125. [Google Scholar] [CrossRef]
- Gopalakrishnan, A.; Ji, L.L.; Cirelli, C. Sleep deprivation and cellular responses to oxidative stress. Sleep 2004, 27, 27–35. [Google Scholar]
- Knapp, L.T.; Klann, E. Role of reactive oxygen species in hippocampal long-term potentiation: Contributory or inhibitory? J. Neurosci. Res. 2002, 70, 1–7. [Google Scholar] [CrossRef]
- Hardeland, R.; Reiter, R.J.; Poeggeler, B.; Tan, D.X. The significance of the metabolism of the neurohormone melatonin: Antioxidative protection and formation of bioactive substances. Neurosci. Biobehav. Rev. 1993, 17, 347–357. [Google Scholar] [CrossRef]
- Poeggeler, B.; Saarela, S.; Reiter, R.J.; Tan, D.X.; Chen, L.D.; Manchester, L.C.; Barlow-Walden, L.R. Melatonin—a highly potent endogenous radical scavenger and electron donor: New aspects of the oxidation chemistry of this indole accessed in vitro. Ann. N. Y. Acad. Sci. 1994, 738, 419–420. [Google Scholar]
- Jan, J.E.; Reiter, R.J.; Bax, M.C.; Ribary, U.; Freeman, R.D.; Wasdell, M.B. Long-term sleep disturbances in children: A cause of neuronal loss. Eur. J. Paediatr. Neurol. 2010, 14, 380–390. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One molecule, many derivatives: A never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal Res. 2007, 42, 28–42. [Google Scholar] [CrossRef]
- Reiter, R.J.; Richardson, B.A. Some perturbations that disturb the circadian melatonin rhythm. Chronobiol. Int. 1992, 9, 314–321. [Google Scholar] [CrossRef]
- Alzoubi, K.H.; Khabour, O.F.; Rashid, B.A.; Damaj, I.M.; Salah, H.A. The neuroprotective effect of vitamin E on chronic sleep deprivation-induced memory impairment: The role of oxidative stress. Behav. Brain Res. 2012, 226, 205–210. [Google Scholar] [CrossRef]
- Shaw, P.J.; Tononi, G.; Greenspan, R.J.; Robinson, D.F. Stress response genes protect against lethal effects of sleep deprivation in Drosophila. Nature 2002, 417, 287–291. [Google Scholar] [CrossRef]
- Brown, M.K.; Naidoo, N. The UPR and the anti-oxidant response: Relevance to sleep and sleep loss. Mol. Neurobiol. 2010, 42, 103–113. [Google Scholar] [CrossRef]
- Biswas, S.; Mishra, P.; Mallick, B.N. Increased apoptosis in rat brain after rapid eye movement sleep loss. Neuroscience 2006, 142, 315–331. [Google Scholar] [CrossRef]
- Artamokhina, I.V.; Belova, V.A.; Romanova, I.V. Immunohistochemical investigation of Bcl-2 and p53 levels in rat hypothalamus after sleep deprivation. J. Evol. Biochem. Physiol. 2011, 47, 458–463. [Google Scholar] [CrossRef]
- Montes-Rodriguez, C.J.; Alavez, S.; Soria-Gomez, E.; Rueda-Orozco, P.E.; Guzman, K.; Moran, J.; Prospero-Garcia, O. BCL-2 and BAX proteins expression throughout the light-dark cycle and modifications induced by sleep deprivation and rebound in adult rat brain. J. Neurosci. Res. 2009, 87, 1602–1609. [Google Scholar] [CrossRef]
- Hipolide, D.C.; D’Almeida, V.; Raymond, R.; Tufik, S.; Nobrega, J.N. Sleep deprivation does not affect indices of necrosis or apoptosis in rat brain. Int. J. Neurosci. 2002, 112, 155–166. [Google Scholar] [CrossRef]
- Adam, K.; Oswald, I. Protein synthesis, bodily renewal and the sleep-wake cycle. Clin. Sci. (Lond.) 1983, 65, 561–567. [Google Scholar]
- Horne, J.A. Human sleep and tissue restitution: Some qualifications and doubts. Clin. Sci. (Lond.) 1983, 65, 569–577. [Google Scholar]
- Ribeiro, S. Sleep and plasticity. Pflugers Arch. 2012, 463, 111–120. [Google Scholar] [CrossRef]
- Drucker-Colin, R.R.; Spanis, C.W.; Hunyadi, J.; Sassin, J.F.; McGaugh, J.L. Growth hormone effects on sleep and wakefulness in the rat. Neuroendocrinology 1975, 18, 1–8. [Google Scholar] [CrossRef]
- Rojas-Ramirez, J.A.; Aguilar-Jimenez, E.; Posadas-Andrews, A.; Bernal-Pedraza, J.G.; Drucker-Colin, R.R. The effects of various protein synthesis inhibitors on the sleep-wake cycle of rats. Psychopharmacology (Berl.) 1977, 53, 147–150. [Google Scholar] [CrossRef]
- Drucker-Colin, R.; Zamora, J.; Bernal-Pedraza, J.; Sosa, B. Modification of REM sleep and associated phasic activities by protein synthesis inhibitors. Exp. Neurol. 1979, 63, 458–467. [Google Scholar] [CrossRef]
- Gutwein, B.M.; Shiromani, P.J.; Fishbein, W. Paradoxical sleep and memory: Long-term disruptive effects of Anisomycin. Pharmacol. Biochem. Behav. 1980, 12, 377–384. [Google Scholar] [CrossRef]
- Methippara, M.M.; Alam, M.N.; Kumar, S.; Bashir, T.; Szymusiak, R.; McGinty, D. Administration of the protein synthesis inhibitor, anisomycin, has distinct sleep-promoting effects in lateral preoptic and perifornical hypothalamic sites in rats. Neuroscience 2008, 151, 1–11. [Google Scholar] [CrossRef]
- Kitahama, K.; Valatx, J.L. Effects of chloramphenicol and thiamphenicol on sleep of the mouse. C. R. Seances Soc. Biol. Fil. 1975, 169, 1522–1525. [Google Scholar]
- Petitjean, F.; Buda, C.; Janin, M.; David, M.; Jouvet, M. The effect of chloramphenicol on sleep in cat—comparison with thiamphenicol, erythromycine, and oxytetracycline. Psychopharmacology (Berl.) 1979, 66, 147–153. [Google Scholar] [CrossRef]
- Drucker-Colin, R.; Espejel, R.M. Chronic administration of chloramphenicol, a protein synthesis inhibitor, selectively decreases REM sleep. Behav. Neural Biol. 1980, 29, 410–413. [Google Scholar] [CrossRef]
- Bowersox, S.S.; Shouse, M.N. Effects of protein synthesis inhibitors on sleep and seizure susceptibility in kindled and nonkindled cats. Behav. Neural Biol. 1982, 35, 432–437. [Google Scholar] [CrossRef]
- Moulin-Sallanon, M.; Millet, P.; Rousset, C.; Zimmer, L.; Debilly, G.; Petit, J.M.; Cespuglio, R.; Magistretti, P.; Ibanez, V. Chloramphenicol decreases brain glucose utilization and modifies the sleep-wake cycle architecture in rats. J. Neurochem. 2005, 93, 1623–1632. [Google Scholar] [CrossRef]
- Stern, W.C.; Morgane, P.J.; Panksepp, J.; Zolovick, A.J.; Jalowiec, J.E. Elevation of REM sleep following inhibition of protein synthesis. Brain Res. 1972, 47, 254–258. [Google Scholar] [CrossRef]
- Pegram, V.; Hammond, D.; Bridgers, W. The effects of protein synthesis inhibition on sleep in mice. Behav. Biol. 1973, 9, 377–382. [Google Scholar] [CrossRef]
- Uezu, E.; Sano, A.; Matsumoto, J. Effects of inhibitor of protein synthesis on sleep in rats. Tokushima J. Exp. Med. 1981, 28, 9–16. [Google Scholar]
- Nonaka, K.; Nakazawa, Y.; Kotorii, T. Effects of antibiotics, minocycline and ampicillin, on human sleep. Brain Res. 1983, 288, 253–259. [Google Scholar] [CrossRef]
- Van Cauter, E.; Copinschi, G. Interrelationships between growth hormone and sleep. Growth Horm. IGF Res. 2000, 10, S57–S62. [Google Scholar] [CrossRef]
- Kirvela, O.; Thorpy, M.; Takala, J.; Askanazi, J.; Singer, P.; Kvetan, V. Respiratory and sleep patterns during nocturnal infusions of branched chain amino acids. Acta Anaesthesiol. Scand. 1990, 34, 645–648. [Google Scholar] [CrossRef]
- Bobillier, P.; Sakai, F.; Seguin, S.; Jouvet, M. The effect of sleep deprivation upon the in vivo and in vitro incorporation of tritiated amino acids into brain proteins in the rat at three different age levels. J. Neurochem. 1974, 22, 21–31. [Google Scholar]
- Lambrey-Sakai, F. Incorporation d’un Melange D’acids Amines Trities dans les Proteines Cerebral du rat, en Rapport avec la PRIVATION de Sommeil; Universite de Lyon: Lyon, France, 1972. [Google Scholar]
- Shapiro, C.; Girdwood, P. Protein synthesis in rat brain during sleep. Neuropharmacology 1981, 20, 457–460. [Google Scholar] [CrossRef]
- Brodskii, V.; Gusatinskii, V.N.; Kogan, A.B.; Nechaeva, N.V. Variations in the intensity of H3-leucine incorporation into proteins during slow-wave and paradoxical phases of natural sleep in the cat associative cortex. Dokl. Akad. Nauk SSSR 1974, 215, 748–750. [Google Scholar]
- Fel’dman, G.L.; Fedorenko, G.M.; Gusatinskii, V.N. Protein synthesis in the large cortical pyramidal neurons during the paradoxical phase of sleep. Tsitologiia 1980, 22, 548–552. [Google Scholar]
- Fel’dman, G.L.; Fedorenko, G.M.; Gusatinskii, V.N. Topography and the protein synthesis level in the large pyramidal neurons during sleep and wakefulness. Tsitologiia 1979, 21, 429–433. [Google Scholar]
- Smith, C.B.; Deibler, G.E.; Eng, N.; Schmidt, K.; Sokoloff, L. Measurement of local cerebral protein synthesis in vivo: Influence of recycling of amino acids derived from protein degradation. Proc. Natl. Acad. Sci. USA 1988, 85, 9341–9345. [Google Scholar] [CrossRef]
- Czikk, M.J.; Sweeley, J.C.; Homan, J.H.; Milley, J.R.; Richardson, B.S. Cerebral leucine uptake and protein synthesis in the near-term ovine fetus: Relation to fetal behavioral state. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R200–R207. [Google Scholar]
- Nakanishi, H.; Sun, Y.; Nakamura, R.K.; Mori, K.; Ito, M.; Suda, S.; Namba, H.; Storch, F.I.; Dang, T.P.; Mendelson, W.; et al. Positive correlations between cerebral protein synthesis rates and deep sleep in Macaca mulatta. Eur. J. Neurosci. 1997, 9, 271–279. [Google Scholar] [CrossRef]
- Ramm, P.; Smith, C.T. Rates of cerebral protein synthesis are linked to slow wave sleep in the rat. Physiol. Behav. 1990, 48, 749–753. [Google Scholar] [CrossRef]
- Hernandez, P.J.; Abel, T. The role of protein synthesis in memory consolidation: Progress amid decades of debate. Neurobiol. Learn. Mem. 2008, 89, 293–311. [Google Scholar] [CrossRef]
- Graves, L.; Pack, A.; Abel, T. Sleep and memory: A molecular perspective. Trends Neurosci. 2001, 24, 237–243. [Google Scholar] [CrossRef]
- Smith, C.; Tenn, C.; Annett, R. Some biochemical and behavioural aspects of the paradoxical sleep window. Can. J. Psychol. 1991, 45, 115–124. [Google Scholar] [CrossRef]
- Rolls, A.; Makam, M.; Kroeger, D.; Colas, D.; de Lecea, L.; Heller, H.C. Sleep to forget: Interference of fear memories during sleep. Mol. Psychiatry 2013, 18, 1166–1170. [Google Scholar] [CrossRef]
- Frank, M.G.; Issa, N.P.; Stryker, M.P. Sleep enhances plasticity in the developing visual cortex. Neuron 2001, 30, 275–287. [Google Scholar] [CrossRef]
- Seibt, J.; Dumoulin, M.C.; Aton, S.J.; Coleman, T.; Watson, A.; Naidoo, N.; Frank, M.G. Protein synthesis during sleep consolidates cortical plasticity in vivo. Curr. Biol. 2012, 22, 676–682. [Google Scholar] [CrossRef]
- Dumoulin, M.C.; Aton, S.J.; Watson, A.J.; Renouard, L.; Coleman, T.; Frank, M.G. Extracellular Signal-Regulated Kinase (ERK) Activity during Sleep Consolidates Cortical Plasticity in Vivo. Cereb. Cortex 2013. [Google Scholar] [CrossRef]
- Durmer, J.S.; Dinges, D.F. Neurocognitive consequences of sleep deprivation. Semin. Neurol. 2005, 25, 117–129. [Google Scholar] [CrossRef]
- Krueger, J.M.; Obal, F. A neuronal group theory of sleep function. J. Sleep Res. 1993, 2, 63–69. [Google Scholar] [CrossRef]
- Frey, U.; Morris, R.G. Synaptic tagging: Implications for late maintenance of hippocampal long-term potentiation. Trends Neurosci. 1998, 21, 181–188. [Google Scholar] [CrossRef]
- Fujita, E.; Dai, H.; Tanabe, Y.; Zhiling, Y.; Yamagata, T.; Miyakawa, T.; Tanokura, M.; Momoi, M.Y.; Momoi, T. Autism spectrum disorder is related to endoplasmic reticulum stress induced by mutations in the synaptic cell adhesion molecule, CADM1. Cell Death Dis. 2010, 1, e47. [Google Scholar] [CrossRef]
- Reith, R.M.; Way, S.; McKenna, J., 3rd; Haines, K.; Gambello, M.J. Loss of the tuberous sclerosis complex protein tuberin causes Purkinje cell degeneration. Neurobiol. Dis. 2011, 43, 113–122. [Google Scholar] [CrossRef]
- Di Nardo, A.; Kramvis, I.; Cho, N.; Sadowski, A.; Meikle, L.; Kwiatkowski, D.J.; Sahin, M. Tuberous sclerosis complex activity is required to control neuronal stress responses in an mTOR-dependent manner. J. Neurosci. 2009, 29, 5926–5937. [Google Scholar] [CrossRef]
- Koh, K.; Evans, J.M.; Hendricks, J.C.; Sehgal, A. A Drosophila model for age-associated changes in sleep:wake cycles. Proc. Natl. Acad. Sci. USA 2006, 103, 13843–13847. [Google Scholar] [CrossRef]
- Frustaci, A.; Neri, M.; Cesario, A.; Adams, J.B.; Domenici, E.; Dalla Bernardina, B.; Bonassi, S. Oxidative stress-related biomarkers in autism: Systematic review and meta-analyses. Free Radic. Biol. Med. 2012, 52, 2128–2141. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. A review of research trends in physiological abnormalities in autism spectrum disorders: Immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol. Psychiatry 2012, 17, 389–401. [Google Scholar] [CrossRef]
- Kern, J.K.; Jones, A.M. Evidence of toxicity, oxidative stress, and neuronal insult in autism. J. Toxicol. Environ. Health B Crit. Rev. 2006, 9, 485–499. [Google Scholar] [CrossRef]
- Romero-Zerbo, Y.; Decara, J.; el Bekay, R.; Sanchez-Salido, L.; Del Arco-Herrera, I.; de Fonseca, F.R.; de Diego-Otero, Y. Protective effects of melatonin against oxidative stress in Fmr1 knockout mice: A therapeutic research model for the fragile X syndrome. J. Pineal Res. 2009, 46, 224–234. [Google Scholar] [CrossRef]
- De Felice, C.; Signorini, C.; Leoncini, S.; Pecorelli, A.; Durand, T.; Valacchi, G.; Ciccoli, L.; Hayek, J. The role of oxidative stress in Rett syndrome: An overview. Ann. N. Y. Acad. Sci. 2012, 1259, 121–135. [Google Scholar] [CrossRef]
- Boddaert, N.; Zilbovicius, M.; Philipe, A.; Robel, L.; Bourgeois, M.; Barthelemy, C.; Seidenwurm, D.; Meresse, I.; Laurier, L.; Desguerre, I.; Bahi-Buisson, N.; Brunelle, F.; Munnich, A.; Samson, Y.; Mouren, M.C.; Chabane, N. MRI findings in 77 children with non-syndromic autistic disorder. PLoS One 2009, 4, e4415. [Google Scholar] [CrossRef]
- Bauman, M.L.; Kemper, T.L. The Neurobiology of Autism, 2nd ed.; The Johns Hopkins University Press: Baltimore, MD, USA, 2005. [Google Scholar]
- Makki, M.I.; Chugani, D.C.; Janisse, J.; Chugani, H.T. Characteristics of abnormal diffusivity in normal-appearing white matter investigated with diffusion tensor MR imaging in tuberous sclerosis complex. AJNR Am. J. Neuroradiol. 2007, 28, 1662–1667. [Google Scholar] [CrossRef]
- Way, S.W.; McKenna, J., 3rd; Mietzsch, U.; Reith, R.M.; Wu, H.C.; Gambello, M.J. Loss of Tsc2 in radial glia models the brain pathology of tuberous sclerosis complex in the mouse. Human Mol. Genet. 2009, 18, 1252–1265. [Google Scholar] [CrossRef]
- Haas, B.W.; Barnea-Goraly, N.; Lightbody, A.A.; Patnaik, S.S.; Hoeft, F.; Hazlett, H.; Piven, J.; Reiss, A.L. Early white-matter abnormalities of the ventral frontostriatal pathway in fragile X syndrome. Dev. Med. Child Neurol. 2009, 51, 593–599. [Google Scholar] [CrossRef]
- Barnea-Goraly, N.; Eliez, S.; Hedeus, M.; Menon, V.; White, C.D.; Moseley, M.; Reiss, A.L. White matter tract alterations in fragile X syndrome: Preliminary evidence from diffusion tensor imaging. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2003, 118B, 81–88. [Google Scholar] [CrossRef]
- Mahmood, A.; Bibat, G.; Zhan, A.L.; Izbudak, I.; Farage, L.; Horska, A.; Mori, S.; Naidu, S. White matter impairment in Rett syndrome: Diffusion tensor imaging study with clinical correlations. AJNR Am. J. Neuroradiol. 2010, 31, 295–299. [Google Scholar] [CrossRef]
- Yamada, K.; Matsuzawa, H.; Uchiyama, M.; Kwee, I.L.; Nakada, T. Brain developmental abnormalities in Prader-Willi syndrome detected by diffusion tensor imaging. Pediatrics 2006, 118, e442–e448. [Google Scholar] [CrossRef]
- Harting, I.; Seitz, A.; Rating, D.; Sartor, K.; Zschocke, J.; Janssen, B.; Ebinger, F.; Wolf, N.I. Abnormal myelination in Angelman syndrome. Eur. J. Paediatr. Neurol. 2009, 13, 271–276. [Google Scholar] [CrossRef]
- Faria, A.V.; Landau, B.; O’Hearn, K.M.; Li, X.; Jiang, H.; Oishi, K.; Zhang, J.; Mori, S. Quantitative analysis of gray and white matter in Williams syndrome. Neuroreport 2012, 23, 283–289. [Google Scholar] [CrossRef]
- Nagel, B.J.; Bathula, D.; Herting, M.; Schmitt, C.; Kroenke, C.D.; Fair, D.; Nigg, J.T. Altered white matter microstructure in children with attention-deficit/hyperactivity disorder. J. Am. Acad. Child Adolesc. Psychiatry 2011, 50, 283–292. [Google Scholar] [CrossRef]
- Liu, Z.H.; Chuang, D.M.; Smith, C.B. Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int. J. Neuropsychopharmacol. 2011, 14, 618–630. [Google Scholar] [CrossRef]
- Hinton, V.J.; Brown, W.T.; Wisniewski, K.; Rudelli, R.D. Analysis of neocortex in three males with the fragile X syndrome. Am. J. Med. Genet. 1991, 41, 289–294. [Google Scholar] [CrossRef]
- Vaags, A.K.; Lionel, A.C.; Sato, D.; Goodenberger, M.; Stein, Q.P.; Curran, S.; Ogilvie, C.; Ahn, J.W.; Drmic, I.; Senman, L.; et al. Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am. J. Human Genet. 2012, 90, 133–141. [Google Scholar] [CrossRef]
- Harrison, V.; Connell, L.; Hayesmoore, J.; McParland, J.; Pike, M.G.; Blair, E. Compound heterozygous deletion of NRXN1 causing severe developmental delay with early onset epilepsy in two sisters. Am. J. Med. Genet. A 2011, 155A, 2826–2831. [Google Scholar]
- Zweier, C.; de Jong, E.K.; Zweier, M.; Orrico, A.; Ousager, L.B.; Collins, A.L.; Bijlsma, E.K.; Oortveld, M.A.; Ekici, A.B.; Reis, A.; et al. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am. J. Human Genet. 2009, 85, 655–666. [Google Scholar] [CrossRef]
- Mignon-Ravix, C.; Mugneret, F.; Stavropoulou, C.; Depetris, D.; Khau Van Kien, P.; Mattei, M.G. Maternally inherited duplication of the possible imprinted 14q31 region. J. Med. Genet. 2001, 38, 343–347. [Google Scholar] [CrossRef]
- Darnell, J.C.; van Driesche, S.J.; Zhang, C.; Hung, K.Y.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef]
- Dahlhaus, R.; El-Husseini, A. Altered neuroligin expression is involved in social deficits in a mouse model of the fragile X syndrome. Behav. Brain Res. 2010, 208, 96–105. [Google Scholar] [CrossRef]
- Geschwind, D.H.; Levitt, P. Autism spectrum disorders: Developmental disconnection syndromes. Curr. Opin. Neurobiol. 2007, 17, 103–111. [Google Scholar] [CrossRef]
- Zoghbi, H.Y. Postnatal neurodevelopmental disorders: Meeting at the synapse? Science 2003, 302, 826–830. [Google Scholar] [CrossRef]
- Bourgeron, T. A synaptic trek to autism. Curr. Opin. Neurobiol. 2009, 19, 231–234. [Google Scholar] [CrossRef]
- Sandsmark, D.K.; Pelletier, C.; Weber, J.D.; Gutmann, D.H. Mammalian target of rapamycin: Master regulator of cell growth in the nervous system. Histol. Histopathol. 2007, 22, 895–903. [Google Scholar]
- Swiech, L.; Perycz, M.; Malik, A.; Jaworski, J. Role of mTOR in physiology and pathology of the nervous system. Biochim. Biophys. Acta 2008, 1784, 116–132. [Google Scholar] [CrossRef]
- Ehninger, D.; Silva, A.J. Rapamycin for treating tuberous sclerosis and autism spectrum disorders. Trends Mol. Med. 2011, 17, 78–87. [Google Scholar] [CrossRef]
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13571–13576. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. United at last: The tuberous sclerosis complex gene products connect the phosphoinositide 3-kinase/Akt pathway to mammalian target of rapamycin (mTOR) signalling. Biochem. Soc. Trans. 2003, 31, 573–578. [Google Scholar] [CrossRef]
- Crino, P.B.; Aronica, E.; Baltuch, G.; Nathanson, K.L. Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurology 2010, 74, 1716–1723. [Google Scholar] [CrossRef]
- Kelleher, R.J., III; Bear, M.F. The autistic neuron: Troubled translation? Cell 2008, 135, 401–406. [Google Scholar] [CrossRef]
- Godbout, R.; Bergeron, C.; Limoges, E.; Stip, E.; Mottron, L. A laboratory study of sleep in Asperger’s syndrome. Neuroreport 2000, 11, 127–130. [Google Scholar] [CrossRef]
- Vitiello, M.V. Recent advances in understanding sleep and sleep disturbances in older adults: Growing older does not mean sleeping poorly. Curr. Dir. Psychol. Sci. 2009, 18, 316–320. [Google Scholar] [CrossRef]
- Petit, D.; Gagnon, J.F.; Fantini, M.L.; Ferini-Strambi, L.; Montplaisir, J. Sleep and quantitative EEG in neurodegenerative disorders. J. Psychosom. Res. 2004, 56, 487–496. [Google Scholar] [CrossRef]
- Feinberg, I. Functional implications of changes in sleep physiology with age. In Neurobiology of Aging; Terry, R.D., Gershon, S., Eds.; Raven Press: New York, NY, USA, 1976; pp. 23–41. [Google Scholar]
- Dement, W.C.; Miles, L.E.; Carskadon, M.A. “White paper” on sleep and aging. J. Am. Geriatr. Soc. 1982, 30, 25–50. [Google Scholar]
- Hayashi, Y.; Otomo, E.; Endo, S.; Watanabe, H. The all-night polygraphies for healthy aged persons. Sleep Res. 1979, 8, 122. [Google Scholar]
- Wauquier, A. Aging and changes in phasic events during sleep. Physiol. Behav. 1993, 54, 803–806. [Google Scholar] [CrossRef]
- Schubert, C.R.; Cruickshanks, K.J.; Dalton, D.S.; Klein, B.E.; Klein, R.; Nondahl, D.M. Prevalence of sleep problems and quality of life in an older population. Sleep 2002, 25, 889–893. [Google Scholar]
- Minino, A.; Xu, J.; Kochanek, K.D. Deaths: Preliminary Data for 2008; National Center for Health Statistics: Hyattsville, MD, USA, 2010. [Google Scholar]
- Artero, S.; Ancelin, M.L.; Portet, F.; Dupuy, A.; Berr, C.; Dartigues, J.F.; Tzourio, C.; Rouaud, O.; Poncet, M.; Pasquier, F.; et al. Risk profiles for mild cognitive impairment and progression to dementia are gender specific. J. Neurol. Neurosurg. Psychiatry 2008, 79, 979–984. [Google Scholar] [CrossRef]
- Boyle, P.A.; Buchman, A.S.; Wilson, R.S.; Kelly, J.F.; Bennett, D.A. The APOE epsilon4 allele is associated with incident mild cognitive impairment among community-dwelling older persons. Neuroepidemiology 2010, 34, 43–49. [Google Scholar] [CrossRef]
- Price, J.L.; Morris, J.C. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann. Neurol. 1999, 45, 358–368. [Google Scholar] [CrossRef]
- Loewenstein, R.J.; Weingartner, H.; Gillin, J.C.; Kaye, W.; Ebert, M.; Mendelson, W.B. Disturbances of sleep and cognitive functioning in patients with dementia. Neurobiol. Aging 1982, 3, 371–377. [Google Scholar] [CrossRef]
- Martin, P.R.; Loewenstein, R.J.; Kaye, W.H.; Ebert, M.H.; Weingartner, H.; Gillin, J.C. Sleep EEG in Korsakoff’s psychosis and Alzheimer’s disease. Neurology 1986, 36, 411–414. [Google Scholar] [CrossRef]
- Prinz, P.N.; Vitaliano, P.P.; Vitiello, M.V.; Bokan, J.; Raskind, M.; Peskind, E.; Gerber, C. Sleep, EEG and mental function changes in senile dementia of the Alzheimer’s type. Neurobiol. Aging 1982, 3, 361–370. [Google Scholar] [CrossRef]
- Reynolds, C.F., III; Kupfer, D.J.; Taska, L.S.; Hoch, C.C.; Spiker, D.G.; Sewitch, D.E.; Zimmer, B.; Marin, R.S.; Nelson, J.P.; Martin, D.; et al. EEG sleep in elderly depressed, demented, and healthy subjects. Biol. Psychiatry 1985, 20, 431–442. [Google Scholar] [CrossRef]
- Vitiello, M.V.; Borson, S. Sleep disturbances in patients with Alzheimer’s disease: Epidemiology, pathophysiology and treatment. CNS Drugs 2001, 15, 777–796. [Google Scholar] [CrossRef]
- Bliwise, D.L.; Tinklenberg, J.; Yesavage, J.A.; Davies, H.; Pursley, A.M.; Petta, D.E.; Widrow, L.; Guilleminault, C.; Zarcone, V.P.; Dement, W.C. REM latency in Alzheimer’s disease. Biol. Psychiatry 1989, 25, 320–328. [Google Scholar] [CrossRef]
- Bliwise, D.L. Sleep disorders in Alzheimer’s disease and other dementias. Clin. Cornerstone 2004, 6 (Suppl. 1A), S16–S28. [Google Scholar] [CrossRef]
- Montplaisir, J.; Petit, D.; Lorrain, D.; Gauthier, S.; Nielsen, T. Sleep in Alzheimer’s disease: Further considerations on the role of brainstem and forebrain cholinergic populations in sleep-wake mechanisms. Sleep 1995, 18, 145–148. [Google Scholar]
- Prinz, P.N.; Peskind, E.R.; Vitaliano, P.P.; Raskind, M.A.; Eisdorfer, C.; Zemcuznikov, N.; Gerber, C.J. Changes in the sleep and waking EEGs of nondemented and demented elderly subjects. J. Am. Geriatr. Soc. 1982, 30, 86–93. [Google Scholar]
- Reynolds, C.F., 3rd; Kupfer, D.J.; Houck, P.R.; Hoch, C.C.; Stack, J.A.; Berman, S.R.; Zimmer, B. Reliable discrimination of elderly depressed and demented patients by electroencephalographic sleep data. Arch. Gen. Psychiatry 1988, 45, 258–264. [Google Scholar] [CrossRef]
- Hita-Yanez, E.; Atienza, M.; Gil-Neciga, E.; Cantero, J.L. Disturbed sleep patterns in elders with mild cognitive impairment: The role of memory decline and ApoE epsilon4 genotype. Curr. Alzheimer Res. 2012, 9, 290–297. [Google Scholar] [CrossRef]
- Beaulieu-Bonneau, S.; Hudon, C. Sleep disturbances in older adults with mild cognitive impairment. Int. Psychogeriatr. 2009, 21, 654–666. [Google Scholar] [CrossRef]
- Geda, Y.E.; Smith, G.E.; Knopman, D.S.; Boeve, B.F.; Tangalos, E.G.; Ivnik, R.J.; Mrazek, D.A.; Edland, S.D.; Petersen, R.C. De novo genesis of neuropsychiatric symptoms in mild cognitive impairment (MCI). Int. Psychogeriatr. 2004, 16, 51–60. [Google Scholar] [CrossRef]
- Lee, K.S.; Cho, H.S.; Hong, C.H.; Kim, D.G.; Oh, B.H. Differences in neuropsychiatric symptoms according to mild cognitive impairment subtypes in the community. Dement. Geriatr. Cogn. Disord. 2008, 26, 212–217. [Google Scholar] [CrossRef]
- Hita-Yanez, E.; Atienza, M.; Cantero, J.L. Polysomnographic and subjective sleep markers of mild cognitive impairment. Sleep 2013, 36, 1327–1334. [Google Scholar]
- Meguro, K.; Ueda, M.; Kobayashi, I.; Yamaguchi, S.; Yamazaki, H.; Oikawa, Y.; Kikuchi, Y.; Sasaki, H. Sleep disturbance in elderly patients with cognitive impairment, decreased daily activity and periventricular white matter lesions. Sleep 1995, 18, 109–114. [Google Scholar]
- Allen, S.R.; Seiler, W.O.; Stahelin, H.B.; Spiegel, R. Seventy-two hour polygraphic and behavioral recordings of wakefulness and sleep in a hospital geriatric unit: Comparison between demented and nondemented patients. Sleep 1987, 10, 143–159. [Google Scholar]
- Bliwise, D.L.; Carroll, J.S.; Dement, W.C. Predictors of observed sleep/wakefulness in residents in long-term care. J. Gerontol. 1990, 45, M126–M130. [Google Scholar] [CrossRef]
- Kang, J.E.; Lim, M.M.; Bateman, R.J.; Lee, J.J.; Smyth, L.P.; Cirrito, J.R.; Fujiki, N.; Nishino, S.; Holtzman, D.M. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 2009, 326, 1005–1007. [Google Scholar] [CrossRef]
- Ju, Y.E.; McLeland, J.S.; Toedebusch, C.D.; Xiong, C.; Fagan, A.M.; Duntley, S.P.; Morris, J.C.; Holtzman, D.M. Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 2013, 70, 587–593. [Google Scholar] [CrossRef]
- Naidoo, N.; Ferber, M.; Master, M.; Zhu, Y.; Pack, A.I. Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J. Neurosci. 2008, 28, 6539–6548. [Google Scholar] [CrossRef]
- Lindholm, D.; Wootz, H.; Korhonen, L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006, 13, 385–392. [Google Scholar] [CrossRef]
- Naidoo, N.; Zhu, J.; Zhu, Y.; Fenik, P.; Lian, J.; Galante, R.; Veasey, S. Endoplasmic reticulum stress in wake-active neurons progresses with aging. Aging Cell 2011, 10, 640–649. [Google Scholar] [CrossRef]
- Naidoo, N. Cellular stress/the unfolded protein response: Relevance to sleep and sleep disorders. Sleep Med. Rev. 2009, 13, 195–204. [Google Scholar] [CrossRef]
- Naidoo, N. Roles of endoplasmic reticulum and energetic stress in disturbed sleep. Neuromol. Med. 2012, 14, 213–219. [Google Scholar] [CrossRef]
- Naidoo, N. The unfolded protein response in mouse cerebral cortex. Methods Enzymol. 2011, 489, 3–21. [Google Scholar] [CrossRef]
- Rao, R.V.; Bredesen, D.E. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr. Opin. Cell Biol. 2004, 16, 653–662. [Google Scholar] [CrossRef]
- Selkoe, D.J. Folding proteins in fatal ways. Nature 2003, 426, 900–904. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Castegna, A.; Lauderback, C.M.; Drake, J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol. Aging 2002, 23, 655–664. [Google Scholar] [CrossRef]
- Mishima, K.; Tozawa, T.; Satoh, K.; Matsumoto, Y.; Hishikawa, Y.; Okawa, M. Melatonin secretion rhythm disorders in patients with senile dementia of Alzheimer’s type with disturbed sleep-waking. Biol. Psychiatry 1999, 45, 417–421. [Google Scholar] [CrossRef]
- Smith, C.B.; Schmidt, K.C.; Qin, M.; Burlin, T.V.; Cook, M.P.; Kang, J.; Saunders, R.C.; Bacher, J.D.; Carson, R.E.; Channing, M.A.; et al. Measurement of regional rates of cerebral protein synthesis with L-[1-11C]leucine and PET with correction for recycling of tissue amino acids: II. Validation in rhesus monkeys. J. Cereb. Blood Flow Metab. 2005, 25, 629–640. [Google Scholar] [CrossRef]
- Smith, C.B.; Schmidt, K.C.; Bishu, S.; Channing, M.A.; Bacon, J.; Burlin, T.V.; Qin, M.; Liu, Z.H.; Xia, Z.; Huang, T.; et al. Use of acute hyperphenylalaninemia in rhesus monkeys to examine sensitivity and stability of the l-[1-11C]leucine method for measurement of regional rates of cerebral protein synthesis with PET. J. Cereb. Blood Flow Metab. 2008, 28, 1388–1398. [Google Scholar] [CrossRef]
- Bishu, S.; Schmidt, K.C.; Burlin, T.; Channing, M.; Conant, S.; Huang, T.; Liu, Z.H.; Qin, M.; Unterman, A.; Xia, Z.; et al. Regional rates of cerebral protein synthesis measured with l-[1-11C]leucine and PET in conscious, young adult men: Normal values, variability, and reproducibility. J. Cereb. Blood Flow Metab. 2008, 28, 1502–1513. [Google Scholar] [CrossRef]
- Karni, A.; Sagi, D. Where practice makes perfect in texture discrimination: Evidence for primary visual cortex plasticity. Proc. Natl. Acad. Sci. USA 1991, 88, 4966–4970. [Google Scholar] [CrossRef]
- Stickgold, R.; Whidbee, D.; Schirmer, B.; Patel, V.; Hobson, J.A. Visual discrimination task improvement: A multi-step process occurring during sleep. J. Cogn. Neurosci. 2000, 12, 246–254. [Google Scholar] [CrossRef]
- Frank, M.G.; Benington, J.H. The role of sleep in memory consolidation and brain plasticity: Dream or reality? Neuroscientist 2006, 12, 477–488. [Google Scholar] [CrossRef]
- Mednick, S.; Nakayama, K.; Stickgold, R. Sleep-dependent learning: A nap is as good as a night. Nat. Neurosci. 2003, 6, 697–698. [Google Scholar] [CrossRef]
- Mednick, S.C.; Nakayama, K.; Cantero, J.L.; Atienza, M.; Levin, A.A.; Pathak, N.; Stickgold, R. The restorative effect of naps on perceptual deterioration. Nat. Neurosci. 2002, 5, 677–681. [Google Scholar]
- Scher, M.S.; Loparo, K.A. Neonatal EEG/sleep state analyses: A complex phenotype of developmental neural plasticity. Dev. Neurosci. 2009, 31, 259–275. [Google Scholar] [CrossRef]
- Laborit, H. Gamma-hydroxybutyrate, succinic semialdehyde and sleep. Prog. Neurobiol. 1973, 1, 255–274. [Google Scholar] [CrossRef]
- Van Cauter, E.; Plat, L.; Scharf, M.B.; Leproult, R.; Cespedes, S.; L’Hermite-Baleriaux, M.; Copinschi, G. Simultaneous stimulation of slow-wave sleep and growth hormone secretion by gamma-hydroxybutyrate in normal young Men. J.Clin. Investig. 1997, 100, 745–753. [Google Scholar] [CrossRef]
- Walker, M.P. The role of sleep in cognition and emotion. Ann. N. Y. Acad. Sci. 2009, 1156, 168–197. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Picchioni, D.; Reith, R.M.; Nadel, J.L.; Smith, C.B. Sleep, Plasticity and the Pathophysiology of Neurodevelopmental Disorders: The Potential Roles of Protein Synthesis and Other Cellular Processes. Brain Sci. 2014, 4, 150-201. https://doi.org/10.3390/brainsci4010150
Picchioni D, Reith RM, Nadel JL, Smith CB. Sleep, Plasticity and the Pathophysiology of Neurodevelopmental Disorders: The Potential Roles of Protein Synthesis and Other Cellular Processes. Brain Sciences. 2014; 4(1):150-201. https://doi.org/10.3390/brainsci4010150
Chicago/Turabian StylePicchioni, Dante, R. Michelle Reith, Jeffrey L. Nadel, and Carolyn B. Smith. 2014. "Sleep, Plasticity and the Pathophysiology of Neurodevelopmental Disorders: The Potential Roles of Protein Synthesis and Other Cellular Processes" Brain Sciences 4, no. 1: 150-201. https://doi.org/10.3390/brainsci4010150
APA StylePicchioni, D., Reith, R. M., Nadel, J. L., & Smith, C. B. (2014). Sleep, Plasticity and the Pathophysiology of Neurodevelopmental Disorders: The Potential Roles of Protein Synthesis and Other Cellular Processes. Brain Sciences, 4(1), 150-201. https://doi.org/10.3390/brainsci4010150