Developmental Neurotoxicity of Alcohol and Anesthetic Drugs Is Augmented by Co-Exposure to Caffeine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Procedures
2.1. Subjects
2.2. Blood CAF Levels
2.3. Individual Experiments
2.3.1. Experiment #1—Apoptogenic Action of CAF + Alcohol
2.3.2. Experiment #2—Apoptogenic Action of CAF + NMDA antagonists
2.3.3. Experiment #3—Apoptogenic Action of CAF + GABAmimetics
2.3.4. Experiment #4—Long-Term Neurobehavioral Effects of CAF + NMDA antagonist or GABAmimetic
2.4. Histopathology
2.5. Quantitative Cell Counts
2.6. Statistical Methods
3. Results
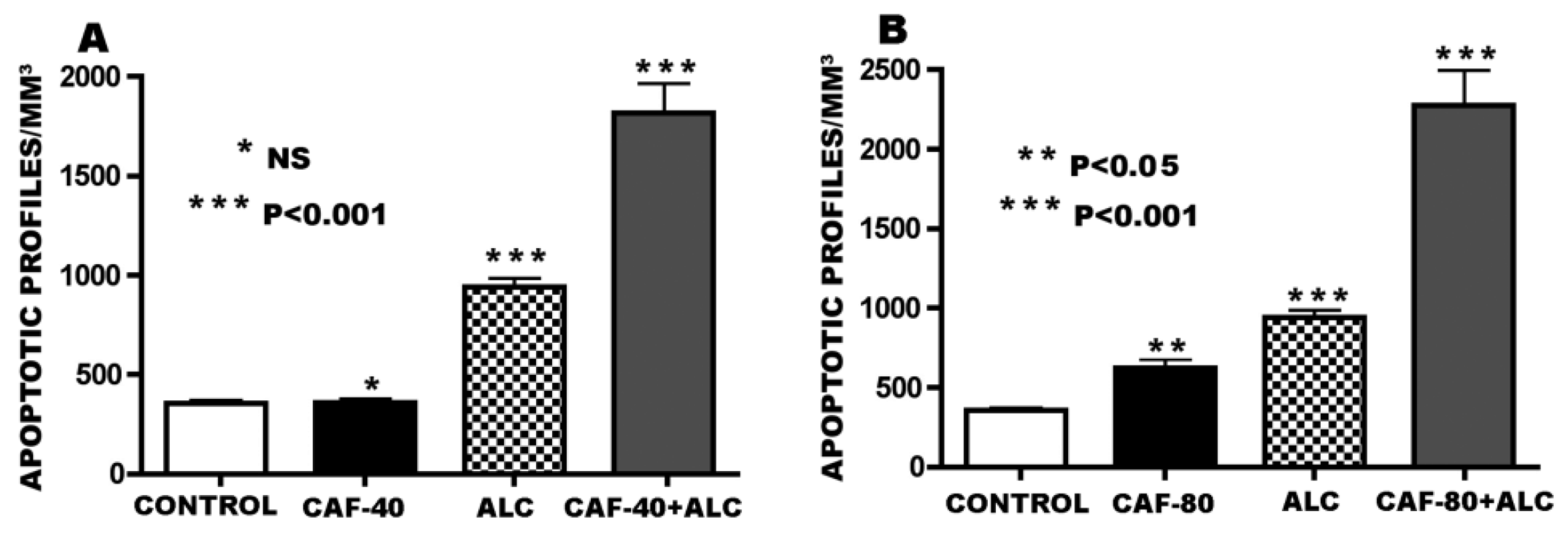
3.1. Apoptogenic Action of CAF + Alcohol

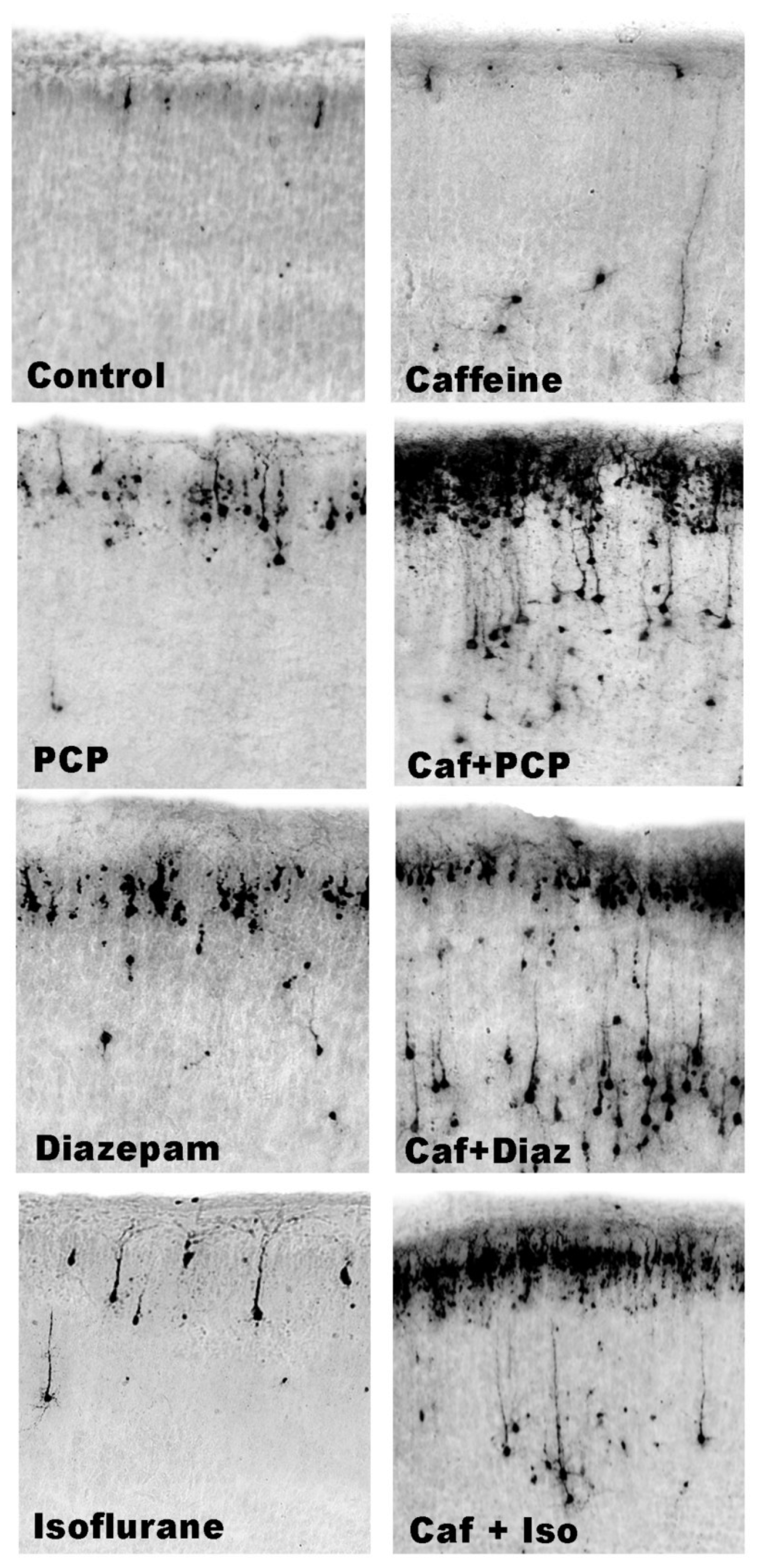
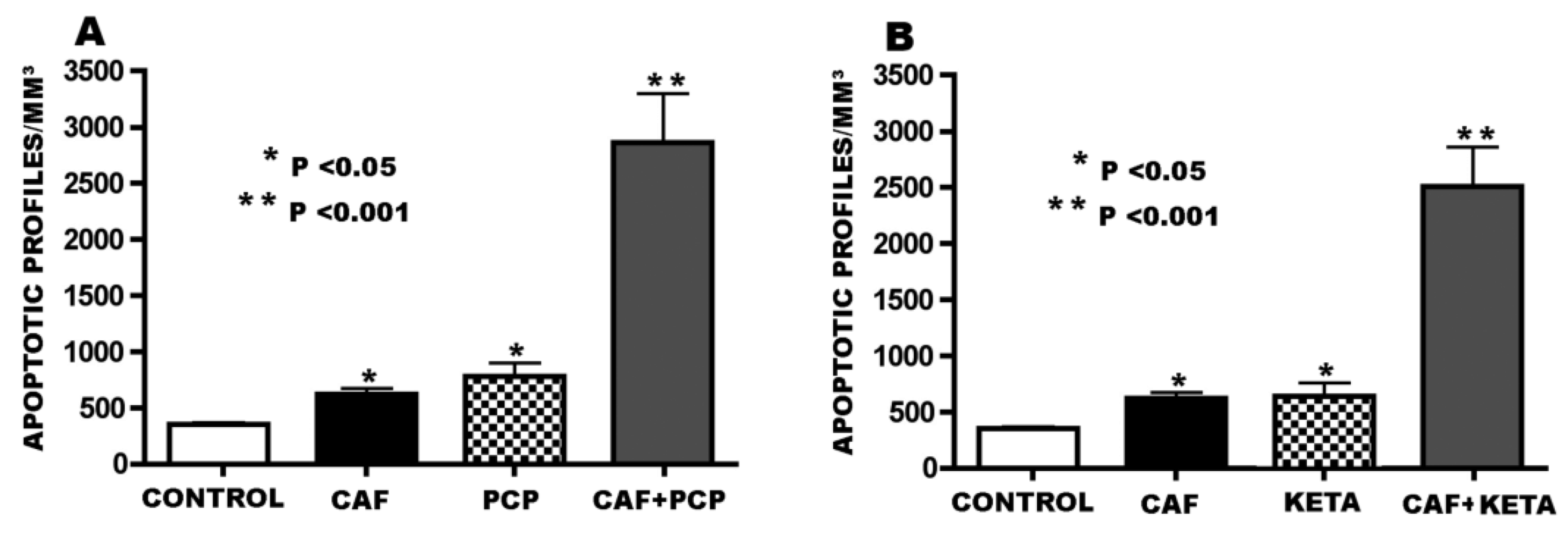
3.2. Apoptogenic Action of CAF + NMDA Antagonists (PCP or Ketamine)
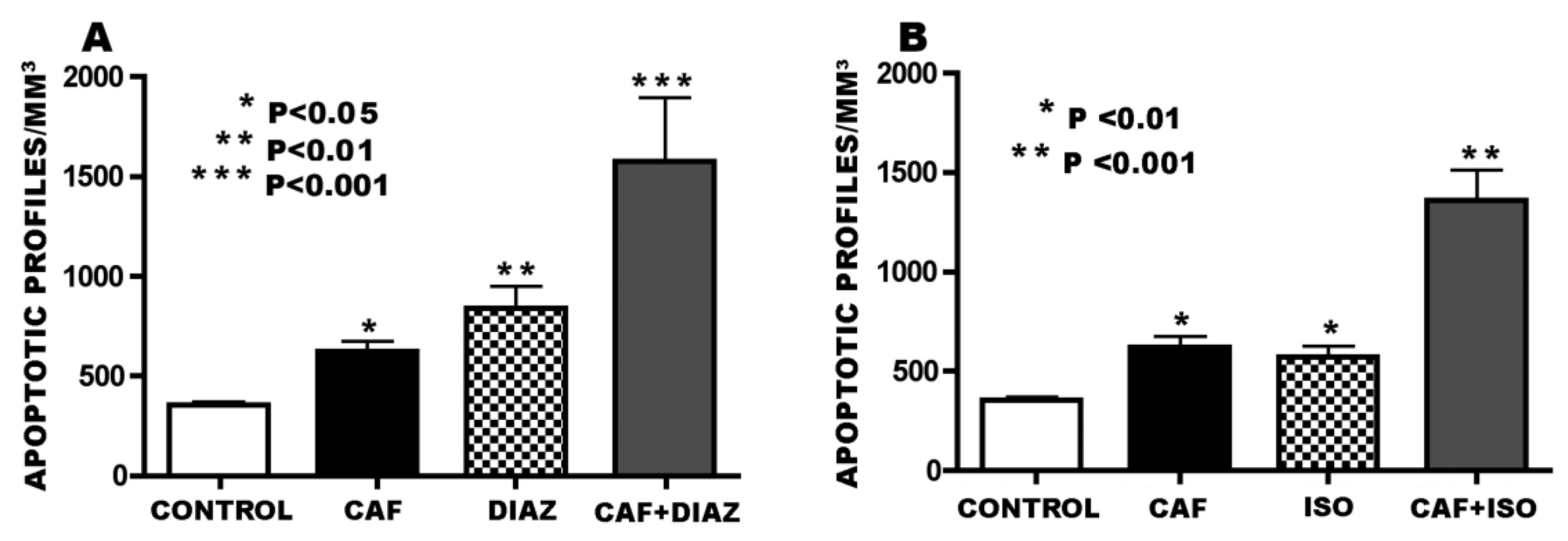
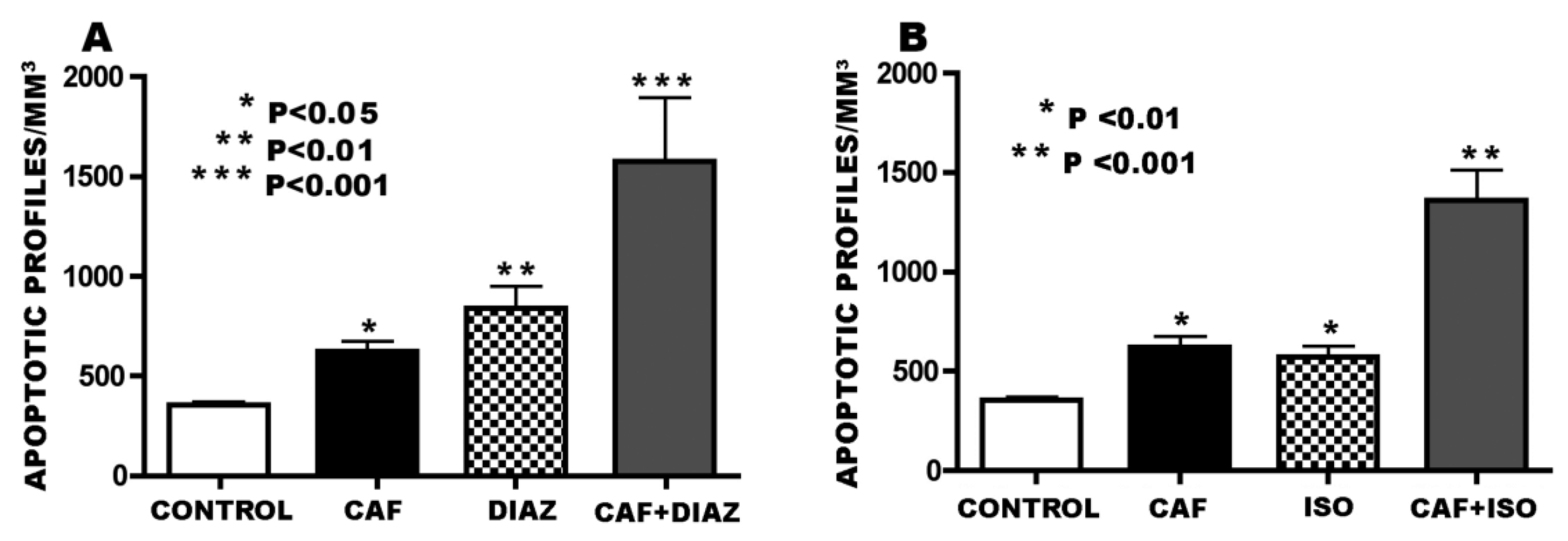
3.3. Apoptogenic Action of CAF + GABAmimetics (Diazepam or Isoflurane)
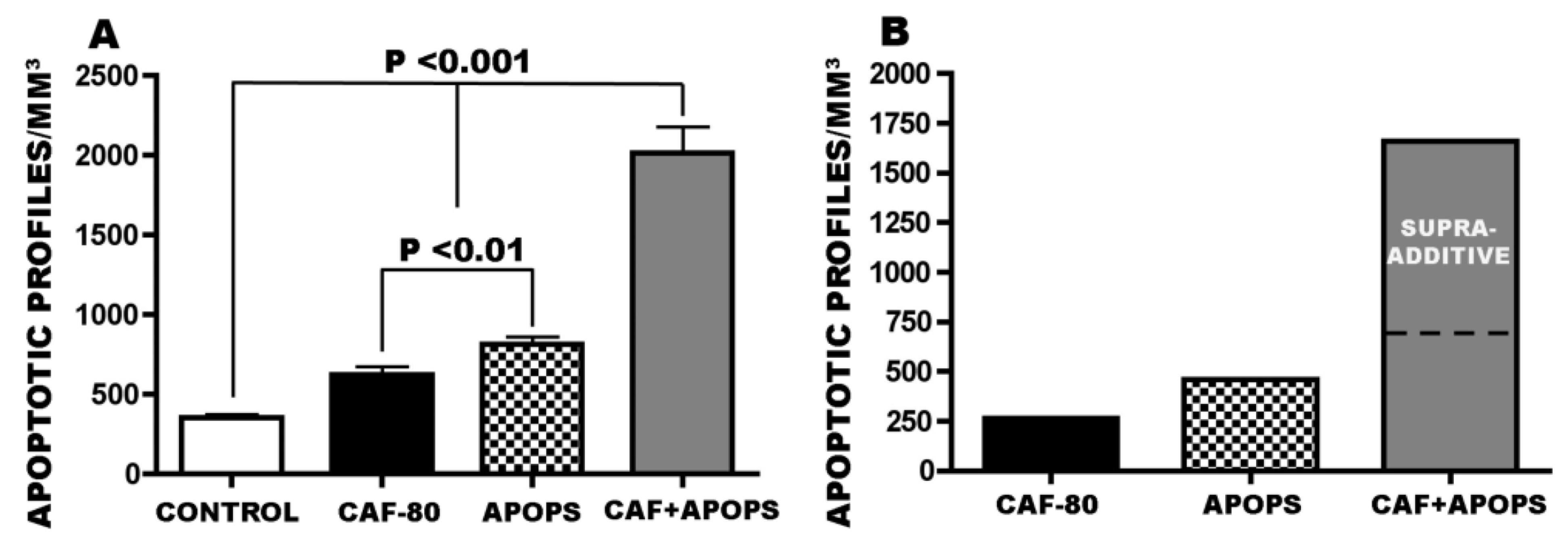
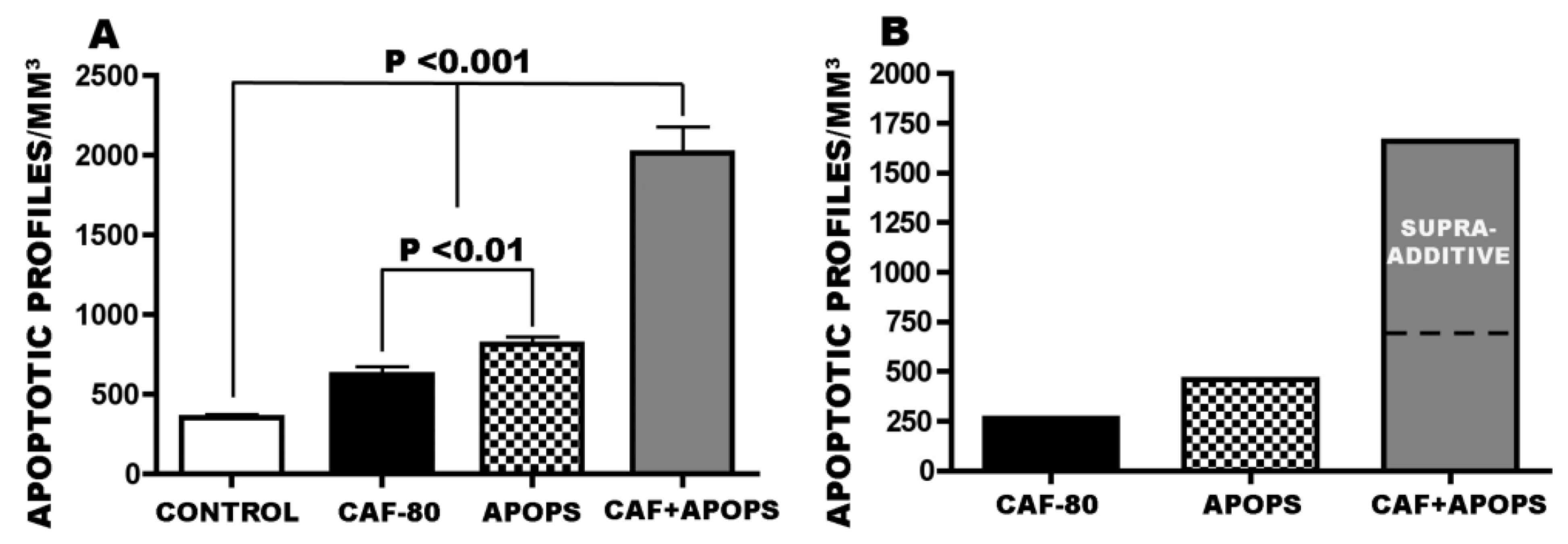
3.4. Overview of CAF’s Pro-Apoptotic Action
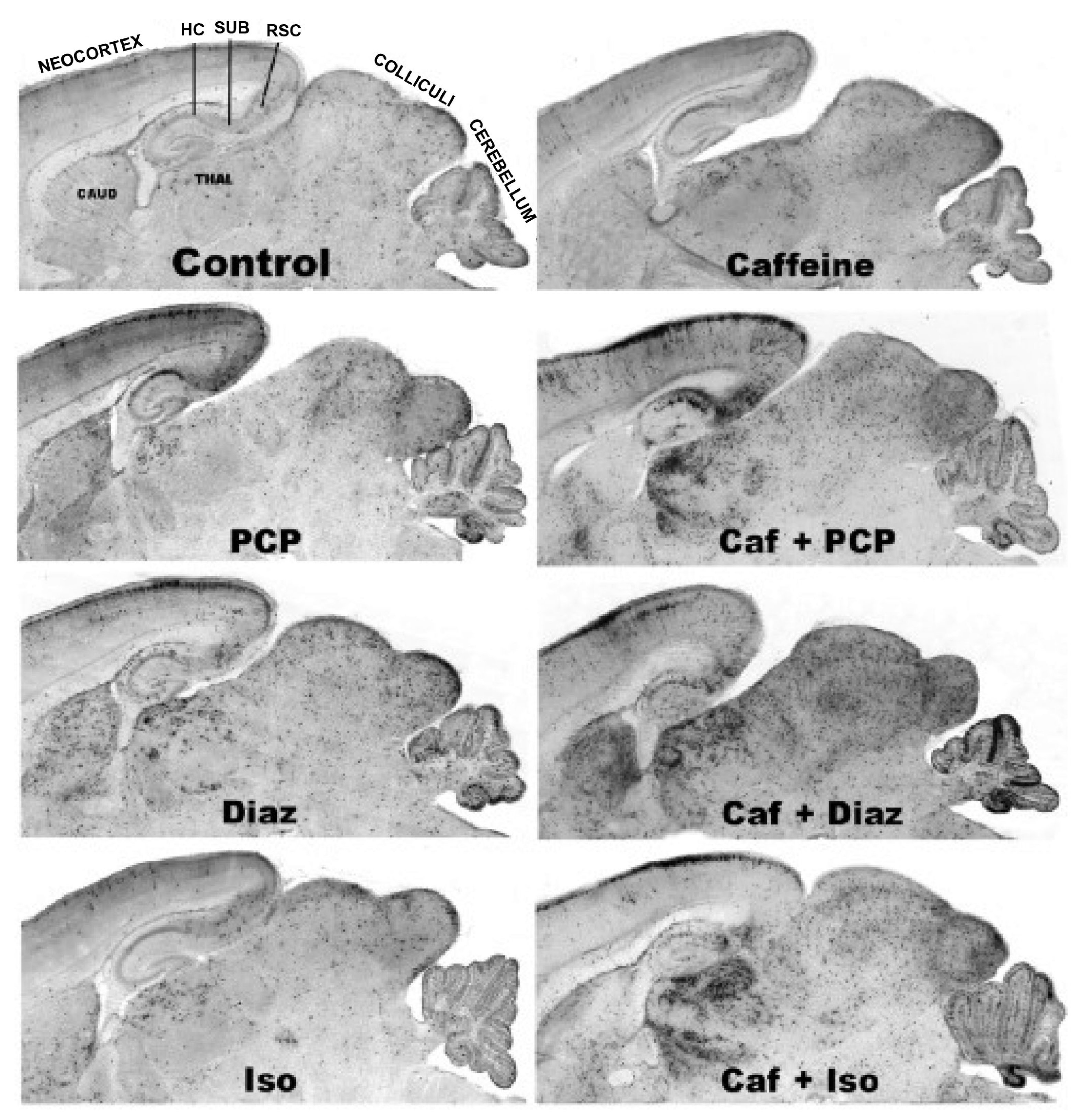
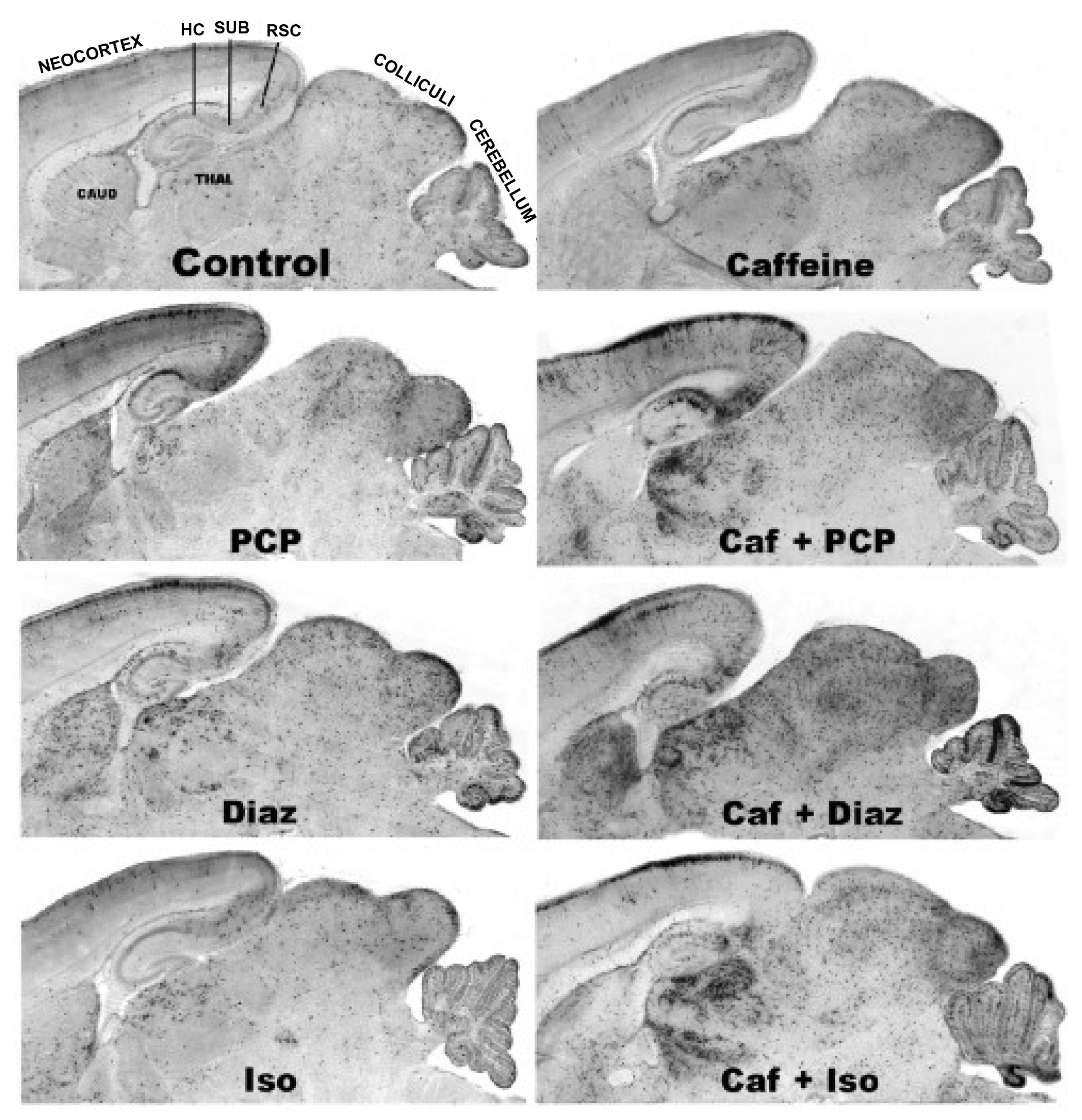
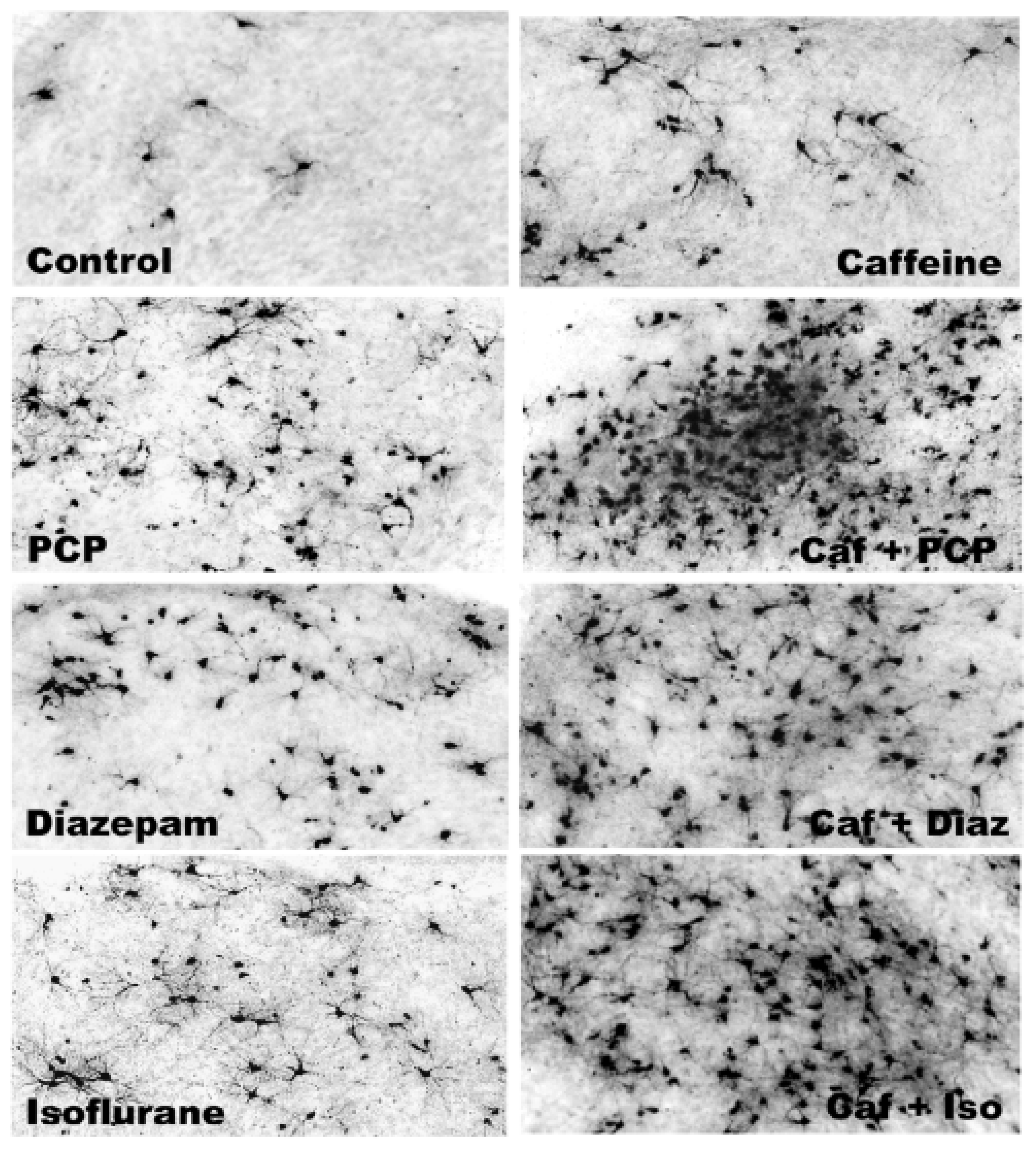
3.5. Regional and Cellular Distribution of CAF’s Pro-Apoptotic Action


3.6. Long-Term Neurobehavioral Effects of CAF + PCP or Diazepam
3.6.1. CAF + PCP
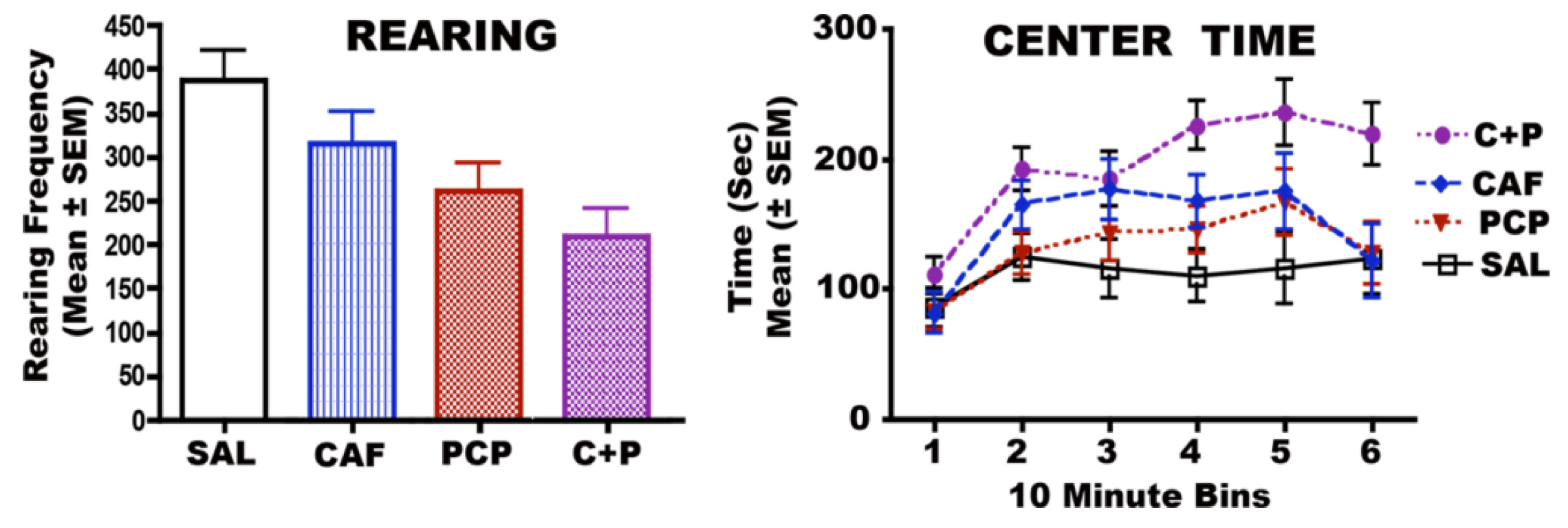
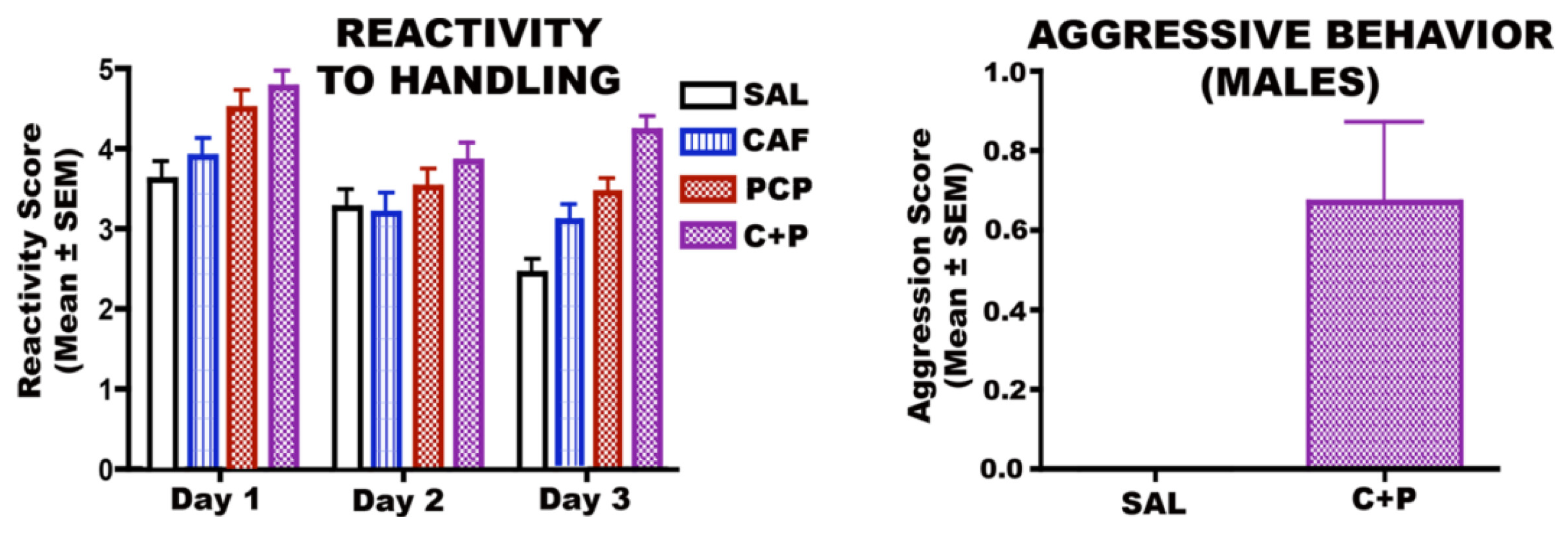
3.6.2. CAF + Diazepam
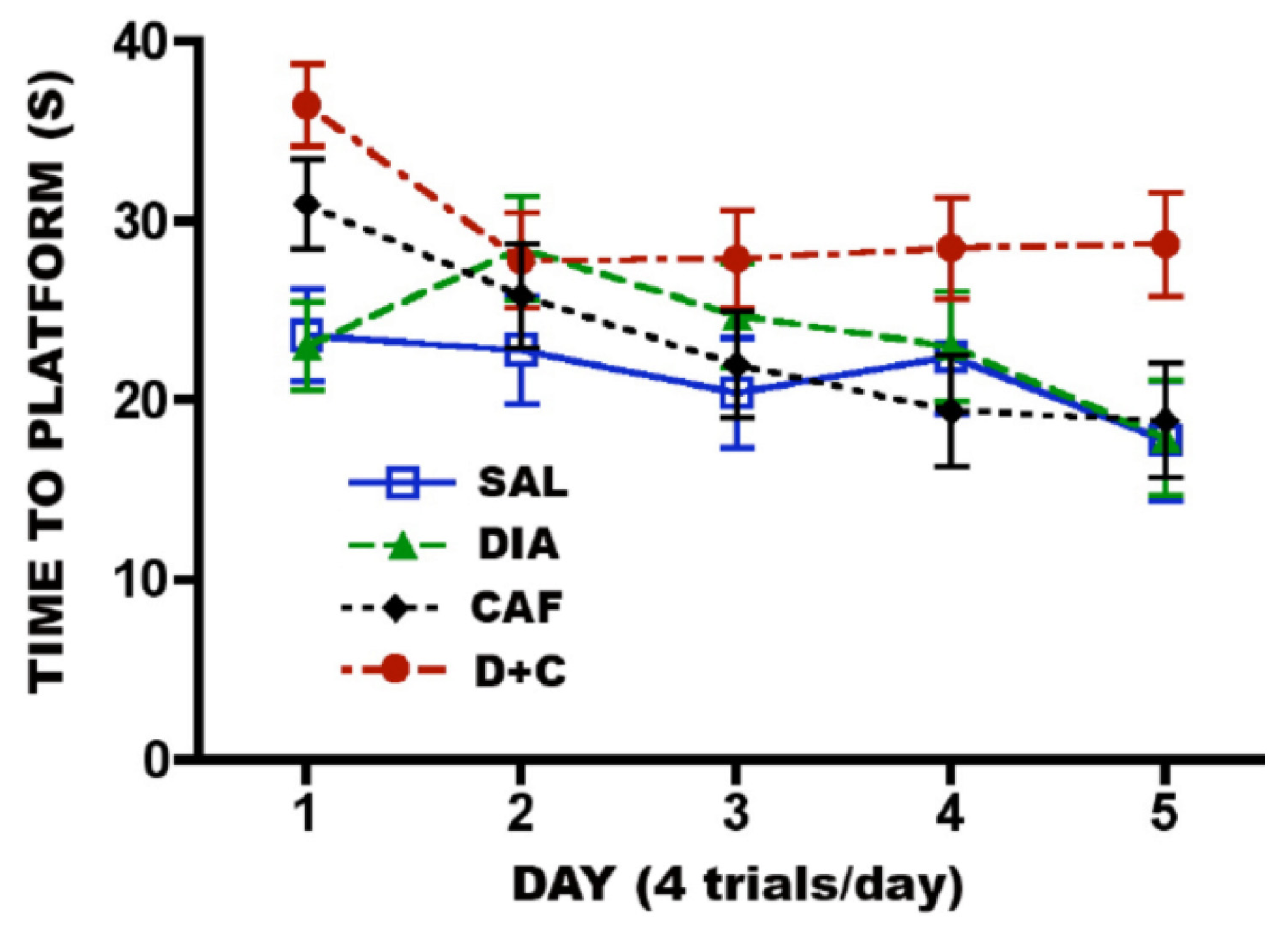
4. Discussion
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Ikonomidou, C.; Bosch, F.; Miksa, M.; Bittigau, P.; Vöckler, J.; Dikranian, K.; Tenkova, T.; Stevoska, V.; Turski, L.; Olney, J.W. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999, 283, 70–74. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Bittigau, P.; Ishimaru, M.J.; Wozniak, D.F.; Koch, C.; Genz, K.; Price, M.T.; Stefovska, V.; Hörster, F.; Tenkova, T.; et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 2000, 287, 1056–1060. [Google Scholar] [CrossRef]
- Bittigau, P.; Sifringer, M.; Genz, K.; Reith, E.; Pospischil, D.; Govindarajalu, S.; Dzietko, M.; Pesditschek, S.; Mai, I.; Dikranian, K.; et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc. Natl. Acad. Sci. USA 2002, 99, 15089–15094. [Google Scholar] [CrossRef]
- Dikranian, K.; Qin, Y.Q.; Labruyere, J.; Nemmers, B.; Olney, J.W. Ethanol-induced neuroapoptosis in the developing rodent cerebellum and related brain stem structures. Dev. Brain Res. 2005, 155, 1–13. [Google Scholar] [CrossRef]
- Istaphanous, G.K.; Howard, J.; Nan, X.; Hughes, E.A.; McCann, J.C.; McAuliffe, J.J.; Danzer, S.C.; Loepke, A.W. Comparison of the neuroapoptotic properties of equipotent anesthetic concentrations of desflurane, isoflurane, or sevoflurane in neonatal mice. Anesthesiology 2011, 114, 578–587. [Google Scholar] [CrossRef]
- Jevtovic-Todorovic, V.; Hartman, R.E.; Izumi, Y.; Benshoff, N.D.; Dikranian, K.; Zorumski, C.F.; Olney, J.W.; Wozniak, D.F. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J. Neurosci. 2003, 23, 876–882. [Google Scholar]
- Olney, J.W.; Tenkova, T.; Dikranian, K.; Qin, Y.Q.; Labruyere, J.; Ikonomidou, C. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Dev. Brain Res. 2002, 133, 115–126. [Google Scholar] [CrossRef]
- Rizzi, S.; Carter, L.B.; Ori, C.; Jevtovic-Todorovic, V. Clinical anesthesia causes permanent damage to the fetal guinea pig brain. Brain Pathol. 2008, 18, 198–210. [Google Scholar] [CrossRef]
- Rizzi, S.; Ori, C.; Jevtovic-Todorovic, V. Timing versus duration: Determinants of anesthesia-induced developmental apoptosis in the young mammalian brain. Ann. N. Y. Acad. Sci. 2010, 1199, 43–51. [Google Scholar] [CrossRef]
- Tenkova, T.; Young, C.; Dikranian, K.; Labruyere, J.; Olney, J.W. Ethanol-induced apoptosis in the developing visual system during synaptogenesis. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2809–2817. [Google Scholar] [CrossRef]
- Young, C.; Jevtovic-Todorovic, V.; Qin, Y.Q.; Tenkova, T.; Wang, H.; Labruyere, J.; Olney, J.W. Potential of ketamine and midazolam, individually or in combination, to induce apoptotic neurodegeneration in the infant mouse brain. Br. J. Pharmacol. 2005, 146, 189–197. [Google Scholar]
- Dikranian, K.; Ishimaru, M.J.; Tenkova, T.; Labruyere, J.; Qin, Y.Q.; Ikonomidou, C.; Olney, J.W. Apoptosis in the in vivo mammalian forebrain. Neurobiol. Dis. 2001, 8, 359–379. [Google Scholar] [CrossRef]
- Nikizad, H.; Yon, J.H.; Carter, L.B.; Jevtovic-Todorovic, V. Early exposure to general anesthesia causes significant neuronal deletion in the developing rat brain. Ann. N. Y. Acad. Sci. 2007, 1122, 69–82. [Google Scholar] [CrossRef]
- Sanders, R.D.; Xu, J.; Shu, Y.; Fidalgo, A.; Ma, D.; Maze, M. General anesthetics induce apoptotic neurodegeneration in the neonatal rat spinal cord. Anesth. Analg. 2008, 106, 1708–1711. [Google Scholar] [CrossRef]
- Young, C.; Klocke, J.; Tenkova, T.; Choi, J.; Labruyere, J.; Qin, Y.Q.; Holtzman, D.M.; Roth, K.A.; Olney, J.W. Ethanol-induced neuronal apoptosis in the in vivo developing mouse brain is BAX dependent. Cell Death Differ. 2003, 10, 1148–1155. [Google Scholar] [CrossRef]
- Young, C.; Straiko, M.M.W.; Johnson, S.A.; Creeley, C.; Olney, J.W. Ethanol causes and lithium prevents neuroapoptosis and suppression of pERK in the infant mouse brain. Neurobiol. Dis. 2008, 31, 355–360. [Google Scholar] [CrossRef]
- Straiko, M.M.W.; Young, C.; Cattano, D.; Creeley, C.E.; Wang, H.; Smith, D.J.; Johnson, S.A.; Li, E.S.; Olney, J.W. Lithium protects against anesthesia-induced developmental neuroapoptosis. Anesthesiology 2009, 110, 662–668. [Google Scholar]
- Sanders, R.D.; Sun, P.; Patel, S.; Li, M.; Maze, M.; Ma, D. Dexmedetomidine provides cortical neuroprotection: Impact on anaesthetic-induced neuroapoptosis in the rat developing brain. ActaAnaesthesiol. Scand. 2010, 54, 710–716. [Google Scholar]
- Yon, J.H.; Carter, L.B.; Jevtovic-Todorovic, V. Melatonin reduces the severity of anesthesia-induced apoptotic neurodegeneration in the developing rat brain. Neurobiol. Dis. 2006, 21, 522–530. [Google Scholar] [CrossRef]
- Olney, J.W.; Tenkova, T.; Dikranian, K.; Muglia, L.J.; Jermakowicz, W.J.; D’Sa, C.; Roth, K.A. Ethanol-induced caspase-3 activation in the in vivo developing mouse brain. Neurobiol. Dis. 2002, 9, 205–219. [Google Scholar] [CrossRef]
- Young, C.; Roth, K.A.; Klocke, B.J.; West, T.; Holtzman, D.M.; Labruyere, J.; Qin, Y.Q.; Dikranian, K.; Olney, J.W. Role of caspase-3 in ethanol-induced developmental neurodegeneration. Neurobiol. Dis. 2005, 20, 608–614. [Google Scholar] [CrossRef]
- Cattano, D.; Young, C.; Olney, J.W. Sub-anesthetic doses of propofol induce neuroapoptosis in the infant mouse brain. Anesth. Analg. 2008, 106, 1712–1714. [Google Scholar] [CrossRef]
- Ma, D.; Williamson, P.; Januszewski, A.; Nogaro, M.C.; Hossain, M.; Ong, L.P.; Shu, Y.; Franks, N.P.; Maze, M. Xenon mitigates isoflurane-induced neuronal apoptosis in the developing rodent brain. Anesthesiology 2007, 106, 746–753. [Google Scholar] [CrossRef]
- Johnson, S.A.; Young, C.; Olney, J.W. Isoflurane-induced neuroapoptosis in the developing brain of non-hypoglycemic mice. J. Neurosurg. Anesth. 2008, 20, 21–28. [Google Scholar] [CrossRef]
- Sanders, R.D.; Xu, J.; Shu, Y.; Januszewski, A.; Halder, S.; Fidalgo, A.; Sun, P.; Hossain, M.; Ma, D.; Maze, M. Dexmedetomidine attenuates isoflurane-induced neurocognitive impairment in neonatal rats. Anesthesiology 2009, 110, 11077–11085. [Google Scholar]
- Zhang, X.; Xue, Z.; Sun, A. Subclinical concentration of sevoflurane potentiates neuronal apoptosis in the developing C57BL/6 mouse brain. Neurosci. Lett. 2008, 447, 109–114. [Google Scholar] [CrossRef]
- Cattano, D.; Williamson, P.; Fukui, K.; Avidan, M.; Evers, A.S.; Olney, J.W.; Young, C. Potential of xenon to induce or to protect against neuroapoptosis in the developing mouse brain. Can. J. Anesth. 2008, 55, 429–436. [Google Scholar] [CrossRef]
- Brambrink, A.M.; Evers, A.S.; Avidan, M.S.; Farber, N.B.; Smith, D.J.; Zhang, X.; Dissen, G.A.; Creeley, C.E.; Olney, J.W. Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology 2010, 112, 834–841. [Google Scholar] [CrossRef]
- Brambrink, A.M.; Evers, A.S.; Avidan, M.S.; Farber, N.B.; Smith, D.J.; Martin, L.D.; Dissen, G.A.; Creeley, C.E.; Olney, J.W. Ketamine-induced neuroapoptosis in the fetal and neonatal rhesus macaque brain. Anesthesiology 2012, 116, 372–384. [Google Scholar] [CrossRef]
- Brambrink, A.M.; Dissen, G.A.; Martin, L.D.; Creeley, C.E.; Olney, J.W. Propofol-Induced Apoptosis of Neurons and Oligodendrocytes in Neonatal Macaque Brain. In Proceedings of the American Society of Anesthesiologists, Washington, DC, USA, 13–17 October 2012. Abstract Number A103.
- Brambrink, A.M.; Back, S.A.; Avidan, M.S.; Creeley, C.E.; Olney, J.W. Ketamine and Isoflurane Anesthesia Triggers Neuronal and Glial Apoptosis in the Neonatal Macaque. In Proceedings of the American Society of Anesthesiologists, San Diego, CA, USA, 16–20 October 2010. Abstract Number A375.
- Brambrink, A.M.; Dissen, G.A.; Martin, L.D.; Creeley, C.E.; Olney, J.W. Neuronal and glial apoptosis observed after intravenous propofol anesthesia in neonatal macaques. J. Neurosurg. Anesthesiol. 2012, 24, 494. [Google Scholar]
- Brambrink, A.M.; Dikranian, K.; Evers, A.S.; Creeley, C.E.; Olney, J.W. Isoflurane-Induced Apoptosis of Neurons and Oligodendrocytes in the Fetal Rhesus Macaque Brain. In Proceedings of the American Society of Anesthesiologists, Washington, DC, USA, 13–17 October 2012. Abstract Number LBB10.
- Brambrink, A.M.; Back, S.A.; Riddle, A.; Gong, X.; Moravec, M.D.; Dissen, G.A.; Creeley, C.E.; Dikranian, K.; Olney, J.W. Isoflurane-induced apoptosis of oligodendrocytes in the neonatal primate brain. Ann. Neurol. 2012, 72, 525–535. [Google Scholar] [CrossRef]
- Creeley, C.E.; Dikranian, K.T.; Johnson, S.A.; Farber, N.B.; Olney, J.W. Alcohol-induced apoptosis of oligodendrocytes in the fetal macaque brain. Acta Neuropathol. Commun. 2013, in press. [Google Scholar]
- Farber, N.B.; Creeley, C.E.; Olney, J.W. Alcohol-induced neuroapoptosis in the fetal macaque brain. Neurobiol. Dis. 2010, 40, 200–206. [Google Scholar] [CrossRef]
- Paule, M.G.; Li, M.; Allen, R.R.; Liu, F.; Zou, X.; Hotchkiss, C.; Hanig, J.P.; Patterson, T.A.; Slikker, W., Jr.; Wang, C. Ketamine anesthesia during the first week of life can cause long-lasting cognitive deficits in rhesus monkeys. Neurotoxicol. Teratol. 2011, 33, 220–230. [Google Scholar] [CrossRef]
- Slikker, W., Jr.; Zou, X.; Hotchkiss, C.E.; Divine, R.L.; Sadovova, N.; Twaddle, N.C.; Doerge, D.R.; Scallet, A.C.; Patterson, T.A.; Hanig, J.P.; et al. Ketamine-induced neuronal cell death in the perinatal rhesus monkey. Toxicol. Sci. 2007, 98, 145–158. [Google Scholar] [CrossRef]
- Zou, X.; Liu, F.; Zhang, X.; Patterson, T.A.; Callicott, R.; Liu, S.; Hanig, J.P.; Paule, M.G.; Slikker, W.; Wang, C. Inhalation anesthetic-induced neuronal damage in the developing rhesus monkey. Neurotoxicol. Teratol. 2011, 33, 592–597. [Google Scholar] [CrossRef]
- Zou, X.; Patterson, T.A.; Divine, R.L.; Sadova, N.; Zhang, X.; Hanig, J.P.; Paule, M.G.; Slikker, W.; Wang, C. Prolonged exposure to ketamine increases neurodegeneration in the developing monkey brain. Int. J. Dev. Neurosci. 2009, 27, 727–731. [Google Scholar] [CrossRef]
- Fredriksson, A.; Archer, T. Neurobehavioural deficits associated with apoptotic neurodegeneration and vulnerability for ADHD. Neurotox. Res. 2004, 6, 435–456. [Google Scholar] [CrossRef]
- Fredriksson, A.; Ponten, E.; Gordh, T.; Eriksson, P. Neonatal exposure to a combination of N-methyl-d-aspartate and γ-aminobutyric acid type A receptor anesthetic agents potentiates apoptotic neurodegeneration and persistent behavioral deficits. Anesthesiology 2007, 107, 427–436. [Google Scholar] [CrossRef]
- Satomoto, M.; Satoh, Y.; Terui, K.; Miyao, H.; Takishima, K.; Ito, M.; Imaki, J. Neonatal exposure to sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. Anesthesiology 2009, 110, 628–637. [Google Scholar] [CrossRef]
- Stratmann, G.; Sall, J.W.; May, L.D.; Bell, J.S.; Magnusson, K.R.; Rau, V.; Visrodia, K.H.; Alvi, R.S.; Ku, B.; Lee, M.T.; et al. Isoflurane differentially affects neurogenesis and long-term neurocognitive function in 60-day-old and 7-day-old rats. Anesthesiology 2009, 110, 834–848. [Google Scholar] [CrossRef]
- Wozniak, D.F.; Hartman, R.E.; Boyle, M.P.; Vogt, S.K.; Brooks, A.R.; Tenkova, T.; Young, C.; Olney, J.W.; Muglia, L.J. Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol. Dis. 2004, 17, 403–414. [Google Scholar] [CrossRef]
- Dobbing, J.; Sands, J. The brain growth spurt in various mammalian species. Early Hum. Dev. 1979, 3, 79–84. [Google Scholar] [CrossRef]
- Streissguth, A.P.; O’Malley, K. Neuropsychiatric implications and long-term consequences of Fetal Alcohol Spectrum Disorders. Semin. Clin. Neuropsych. 2000, 5, 177–190. [Google Scholar]
- Riley, E.P.; McGee, C.L. Fetal alcohol spectrum disorders: An overview with emphasis on changes in brain and behavior. Exp. Biol. Med. 2005, 230, 357–365. [Google Scholar]
- Famy, C.; Streissguth, A.P.; Unis, A.S. Mental illness in adults with fetal alcohol syndrome or fetal alcohol effects. Am. J. Psychiatry 1998, 155, 552–554. [Google Scholar]
- Meador, K.J. NEAD Study Group. Cognitive function at 3 years of age after fetal exposure to antiepileptic drugs. N. Engl. J. Med. 2009, 360, 1597–1605. [Google Scholar] [CrossRef]
- Meador, K.J. NEAD Study Group. Effects of fetal antiepileptic drug exposure: Outcomes at age 4.5 years. Neurology 2012, 78, 1207–1214. [Google Scholar] [CrossRef]
- Banach, R.; Boskovic, R.; Einarson, T.; Koren, G. Long-term developmental outcome of children of women with epilepsy, unexposed or exposed prenatally to antiepileptic drugs: A meta-analysis of cohort studies. Drug Saf. 2010, 33, 73–79. [Google Scholar] [CrossRef]
- DiMaggio, C.; Sun, L.S.; Kakavouli, A.; Burne, M.W.; Li, G. A retrospective cohort study of the association of anesthesia and hernia repair surgery with behavioral and developmental disorders in young children. J. Neurosurg. Anesthesiol. 2009, 4, 286–291. [Google Scholar]
- DiMaggio, C.; Sun, L.; Li, G. Early childhood exposure to anesthesia and risk of developmental and behavioral disorders in a sibling birth cohort. Anesth. Analg. 2011, 113, 1143–1151. [Google Scholar] [CrossRef]
- Wilder, R.T.; Flick, R.P.; Sprung, J.; Katusic, S.K.; Barbaresi, W.J.; Mickelson, C.; Gleich, S.J.; Schroeder, D.R.; Weaver, A.L.; Warner, D.O. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology 2009, 110, 796–804. [Google Scholar] [CrossRef]
- Flick, R.P.; Katusic, S.K.; Colligan, R.C.; Wilder, R.T.; Voigt, R.G.; Olson, M.D.; Sprung, J.; Weaver, A.L.; Schroeder, D.R.; Warner, D.O. Cognitive and behavioral outcomes after early exposure to anesthesia and surgery. Pediatrics 2011, 128, 1053–1061. [Google Scholar] [CrossRef]
- Sprung, J.; Flick, R.P.; Katusic, S.K.; Colligan, R.C.; Barbaresi, W.J.; Bojanic, K.; Welch, T.L.; Olson, M.D.; Hanson, A.C.; Schroeder, D.R.; et al. Attention-deficit/hyperactivity disorder after early exposure to procedures requiring general anesthesia. Mayo Clin. Proc. 2012, 87, 120–129. [Google Scholar] [CrossRef]
- Ing, C.; DiMaggio, C.; Whitehouse, A.; Hegarty, M.K.; Brady, J.; von Ungern-Sternberg, B.S.; Davidson, A.; Wood, A.J.J.; Li, G.; Sun, L.S. Long-term differences in language and cognitive function after childhood exposure to anesthesia. Pediatrics 2012, 130, 476–485. [Google Scholar] [CrossRef]
- Block, R.I.; Thomas, J.J.; Bayman, E.O.; Choi, J.W.; Kimble, K.K.; Todd, M.M. Are anesthesia and surgery during infancy associated with altered academic performance during childhood? Anesthesiology 2012, 117, 494–503. [Google Scholar]
- Henderson-Smart, D.J.; Steer, P.A. Prophylactic caffeine to prevent postoperative apnea following general anesthesia in preterm infants. Cochrane Database Syst. Rev. 2001. [Google Scholar] [CrossRef]
- Charles, B.G.; Townsend, S.R.; Steer, P.A.; Flenady, V.J.; Gray, P.H.; Shearman, A. Caffeine citrate treatment for extremely premature infants with apnea: Population pharmacokinetics, absolute bioavailability, and implications for therapeutic drug monitoring. Ther. Drug Monit. 2008, 30, 709–716. [Google Scholar] [CrossRef]
- Natarajan, G.; Botica, M.-L.; Thomas, R.; Aranda, J.V. Therapeutic drug monitoring for caffeine in preterm neonates: An unnecessary exercise? Pediatrics 2007, 119, 936–940. [Google Scholar]
- Aranda, J.V.; Beharry, K.; Valencia, G.B.; Natarajan, G.; Davis, J. Caffeine impact on neonatal morbidities. J. Matern. Fetal Neonatal Med. Suppl. 2010, 3, 20–23. [Google Scholar]
- Back, S.A.; Craig, A.; Suo, N.L.; Ren, J.; Akundi, R.S.; Ribeira, I.; Rivkees, S.A. Protective effects of caffeine on chronic hypoxia-induced perinatal white matter injury. Ann. Neurol. 2006, 60, 696–705. [Google Scholar] [CrossRef]
- Schmidt, B.; Roberts, R.S.; Davis, P.; Doyle, L.W.; Barrington, K.J.; Ohlsson, A.; Solimano, A.; Tin, W. Long-term effects of caffeine therapy for apnea of prematurity. N. Engl. J. Med. 2007, 357, 1893–1902. [Google Scholar] [CrossRef]
- Kang, S.H.; Lee, Y.A.; Won, S.J.; Rhee, K.-H.; Gwag, B.J. Caffeine-induced neuronal death in neonatal rat brain and cortical cell cultures. Neuroreport 2002, 13, 1945–1950. [Google Scholar] [CrossRef]
- Ishimaru, M.J.; Ikonomidou, C.; Tenkova, T.I.; Der, T.C.; Dikranian, K.; Sesma, M.; Olney, J.W. Distinguishing excitotoxic from apoptotic neurodegeneration in the developing rat brain. J. Comp. Neurol. 1999, 408, 461–476. [Google Scholar] [CrossRef]
- Khanna, N.N.; Somani, S.M. Maternal coffee drinking and unusually high concentrations of caffeine in the newborn. J. Toxicol. Clin. Toxicol. 1984, 22, 473–483. [Google Scholar] [CrossRef]
- Helfer, J.L.; Goodlett, C.R.; Greenough, W.T.; Klintsove, A.Y. The effects of exercise on adolescent hippocampal neurogenesis in a rat model of binge alcohol exposure during the brain growth spurt. Brain Res. 2009, 1294, 1–11. [Google Scholar] [CrossRef]
- Loepke, A.W.; McCann, J.C.; Kurth, C.D.; McAuliffe, J.J. The physiologic effects of isoflurane anesthesia in neonatal mice. Anesth. Analg. 2006, 102, 75–80. [Google Scholar] [CrossRef]
- Wyllie, A.H.; Kerr, J.F.R.; Currie, A.R. Cell death: The significance of apoptosis. Int. Rev. Cytol. 1980, 68, 251–306. [Google Scholar] [CrossRef]
- Brown, J.A.; Emnett, R.J.; White, C.R.; Yuede, C.M.; Conyers, S.B.; O’Malley, K.L.; Wozniak, D.F.; Gutmann, D.H. Reduced striatal dopamine underlies the attention system dysfunction in neurofibromatosis-1 mutant mice. Hum. Mol. Genet. 2010, 19, 4515–4528. [Google Scholar] [CrossRef]
- Leon, A.E.; Michienzi, K.; Ma, C.X.; Hutchison, A.A. Serum caffeine concentrations in preterm neonates. Am. J. Perinatol. 2007, 24, 39–47. [Google Scholar] [CrossRef]
- Sayal, K.; Heron, J.; Golding, J.; Alati, R.; Smith, G.D.; Gray, R.; Emond, A. Binge pattern of alcohol consumption during pregnancy and childhood mental health outcomes: Longitudinal population-based study. Pediatrics 2009, 123, 289–296. [Google Scholar] [CrossRef]
- Bailey, B.N.; Delaney-Black, V.; Covington, C.Y.; Ager, J.; Janisse, J.; Hannigan, J.H.; Sokol, R.J. Prenatal exposure to binge drinking and cognitive and behavioral outcomes at age 7 years. Am. J. Obstet. Gynecol. 2004, 191, 1037–1043. [Google Scholar] [CrossRef]
- Daly, J.W.; Holmen, J.; Fredholm, B.B. Is caffeine addictive? The most widely used psychoactive substance in the world affects same parts of the brain as cocaine. Lakartidningen 1998, 95, 5878–5883. [Google Scholar]
- Svikis, D.S.; Berger, N.; Haug, N.A.; Griffiths, R.R. Caffeine dependence in combination with a family history of alcoholism as a predictor of continued use of caffeine during pregnancy. Am. J. Psychiatry 2005, 162, 2344–2351. [Google Scholar] [CrossRef]
- Knutti, R.; Rothweiler, H.; Schlatter, C. Effect of pregnancy on the pharmacokinetics of caffeine. Eur. J. Clin. Pharmacol. 1981, 21, 121–126. [Google Scholar]
- Mitchell, M.C.; Hoyumpa, A.M.; Schenker, S.; Johnson, R.F.; Nichols, S.; Patwardhan, R.V. Inhibition of caffeine elimination by short-term ethanol administration. J. Lab. Clin. Med. 1983, 101, 826–834. [Google Scholar]
- Zhao, X.; Strong, R.; Piriyawat, P.; Palusinski, R.; Grotta, J.C.; Aronowski, J. Caffeinol at the receptor level: Anti-ischemic effect of N-methyl-d-aspartate receptor blockade is potentiated by caffeine. Stroke 2010, 41, 263–267. [Google Scholar]
- Bespalov, A.; Dravolina, O.; Belozertseva, I.; Adamcio, B.; Zvartau, E. Lowered brain stimulation reward thresholds in rats treated with a combination of caffeine and N-methyl-d-aspartate but not AMPA or metabotropic glutamate receptor-5 antagonists. Behav. Pharmacol. 2006, 17, 295–302. [Google Scholar]
- Olney, J.W. Excitotoxicity and NMDA receptors. Drug Dev. Res. 1989, 17, 299–319. [Google Scholar] [CrossRef]
- Olney, J.W. Excitotoxicity, apoptosis and neuropsychiatric disorders. Curr. Opin. Pharmacol. 2003, 3, 101–109. [Google Scholar]
- Lohaugen, G.C.; Gramstad, A.; Evensen, K.A.; Martinussen, M.; Lindqvist, S.; Indredavik, M.; Vik, T.; Brubakk, A.M.; Skranes, J. Cognitive profile in young adults born preterm at very low birthweight. Dev. Med. Child Neurol. 2010, 52, 1078–1079. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yuede, C.M.; Olney, J.W.; Creeley, C.E. Developmental Neurotoxicity of Alcohol and Anesthetic Drugs Is Augmented by Co-Exposure to Caffeine. Brain Sci. 2013, 3, 1128-1152. https://doi.org/10.3390/brainsci3031128
Yuede CM, Olney JW, Creeley CE. Developmental Neurotoxicity of Alcohol and Anesthetic Drugs Is Augmented by Co-Exposure to Caffeine. Brain Sciences. 2013; 3(3):1128-1152. https://doi.org/10.3390/brainsci3031128
Chicago/Turabian StyleYuede, Carla M., John W. Olney, and Catherine E. Creeley. 2013. "Developmental Neurotoxicity of Alcohol and Anesthetic Drugs Is Augmented by Co-Exposure to Caffeine" Brain Sciences 3, no. 3: 1128-1152. https://doi.org/10.3390/brainsci3031128