NADPH Oxidase as a Therapeutic Target for Neuroprotection against Ischaemic Stroke: Future Perspectives

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Oxidative Stress
3. Antioxidants for Stroke
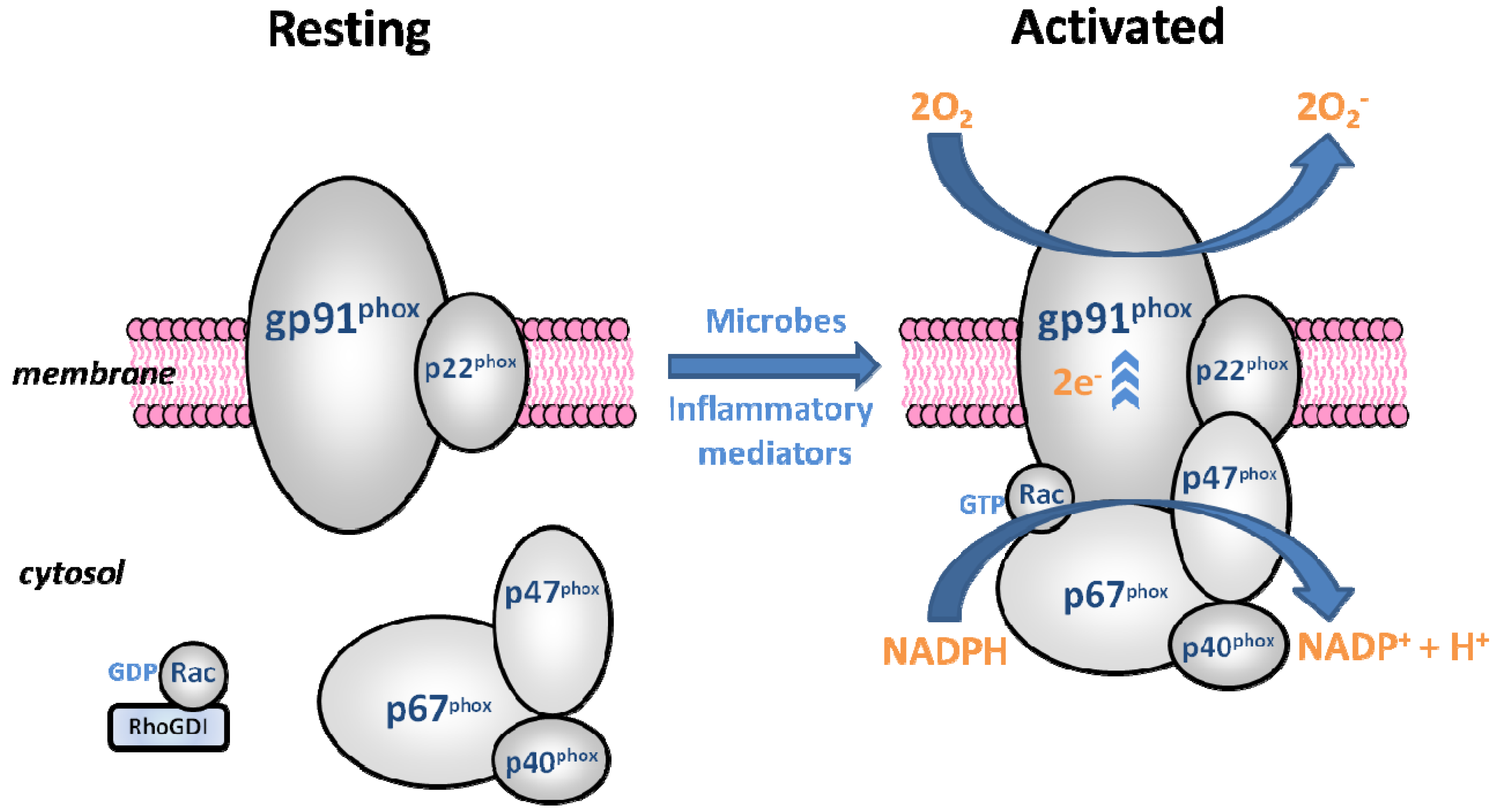
4. NADPH Oxidase
4.1. The Phagocyte NADPH Oxidase

4.2. Nox2 Homologues
5. NADPH Oxidase-Derived ROS

6. Distribution of NADPH Oxidases in the Brain
6.1. Neuronal Nox
6.2. Glial Nox
6.3. Cerebrovascular Nox
6.4. Immune Cell Nox
7. NADPH Oxidase Expression and Activity after Stroke
7.1. Nox2
7.2. Nox1
7.3. Nox4
8. Opposing Roles of NADPH Oxidase in Stroke
8.1. Nox and Cerebrovascular Dysregulation after Stroke
8.2. Nox and Oxygen Sensing in the Ischaemic Brain
8.3. NADPH Oxidase and Brain Tissue Regeneration
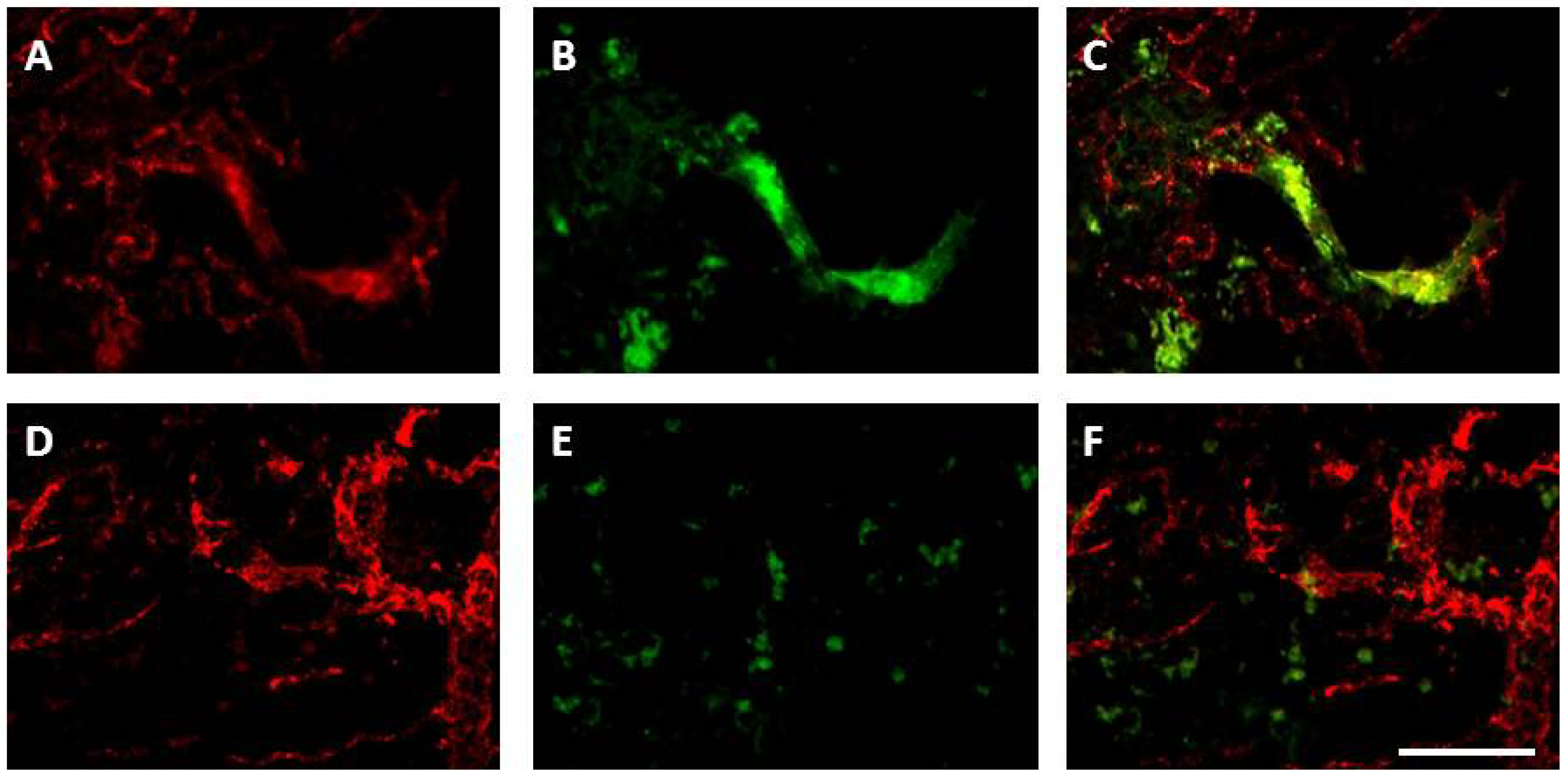
8.3.1. Angiogenesis
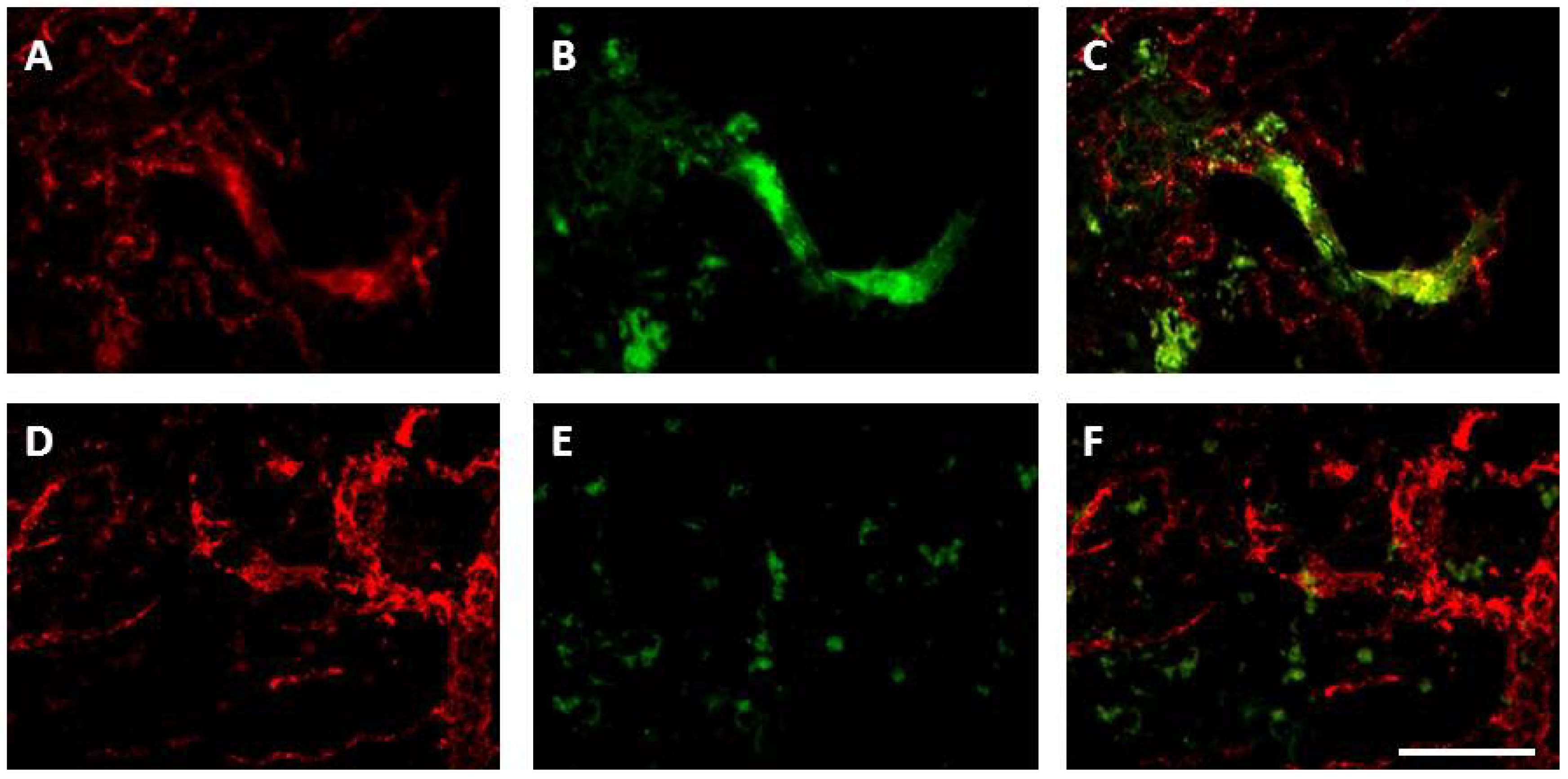
8.3.2. Neurogenesis
9. Inhibitors of NADPH Oxidase
9.1. Apocynin
9.2. DPI
9.3. gp91ds-tat
9.4. VAS2870
10. Future Perspectives
11. Conclusions
Conflict of Interest
References
- Crossley, N.A.; Sena, E.; Goehler, J.; Horn, J.; van der Worp, B.; Bath, P.M.; Macleod, M.; Dirnagl, U. Empirical evidence of bias in the design of experimental stroke studies: A metaepidemiologic approach. Stroke 2008, 39, 929–934. [Google Scholar] [CrossRef]
- Dirnagl, U. Bench to bedside: The quest for quality in experimental stroke research. J. Cereb. Blood Flow Metab. 2006, 26, 1465–1478. [Google Scholar] [CrossRef]
- Green, A.R. Why do neuroprotective drugs that are so promising in animals fail in the clinic? An industry perspective. Clin. Exp. Pharmacol. Physiol. 2002, 29, 1030–1034. [Google Scholar]
- Lo, E.H. Experimental models, neurovascular mechanisms and translational issues in stroke research. Br. J. Pharmacol. 2008, 153, S396–S405. [Google Scholar] [CrossRef]
- Macleod, M.R.; van der Worp, H.B.; Sena, E.S.; Howells, D.W.; Dirnagl, U.; Donnan, G.A. Evidence for the efficacy of NXY-059 in experimental focal cerebral ischaemia is confounded by study quality. Stroke 2008, 39, 2824–2829. [Google Scholar]
- O’Collins, V.E.; Macleod, M.R.; Donnan, G.A.; Horky, L.L.; van der Worp, B.H.; Howells, D.W. 1,026 experimental treatments in acute stroke. Ann. Neurol. 2006, 59, 467–477. [Google Scholar] [CrossRef]
- Feuerstein, G.Z.; Chavez, J. Translational medicine for stroke drug discovery: The pharmaceutical industry perspective. Stroke 2009, 40, S121–S125. [Google Scholar] [CrossRef]
- Feuerstein, G.Z.; Zaleska, M.M.; Krams, M.; Wang, X.; Day, M.; Rutkowski, J.L.; Finklestein, S.P.; Pangalos, M.N.; Poole, M.; Stiles, G.L.; et al. Missing steps in the stair case: A translational medicine perspective on the development of NXY-059 for treatment of acute ischemic stroke. J. Cereb. Blood Flow Metab. 2008, 28, 217–219. [Google Scholar] [CrossRef]
- Proctor, P.H.; Tamborello, L.P. SAINT-I worked, but the neuroprotectant is not NXY-059. Stroke 2007, 38, e109. [Google Scholar] [CrossRef]
- Shuaib, A.; Lees, K.R.; Lyden, P.; Grotta, J.; Davalos, A.; Davis, S.M.; Diener, H.C.; Ashwood, T.; Wasiewski, W.W.; Emeribe, U. NXY-059 for the treatment of acute ischemic stroke. N. Engl. J. Med. 2007, 357, 562–571. [Google Scholar]
- Bal-Price, A.; Matthias, A.; Brown, G.C. Stimulation of the NAD(P)H oxidase in activated rat microglia removes nitric oxide but induces peroxynitrite production. J. Neurochem. 2002, 80, 73–80. [Google Scholar] [CrossRef]
- Schaller, B.; Graf, R. Cerebral ischemia and reperfusion: The pathophysiologic concept as a basis for clinical therapy. J. Cereb. Blood Flow Metab. 2004, 24, 351–371. [Google Scholar]
- Pawate, S.; Shen, Q.; Fan, F.; Bhat, N.R. Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J. Neurosci. Res. 2004, 77, 540–551. [Google Scholar] [CrossRef]
- Miller, A.A.; Dusting, G.J.; Roulston, C.L.; Sobey, C.G. NADPH-oxidase activity is elevated in penumbral and non-ischemic cerebral arteries following stroke. Brain Res. 2006, 1111, 111–116. [Google Scholar] [CrossRef]
- Park, L.; Anrather, J.; Zhou, P.; Frys, K.; Pitstick, R.; Younkin, S.; Carlson, G.A.; Iadecola, C. NADPH oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J. Neurosci. 2005, 25, 1769–1777. [Google Scholar]
- Kleinschnitz, C.; Grund, H.; Wingler, K.; Armitage, M.E.; Jones, E.; Mittal, M.; Barit, D.; Schwarz, T.; Geis, C.; Kraft, P.; et al. Post-stroke inhibition of induced NAD(P)H oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010, 8, e1000479. [Google Scholar]
- McCann, S.K.; Dusting, G.J.; Roulston, C.L. Early increase of Nox4 NAD(P)H oxidase and superoxide generation following endothelin-1-induced stroke in conscious rats. J. Neurosci. Res. 2008, 86, 2524–2534. [Google Scholar]
- Vallet, P.; Charnay, Y.; Steger, K.; Ogier-Denis, E.; Kovari, E.; Herrmann, F.; Michel, J.P.; Szanto, I. Neuronal expression of the NAD(P)H oxidase Nox4, and its regulation in mouse experimental brain ischemia. Neuroscience 2005, 132, 233–238. [Google Scholar] [CrossRef]
- Warner, D.S.; Sheng, H.; Batinic-Haberle, I. Oxidants, antioxidants and the ischemic brain. J. Exp. Biol. 2004, 207, 3221–3231. [Google Scholar] [CrossRef]
- Zalba, G.; Fortuno, A.; San Jose, G.; Moreno, M.U.; Beloqui, O.; Diez, J. Oxidative stress, endothelial dysfunction and cerebrovascular disease. Cerebrovasc. Dis. 2007, 24, 24–29. [Google Scholar]
- Berry, C.E.; Hare, J.M. Xanthine oxidoreductase and cardiovascular disease: Molecular mechanisms and pathophysiological implications. J. Physiol. 2004, 555, 589–606. [Google Scholar] [CrossRef]
- Paravicini, T.M.; Sobey, C.G. Cerebral vascular effects of reactive oxygen species: Recent evidence for a role of NAD(P)H-oxidase. Clin. Exp. Pharmacol. Physiol. 2003, 30, 855–859. [Google Scholar] [CrossRef]
- Im, J.Y.; Kim, D.; Paik, S.G.; Han, P.L. Cyclooxygenase-2-dependent neuronal death proceeds via superoxide anion generation. Free Radic. Biol. Med. 2006, 41, 960–972. [Google Scholar] [CrossRef]
- Fleming, I.; Michaelis, U.R.; Bredenkotter, D.; Fisslthaler, B.; Dehghani, F.; Brandes, R.P.; Busse, R. Endothelium-derived hyperpolarizing factor synthase (cytochrome p450 2c9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ. Res. 2001, 88, 44–51. [Google Scholar]
- Abramov, A.Y.; Scorziello, A.; Duchen, M.R. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J. Neurosci. 2007, 27, 1129–1138. [Google Scholar]
- Beske, P.H.; Jackson, D.A. NADPH oxidase mediates the oxygen-glucose deprivation/ reperfusion-induced increase in the tyrosine phosphorylation of the n-methyl-d-aspartate receptor NR2A subunit in retinoic acid differentiated SH-SY5Y cells. J. Mol. Signal. 2012, 7, 15. [Google Scholar] [CrossRef]
- Suh, S.W.; Shin, B.S.; Ma, H.; van Hoecke, M.; Brennan, A.M.; Yenari, M.A.; Swanson, R.A. Glucose and NAD(P)H oxidase drive neuronal superoxide formation in stroke. Ann. Neurol. 2008, 64, 654–663. [Google Scholar] [CrossRef]
- Durukan, A.; Tatlisumak, T. Acute ischemic stroke: Overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol. Biochem. Behav. 2007, 87, 179–197. [Google Scholar] [CrossRef]
- Manea, A. NADPH oxidase-derived reactive oxygen species: Involvement in vascular physiology and pathology. Cell Tissue Res. 2010, 342, 325–339. [Google Scholar] [CrossRef]
- Mills, E.; Dong, X.P.; Wang, F.; Xu, H. Mechanisms of brain iron transport: Insight into neurodegeneration and cns disorders. Future Med. Chem. 2010, 2, 51–64. [Google Scholar] [CrossRef]
- Reiter, R.J. Oxidative processes and antioxidative defense mechanisms in the aging brain. FASEB J. 1995, 9, 526–533. [Google Scholar]
- Liu, J.; Yeo, H.C.; Overvik-Douki, E.; Hagen, T.; Doniger, S.J.; Chyu, D.W.; Brooks, G.A.; Ames, B.N. Chronically and acutely exercised rats: Biomarkers of oxidative stress and endogenous antioxidants. J. Appl. Physiol. 2000, 89, 21–28. [Google Scholar]
- Matsuo, M.; Gomi, F.; Dooley, M.M. Age-related alterations in antioxidant capacity and lipid peroxidation in brain, liver, and lung homogenates of normal and vitamin E-deficient rats. Mech. Ageing Dev. 1992, 64, 273–292. [Google Scholar] [CrossRef]
- Chan, P.H. Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metab. 2001, 21, 2–14. [Google Scholar]
- Kamii, H.; Mikawa, S.; Murakami, K.; Kinouchi, H.; Yoshimoto, T.; Reola, L.; Carlson, E.; Epstein, C.J.; Chan, P.H. Effects of nitric oxide synthase inhibition on brain infarction in sod-1-transgenic mice following transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1996, 16, 1153–1157. [Google Scholar]
- Keller, J.N.; Kindy, M.S.; Holtsberg, F.W.; St Clair, D.K.; Yen, H.C.; Germeyer, A.; Steiner, S.M.; Bruce-Keller, A.J.; Hutchins, J.B.; Mattson, M.P. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: Suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J. Neurosci. 1998, 18, 687–697. [Google Scholar]
- Sheng, H.; Bart, R.D.; Oury, T.D.; Pearlstein, R.D.; Crapo, J.D.; Warner, D.S. Mice overexpressing extracellular superoxide dismutase have increased resistance to focal cerebral ischemia. Neuroscience 1999, 88, 185–191. [Google Scholar] [CrossRef]
- Yang, G.; Chan, P.H.; Chen, J.; Carlson, E.; Chen, S.F.; Weinstein, P.; Epstein, C.J.; Kamii, H. Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke 1994, 25, 165–170. [Google Scholar]
- Murakami, K.; Kondo, T.; Epstein, C.J.; Chan, P.H. Overexpression of CuZn-superoxide dismutase reduces hippocampal injury after global ischemia in transgenic mice. Stroke 1997, 28, 1797–1804. [Google Scholar] [CrossRef]
- Kondo, T.; Reaume, A.G.; Huang, T.T.; Carlson, E.; Murakami, K.; Chen, S.F.; Hoffman, E.K.; Scott, R.W.; Epstein, C.J.; Chan, P.H. Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J. Neurosci. 1997, 17, 4180–4189. [Google Scholar]
- Murakami, K.; Kondo, T.; Kawase, M.; Li, Y.; Sato, S.; Chen, S.F.; Chan, P.H. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J. Neurosci. 1998, 18, 205–213. [Google Scholar]
- Sheng, H.; Brady, T.C.; Pearlstein, R.D.; Crapo, J.D.; Warner, D.S. Extracellular superoxide dismutase deficiency worsens outcome from focal cerebral ischemia in the mouse. Neurosci. Lett. 1999, 267, 13–16. [Google Scholar]
- Crack, P.J.; Taylor, J.M.; Flentjar, N.J.; de Haan, J.; Hertzog, P.; Iannello, R.C.; Kola, I. Increased infarct size and exacerbated apoptosis in the glutathione peroxidase-1 (Gpx-1) knockout mouse brain in response to ischemia/reperfusion injury. J. Neurochem. 2001, 78, 1389–1399. [Google Scholar]
- Weisbrot-Lefkowitz, M.; Reuhl, K.; Perry, B.; Chan, P.H.; Inouye, M.; Mirochnitchenko, O. Overexpression of human glutathione peroxidase protects transgenic mice against focal cerebral ischemia/reperfusion damage. Brain Res. Mol. Brain Res. 1998, 53, 333–338. [Google Scholar] [CrossRef]
- Crack, P.J.; Taylor, J.M.; de Haan, J.B.; Kola, I.; Hertzog, P.; Iannello, R.C. Glutathione peroxidase-1 contributes to the neuroprotection seen in the superoxide dismutase-1 transgenic mouse in response to ischemia/reperfusion injury. J. Cereb. Blood Flow Metab. 2003, 23, 19–22. [Google Scholar]
- De Haan, J.B.; Crack, P.J.; Flentjar, N.; Iannello, R.C.; Hertzog, P.J.; Kola, I. An imbalance in antioxidant defense affects cellular function: The pathophysiological consequences of a reduction in antioxidant defense in the glutathione peroxidase-1 (Gpx1) knockout mouse. Redox. Rep. 2003, 8, 69–79. [Google Scholar] [CrossRef]
- Baker, K.; Marcus, C.B.; Huffman, K.; Kruk, H.; Malfroy, B.; Doctrow, S.R. Synthetic combined superoxide dismutase/catalase mimetics are protective as a delayed treatment in a rat stroke model: A key role for reactive oxygen species in ischemic brain injury. J. Pharmacol. Exp. Ther. 1998, 284, 215–221. [Google Scholar]
- Chaudhary, G.; Sinha, K.; Gupta, Y.K. Protective effect of exogenous administration of alpha-tocopherol in middle cerebral artery occlusion model of cerebral ischemia in rats. Fundam. Clin. Pharmacol. 2003, 17, 703–707. [Google Scholar] [CrossRef]
- Henry, P.T.; Chandy, M.J. Effect of ascorbic acid on infarct size in experimental focal cerebral ischaemia and reperfusion in a primate model. Acta Neurochir. (Wien) 1998, 140, 977–980. [Google Scholar] [CrossRef]
- Lagowska-Lenard, M.; Stelmasiak, Z.; Bartosik-Psujek, H. Influence of vitamin C on markers of oxidative stress in the earliest period of ischemic stroke. Pharmacol. Rep. 2010, 62, 751–756. [Google Scholar]
- Ullegaddi, R.; Powers, H.J.; Gariballa, S.E. Antioxidant supplementation enhances antioxidant capacity and mitigates oxidative damage following acute ischaemic stroke. Eur. J. Clin. Nutr. 2005, 59, 1367–1373. [Google Scholar] [CrossRef]
- Amaro, S.; Chamorro, A. Translational stroke research of the combination of thrombolysis and antioxidant therapy. Stroke 2011, 42, 1495–1499. [Google Scholar] [CrossRef]
- Margaill, I.; Plotkine, M.; Lerouet, D. Antioxidant strategies in the treatment of stroke. Free Radic. Biol. Med. 2005, 39, 429–443. [Google Scholar] [CrossRef]
- Fisher, M.; Feuerstein, G.; Howells, D.W.; Hurn, P.D.; Kent, T.A.; Savitz, S.I.; Lo, E.H. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke 2009, 40, 2244–2250. [Google Scholar] [CrossRef]
- Stroke Therapy Academic Industry Roundtable (STAIR). Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke 1999, 30, 2752–2758. [CrossRef]
- Bath, P.M.; Gray, L.J.; Bath, A.J.; Buchan, A.; Miyata, T.; Green, A.R. Effects of NXY-059 in experimental stroke: An individual animal meta-analysis. Br. J. Pharmacol. 2009, 157, 1157–1171. [Google Scholar] [CrossRef]
- Nakase, T.; Yoshioka, S.; Suzuki, A. Free radical scavenger, edaravone, reduces the lesion size of lacunar infarction in human brain ischemic stroke. BMC Neurol. 2011, 11, 39. [Google Scholar] [CrossRef]
- Lapchak, P.A. A critical assessment of edaravone acute ischemic stroke efficacy trials: Is edaravone an effective neuroprotective therapy? Expert Opin. Pharmacother. 2010, 11, 1753–1763. [Google Scholar] [CrossRef]
- Lapchak, P.A.; Zivin, J.A. The lipophilic multifunctional antioxidant edaravone (radicut) improves behavior following embolic strokes in rabbits: A combination therapy study with tissue plasminogen activator. Exp. Neurol. 2009, 215, 95–100. [Google Scholar] [CrossRef]
- Isahaya, K.; Yamada, K.; Yamatoku, M.; Sakurai, K.; Takaishi, S.; Kato, B.; Hirayama, T.; Hasegawa, Y. Effects of edaravone, a free radical scavenger, on serum levels of inflammatory biomarkers in acute brain infarction. J. Stroke Cerebrovasc. Dis. 2012, 21, 102–107. [Google Scholar] [CrossRef]
- Roulston, C.L.; McCann, S.; Weston, R.M.; Jarrott, B. Animal Models of Stroke for Preclinical Drug Development: A Comparative Study of Flavonols for Cytoprotection. In Translational Stroke Research; Springer: New York, NY, USA, 2012; pp. 493–524. [Google Scholar]
- Halliwell, B. Free radicals, antioxidants, and human disease: Curiosity, cause, or consequence? Lancet 1994, 344, 721–724. [Google Scholar] [CrossRef]
- Rimm, E.B.; Katan, M.B.; Ascherio, A.; Stampfer, M.J.; Willett, W.C. Relation between intake of flavonoids and risk for coronary heart disease in male health professionals. Ann. Intern. Med. 1996, 125, 384–389. [Google Scholar] [CrossRef]
- Rivera, F.; Urbanavicius, J.; Gervaz, E.; Morquio, A.; Dajas, F. Some aspects of the in vivo neuroprotective capacity of flavonoids: Bioavailability and structure-activity relationship. Neurotox. Res. 2004, 6, 543–553. [Google Scholar] [CrossRef]
- Roulston, C.L.; Callaway, J.K.; Jarrott, B.; Woodman, O.L.; Dusting, G.J. Using behaviour to predict stroke severity in conscious rats: Post-stroke treatment with 3′,4′-dihydroxyflavonol improves recovery. Eur. J. Pharmacol. 2008, 584, 100–110. [Google Scholar] [CrossRef]
- Firuzi, O.; Miri, R.; Tavakkoli, M.; Saso, L. Antioxidant therapy: Current status and future prospects. Curr. Med. Chem. 2011, 18, 3871–3888. [Google Scholar] [CrossRef]
- Kamat, C.D.; Gadal, S.; Mhatre, M.; Williamson, K.S.; Pye, Q.N.; Hensley, K. Antioxidants in central nervous system diseases: Preclinical promise and translational challenges. J. Alzheimers Dis. 2008, 15, 473–493. [Google Scholar]
- Andriantsitohaina, R.; Duluc, L.; Garcia-Rodriguez, J.C.; Gil-del Valle, L.; Guevara-Garcia, M.; Simard, G.; Soleti, R.; Su, D.F.; Velasquez-Perez, L.; Wilson, J.X.; Laher, I. Systems biology of antioxidants. Clin. Sci. (Lond.) 2012, 123, 173–192. [Google Scholar] [CrossRef]
- Pandya, R.S.; Mao, L.; Zhou, H.; Zhou, S.; Zeng, J.; Popp, A.J.; Wang, X. Central nervous system agents for ischemic stroke: Neuroprotection mechanisms. Cent. Nerv. Syst. Agents Med. Chem. 2011, 11, 81–97. [Google Scholar] [CrossRef]
- Slemmer, J.E.; Shacka, J.J.; Sweeney, M.I.; Weber, J.T. Antioxidants and free radical scavengers for the treatment of stroke, traumatic brain injury and aging. Curr. Med. Chem. 2008, 15, 404–414. [Google Scholar] [CrossRef]
- Cherubini, A.; Ruggiero, C.; Morand, C.; Lattanzio, F.; Dell’aquila, G.; Zuliani, G.; Di Iorio, A.; Andres-Lacueva, C. Dietary antioxidants as potential pharmacological agents for ischemic stroke. Curr. Med. Chem. 2008, 15, 1236–1248. [Google Scholar] [CrossRef]
- Wang, C.X.; Shuaib, A. Neuroprotective effects of free radical scavengers in stroke. Drugs Aging 2007, 24, 537–546. [Google Scholar] [CrossRef]
- Doeppner, T.R.; Hermann, D.M. Free radical scavengers and spin traps—Therapeutic implications for ischemic stroke. Best Pract. Res. Clin. Anaesthesiol. 2010, 24, 511–520. [Google Scholar] [CrossRef]
- Kunz, A.; Anrather, J.; Zhou, P.; Orio, M.; Iadecola, C. Cyclooxygenase-2 does not contribute to postischemic production of reactive oxygen species. J. Cereb. Blood Flow Metab. 2007, 27, 545–551. [Google Scholar] [CrossRef]
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free Radic. Biol. Med. 2009, 47, 1239–1253. [Google Scholar] [CrossRef]
- Infanger, D.W.; Sharma, R.V.; Davisson, R.L. NADPH oxidases of the brain: Distribution, regulation, and function. Antioxid. Redox Signal. 2006, 8, 1583–1596. [Google Scholar] [CrossRef]
- Sorce, S.; Krause, K.H. Nox enzymes in the central nervous system: From signaling to disease. Antioxid. Redox Signal. 2009, 11, 2481–2504. [Google Scholar] [CrossRef]
- Lambeth, J.D.; Krause, K.H.; Clark, R.A. Nox enzymes as novel targets for drug development. Semin. Immunopathol. 2008, 30, 339–363. [Google Scholar] [CrossRef]
- Baldridge, C.W.; Gerard, R.W. The extra respiration of phagocytosis. Am. J. Physiol. 1933, 103, 235–236. [Google Scholar]
- Baehner, R.L.; Nathan, D.G. Leukocyte oxidase: Defective activity in chronic granulomatous disease. Science 1967, 155, 835–836. [Google Scholar]
- Berendes, H.; Bridges, R.A.; Good, R.A. A fatal granulomatosus of childhood: The clinical study of a new syndrome. Minn. Med. 1957, 40, 309–312. [Google Scholar]
- Landing, B.H.; Shirkey, H.S. A syndrome of recurrent infection and infiltration of viscera by pigmented lipid histiocytes. Pediatrics 1957, 20, 431–438. [Google Scholar]
- Babior, B.M.; Kipnes, R.S.; Curnutte, J.T. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J. Clin. Invest. 1973, 52, 741–744. [Google Scholar] [CrossRef]
- Curnutte, J.T.; Whitten, D.M.; Babior, B.M. Defective superoxide production by granulocytes from patients with chronic granulomatous disease. N. Engl. J. Med. 1974, 290, 593–597. [Google Scholar] [CrossRef]
- Hohn, D.C.; Lehrer, R.I. NADPH oxidase deficiency in X-linked chronic granulomatous disease. J. Clin. Invest. 1975, 55, 707–713. [Google Scholar] [CrossRef]
- Curnutte, J.T. Chronic granulomatous disease: The solving of a clinical riddle at the molecular level. Clin. Immunol. Immunopathol. 1993, 67, S2–S15. [Google Scholar]
- Babior, B.M.; Lambeth, J.D.; Nauseef, W. The neutrophil NAD(P)H oxidase. Arch. Biochem. Biophys. 2002, 397, 342–344. [Google Scholar] [CrossRef]
- Vignais, P.V. The superoxide-generating NAD(P)H oxidase: Structural aspects and activation mechanism. Cell. Mol. Life Sci. 2002, 59, 1428–1459. [Google Scholar] [CrossRef]
- Nauseef, W.M. Biological roles for the nox family NAD(P)H oxidases. J. Biol. Chem. 2008, 283, 16961–16965. [Google Scholar] [CrossRef]
- Rada, B.; Leto, T.L. Oxidative innate immune defenses by Nox/Duox family NAD(P)H oxidases. Contrib. Microbiol. 2008, 15, 164–187. [Google Scholar] [CrossRef]
- Gorlach, A.; Holtermann, G.; Jelkmann, W.; Hancock, J.T.; Jones, S.A.; Jones, O.T.; Acker, H. Photometric characteristics of haem proteins in erythropoietin-producing hepatoma cells (HepG2). Biochem. J. 1993, 290, 771–776. [Google Scholar]
- Meier, B.; Radeke, H.H.; Selle, S.; Younes, M.; Sies, H.; Resch, K.; Habermehl, G.G. Human fibroblasts release reactive oxygen species in response to interleukin-1 or tumour necrosis factor-alpha. Biochem. J. 1989, 263, 539–545. [Google Scholar]
- Suh, Y.A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar]
- Kikuchi, H.; Hikage, M.; Miyashita, H.; Fukumoto, M. NADPH oxidase subunit, gp91(phox) homologue, preferentially expressed in human colon epithelial cell. Gene 2000, 254, 237–243. [Google Scholar] [CrossRef]
- Banfi, B.; Malgrange, B.; Knisz, J.; Steger, K.; Dubois-Dauphin, M.; Krause, K.H. Nox3, a superoxide-generating NAD(P)H oxidase of the inner ear. J. Biol. Chem. 2004, 279, 46065–46072. [Google Scholar]
- Geiszt, M.; Kopp, J.B.; Varnai, P.; Leto, T.L. Identification of renox, an NAD(P)H oxidase in kidney. Proc. Natl. Acad. Sci. USA 2000, 97, 8010–8014. [Google Scholar] [CrossRef]
- Lassegue, B.; Griendling, K.K. NADPH oxidases: Functions and pathologies in the vasculature. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 653–661. [Google Scholar] [CrossRef]
- Banfi, B.; Molnar, G.; Maturana, A.; Steger, K.; Hegedus, B.; Demaurex, N.; Krause, K.H. A Ca2+-activated NAD(P)H oxidase in testis, spleen, and lymph nodes. J. Biol. Chem. 2001, 276, 37594–37601. [Google Scholar]
- De Deken, X.; Wang, D.; Many, M.C.; Costagliola, S.; Libert, F.; Vassart, G.; Dumont, J.E.; Miot, F. Cloning of two human thyroid cdnas encoding new members of the NAD(P)H oxidase family. J. Biol. Chem. 2000, 275, 23227–23233. [Google Scholar]
- Dupuy, C.; Ohayon, R.; Valent, A.; Noel-Hudson, M.S.; Deme, D.; Virion, A. Purification of a novel flavoprotein involved in the thyroid NAD(P)H oxidase. Cloning of the porcine and human cdnas. J. Biol. Chem. 1999, 274, 37265–37269. [Google Scholar]
- Edens, W.A.; Sharling, L.; Cheng, G.; Shapira, R.; Kinkade, J.M.; Lee, T.; Edens, H.A.; Tang, X.; Sullards, C.; Flaherty, D.B.; et al. Tyrosine cross-linking of extracellular matrix is catalyzed by Duox, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. J. Cell Biol. 2001, 154, 879–891. [Google Scholar] [CrossRef]
- Kawahara, T.; Ritsick, D.; Cheng, G.; Lambeth, J.D. Point mutations in the proline-rich region of p22phox are dominant inhibitors of Nox1- and Nox2-dependent reactive oxygen generation. J. Biol. Chem. 2005, 280, 31859–31869. [Google Scholar] [CrossRef]
- Selemidis, S.; Sobey, C.G.; Wingler, K.; Schmidt, H.H.; Drummond, G.R. NADPH oxidases in the vasculature: Molecular features, roles in disease and pharmacological inhibition. Pharmacol. Ther. 2008, 120, 254–291. [Google Scholar] [CrossRef]
- Cheng, G.; Ritsick, D.; Lambeth, J.D. Nox3 regulation by NOXO1, p47phox, and p67phox. J. Biol. Chem. 2004, 279, 34250–34255. [Google Scholar] [CrossRef]
- Lambeth, J.D.; Kawahara, T.; Diebold, B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic. Biol. Med. 2007, 43, 319–331. [Google Scholar] [CrossRef]
- Ueno, N.; Takeya, R.; Miyano, K.; Kikuchi, H.; Sumimoto, H. The NAD(P)H oxidase Nox3 constitutively produces superoxide in a p22phox-dependent manner: Its regulation by oxidase organizers and activators. J. Biol. Chem. 2005, 280, 23328–23339. [Google Scholar]
- Lyle, A.N.; Deshpande, N.N.; Taniyama, Y.; Seidel-Rogol, B.; Pounkova, L.; Du, P.; Papaharalambus, C.; Lassegue, B.; Griendling, K.K. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ. Res. 2009, 105, 249–259. [Google Scholar] [CrossRef]
- Martyn, K.D.; Frederick, L.M.; von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional analysis of Nox4 reveals unique characteristics compared to other NAD(P)H oxidases. Cell. Signal. 2006, 18, 69–82. [Google Scholar] [CrossRef]
- Sumimoto, H. Structure, regulation and evolution of Nox-family NAD(P)H oxidases that produce reactive oxygen species. FEBS J. 2008, 275, 3249–3277. [Google Scholar] [CrossRef]
- Cheng, G.; Lambeth, J.D. NOXO1, regulation of lipid binding, localization, and activation of Nox1 by the Phox homology (PX) domain. J. Biol. Chem. 2004, 279, 4737–4742. [Google Scholar] [CrossRef]
- Lam, G.Y.; Huang, J.; Brumell, J.H. The many roles of Nox2 NAD(P)H oxidase-derived ROS in immunity. Semin. Immunopathol. 2010, 32, 415–430. [Google Scholar] [CrossRef]
- Lambeth, J.D. Nox enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Touyz, R.M. Reactive oxygen species in vascular biology: Role in arterial hypertension. Expert Rev. Cardiovasc. Ther. 2003, 1, 91–106. [Google Scholar] [CrossRef]
- Dale, D.C.; Boxer, L.; Liles, W.C. The phagocytes: Neutrophils and monocytes. Blood 2008, 112, 935–945. [Google Scholar] [CrossRef]
- Reinehr, R.; Gorg, B.; Becker, S.; Qvartskhava, N.; Bidmon, H.J.; Selbach, O.; Haas, H.L.; Schliess, F.; Haussinger, D. Hypoosmotic swelling and ammonia increase oxidative stress by NAD(P)H oxidase in cultured astrocytes and vital brain slices. Glia 2007, 55, 758–771. [Google Scholar] [CrossRef]
- Mizuki, K.; Kadomatsu, K.; Hata, K.; Ito, T.; Fan, Q.W.; Kage, Y.; Fukumaki, Y.; Sakaki, Y.; Takeshige, K.; Sumimoto, H. Functional modules and expression of mouse p40(phox) and p67(phox), SH3-domain-containing proteins involved in the phagocyte NAD(P)H oxidase complex. Eur. J. Biochem. 1998, 251, 573–582. [Google Scholar]
- Dvorakova, M.; Hohler, B.; Richter, E.; Burritt, J.B.; Kummer, W. Rat sensory neurons contain cytochrome b558 large subunit immunoreactivity. Neuroreport 1999, 10, 2615–2617. [Google Scholar] [CrossRef]
- Tammariello, S.P.; Quinn, M.T.; Estus, S. NADPH oxidase contributes directly to oxidative stress and apoptosis in nerve growth factor-deprived sympathetic neurons. J. Neurosci. 2000, 20, RC53. [Google Scholar]
- Noh, K.M.; Koh, J.Y. Induction and activation by zinc of NAD(P)H oxidase in cultured cortical neurons and astrocytes. J. Neurosci. 2000, 20, RC111. [Google Scholar]
- Park, K.W.; Jin, B.K. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons: Role of neuronal NAD(P)H oxidase. J. Neurosci. Res. 2008, 86, 1053–1063. [Google Scholar] [CrossRef]
- Tejada-Simon, M.V.; Serrano, F.; Villasana, L.E.; Kanterewicz, B.I.; Wu, G.Y.; Quinn, M.T.; Klann, E. Synaptic localization of a functional NAD(P)H oxidase in the mouse hippocampus. Mol. Cell. Neurosci. 2005, 29, 97–106. [Google Scholar] [CrossRef]
- Kim, M.J.; Shin, K.S.; Chung, Y.B.; Jung, K.W.; Cha, C.I.; Shin, D.H. Immunohistochemical study of p47phox and gp91phox distributions in rat brain. Brain Res. 2005, 1040, 178–186. [Google Scholar] [CrossRef]
- Serrano, F.; Kolluri, N.S.; Wientjes, F.B.; Card, J.P.; Klann, E. NADPH oxidase immunoreactivity in the mouse brain. Brain Res. 2003, 988, 193–198. [Google Scholar] [CrossRef]
- Green, S.P.; Cairns, B.; Rae, J.; Errett-Baroncini, C.; Hongo, J.A.; Erickson, R.W.; Curnutte, J.T. Induction of gp91-phox, a component of the phagocyte NAD(P)H oxidase, in microglial cells during central nervous system inflammation. J. Cereb. Blood Flow Metab. 2001, 21, 374–384. [Google Scholar]
- Ha, J.S.; Lee, J.E.; Lee, J.R.; Lee, C.S.; Maeng, J.S.; Bae, Y.S.; Kwon, K.S.; Park, S.S. Nox4-dependent H2O2 production contributes to chronic glutamate toxicity in primary cortical neurons. Exp. Cell. Res. 2010, 316, 1651–1661. [Google Scholar] [CrossRef]
- Kahles, T.; Kohnen, A.; Heumueller, S.; Rappert, A.; Bechmann, I.; Liebner, S.; Wittko, I.M.; Neumann-Haefelin, T.; Steinmetz, H.; Schroeder, K.; et al. NADPH oxidase Nox1 contributes to ischemic injury in experimental stroke in mice. Neurobiol. Dis. 2010, 40, 185–192. [Google Scholar] [CrossRef]
- Ibi, M.; Katsuyama, M.; Fan, C.; Iwata, K.; Nishinaka, T.; Yokoyama, T.; Yabe-Nishimura, C. Nox1/NAD(P)H oxidase negatively regulates nerve growth factor-induced neurite outgrowth. Free Radic. Biol. Med. 2006, 40, 1785–1795. [Google Scholar] [CrossRef]
- Suzukawa, K.; Miura, K.; Mitsushita, J.; Resau, J.; Hirose, K.; Crystal, R.; Kamata, T. Nerve growth factor-induced neuronal differentiation requires generation of Rac1-regulated reactive oxygen species. J. Biol. Chem. 2000, 275, 13175–13178. [Google Scholar]
- Liu, Q.; Kang, J.H.; Zheng, R.L. NADPH oxidase produces reactive oxygen species and maintains survival of rat astrocytes. Cell Biochem. Funct. 2005, 23, 93–100. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Jacobson, J.; Wientjes, F.; Hothersall, J.; Canevari, L.; Duchen, M.R. Expression and modulation of an NAD(P)H oxidase in mammalian astrocytes. J. Neurosci. 2005, 25, 9176–9184. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Sankarapandi, S.; Zweier, J.L.; Mukherjee, G.; Quinn, M.T.; Huso, D.L. Measurement and characterization of superoxide generation in microglial cells: Evidence for an NAD(P)H oxidase-dependent pathway. Arch. Biochem. Biophys. 1998, 353, 312–321. [Google Scholar] [CrossRef]
- Choi, S.H.; Lee, D.Y.; Kim, S.U.; Jin, B.K. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: Role of microglial NAD(P)H oxidase. J. Neurosci. 2005, 25, 4082–4090. [Google Scholar] [CrossRef]
- Lavigne, M.C.; Malech, H.L.; Holland, S.M.; Leto, T.L. Genetic requirement of p47phox for superoxide production by murine microglia. FASEB J. 2001, 15, 285–287. [Google Scholar]
- Li, B.; Bedard, K.; Sorce, S.; Hinz, B.; Dubois-Dauphin, M.; Krause, K.H. Nox4 expression in human microglia leads to constitutive generation of reactive oxygen species and to constitutive IL-6 expression. J. Innate Immun. 2009, 1, 570–581. [Google Scholar] [CrossRef]
- Cheret, C.; Gervais, A.; Lelli, A.; Colin, C.; Amar, L.; Ravassard, P.; Mallet, J.; Cumano, A.; Krause, K.H.; Mallat, M. Neurotoxic activation of microglia is promoted by a Nox1-dependent NAD(P)H oxidase. J. Neurosci. 2008, 28, 12039–12051. [Google Scholar] [CrossRef]
- Harrigan, T.J.; Abdullaev, I.F.; Jourd’heuil, D.; Mongin, A.A. Activation of microglia with zymosan promotes excitatory amino acid release via volume-regulated anion channels: The role of NAD(P)H oxidases. J. Neurochem. 2008, 106, 2449–2462. [Google Scholar]
- Mander, P.K.; Jekabsone, A.; Brown, G.C. Microglia proliferation is regulated by hydrogen peroxide from NAD(P)H oxidase. J. Immunol. 2006, 176, 1046–1052. [Google Scholar]
- Miller, A.A.; Budzyn, K.; Sobey, C.G. Vascular dysfunction in cerebrovascular disease: Mechanisms and therapeutic intervention. Clin. Sci. (Lond.) 2010, 119, 1–17. [Google Scholar] [CrossRef]
- Chrissobolis, S.; Faraci, F.M. The role of oxidative stress and NAD(P)H oxidase in cerebrovascular disease. Trends Mol. Med. 2008, 14, 495–502. [Google Scholar] [CrossRef]
- Miller, A.A.; Drummond, G.R.; de Silva, T.M.; Mast, A.E.; Hickey, H.; Williams, J.P.; Broughton, B.R.; Sobey, C.G. NADPH oxidase activity is higher in cerebral versus systemic arteries of four animal species: Role of Nox2. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H220–H225. [Google Scholar]
- Miller, A.A.; Drummond, G.R.; Schmidt, H.H.; Sobey, C.G. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ. Res. 2005, 97, 1055–1062. [Google Scholar] [CrossRef]
- Ago, T.; Kitazono, T.; Kuroda, J.; Kumai, Y.; Kamouchi, M.; Ooboshi, H.; Wakisaka, M.; Kawahara, T.; Rokutan, K.; Ibayashi, S.; et al. NAD(P)H oxidases in rat basilar arterial endothelial cells. Stroke 2005, 36, 1040–1046. [Google Scholar] [CrossRef]
- Jackman, K.A.; Miller, A.A.; Drummond, G.R.; Sobey, C.G. Importance of NOX1 for angiotensin II-induced cerebrovascular superoxide production and cortical infarct volume following ischemic stroke. Brain Res. 2009, 1286, 215–220. [Google Scholar] [CrossRef]
- Cheng, G.; Cao, Z.; Xu, X.; van Meir, E.G.; Lambeth, J.D. Homologs of gp91phox: Cloning and tissue expression of Nox3, Nox4, and Nox. Gene 2001, 269, 131–140. [Google Scholar] [CrossRef]
- Paravicini, T.M.; Chrissobolis, S.; Drummond, G.R.; Sobey, C.G. Increased NAD(P)H-oxidase activity and Nox4 expression during chronic hypertension is associated with enhanced cerebral vasodilatation to NAD(P)H in vivo. Stroke 2004, 35, 584–589. [Google Scholar] [CrossRef]
- Park, L.; Anrather, J.; Zhou, P.; Frys, K.; Wang, G.; Iadecola, C. Exogenous NAD(P)H increases cerebral blood flow through NAD(P)H oxidase-dependent and -independent mechanisms. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1860–1865. [Google Scholar] [CrossRef]
- Kim, J.S.; Yeo, S.; Shin, D.G.; Bae, Y.S.; Lee, J.J.; Chin, B.R.; Lee, C.H.; Baek, S.H. Glycogen synthase kinase 3beta and beta-catenin pathway is involved in toll-like receptor 4-mediated NAD(P)H oxidase 1 expression in macrophages. FEBS J. 2010, 277, 2830–2837. [Google Scholar] [CrossRef]
- Sorescu, D.; Weiss, D.; Lassegue, B.; Clempus, R.E.; Szocs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar]
- Lee, C.F.; Qiao, M.; Schroder, K.; Zhao, Q.; Asmis, R. Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circ. Res. 2010, 106, 1489–1497. [Google Scholar] [CrossRef]
- Hong, H.; Zeng, J.S.; Kreulen, D.L.; Kaufman, D.I.; Chen, A.F. Atorvastatin protects against cerebral infarction via inhibition of NAD(P)H oxidase-derived superoxide in ischemic stroke. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2210–H2215. [Google Scholar] [CrossRef]
- Lu, Q.; Xia, N.; Xu, H.; Guo, L.; Wenzel, P.; Daiber, A.; Munzel, T.; Forstermann, U.; Li, H. Betulinic acid protects against cerebral ischemia-reperfusion injury in mice by reducing oxidative and nitrosative stress. Nitric Oxide 2011, 24, 132–138. [Google Scholar] [CrossRef]
- Murotomi, K.; Takagi, N.; Takeo, S.; Tanonaka, K. NADPH oxidase-mediated oxidative damage to proteins in the postsynaptic density after transient cerebral ischemia and reperfusion. Mol. Cell. Neurosci. 2011, 46, 681–688. [Google Scholar] [CrossRef]
- Jackman, K.A.; Miller, A.A.; de Silva, T.M.; Crack, P.J.; Drummond, G.R.; Sobey, C.G. Reduction of cerebral infarct volume by apocynin requires pretreatment and is absent in Nox2-deficient mice. Br. J. Pharmacol. 2009, 156, 680–688. [Google Scholar]
- Walder, C.E.; Green, S.P.; Darbonne, W.C.; Mathias, J.; Rae, J.; Dinauer, M.C.; Curnutte, J.T.; Thomas, G.R. Ischemic stroke injury is reduced in mice lacking a functional NAD(P)H oxidase. Stroke 1997, 28, 2252–2258. [Google Scholar] [CrossRef]
- Schroeter, M.; Jander, S.; Witte, O.W.; Stoll, G. Local immune responses in the rat cerebral cortex after middle cerebral artery occlusion. J. Neuroimmunol. 1994, 55, 195–203. [Google Scholar] [CrossRef]
- Gelderblom, M.; Leypoldt, F.; Lewerenz, J.; Birkenmayer, G.; Orozco, D.; Ludewig, P.; Thundyil, J.; Arumugam, T.V.; Gerloff, C.; Tolosa, E.; et al. The flavonoid fisetin attenuates postischemic immune cell infiltration, activation and infarct size after transient cerebral middle artery occlusion in mice. J. Cereb. Blood Flow Metab. 2012, 32, 835–843. [Google Scholar] [CrossRef]
- Brait, V.H.; Jackman, K.A.; Walduck, A.K.; Selemidis, S.; Diep, H.; Mast, A.E.; Guida, E.; Broughton, B.R.; Drummond, G.R.; Sobey, C.G. Mechanisms contributing to cerebral infarct size after stroke: Gender, reperfusion, T lymphocytes, and Nox2-derived superoxide. J. Cereb. Blood Flow Metab. 2010, 30, 1306–1317. [Google Scholar] [CrossRef]
- Weston, R.M.; Jarrott, B.; Ishizuka, Y.; Callaway, J.K. AM-36 modulates the neutrophil inflammatory response and reduces breakdown of the blood brain barrier after endothelin-1 induced focal brain ischaemia. Br. J. Pharmacol. 2006, 149, 712–723. [Google Scholar] [CrossRef]
- Tang, X.N.; Zheng, Z.; Giffard, R.G.; Yenari, M.A. Significance of marrow-derived nicotinamide adenine dinucleotide phosphate oxidase in experimental ischemic stroke. Ann. Neurol. 2011, 70, 606–615. [Google Scholar] [CrossRef]
- Jackson, S.H.; Devadas, S.; Kwon, J.; Pinto, L.A.; Williams, M.S. T cells express a phagocyte-type NAD(P)H oxidase that is activated after T cell receptor stimulation. Nat. Immunol. 2004, 5, 818–827. [Google Scholar] [CrossRef]
- Coimbra, C.; Drake, M.; Boris-Moller, F.; Wieloch, T. Long-lasting neuroprotective effect of postischemic hypothermia and treatment with an anti-inflammatory/antipyretic drug. Evidence for chronic encephalopathic processes following ischemia. Stroke 1996, 27, 1578–1585. [Google Scholar] [CrossRef]
- Fox, C.; Dingman, A.; Derugin, N.; Wendland, M.F.; Manabat, C.; Ji, S.; Ferriero, D.M.; Vexler, Z.S. Minocycline confers early but transient protection in the immature brain following focal cerebral ischemia-reperfusion. J. Cereb. Blood Flow Metab. 2005, 25, 1138–1149. [Google Scholar]
- Valtysson, J.; Hillered, L.; Andine, P.; Hagberg, H.; Persson, L. Neuropathological endpoints in experimental stroke pharmacotherapy: The importance of both early and late evaluation. Acta Neurochir. (Wien) 1994, 129, 58–63. [Google Scholar] [CrossRef]
- Kahles, T.; Luedike, P.; Endres, M.; Galla, H.J.; Steinmetz, H.; Busse, R.; Neumann-Haefelin, T.; Brandes, R.P. NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke 2007, 38, 3000–3006. [Google Scholar]
- Chen, H.; Kim, G.S.; Okami, N.; Narasimhan, P.; Chan, P.H. NADPH oxidase is involved in post-ischemic brain inflammation. Neurobiol. Dis. 2011, 42, 341–348. [Google Scholar] [CrossRef]
- Chen, H.; Song, Y.S.; Chan, P.H. Inhibition of NAD(P)H oxidase is neuroprotective after ischemia-reperfusion. J. Cereb. Blood Flow Metab. 2009, 29, 1262–1272. [Google Scholar] [CrossRef]
- Kim, H.A.; Brait, V.H.; Lee, S.; de Silva, T.M.; Diep, H.; Eisenhardt, A.; Drummond, G.R.; Sobey, C.G. Brain infarct volume after permanent focal ischemia is not dependent on Nox2 expression. Brain Res. 2012, 1483, 105–111. [Google Scholar] [CrossRef]
- Hur, J.; Lee, P.; Kim, M.J.; Kim, Y.; Cho, Y.W. Ischemia-activated microglia induces neuronal injury via activation of gp91phox NAD(P)H oxidase. Biochem. Biophys. Res. Commun. 2010, 391, 1526–1530. [Google Scholar] [CrossRef]
- Spranger, M.; Kiprianova, I.; Krempien, S.; Schwab, S. Reoxygenation increases the release of reactive oxygen intermediates in murine microglia. J. Cereb. Blood Flow Metab. 1998, 18, 670–674. [Google Scholar]
- Mander, P.; Brown, G.C. Activation of microglial NAD(P)H oxidase is synergistic with glial iNOS expression in inducing neuronal death: A dual-key mechanism of inflammatory neurodegeneration. J. Neuroinflammation 2005, 2, 20. [Google Scholar] [CrossRef]
- Knorpp, T.; Robinson, S.R.; Crack, P.J.; Dringen, R. Glutathione peroxidase-1 contributes to the protection of glutamine synthetase in astrocytes during oxidative stress. J. Neural. Transm. 2006, 113, 1145–1155. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Duchen, M.R. The role of an astrocytic NAD(P)H oxidase in the neurotoxicity of amyloid beta peptides. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2309–2314. [Google Scholar] [CrossRef]
- Desagher, S.; Glowinski, J.; Premont, J. Astrocytes protect neurons from hydrogen peroxide toxicity. J. Neurosci. 1996, 16, 2553–2562. [Google Scholar]
- Rosenberg, P.A.; Aizenman, E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci. Lett. 1989, 103, 162–168. [Google Scholar] [CrossRef]
- Tang, L.L.; Ye, K.; Yang, X.F.; Zheng, J.S. Apocynin attenuates cerebral infarction after transient focal ischaemia in rats. J. Int. Med. Res. 2007, 35, 517–522. [Google Scholar]
- Tang, X.N.; Cairns, B.; Cairns, N.; Yenari, M.A. Apocynin improves outcome in experimental stroke with a narrow dose range. Neuroscience 2008, 154, 556–562. [Google Scholar] [CrossRef]
- Genovese, T.; Mazzon, E.; Paterniti, I.; Esposito, E.; Bramanti, P.; Cuzzocrea, S. Modulation of NAD(P)H oxidase activation in cerebral ischemia/reperfusion injury in rats. Brain Res. 2011, 1372, 92–102. [Google Scholar] [CrossRef]
- Kelly, K.A.; Li, X.; Tan, Z.; vanGilder, R.L.; Rosen, C.L.; Huber, J.D. NOX2 inhibition with apocynin worsens stroke outcome in aged rats. Brain Res. 2009, 1292, 165–172. [Google Scholar]
- Doverhag, C.; Keller, M.; Karlsson, A.; Hedtjarn, M.; Nilsson, U.; Kapeller, E.; Sarkozy, G.; Klimaschewski, L.; Humpel, C.; Hagberg, H.; et al. Pharmacological and genetic inhibition of NAD(P)H oxidase does not reduce brain damage in different models of perinatal brain injury in newborn mice. Neurobiol. Dis. 2008, 31, 133–144. [Google Scholar] [CrossRef]
- Arimura, K.; Ago, T.; Kuroda, J.; Ishitsuka, K.; Nishimura, A.; Sugimori, H.; Kamouchi, M.; Sasaki, T.; Kitazono, T. Role of NAD(P)H oxidase 4 in brain endothelial cells after ischemic stroke. Stroke 2012, 43, A2514. [Google Scholar]
- De Silva, T.M.; Brait, V.H.; Drummond, G.R.; Sobey, C.G.; Miller, A.A. Nox2 oxidase activity accounts for the oxidative stress and vasomotor dysfunction in mouse cerebral arteries following ischemic stroke. PLoS One 2011, 6, e28393. [Google Scholar]
- Acker, T.; Acker, H. Cellular oxygen sensing need in cns function: Physiological and pathological implications. J. Exp. Biol. 2004, 207, 3171–3188. [Google Scholar] [CrossRef]
- Goyal, P.; Weissmann, N.; Grimminger, F.; Hegel, C.; Bader, L.; Rose, F.; Fink, L.; Ghofrani, H.A.; Schermuly, R.T.; Schmidt, H.H.; et al. Upregulation of NAD(P)H oxidase 1 in hypoxia activates hypoxia-inducible factor 1 via increase in reactive oxygen species. Free Radic. Biol. Med. 2004, 36, 1279–1288. [Google Scholar] [CrossRef]
- Diebold, I.; Petry, A.; Hess, J.; Gorlach, A. The NAD(P)H oxidase subunit Nox4 is a new target gene of the hypoxia-inducible factor-1. Mol. Biol. Cell 2010, 21, 2087–2096. [Google Scholar] [CrossRef]
- Diebold, I.; Petry, A.; Sabrane, K.; Djordjevic, T.; Hess, J.; Gorlach, A. The HIF1 target gene NOX2 promotes angiogenesis through urotensin-II. J. Cell Sci. 2012, 125, 956–964. [Google Scholar] [CrossRef]
- Yuan, G.; Khan, S.A.; Luo, W.; Nanduri, J.; Semenza, G.L.; Prabhakar, N.R. Hypoxia-inducible factor 1 mediates increased expression of NAD(P)H oxidase-2 in response to intermittent hypoxia. J. Cell. Physiol. 2011, 226, 2925–2933. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Chopp, M. Neurorestorative therapies for stroke: Underlying mechanisms and translation to the clinic. Lancet Neurol. 2009, 8, 491–500. [Google Scholar] [CrossRef]
- Mancuso, M.R.; Kuhnert, F.; Kuo, C.J. Developmental angiogenesis of the central nervous system. Lymphat. Res. Biol. 2008, 6, 173–180. [Google Scholar]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH oxidase-mediated redox signaling: Roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef]
- Ushio-Fukai, M. Redox signaling in angiogenesis: Role of NAD(P)H oxidase. Cardiovasc. Res. 2006, 71, 226–235. [Google Scholar] [CrossRef]
- Ushio-Fukai, M. VEGF signaling through NAD(P)H oxidase-derived ROS. Antioxid. Redox Signal. 2007, 9, 731–739. [Google Scholar] [CrossRef]
- Krupinski, J.; Kaluza, J.; Kumar, P.; Kumar, S.; Wang, J.M. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke 1994, 25, 1794–1798. [Google Scholar] [CrossRef]
- Wei, L.; Erinjeri, J.P.; Rovainen, C.M.; Woolsey, T.A. Collateral growth and angiogenesis around cortical stroke. Stroke 2001, 32, 2179–2184. [Google Scholar] [CrossRef]
- Hayashi, T.; Noshita, N.; Sugawara, T.; Chan, P.H. Temporal profile of angiogenesis and expression of related genes in the brain after ischemia. J. Cereb. Blood Flow Metab. 2003, 23, 166–180. [Google Scholar]
- Tojo, T.; Ushio-Fukai, M.; Yamaoka-Tojo, M.; Ikeda, S.; Patrushev, N.; Alexander, R.W. Role of gp91phox (Nox2)-containing NAD(P)H oxidase in angiogenesis in response to hindlimb ischemia. Circulation 2005, 111, 2347–2355. [Google Scholar] [CrossRef]
- Urao, N.; Inomata, H.; Razvi, M.; Kim, H.W.; Wary, K.; McKinney, R.; Fukai, T.; Ushio-Fukai, M. Role of Nox2-based NAD(P)H oxidase in bone marrow and progenitor cell function involved in neovascularization induced by hindlimb ischemia. Circ. Res. 2008, 103, 212–220. [Google Scholar] [CrossRef]
- Datla, S.R.; Peshavariya, H.; Dusting, G.J.; Mahadev, K.; Goldstein, B.J.; Jiang, F. Important role of Nox4 type NAD(P)H oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2319–2324. [Google Scholar] [CrossRef]
- Chan, E.C.; Liu, G.S.; Roulston, C.L.; Lim, S.Y.; Dusting, G.J. NADPH Oxidase in Tissue Repair and Regeneration. In Systems of Biology and Oxidative Stress; Springer: New York, NY, USA, 2012. [Google Scholar]
- Taylor, C.J.; Weston, R.M.; Dusting, G.J.; Roulston, C.L. NADPH oxidase and angiogenesis following endothelin-1 induced stroke in rats: Role for Nox2 in brain repair. Brain Sci. 2013, 3, 294–317. [Google Scholar] [CrossRef]
- Zhang, R.L.; Zhang, Z.G.; Chopp, M. Ischemic stroke and neurogenesis in the subventricular zone. Neuropharmacology 2008, 55, 345–352. [Google Scholar] [CrossRef]
- Manoonkitiwongsa, P.S.; Jackson-Friedman, C.; McMillan, P.J.; Schultz, R.L.; Lyden, P.D. Angiogenesis after stroke is correlated with increased numbers of macrophages: The clean-up hypothesis. J. Cereb. Blood Flow Metab. 2001, 21, 1223–1231. [Google Scholar]
- Yu, S.W.; Friedman, B.; Cheng, Q.; Lyden, P.D. Stroke-evoked angiogenesis results in a transient population of microvessels. J. Cereb. Blood Flow Metab. 2007, 27, 755–763. [Google Scholar]
- Tsatmali, M.; Walcott, E.C.; Makarenkova, H.; Crossin, K.L. Reactive oxygen species modulate the differentiation of neurons in clonal cortical cultures. Mol. Cell. Neurosci. 2006, 33, 345–357. [Google Scholar] [CrossRef]
- Knapp, L.T.; Klann, E. Role of reactive oxygen species in hippocampal long-term potentiation: Contributory or inhibitory? J. Neurosci. Res. 2002, 70, 1–7. [Google Scholar] [CrossRef]
- Zhao, C.; Deng, W.; Gage, F.H. Mechanisms and functional implications of adult neurogenesis. Cell 2008, 132, 645–660. [Google Scholar] [CrossRef]
- Pao, M.; Wiggs, E.A.; Anastacio, M.M.; Hyun, J.; DeCarlo, E.S.; Miller, J.T.; Anderson, V.L.; Malech, H.L.; Gallin, J.I.; Holland, S.M. Cognitive function in patients with chronic granulomatous disease: A preliminary report. Psychosomatics 2004, 45, 230–234. [Google Scholar] [CrossRef]
- Kishida, K.T.; Hoeffer, C.A.; Hu, D.; Pao, M.; Holland, S.M.; Klann, E. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol. Cell. Biol 2006, 26, 5908–5920. [Google Scholar] [CrossRef]
- Dickinson, B.C.; Peltier, J.; Stone, D.; Schaffer, D.V.; Chang, C.J. Nox2 redox signaling maintains essential cell populations in the brain. Nat. Chem. Biol. 2011, 7, 106–112. [Google Scholar] [CrossRef]
- Kim, J.A.; Neupane, G.P.; Lee, E.S.; Jeong, B.S.; Park, B.C.; Thapa, P. NADPH oxidase inhibitors: A patent review. Expert Opin. Ther. Pat. 2011, 21, 1147–1158. [Google Scholar] [CrossRef]
- Schroder, K.; Wandzioch, K.; Helmcke, I.; Brandes, R.P. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 239–245. [Google Scholar] [CrossRef]
- Peshavariya, H.; Jiang, F.; Taylor, C.J.; Selemidis, S.; Chang, C.W.; Dusting, G.J. Translation-linked mRNA destabilization accompanying serum-induced Nox4 expression in human endothelial cells. Antioxid. Redox Signal. 2009, 11, 2399–2408. [Google Scholar] [CrossRef]
- Van Den Worm, E.; Beukelman, C.J.; van den Berg, A.J.; Kroes, B.H.; Labadie, R.P.; van Dijk, H. Effects of methoxylation of apocynin and analogs on the inhibition of reactive oxygen species production by stimulated human neutrophils. Eur. J. Pharmacol. 2001, 433, 225–230. [Google Scholar] [CrossRef]
- Stolk, J.; Hiltermann, T.J.; Dijkman, J.H.; Verhoeven, A.J. Characteristics of the inhibition of NAD(P)H oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am. J. Respir. Cell Mol. Biol. 1994, 11, 95–102. [Google Scholar]
- Heumuller, S.; Wind, S.; Barbosa-Sicard, E.; Schmidt, H.H.; Busse, R.; Schroder, K.; Brandes, R.P. Apocynin is not an inhibitor of vascular NAD(P)H oxidases but an antioxidant. Hypertension 2008, 51, 211–217. [Google Scholar] [CrossRef]
- Touyz, R.M. Apocynin, NAD(P)H oxidase, and vascular cells: A complex matter. Hypertension 2008, 51, 172–174. [Google Scholar] [CrossRef]
- Weston, R.M.; Lin, B.; Dusting, G.J.; Roulston, C.L. Targeting oxidative stress injury after ischemic stroke in conscious rats: Limited benefits with apocynin highlights the need to incorporate long term recovery. Stroke Res. Treat. 2013, in press. [Google Scholar]
- Weston, R.M.; Jones, N.M.; Jarrott, B.; Callaway, J.K. Inflammatory cell infiltration after endothelin-1-induced cerebral ischemia: Histochemical and myeloperoxidase correlation with temporal changes in brain injury. J. Cereb. Blood Flow Metab. 2007, 27, 100–114. [Google Scholar] [CrossRef]
- Simons, J.M.; Hart, B.A.; Ip Vai Ching, T.R.; van Dijk, H.; Labadie, R.P. Metabolic activation of natural phenols into selective oxidative burst agonists by activated human neutrophils. Free Radic. Biol. Med. 1990, 8, 251–258. [Google Scholar]
- Aldieri, E.; Riganti, C.; Polimeni, M.; Gazzano, E.; Lussiana, C.; Campia, I.; Ghigo, D. Classical inhibitors of NOX NAD(P)H oxidases are not specific. Curr. Drug Metab. 2008, 9, 686–696. [Google Scholar] [CrossRef]
- Nagel, S.; Genius, J.; Heiland, S.; Horstmann, S.; Gardner, H.; Wagner, S. Diphenyleneiodonium and dimethylsulfoxide for treatment of reperfusion injury in cerebral ischemia of the rat. Brain Res. 2007, 1132, 210–217. [Google Scholar] [CrossRef]
- Rey, F.E.; Cifuentes, M.E.; Kiarash, A.; Quinn, M.T.; Pagano, P.J. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2− and systolic blood pressure in mice. Circ. Res. 2001, 89, 408–414. [Google Scholar]
- Park, L.; Anrather, J.; Girouard, H.; Zhou, P.; Iadecola, C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J. Cereb. Blood Flow Metab. 2007, 27, 1908–1918. [Google Scholar] [CrossRef]
- Cayatte, A.J.; Rupin, A.; Oliver-Krasinski, J.; Maitland, K.; Sansilvestri-Morel, P.; Boussard, M.F.; Wierzbicki, M.; Verbeuren, T.J.; Cohen, R.A. S17834, a new inhibitor of cell adhesion and atherosclerosis that targets NAD(P)H oxidase. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1577–1584. [Google Scholar] [CrossRef]
- Xu, S.; Jiang, B.; Hou, X.; Shi, C.; Bachschmid, M.M.; Zang, M.; Verbeuren, T.J.; Cohen, R.A. High-fat diet increases and the polyphenol, s17834, decreases acetylation of the sirtuin-1-dependent lysine-382 on p53 and apoptotic signaling in atherosclerotic lesion-prone aortic endothelium of normal mice. J. Cardiovasc. Pharmacol. 2011, 58, 263–271. [Google Scholar]
- Wang, Q.; Tompkins, K.D.; Simonyi, A.; Korthuis, R.J.; Sun, A.Y.; Sun, G.Y. Apocynin protects against global cerebral ischemia-reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain Res. 2006, 1090, 182–189. [Google Scholar] [CrossRef]
- Hultqvist, M.; Olsson, L.M.; Gelderman, K.A.; Holmdahl, R. The protective role of ROS in autoimmune disease. Trends Immunol. 2009, 30, 201–208. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
McCann, S.K.; Roulston, C.L. NADPH Oxidase as a Therapeutic Target for Neuroprotection against Ischaemic Stroke: Future Perspectives. Brain Sci. 2013, 3, 561-598. https://doi.org/10.3390/brainsci3020561
McCann SK, Roulston CL. NADPH Oxidase as a Therapeutic Target for Neuroprotection against Ischaemic Stroke: Future Perspectives. Brain Sciences. 2013; 3(2):561-598. https://doi.org/10.3390/brainsci3020561
Chicago/Turabian StyleMcCann, Sarah K., and Carli L. Roulston. 2013. "NADPH Oxidase as a Therapeutic Target for Neuroprotection against Ischaemic Stroke: Future Perspectives" Brain Sciences 3, no. 2: 561-598. https://doi.org/10.3390/brainsci3020561
APA StyleMcCann, S. K., & Roulston, C. L. (2013). NADPH Oxidase as a Therapeutic Target for Neuroprotection against Ischaemic Stroke: Future Perspectives. Brain Sciences, 3(2), 561-598. https://doi.org/10.3390/brainsci3020561