Physical Activity and Brain Function in Older Adults at Increased Risk for Alzheimer’s Disease

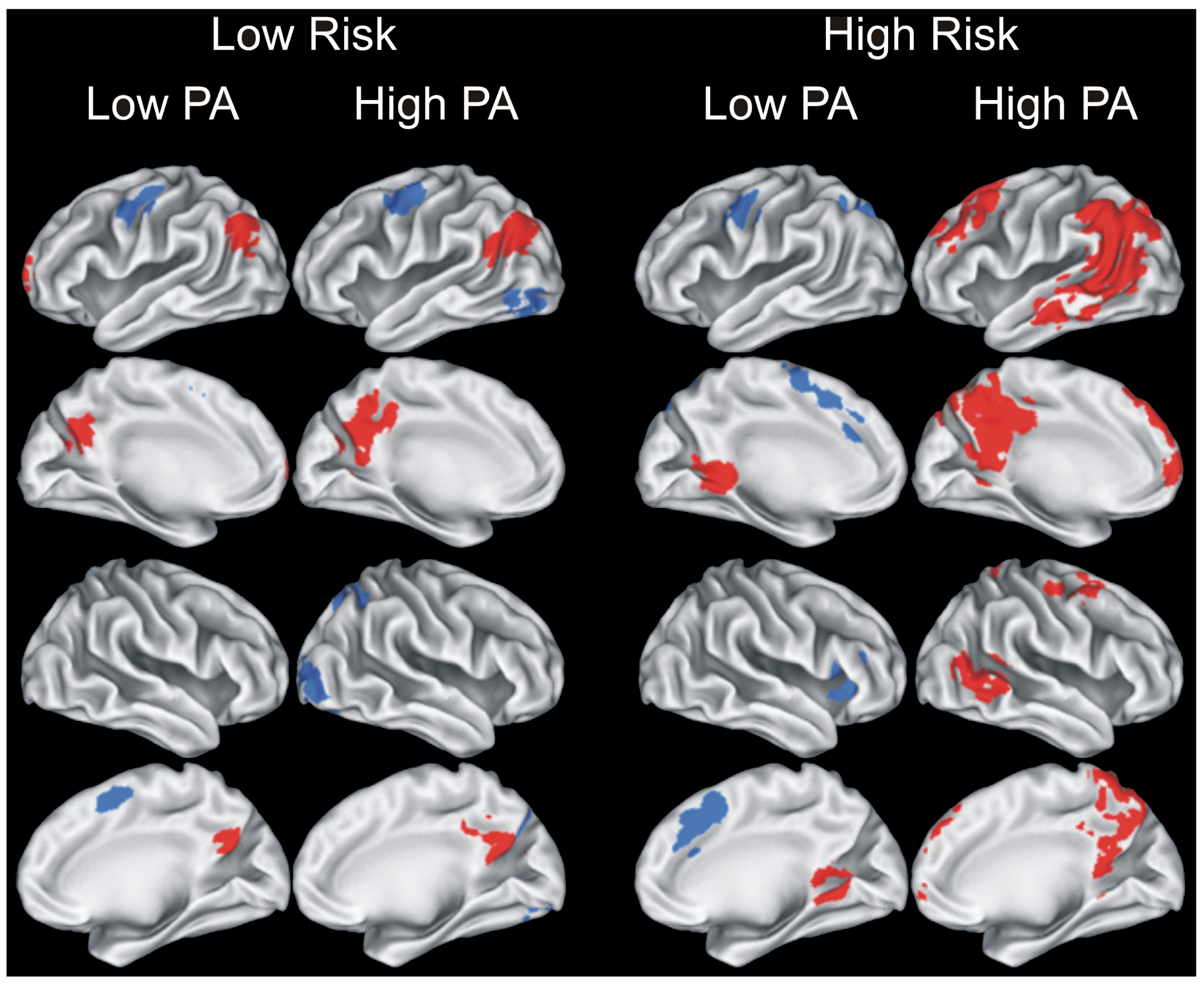{kind=link}
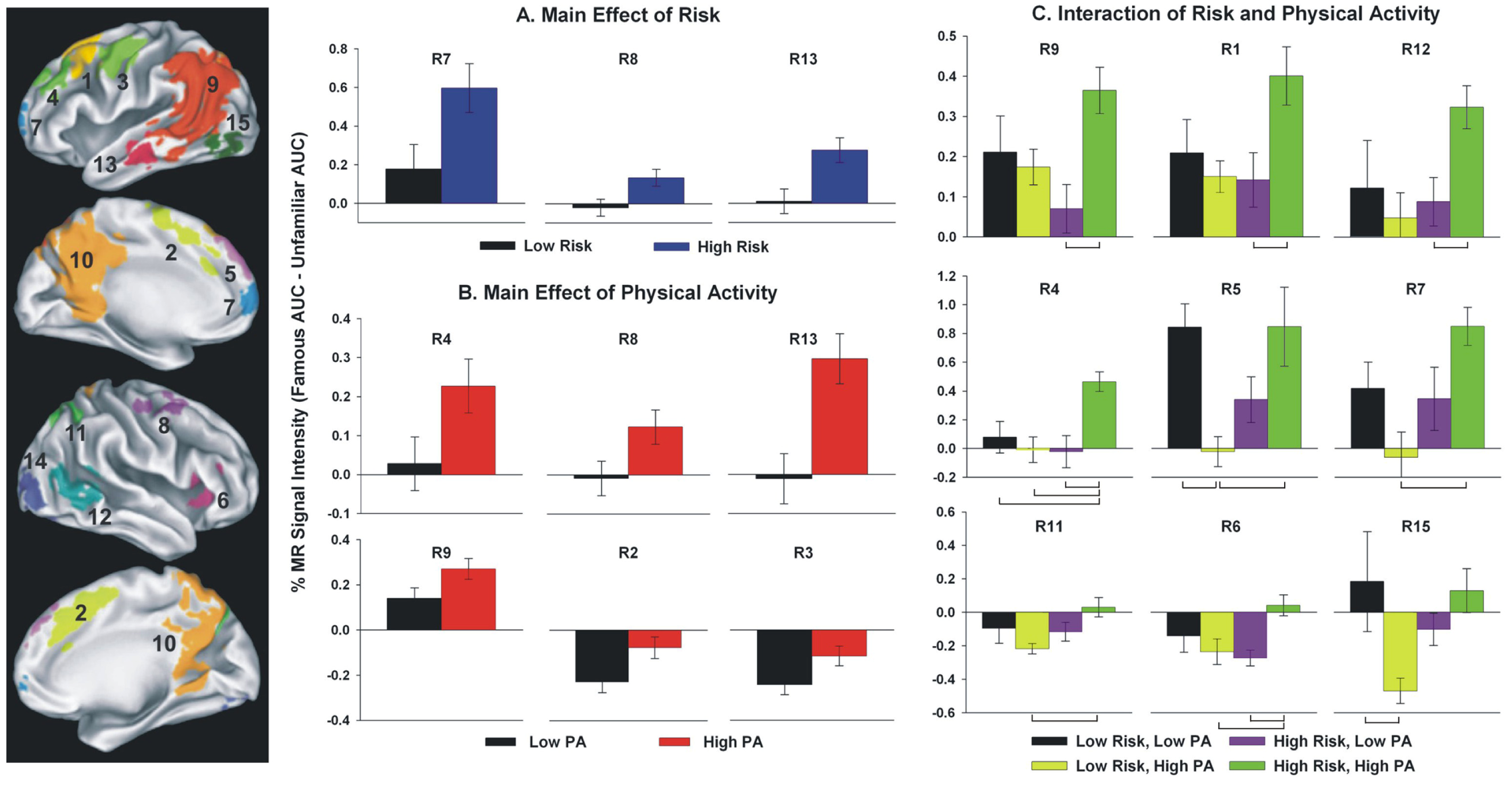{kind=link}
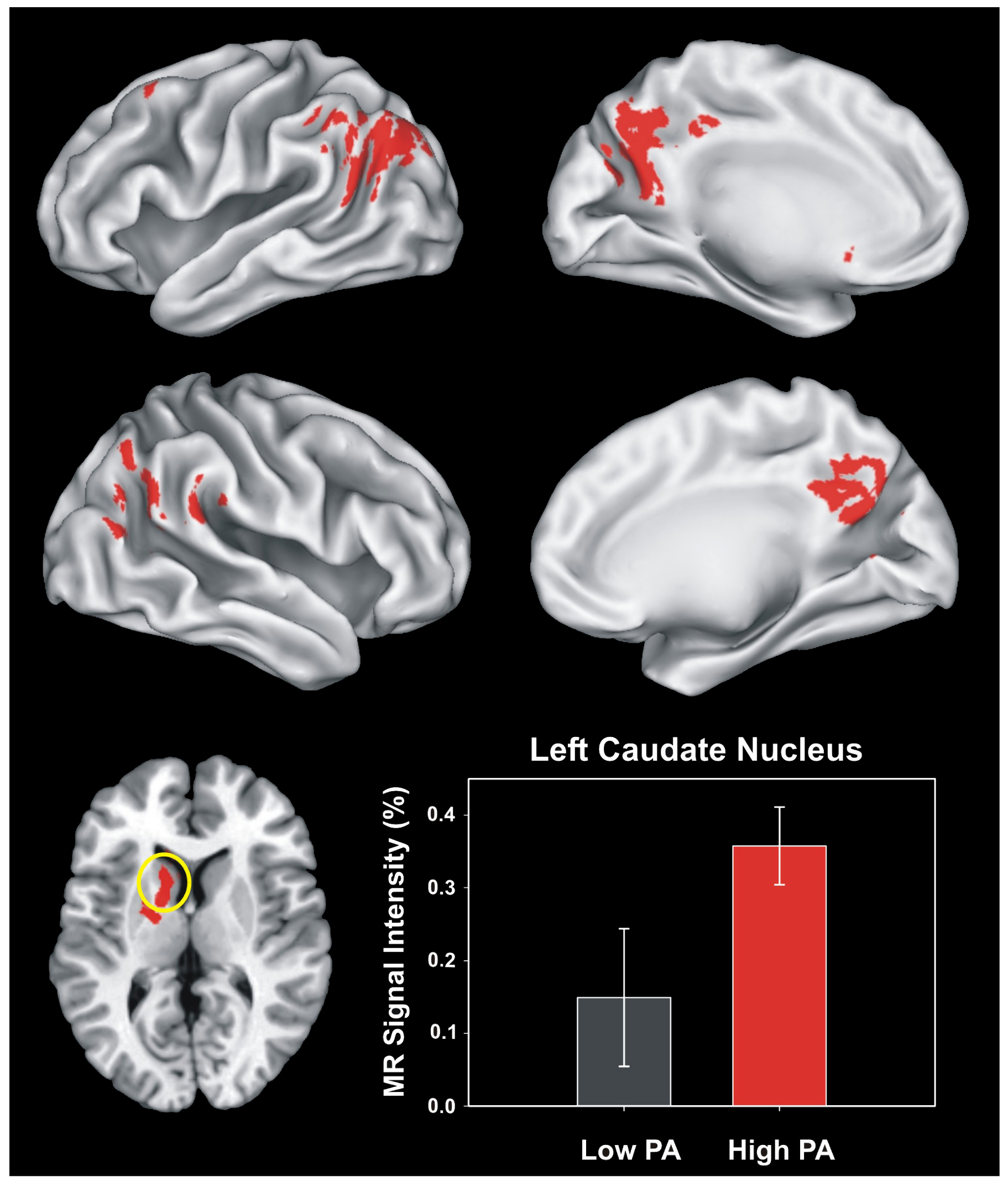{kind=link}
Abstract
:1. Introduction
2. Risk for AD
3. Physical Activity, Cognitive Function, and Increased Risk for AD
4. Exercise Training in Mild Cognitive Impairment
5. Exercise Training with Increased Metabolic Risk for AD
6. Physical Activity and Brain Structure
7. Physical Activity and Brain Function

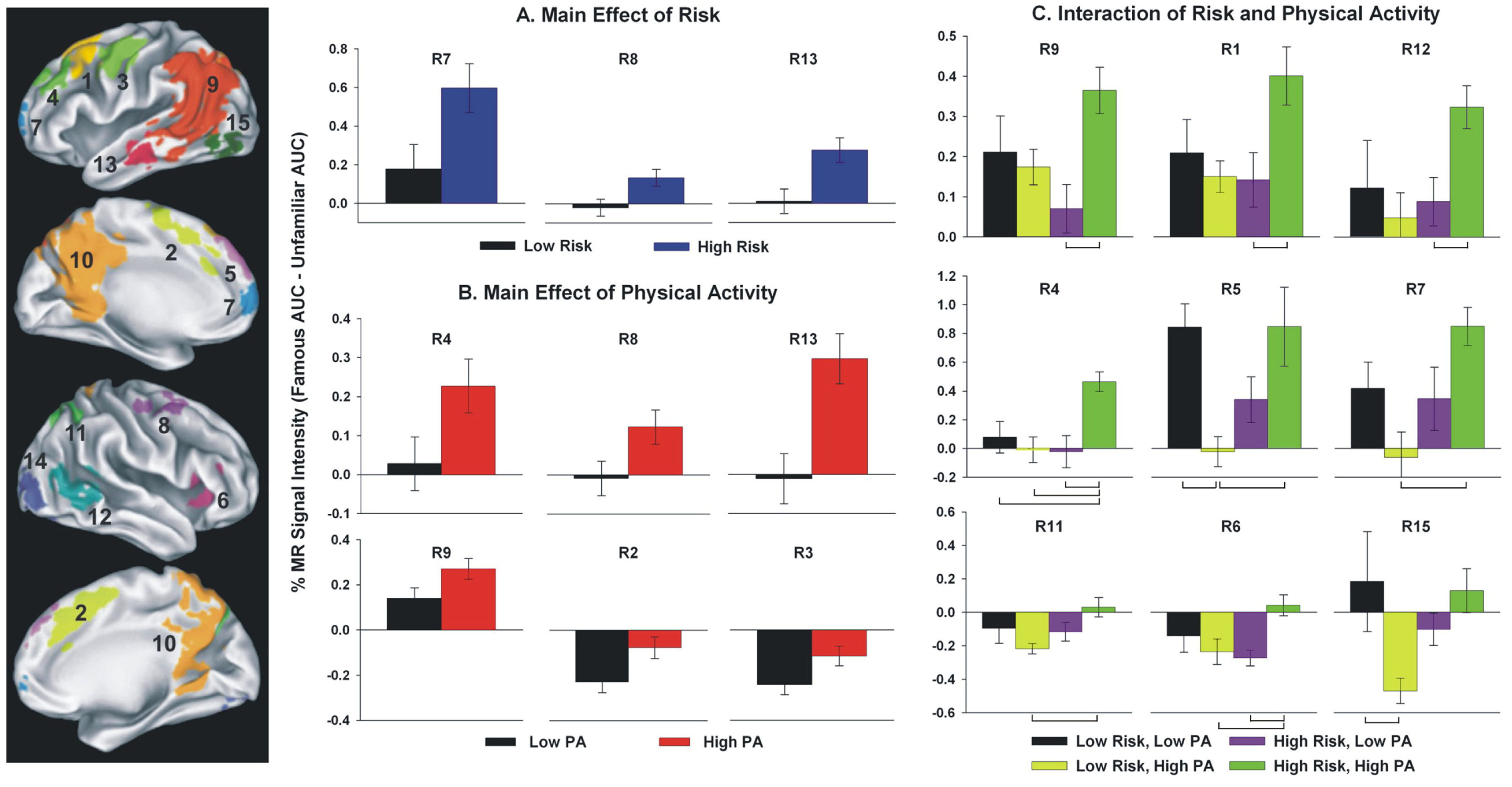
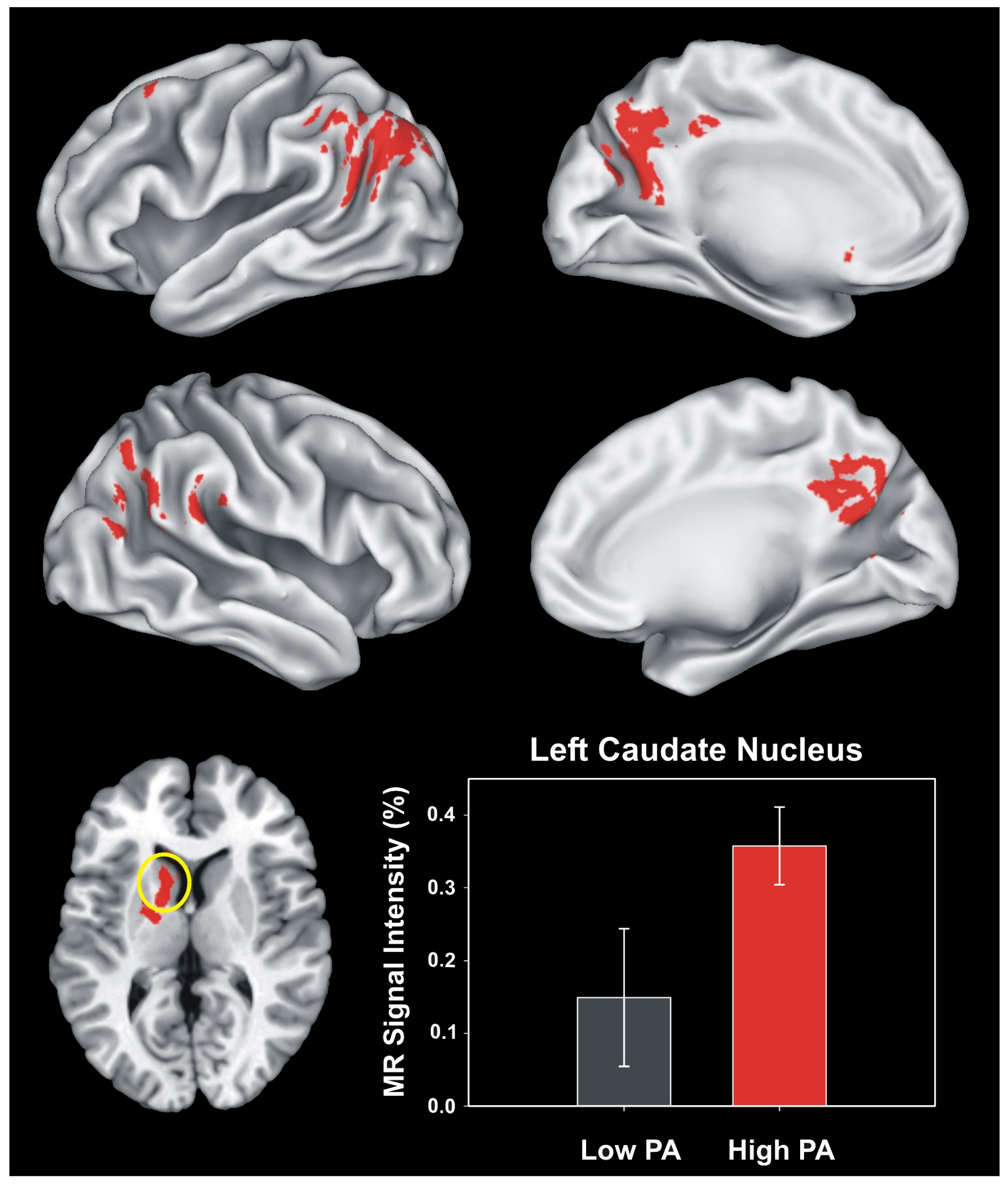
8. Potential Mechanisms for Physical Activity Benefits to Those at Greater Risk for Alzheimer’s Disease
9. Recommendations for Future Research
10. Summary and Conclusions
Acknowledgments
Conflict of Interest
References
- Sands, L.P.; Yaffe, K.; Lui, L.Y.; Stewart, A.; Eng, C.; Covinsky, K. The effects of acute illness on ADL decline over 1 year in frail older adults with and without cognitive impairment. J. Gerontol. 2002, 57, M449–M454. [Google Scholar] [CrossRef]
- Yaffe, K.; Fox, P.; Newcomer, R.; Sands, L.; Lindquist, K.; Dane, K.; Covinsky, K.E. Patient and caregiver characteristics and nursing home placement in patients with dementia. JAMA 2002, 287, 2090–2097. [Google Scholar]
- Yaffe, K.; Petersen, R.C.; Lindquist, K.; Kramer, J.; Miller, B. Subtype of mild cognitive impairment and progression to dementia and death. Dement. Geriatr. Cogn. Dis. 2006, 22, 312–319. [Google Scholar]
- Alzheimer’s Association. 2012 Alzheimer’s disease facts and figures. Alzheimers Dement. 2012, 8, 131–168. [Google Scholar]
- Daviglus, M.L.; Bell, C.C.; Berrettini, W.; Bowen, P.E.; Connolly, E.S.; Cox, N.J.; Dunbar-Jacob, J.M.; Granieri, E.C.; Hunt, G.; McGarry, K.; et al. NIH State-of-the-Science Conference Statement: Preventing Alzheimer’s Disease and Cognitive Decline. NIH Consens. State Sci. Statements 2010, 27, 1–30. [Google Scholar]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar]
- Woodard, J.; Seidenberg, M.; Nielson, K.A.; Smith, J.C.; Antuono, P.; Durgerian, S.; Guidotti, L.; Zhang, Q.; Butts, A.; Hantke, N.; et al. Prediction of cognitive decline in healthy older adults using fMRI. J. Alzheimers Dis. 2010, 21, 871–885. [Google Scholar]
- Clark, C.M.; Davatzikos, C.; Borthakur, A.; Newberg, A.; Leight, S.; Lee, V.M.; Trojanowski, J.Q. Biomarkers for early detection of Alzheimer pathology. Neurosignals 2008, 16, 11–18. [Google Scholar] [CrossRef]
- Reiman, E.M.; Langbaum, J.B.; Tariot, P.N. Alzheimer’s prevention initiative: A proposal to evaluate presymptomatic treatments as quickly as possible. Biomark. Med. 2010, 4, 3–14. [Google Scholar] [CrossRef]
- Woodard, J.L.; Sugarman, M.A.; Nielson, K.A.; Smith, J.C.; Seidenberg, M.; Durgerian, S.; Butts, A.; Hantke, N.; Lancaster, M.; Matthews, M.A.; Rao, S.M. Lifestyle and genetic contributions to cognitive decline and hippocampal structure and function in healthy aging. Curr. Alzheimer Res. 2012, 9, 436–446. [Google Scholar] [CrossRef]
- Samitz, G.; Egger, M.; Zwahlen, M. Domains of physical activity and all-cause mortality: Systematic review and dose-response meta-analysis of cohort studies. Int. J. Epidemiol. 2011, 40, 1382–1400. [Google Scholar] [CrossRef]
- Blair, S.N.; Kohl, H.W., III; Paffenbarger, R.S., Jr.; Clark, D.G.; Cooper, K.H.; Gibbons, L.W. Physical fitness and all-cause mortality. A prospective study of healthy men and women. JAMA 1989, 262, 2395–2401. [Google Scholar]
- Kujala, U.M.; Kaprio, J.; Sarna, S.; Koskenvuo, M. Relationship of leisure-time physical activity and mortality: The Finnish twin cohort. JAMA 1998, 279, 440–444. [Google Scholar]
- Liu, R.; Sui, X.; Laditka, J.N.; Church, T.S.; Colabianchi, N.; Hussey, J.; Blair, S.N. Cardiorespiratory fitness as a predictor of dementia mortality in men and women. Med. Sci. Sports Exerc. 2012, 44, 253–259. [Google Scholar]
- Scarmeas, N.; Luchsinger, J.A.; Brickman, A.M.; Cosentino, S.; Schupf, N.; Xin-Tang, M.; Gu, Y.; Stern, Y. Physical activity and Alzheimer disease course. Am. J. Geriatr. Psychiatry 2011, 19, 471–481. [Google Scholar] [CrossRef]
- Pereira, A.C.; Huddleston, D.E.; Brickman, A.M.; Sosunov, A.A.; Hen, R.; McKhann, G.M.; Sloan, R.; Gage, F.H.; Brown, T.R.; Small, S.A. An in vivo correlate of exercise-induced neurogenesis in the adult dentate gyrus. Proc. Natl. Acad. Sci. USA 2007, 104, 5638–5643. [Google Scholar]
- Cotman, C.W.; Berchtold, N.C. Exercise: A behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 2002, 25, 295–301. [Google Scholar] [CrossRef]
- Nichol, K.; Deeny, S.P.; Seif, J.; Camaclang, K.; Cotman, C.W. Exercise improves cognition and hippocampal plasticity in APOE epsilon4 mice. Alzheimers Dement. 2009, 5, 287–294. [Google Scholar] [CrossRef]
- Colcombe, S.; Kramer, A.F. Fitness effects on the cognitive function of older adults: A meta-analytic study. Psychol. Sci. 2003, 14, 125–130. [Google Scholar] [CrossRef]
- Etnier, J.L.; Nowell, P.M.; Landers, D.M.; Sibley, B.A. A meta-regression to examine the relationship between aerobic fitness and cognitive performance. Brain Res. Rev. 2006, 52, 119–130. [Google Scholar] [CrossRef]
- Kramer, A.F.; Erickson, K.I.; Colcombe, S.J. Exercise, cognition, and the aging brain. J. Appl. Physiol. 2006, 101, 1237–1242. [Google Scholar] [CrossRef]
- Van Praag, H.; Kempermann, G.; Gage, F.H. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat. Neurosci. 1999, 2, 266–270. [Google Scholar]
- Erickson, K.I.; Voss, M.W.; Prakash, R.S.; Basak, C.; Szabo, A.; Chaddock, L.; Kim, J.S.; Heo, S.; Alves, H.; White, S.M.; et al. Exercise training increases size of hippocampus and improves memory. Proc. Natl. Acad. Sci. USA 2011, 108, 3017–3022. [Google Scholar]
- Alzheimer’s Association. 2011 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2011, 7, 208–244. [Google Scholar]
- Geda, Y.E.; Roberts, R.O.; Knopman, D.S.; Christianson, T.J.; Pankratz, V.S.; Ivnik, R.J.; Boeve, B.F.; Tangalos, E.G.; Petersen, R.C.; Rocca, W.A. Physical exercise, aging, and mild cognitive impairment: A population-based study. Arch. Neurol. 2010, 67, 80–86. [Google Scholar] [CrossRef]
- Etgen, T.; Sander, D.; Huntgeburth, U.; Poppert, H.; Forstl, H.; Bickel, H. Physical activity and incident cognitive impairment in elderly persons: The INVADE study. Arch. Intern. Med. 2010, 170, 186–193. [Google Scholar] [CrossRef]
- Laurin, D.; Verreault, R.; Lindsay, J.; MacPherson, K.; Rockwood, K. Physical activity and risk of cognitive impairment and dementia in elderly persons. Arch. Neurol. 2001, 58, 498–504. [Google Scholar] [CrossRef]
- Etnier, J.L. Chronic exercise and cognition in older adults. In Exercise and Cognitive Function; McMorris, T., Tomporowski, P.D., Audiffren, M., Eds.; Wiley-Blackwell: Chichester, UK, 2009; pp. 227–248. [Google Scholar]
- Sofi, F.; Valecchi, D.; Bacci, D.; Abbate, R.; Gensini, G.F.; Casini, A.; Macchi, C. Physical activity and risk of cognitive decline: A meta-analysis of prospective studies. J. Intern. Med. 2011, 269, 107–117. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Chui, H.C.; Zheng, L.; Reed, B.R.; Vinters, H.V.; Mack, W.J. Vascular risk factors and Alzheimer’s disease: Are these risk factors for plaques and tangles or for concomitant vascular pathology that increases the likelihood of dementia? An evidence-based review. Alzheimers Res. Ther. 2012, 4. [Google Scholar] [CrossRef]
- Jack, C.R., Jr. Alzheimer disease: New concepts on its neurobiology and the clinical role imaging will play. Radiology 2012, 263, 344–361. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar]
- Balasubramanian, A.B.; Kawas, C.H.; Peltz, C.B.; Brookmeyer, R.; Corrada, M.M. Alzheimer disease pathology and longitudinal cognitive performance in the oldest-old with no dementia. Neurology 2012, 79, 915–921. [Google Scholar]
- Ward, A.; Crean, S.; Mercaldi, C.J.; Collins, J.M.; Boyd, D.; Cook, M.N.; Arrighi, H.M. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer’s disease: A systematic review and meta-analysis. Neuroepidemiology 2012, 38, 1–17. [Google Scholar] [CrossRef]
- Raber, J.; Huang, Y.; Ashford, J.W. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol. Aging 2004, 25, 641–650. [Google Scholar] [CrossRef]
- Cabeza, R.; Grady, C.L.; Nyberg, L.; McIntosh, A.R.; Tulving, E.; Kapur, S.; Jennings, J.M.; Houle, S.; Craik, F.I. Age-related differences in neural activity during memory encoding and retrieval: A positron emission tomography study. J. Neurosci. 1997, 17, 391–400. [Google Scholar]
- Petersen, R.C.; Stevens, J.C.; Ganguli, M.; Tangalos, E.G.; Cummings, J.L.; Dekosky, S.T. Practice parameter: Early detection of dementia: Mild cognitive impairment (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2001, 56, 1133–1142. [Google Scholar] [CrossRef]
- Petersen, R.C. Mild cognitive impairment: Transition between aging and Alzheimer’s disease. Neurologia 2000, 15, 93–101. [Google Scholar]
- DeKosky, S.T.; Marek, K. Looking backward to move forward: Early detection of neurodegenerative disorders. Science 2003, 302, 830–834. [Google Scholar]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 270–279. [Google Scholar]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar]
- Luchsinger, J.A.; Tang, M.X.; Stern, Y.; Shea, S.; Mayeux, R. Diabetes mellitus and risk of Alzheimer’s disease and dementia with stroke in a multiethnic cohort. Am. J. Epidemiol. 2001, 154, 635–641. [Google Scholar]
- Grossman, H. Does diabetes protect or provoke Alzheimer’s disease? Insights into the pathobiology and future treatment of Alzheimer’s disease. CNS Spectr. 2003, 8, 815–823. [Google Scholar]
- Yaffe, K.; Kanaya, A.; Lindquist, K.; Simonsick, E.M.; Harris, T.; Shorr, R.I.; Tylavsky, F.A.; Newman, A.B. The metabolic syndrome, inflammation, and risk of cognitive decline. JAMA 2004, 292, 2237–2242. [Google Scholar]
- Cukierman, T.; Gerstein, H.C.; Williamson, J.D. Cognitive decline and dementia in diabetes—systematic overview of prospective observational studies. Diabetologia 2005, 48, 2460–2469. [Google Scholar]
- Goveas, J.S.; Espeland, M.A.; Woods, N.F.; Wassertheil-Smoller, S.; Kotchen, J.M. Depressive symptoms and incidence of mild cognitive impairment and probable dementia in elderly women: The Women’s Health Initiative Memory Study. J. Am. Geriatr. Soc. 2011, 59, 57–66. [Google Scholar]
- Purnell, C.; Gao, S.; Callahan, C.M.; Hendrie, H.C. Cardiovascular risk factors and incident Alzheimer disease: A systematic review of the literature. Alzheimer Dis. Assoc. Disord. 2009, 23, 1–10. [Google Scholar] [CrossRef]
- Etnier, J.L.; Caselli, R.J.; Reiman, E.M.; Alexander, G.E.; Sibley, B.A.; Tessier, D.; McLemore, E.C. Cognitive performance in older women relative to ApoE-epsilon4 genotype and aerobic fitness. Med. Sci. Sports Exerc. 2007, 39, 199–207. [Google Scholar]
- Schuit, A.J.; Feskens, E.J.; Launer, L.J.; Kromhout, D. Physical activity and cognitive decline, the role of the apolipoprotein e4 allele. Med. Sci. Sports Exerc. 2001, 33, 772–777. [Google Scholar]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Rovio, S.; Kareholt, I.; Helkala, E.L.; Viitanen, M.; Winblad, B.; Tuomilehto, J.; Soininen, H.; Nissinen, A.; Kivipelto, M. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet Neurol. 2005, 4, 705–711. [Google Scholar] [CrossRef]
- Lindsay, J.; Laurin, D.; Verreault, R.; Hebert, R.; Helliwell, B.; Hill, G.B.; McDowell, I. Risk factors for Alzheimer’s disease: A prospective analysis from the Canadian Study of Health and Aging. Am. J. Epidemiol. 2002, 156, 445–453. [Google Scholar] [CrossRef]
- Podewils, L.J.; Guallar, E.; Kuller, L.H.; Fried, L.P.; Lopez, O.L.; Carlson, M.; Lyketsos, C.G. Physical activity, APOE genotype, and dementia risk: Findings from the Cardiovascular Health Cognition Study. Am. J. Epidemiol. 2005, 161, 639–651. [Google Scholar] [CrossRef]
- Rey, A. L’examen Clinique en Psychologie; Presses Universitaires de France: Paris, France, 1958. [Google Scholar]
- Corwin, J.; Bylsma, F.W. “Psychological Examination Traumatic Encephalopathy” by A. Rey and “The Complex Figure Copy Test” by P.A. Osterrieth. Clin. Neuropsychol. 1993, 7, 3–21. [Google Scholar] [CrossRef]
- Gromwall, D.M. Paced Auditory Serial Addition Task: A measure of recovery from concussion. Percept. Mot. Skills 1977, 44, 367–373. [Google Scholar] [CrossRef]
- Eggermont, L.H.; Swaab, D.F.; Hol, E.M.; Scherder, E.J. Walking the line: A randomised trial on the effects of a short term walking programme on cognition in dementia. J. Neurol. Neurosurg. Psychiatry 2009, 80, 802–804. [Google Scholar] [CrossRef]
- Namazi, K.H.; Gwinnup, P.B.; Zadorozny, C.A. A low intensity exercise-movement program for patients with Alzheimer’s disease: The TEMP-AD protocol. J. Aging Phys. Act. 1994, 2, 80–92. [Google Scholar]
- Rolland, Y.; Pillard, F.; Klapouszczak, A.; Reynish, E.; Thomas, D.; Andrieu, S.; Riviere, D.; Vellas, B. Exercise program for nursing home residents with Alzheimer’s disease: A 1-year randomized, controlled trial. J. Am. Geriatr. Soc. 2007, 55, 158–165. [Google Scholar] [CrossRef]
- Venturelli, M.; Scarsini, R.; Schena, F. Six-month walking program changes cognitive and ADL performance in patients with Alzheimer. Am. J. Alzheimers Dis. Dement. 2011, 26, 381–388. [Google Scholar] [CrossRef]
- Yaguez, L.; Shaw, K.N.; Morris, R.; Matthews, D. The effects on cognitive functions of a movement-based intervention in patients with Alzheimer’s type dementia: A pilot study. Int. J. Geriatr. Psychiatry 2011, 26, 173–181. [Google Scholar] [CrossRef]
- Littbrand, H.; Stenvall, M.; Rosendahl, E. Applicability and effects of physical exercise on physical and cognitive functions and activities of daily living among people with dementia: A systematic review. Am. J. Phys. Med. Rehabil. 2011, 90, 495–518. [Google Scholar] [CrossRef]
- Kramer, A.F.; Hahn, S.; Cohen, N.J.; Banich, M.T.; McAuley, E.; Harrison, C.R.; Chason, J.; Vakil, E.; Bardell, L.; Boileau, R.A.; Colcombe, A. Ageing, fitness and neurocognitive function. Nature 1999, 400, 418–419. [Google Scholar]
- Erickson, K.I.; Miller, D.L.; Weinstein, A.M.; Akl, S.L.; Banducci, S.E. Physical activity and brain plasticity in late adulthood: A conceptual review. Ageing Res. 2012, 3. [Google Scholar] [CrossRef]
- Smith, P.J.; Blumenthal, J.A.; Hoffman, B.M.; Cooper, H.; Strauman, T.A.; Welsh-Bohmer, K.; Browndyke, J.N.; Sherwood, A. Aerobic exercise and neurocognitive performance: A meta-analytic review of randomized controlled trials. Psychosom. Med. 2010, 72, 239–252. [Google Scholar]
- Angevaren, M.; Aufdemkampe, G.; Verhaar, H.J.; Aleman, A.; Vanhees, L. Physical activity and enhanced fitness to improve cognitive function in older people without known cognitive impairment. Cochrane Database Syst. Rev. 2008. [Google Scholar] [CrossRef]
- Scherder, E.J.; Van Paasschen, J.; Deijen, J.B.; Van Der Knokke, S.; Orlebeke, J.F.; Burgers, I.; Devriese, P.P.; Swaab, D.F.; Sergeant, J.A. Physical activity and executive functions in the elderly with mild cognitive impairment. Aging Ment. Health 2005, 9, 272–280. [Google Scholar]
- Boule, N.G.; Kenny, G.P.; Haddad, E.; Wells, G.A.; Sigal, R.J. Meta-analysis of the effect of structured exercise training on cardiorespiratory fitness in Type 2 diabetes mellitus. Diabetologia 2003, 46, 1071–1081. [Google Scholar] [CrossRef]
- Bouchard, C.; Sarzynski, M.A.; Rice, T.K.; Kraus, W.E.; Church, T.S.; Sung, Y.J.; Rao, D.C.; Rankinen, T. Genomic predictors of the maximal O2 uptake response to standardized exercise training programs. J. Appl. Physiol. 2011, 110, 1160–1170. [Google Scholar] [CrossRef]
- Lautenschlager, N.T.; Cox, K.L.; Flicker, L.; Foster, J.K.; van Bockxmeer, F.M.; Xiao, J.; Greenop, K.R.; Almeida, O.P. Effect of physical activity on cognitive function in older adults at risk for Alzheimer disease: A randomized trial. J. Am. Med. Assoc. 2008, 300, 1027–1037. [Google Scholar] [CrossRef]
- Miller, L.A.; Spitznagel, M.B.; Busko, S.; Potter, V.; Juvancic-Heltzel, J.; Istenes, N.; Glickman, E.; Gunstad, J. Structured exercise does not stabilize cognitive function in individuals with mild cognitive impairment residing in a structured living facility. Int. J. Neurosci. 2011, 121, 218–223. [Google Scholar] [CrossRef]
- Baker, L.D.; Frank, L.L.; Foster-Schubert, K.; Green, P.S.; Wilkinson, C.W.; McTiernan, A.; Plymate, S.R.; Fishel, M.A.; Watson, G.S.; Cholerton, B.A.; et al. Effects of aerobic exercise on mild cognitive impairment: A controlled trial. Arch. Neurol. 2010, 67, 71–79. [Google Scholar]
- Petersen, R.C.; Smith, G.E.; Waring, S.C.; Ivnik, R.J.; Tangalos, E.G.; Kokmen, E. Mild cognitive impairment: Clinical characterization and outcome. Arch. Neurol. 1999, 56, 303–308. [Google Scholar] [CrossRef]
- Libon, D.J.; Xie, S.X.; Eppig, J.; Wicas, G.; Lamar, M.; Lippa, C.; Bettcher, B.M.; Price, C.C.; Giovannetti, T.; Swenson, R.; Wambach, D.M. The heterogeneity of mild cognitive impairment: A neuropsychological analysis. J. Int. Neuropsychol. Soc. 2010, 16, 84–93. [Google Scholar] [CrossRef]
- Morris, J.C. Revised criteria for mild cognitive impairment may compromise the diagnosis of Alzheimer disease dementia. Arch. Neurol. 2012, 69, 700–708. [Google Scholar]
- Morris, J.C.; McKeel, D.W., Jr.; Storandt, M.; Rubin, E.H.; Price, J.L.; Grant, E.A.; Ball, M.J.; Berg, L. Very mild Alzheimer’s disease: Informant-based clinical, psychometric, and pathologic distinction from normal aging. Neurology 1991, 41, 469–478. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar]
- Li, J.; Wang, Y.J.; Zhang, M.; Xu, Z.Q.; Gao, C.Y.; Fang, C.Q.; Yan, J.C.; Zhou, H.D. Vascular risk factors promote conversion from mild cognitive impairment to Alzheimer disease. Neurology 2011, 76, 1485–1491. [Google Scholar]
- Baker, L.D.; Frank, L.L.; Foster-Schubert, K.; Green, P.S.; Wilkinson, C.W.; McTiernan, A.; Cholerton, B.A.; Plymate, S.R.; Fishel, M.A.; Watson, G.S.; et al. Aerobic exercise improves cognition for older adults with glucose intolerance, a risk factor for Alzheimer’s disease. J. Alzheimers Dis. 2010, 22, 569–579. [Google Scholar]
- Burns, J.M.; Cronk, B.B.; Anderson, H.S.; Donnelly, J.E.; Thomas, G.P.; Harsha, A.; Brooks, W.M.; Swerdlow, R.H. Cardiorespiratory fitness and brain atrophy in early Alzheimer disease. Neurology 2008, 71, 210–216. [Google Scholar] [CrossRef]
- Honea, R.A.; Thomas, G.P.; Harsha, A.; Anderson, H.S.; Donnelly, J.E.; Brooks, W.M.; Burns, J.M. Cardiorespiratory fitness and preserved medial temporal lobe volume in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2009, 23, 188–197. [Google Scholar] [CrossRef]
- Vidoni, E.D.; Honea, R.A.; Billinger, S.A.; Swerdlow, R.H.; Burns, J.M. Cardiorespiratory fitness is associated with atrophy in Alzheimer’s and aging over 2 years. Neurobiol. Aging 2012, 33, 1624–1632. [Google Scholar] [CrossRef]
- Head, D.; Bugg, J.M.; Goate, A.M.; Fagan, A.M.; Mintun, M.A.; Benzinger, T.; Holtzman, D.M.; Morris, J.C. Exercise Engagement as a Moderator of the Effects of APOE Genotype on Amyloid Deposition. Arch. Neurol. 2012, 69, 636–643. [Google Scholar] [CrossRef]
- Deeny, S.P.; Poeppel, D.; Zimmerman, J.B.; Roth, S.M.; Brandauer, J.; Witkowski, S.; Hearn, J.W.; Ludlow, A.T.; Contreras-Vidal, J.L.; Brandt, J.; Hatfield, B.D. Exercise, APOE, and working memory: MEG and behavioral evidence for benefit of exercise in epsilon4 carriers. Biol. Psychol. 2008, 78, 179–187. [Google Scholar] [CrossRef]
- Deeny, S.P.; Winchester, J.; Nichol, K.; Roth, S.M.; Wu, J.C.; Dick, M.; Cotman, C.W. Cardiovascular fitness is associated with altered cortical glucose metabolism during working memory in varepsilon4 carriers. Alzheimers Dement. 2012, 8, 352–356. [Google Scholar]
- Aggarwal, N.T.; Wilson, R.S.; Beck, T.L.; Bienias, J.L.; Bennett, D.A. Mild cognitive impairment in different functional domains and incident Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1479–1484. [Google Scholar]
- Trachtenberg, A.J.; Filippini, N.; Mackay, C.E. The effects of APOE-epsilon4 on the BOLD response. Neurobiol. Aging 2010, 33, 323–334. [Google Scholar]
- Bondi, M.W.; Kaszniak, A.W. Implicit and explicit memory in Alzheimer’s disease and Parkinson’s disease. J. Clin. Exp. Neuropsychol. 1991, 13, 339–358. [Google Scholar] [CrossRef]
- Irle, E.; Kaiser, P.; Naumann-Stoll, G. Differential patterns of memory loss in patients with Alzheimer’s disease and Korsakoff’s disease. Int. J. Neurosci. 1990, 52, 67–77. [Google Scholar] [CrossRef]
- Petersen, R.C.; Smith, G.E.; Ivnik, R.J.; Kokmen, E.; Tangalos, E.G. Memory function in very early Alzheimer’s disease. Neurology 1994, 44, 867–872. [Google Scholar] [CrossRef]
- Nilsson, L.G. Memory function in normal aging. Acta Neurol. Scand. Suppl. 2003, 179, 7–13. [Google Scholar] [CrossRef]
- Nielson, K.A.; Langenecker, S.A.; Garavan, H. Differences in the functional neuroanatomy of inhibitory control across the adult lifespan. Psychol. Aging 2002, 17, 56–57. [Google Scholar]
- Persson, J.; Nyberg, L.; Lind, J.; Larsson, A.; Nilsson, L.G.; Ingvar, M.; Buckner, R.L. Structure-function correlates of cognitive decline in aging. Cereb. Cortex 2006, 16, 907–915. [Google Scholar]
- Bookheimer, S.Y.; Strojwas, M.H.; Cohen, M.S.; Saunders, A.M.; Pericak-Vance, M.A.; Mazziotta, J.C.; Small, G.W. Patterns of brain activation in people at risk for Alzheimer’s Disease. N. Engl. J. Med. 2000, 343, 450–456. [Google Scholar] [CrossRef]
- O’Brien, J.L.; O’Keefe, K.M.; LaViolette, P.S.; DeLuca, A.N.; Blacker, D.; Dickerson, B.C.; Sperling, R.A. Longitudinal fMRI in elderly reveals loss of hippocampal activation with clinical decline. Neurology 2010, 74, 1969–1976. [Google Scholar]
- Lonie, J.A.; Herrmann, L.L.; Tierney, K.M.; Donaghey, C.; O’Carroll, R.; Lee, A.; Ebmeier, K.P. Lexical and semantic fluency discrepancy scores in aMCI and early Alzheimer’s disease. J. Neuropsychol. 2009, 3, 79–92. [Google Scholar] [CrossRef]
- Henry, J.D.; Crawford, J.R.; Phillips, L.H. Verbal fluency performance in dementia of the Alzheimer’s type: A meta-analysis. Neuropsychologia 2004, 42, 1212–1222. [Google Scholar] [CrossRef]
- Hodges, J.R.; Salmon, D.P.; Butters, N. Semantic memory impairment in Alzheimer’s disease: Failure of access or degraded knowledge? Neuropsychologia 1992, 30, 301–314. [Google Scholar] [CrossRef]
- Hodges, J.R.; Salmon, D.P.; Butters, N. Differential impairment of semantic and episodic memory in Alzheimer’s and Huntington’s diseases: A controlled prospective study. J. Neurol. Neurosurg. Psychiatry 1990, 53, 1089–1095. [Google Scholar] [CrossRef]
- Nebes, R.D. Semantic memory in Alzheimer’s disease. Psychol. Bull. 1989, 106, 377–394. [Google Scholar] [CrossRef]
- Lind, J.; Ingvar, M.; Persson, J.; Sleegers, K.; van Broeckhoven, C.; Adolfsson, R.; Nilsson, L.G.; Nyberg, L. Parietal cortex activation predicts memory decline in apolipoprotein E-epsilon4 carriers. Neuroreport 2006, 17, 1683–1686. [Google Scholar] [CrossRef]
- Binder, J.R.; Desai, R.H.; Graves, W.W.; Conant, L.L. Where is the semantic system? A critical review and meta-analysis of 120 functional neuroimaging studies. Cereb. Cortex 2009, 19, 2767–2796. [Google Scholar] [CrossRef]
- Buckner, R.L.; Snyder, A.Z.; Shannon, B.J.; LaRossa, G.; Sachs, R.; Fotenos, A.F.; Sheline, Y.I.; Klunk, W.E.; Mathis, C.A.; Morris, J.C.; Mintun, M.A. Molecular, structural, and functional characterization of Alzheimer’s disease: Evidence for a relationship between default activity, amyloid, and memory. J. Neurosci. 2005, 25, 7709–7717. [Google Scholar]
- Hantke, N.; Nielson, K.A.; Woodard, J.L.; Breting, L.M.G.; Butts, A.; Seidenberg, M.; Smith, J.C.; Durgerian, S.; Lancaster, M.; Matthews, M.; et al. Comparison of semantic and episodic memory BOLD fMRI activation in predicting cognitive decline in older adults. J. Int. Neuropsychol. Soc. 2012. [Google Scholar] [CrossRef]
- Sugarman, M.A.; Woodard, J.L.; Nielson, K.A.; Seidenberg, M.; Smith, J.C.; Durgerian, S.; Rao, S.M. Functional magnetic resonance imaging of semantic memory as a presymptomatic biomarker of Alzheimer's disease risk. Biochim. Biophys. Acta 2012, 1822, 442–456. [Google Scholar]
- Smith, J.C.; Nielson, K.A.; Woodard, J.L.; Seidenberg, M.; Durgerian, S.; Antuono, P.; Butts, A.M.; Hantke, N.C.; Lancaster, M.A.; Rao, S.M. Interactive effects of physical activity and APOE-epsilon4 on BOLD semantic memory activation in healthy elders. Neuroimage 2011, 54, 635–644. [Google Scholar] [CrossRef]
- Taylor-Piliae, R.E.; Haskell, W.L.; Iribarren, C.; Norton, L.C.; Mahbouba, M.H.; Fair, J.M.; Hlatky, M.A.; Go, A.S.; Fortmann, S.P. Clinical utility of the Stanford brief activity survey in men and women with early-onset coronary artery disease. J. Cardiopulm. Rehabil. Prev. 2007, 27, 227–232. [Google Scholar]
- Taylor-Piliae, R.E.; Norton, L.C.; Haskell, W.L.; Mahbouda, M.H.; Fair, J.M.; Iribarren, C.; Hlatky, M.A.; Go, A.S.; Fortmann, S.P. Validation of a new brief physical activity survey among men and women aged 60-69 years. Am. J. Epidemiol. 2006, 164, 598–606. [Google Scholar] [CrossRef]
- Park, D.C.; Reuter-Lorenz, P. The adaptive brain: Aging and neurocognitive scaffolding. Annu. Rev. Psychol. 2009, 60, 173–196. [Google Scholar] [CrossRef]
- Stern, Y. What is cognitive reserve? Theory and research application of the reserve concept. J. Int. Neuropsychol. Soc. 2002, 8, 448–460. [Google Scholar] [CrossRef]
- Smith, J.C.; Nielson, K.A.; Woodard, J.L.; Seidenberg, M.; Verber, M.D.; Durgerian, S.; Antuono, P.; Butts, A.M.; Hantke, N.C.; Lancaster, M.A.; Rao, S.M. Does physical activity influence semantic memory activation in amnestic mild cognitive impairment? Psychiatry Res. 2011, 193, 60–62. [Google Scholar]
- Hakamata, Y.; Lissek, S.; Bar-Haim, Y.; Britton, J.C.; Fox, N.A.; Leibenluft, E.; Ernst, M.; Pine, D.S. Attention bias modification treatment: A meta-analysis toward the establishment of novel treatment for anxiety. Biol. Psychiatry 2010, 68, 982–990. [Google Scholar] [CrossRef]
- Smith, A.G.; Russell, J.; Feldman, E.L.; Goldstein, J.; Peltier, A.; Smith, S.; Hamwi, J.; Pollari, D.; Bixby, B.; Howard, J.; Singleton, J.R. Lifestyle intervention for pre-diabetic neuropathy. Diabetes Care 2006, 29, 1294–1299. [Google Scholar] [CrossRef]
- Crosson, B.; Benjamin, M.; Levy, I. Role of the basal ganglia in language and semantics: Supporting cast. In Neural Basis of Semantic Memory; Hart, J., Jr., Kraut, M.A., Eds.; Cambridge University Press: New York, NY, USA, 2007; pp. 219–243. [Google Scholar]
- De Castro, J.M.; Duncan, G. Operantly conditioned running: Effects on brain catecholamine concentrations and receptor densities in the rat. Pharmacol. Biochem. Behav. 1985, 23, 495–500. [Google Scholar] [CrossRef]
- Intlekofer, K.A.; Cotman, C.W. Exercise counteracts declining hippocampal function in aging and Alzheimer’s disease. Neurobiol. Dis. 2012. [Google Scholar] [CrossRef]
- Van Praag, H.; Shubert, T.; Zhao, C.; Gage, F.H. Exercise enhances learning and hippocampal neurogenesis in aged mice. J. Neurosci. 2005, 25, 8680–8685. [Google Scholar] [CrossRef]
- Trejo, J.L.; Carro, E.; Torres-Aleman, I. Circulating insulin-like growth factor I mediates exercise-induced increases in the number of new neurons in the adult hippocampus. J. Neurosci. 2001, 21, 1628–1634. [Google Scholar]
- Adlard, P.A.; Perreau, V.M.; Pop, V.; Cotman, C.W. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer’s disease. J. Neurosci. 2005, 25, 4217–4221. [Google Scholar]
- Poirier, J. Apolipoprotein E and Alzheimer’s disease. A role in amyloid catabolism. Ann. N. Y. Acad. Sci. 2000, 924, 81–90. [Google Scholar] [CrossRef]
- Lane, R.M.; Farlow, M.R. Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimer’s disease. J. Lipid Res. 2005, 46, 949–968. [Google Scholar] [CrossRef]
- Leduc, V.; Domenger, D.; de Beaumont, L.; Lalonde, D.; Belanger-Jasmin, S.; Poirier, J. Function and comorbidities of apolipoprotein e in Alzheimer’s disease. Int. J. Alzheimers Dis. 2011, 2011. [Google Scholar] [CrossRef]
- Ben, J.; Soares, F.M.; Cechetti, F.; Vuaden, F.C.; Bonan, C.D.; Netto, C.A.; Wyse, A.T. Exercise effects on activities of Na+,K+-ATPase, acetylcholinesterase and adenine nucleotides hydrolysis in ovariectomized rats. Brain Res. 2009, 1302, 248–255. [Google Scholar]
- Fukao, K.; Shimada, K.; Naito, H.; Sumiyoshi, K.; Inoue, N.; Iesaki, T.; Kume, A.; Kiyanagi, T.; Hiki, M.; Hirose, K.; et al. Voluntary exercise ameliorates the progression of atherosclerotic lesion formation via anti-inflammatory effects in apolipoprotein E-deficient mice. J. Atheroscler. Thromb. 2010, 17, 1226–1236. [Google Scholar]
- Rankinen, T.; Roth, S.M.; Bray, M.S.; Loos, R.; Perusse, L.; Wolfarth, B.; Hagberg, J.M.; Bouchard, C. Advances in exercise, fitness, and performance genomics. Med. Sci. Sports Exerc. 2010, 42, 835–846. [Google Scholar]
- Leon, A.S.; Togashi, K.; Rankinen, T.; Despres, J.P.; Rao, D.C.; Skinner, J.S.; Wilmore, J.H.; Bouchard, C. Association of apolipoprotein E polymorphism with blood lipids and maximal oxygen uptake in the sedentary state and after exercise training in the HERITAGE family study. Metabolism 2004, 53, 108–116. [Google Scholar] [CrossRef]
- Obisesan, T.O.; Ferrell, R.E.; Goldberg, A.P.; Phares, D.A.; Ellis, T.J.; Hagberg, J.M. APOE genotype affects black-white responses of high-density lipoprotein cholesterol subspecies to aerobic exercise training. Metabolism 2008, 57, 1669–1676. [Google Scholar] [CrossRef]
- Hagberg, J.M.; Ferrell, R.E.; Katzel, L.I.; Dengel, D.R.; Sorkin, J.D.; Goldberg, A.P. Apolipoprotein E genotype and exercise training-induced increases in plasma high-density lipoprotein (HDL)- and HDL2-cholesterol levels in overweight men. Metabolism 1999, 48, 943–945. [Google Scholar] [CrossRef]
- Seip, R.L.; Otvos, J.; Bilbie, C.; Tsongalis, G.J.; Miles, M.; Zoeller, R.; Visich, P.; Gordon, P.; Angelopoulos, T.J.; Pescatello, L.; et al. The effect of apolipoprotein E genotype on serum lipoprotein particle response to exercise. Atherosclerosis 2006, 188, 126–133. [Google Scholar]
- Crabbe, J.B.; Dishman, R.K. Brain electrocortical activity during and after exercise: A quantitative synthesis. Psychophysiology 2004, 41, 563–574. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Smith, J.C.; Nielson, K.A.; Woodard, J.L.; Seidenberg, M.; Rao, S.M. Physical Activity and Brain Function in Older Adults at Increased Risk for Alzheimer’s Disease. Brain Sci. 2013, 3, 54-83. https://doi.org/10.3390/brainsci3010054
Smith JC, Nielson KA, Woodard JL, Seidenberg M, Rao SM. Physical Activity and Brain Function in Older Adults at Increased Risk for Alzheimer’s Disease. Brain Sciences. 2013; 3(1):54-83. https://doi.org/10.3390/brainsci3010054
Chicago/Turabian StyleSmith, J. Carson, Kristy A. Nielson, John L. Woodard, Michael Seidenberg, and Stephen M. Rao. 2013. "Physical Activity and Brain Function in Older Adults at Increased Risk for Alzheimer’s Disease" Brain Sciences 3, no. 1: 54-83. https://doi.org/10.3390/brainsci3010054
APA StyleSmith, J. C., Nielson, K. A., Woodard, J. L., Seidenberg, M., & Rao, S. M. (2013). Physical Activity and Brain Function in Older Adults at Increased Risk for Alzheimer’s Disease. Brain Sciences, 3(1), 54-83. https://doi.org/10.3390/brainsci3010054