Neuroprotective Therapies after Perinatal Hypoxic-Ischemic Brain Injury

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pathogenesis of Perinatal Brain

3. Calcium Influx and Free Radical Formation
4. Nitric Oxide Synthases Activation
5. Inflammation
6. Apoptosis Activation
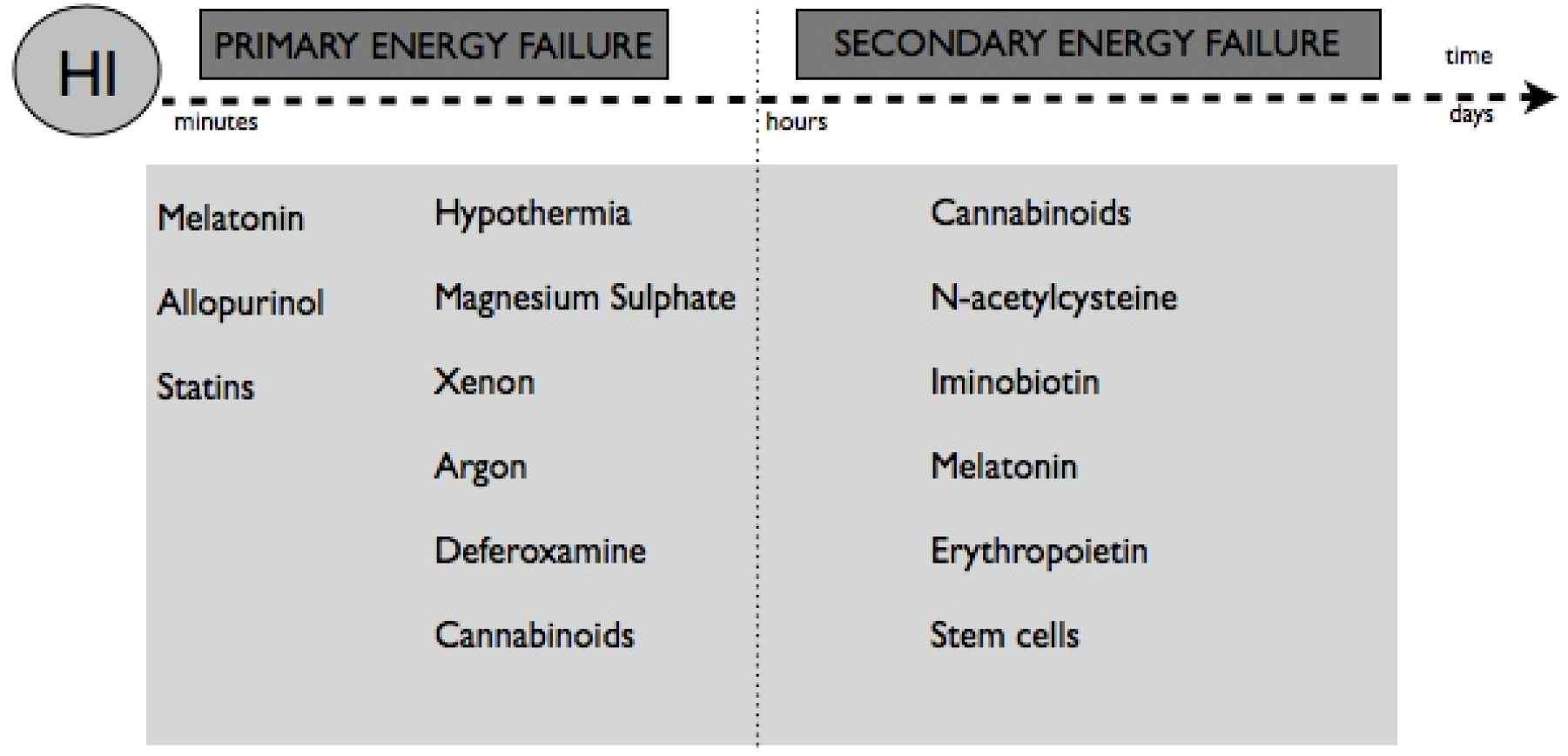
7. Neuroprotective Therapies

8. Non-Pharmacological Therapies
9. Pharmacological Therapies
9.1. Therapeutical Strategies Related to Antioxidants
9.2. Therapeutical Strategies Related to Anti-Inflammation and Anti-Apoptosis
10. Delayed Possibilities: Regeneration
Acknowledgements
References
- Kurinczuk, J.J.; White-Koning, M.; Badawi, N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum. Dev. 2010, 86, 329–338. [Google Scholar] [CrossRef]
- Lawn, J.E.; Cousens, S.; Zupan, J.; Lancet Neonatal Survival Steering Team. 4 million neonatal deaths: When? Where? Why? Lancet 2005, 365, 891–900. [Google Scholar]
- Pierrat, V.; Haouari, N.; Liska, A.; Thomas, D.; Subtil, D.; Truffert, P.; Groupe d’Etudes en Epidemiologie Perinatale. Prevalence, causes, and outcome at 2 years of age of newborn encephalopathy: Population based study. Arch. Dis. Child. Fetal Neonatal Ed. 2005, 90, F257–F261. [Google Scholar] [CrossRef]
- Marlow, N.; Budge, H. Prevalence, causes, and outcome at 2 years of age of newborn encephalopathy. Arch. Dis. Child. Fetal Neonatal Ed. 2005, 90, F193–F194. [Google Scholar] [CrossRef]
- Cowan, F. Outcome after intrapartum asphyxia in term infants. Semin. Neonatol. 2000, 5, 127–140. [Google Scholar] [CrossRef]
- Al-Macki, N.; Miller, S.P.; Hall, N.; Shevell, M. The spectrum of abnormal neurologic outcomes subsequent to term intrapartum asphyxia. Pediatr. Neurol. 2009, 41, 399–405. [Google Scholar] [CrossRef]
- Barnett, A.; Mercuri, E.; Rutherford, M.; Haataja, L.; Frisone, M.F.; Henderson, S.; Cowan, F.; Dubowitz, L. Neurological and perceptual-motor outcome at 5–6 years of age in children with neonatal encephalopathy: Relationship with neonatal brain MRI. Neuropediatrics 2002, 33, 242–248. [Google Scholar] [CrossRef]
- Glass, H.C.; Ferriero, D.M. Treatment of hypoxic-ischemic encephalopathy in newborns. Curr. Treat. Options Neurol. 2007, 9, 414–423. [Google Scholar] [CrossRef]
- Carl, G.; Reiger, I.; Evans, N. One-year neurodevelopmental outcome after moderate newborn hypoxic ischemic encephalopathy. J. Paediatr. Child Health 2004, 40, 217–220. [Google Scholar] [CrossRef]
- Du Plessis, A.J.; Volpe, J.J. Perinatal brain injury in the preterm and term newborn. Curr. Opin. Neurol. 2002, 15, 151–157. [Google Scholar] [CrossRef]
- Gonzalez, F.F.; Ferriero, D.M. Therapeutics for neonatal brain injury. Pharmacol. Ther. 2008, 120, 43–53. [Google Scholar] [CrossRef]
- Fan, X.; Kavelaars, A.; Heijnen, C.J.; Groenendaal, F.; van Bel, F. Pharmacological neuroprotection after perinatal hypoxic-ischemic brain injury. Curr. Neuropharmacol. 2010, 8, 324–334. [Google Scholar] [CrossRef]
- Ferriero, D. Neonatal brain injury. N. Engl. J. Med. 2004, 351, 1985–1995. [Google Scholar] [CrossRef]
- Walton, M.; Connor, B.; Lawlor, P.; Young, D.; Sirimanne, E.; Gluckman, P.; Cole, G.; Dragunow, M. Neuronal death and survival in two models of hypoxic-ischemic brain damage. Brain Res. Rev. 1999, 29, 137–168. [Google Scholar] [CrossRef]
- Sarnat, H.B.; Sarnat, M.S. Neonatal encephalopathy following fetal distress. A clinical and electroencephalographic study. Arch. Neurol. 1976, 33, 696–705. [Google Scholar] [CrossRef]
- Vannucci, R.C.; Vannucci, S.J. A model of perinatal hypoxic-ischemic brain damage. Ann. N. Y. Acad. Sci. 1997, 835, 234–249. [Google Scholar] [CrossRef]
- Yager, J.Y.; Thornhill, J.A. The effect of age on susceptibility to hypoxic-ischemic brain damage. Neurosci. Biobehav. Rev. 1997, 21, 167–174. [Google Scholar] [CrossRef]
- Shalak, L.F.; Laptook, A.R.; Velaphi, S.C.; Perlman, J.M. Amplitude-integrated electroencephalography coupled with an early neurologic examination enhances prediction of term infants at risk for persistent encephalopathy. Pediatrics 2003, 111, 351–357. [Google Scholar] [CrossRef]
- Sanders, R.D.; Manning, H.J.; Robertson, N.J.; Ma, D.; Edwards, A.D.; Hagberg, H.; Maze, M. Preconditioning and postinsult therapies for perinatal hypoxic-ischemic injury at term. Anesthesiology 2010, 113, 233–249. [Google Scholar] [CrossRef]
- Perlman, J.M. Summary proceedings from the neurology group on hypoxic-ischemic encephalopathy. Pediatrics 2006, 117, 28–33. [Google Scholar] [CrossRef]
- Volpe, J.J. Neurology of the Newborn, 5th ed; Elsevier Health Sciences: New York, NY, USA, 2008. [Google Scholar]
- Wyatt, J.S.; Edwards, A.D.; Azzopardi, D.; Reynolds, E.O. Magnetic resonance and near infrared spectroscopy for investigation of perinatal hypoxic-ischaemic brain injury. Arch. Dis. Child. 1989, 64, 953–963. [Google Scholar] [CrossRef]
- Yager, J.Y.; Brucklacher, R.M.; Vannucci, R.C. Cerebral energy metabolism during hypoxia-ischemia and early recovery in immature rats. Am. J. Physiol. 1992, 262, 672–677. [Google Scholar]
- Inder, T.E.; Huppi, P.S. In vivo studies of brain development by magnetic resonance techniques. Ment. Retard. Dev. Disabil. Res. Rev. 2000, 6, 59–67. [Google Scholar] [CrossRef]
- Girard, S.; Kadhim, H.; Larouche, A.; Roy, M.; Gobeil, F.; Sébire, G. Pro-inflammatory disequilibrium of the IL-1 beta/IL-1ra ratio in an experimental model of perinatal brain damages induced by lipopolysaccharide and hypoxia-ischemia. Cytokine 2008, 43, 54–62. [Google Scholar] [CrossRef]
- Hilton, G.D.; Nunez, J.L.; Bambrick, L.; Thompson, S.M.; McCarthy, M.M. Glutamate-mediated excitotoxicity in neonatal hippocampal neurons is mediated by mGluR-induced release of Ca2+ from intracellular stores and is prevented by estradiol. Eur. J. Neurosci. 2006, 24, 3008–3016. [Google Scholar] [CrossRef]
- Kumar, A.; Mittal, R.; Khanna, H.D.; Basu, S. Free radical injury and blood-brain barrier permeability in hypoxic-ischemic encephalopathy. Pediatrics 2008, 122, 722–727. [Google Scholar] [CrossRef]
- Cross, J.L.; Meloni, B.P.; Bakker, A.J.; Lee, S.; Knuckey, N.W. Modes of Neuronal Calcium Entry and Homeostasis following Cerebral Ischemia. Stroke Res. Treat. 2010, 2010. [Google Scholar] [CrossRef]
- McDonald, J.W.; Johnston, M.V. Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res. Rev. 1990, 15, 41–70. [Google Scholar] [CrossRef]
- Monyer, H.; Brunashev, N.; Laurie, D.J. Development and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 1993, 12, 529–540. [Google Scholar] [CrossRef]
- Van den Tweel, E.R.; Nijboer, C.; Kavelaars, A.; Heijnen, C.J.; Groenendaal, F.; van Bel, F. Expression of nitric oxide synthase isoforms and nitrotyrosine formation after hypoxia-ischemia in the neonatal rat brain. J. Neuroimmunol. 2005, 167, 64–71. [Google Scholar] [CrossRef]
- Blomgren, K.; Hagberg, H. Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radic. Biol. Med. 2006, 40, 388–397. [Google Scholar] [CrossRef]
- Chang, Y.; Hsieh, C.Y.; Peng, Z.A.; Yen, T.L.; Hsiao, G.; Chou, D.S.; Chen, C.M.; Sheu, J.R. Neuroprotective mechanisms of puerarin in middle cerebral artery occlusion-induced brain infarction in rats. J. Biomed. Sci. 2009, 16. [Google Scholar] [CrossRef]
- Fabian, R.H.; Perez-Polo, J.R.; Kent, T.A. Perivascular nitric oxide and superoxide in neonatal cerebral hypoxia-ischemia. Am. J. Physiol. Heart. Circ. Physiol. 2008, 295, 1809–1814. [Google Scholar] [CrossRef]
- Suzuki, M.; Tabuchi, M.; Ikeda, M.; Tomita, T. Concurrent formation of peroxynitrite with the expression of inducible nitric oxide synthase in the brain during middle cerebral artery occlusion and reperfusion in rats. Brain Res. 2002, 951, 113–120. [Google Scholar] [CrossRef]
- Yang, L.; Sameshima, H.; Yamaguchi, M.; Ikenoue, T. Expression of inducible nitric oxide synthase and cyclooxygenase-2 mRNA in brain damage induced by lipopolysaccharide and intermittent hypoxia-ischemia in neonatal rats. J. Obstet. Gynaecol. Res. 2005, 31, 185–191. [Google Scholar] [CrossRef]
- Robertson, C.L.; Scafidi, S.; McKenna, M.C.; Fiskum, G. Mitochondrial mechanisms of cell death and neuroprotection in pediatric ischemic and traumatic brain injury. Exp. Neurol. 2009, 218, 371–380. [Google Scholar] [CrossRef]
- Hagberg, H.; Mallard, C.; Rousset, C.I.; Wang, X.Y. Apoptotic mechanisms in the immature brain: Involvement of mitochondria. J. Child Neurol. 2009, 24, 1141–1146. [Google Scholar] [CrossRef]
- Leonardo, C.C.; Pennypacker, K.R. Neuroinflammation and MMPs: Potential therapeutic targets in neonatal hypoxic-ischemic injury. J. Neuroinflamm. 2009, 6, 13. [Google Scholar] [CrossRef]
- Nijboer, C.H.; Heijnen, C.J.; Groenendaal, F.; May, M.J.; van Bel, F.; Kavelaars, A. Strong neuroprotection by inhibition of NF-kappaB after neonatal hypoxia-ischemia involves apoptotic mechanisms but is independent of cytokines. Stroke 2008, 39, 2129–2137. [Google Scholar] [CrossRef]
- Nijboer, C.H.; Heijnen, C.J.; Groenendaal, F.; May, M.J.; van Bel, F.; Kavelaars, A. A dual role of the NF-kappaB pathway in neonatal hypoxic-ischemic brain damage. Stroke 2008, 39, 2578–2586. [Google Scholar] [CrossRef]
- Waetzig, V.; Czeloth, K.; Hidding, U.; Mielke, K.; Kanzow, M.; Brecht, S.; Goetz, M.; Lucius, R.; Herdegen, T.; Hanisch, U.K. c-Jun N-terminal kinases (JNKs) mediate pro-inflammatory actions of microglia. Glia 2005, 50, 235–246. [Google Scholar] [CrossRef]
- Northington, F.J.; Ferriero, D.M.; Graham, E.M.; Traystman, R.J.; Martin, L.J. Early neurodegeneration after hypoxia-ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol. Dis. 2001, 8, 207–219. [Google Scholar] [CrossRef]
- Blomgren, K.; Leist, M.; Groc, L. Pathological apoptosis in the developing brain. Apoptosis 2007, 12, 993–1010. [Google Scholar] [CrossRef]
- Wang, X.; Karlsson, J.O.; Zhu, C.; Bahr, B.A.; Hagberg, H.; Blomgren, K. Caspase-3 activation after neonatal rat cerebral hypoxia-ischemia. Biol. Neonate 2001, 79, 172–179. [Google Scholar] [CrossRef]
- Gill, R.; Soriano, M.; Blomgren, K.; Hagberg, H.; Wybrecht, R.; Miss, M.T.; Hoefer, S.; Adam, G.; Niederhauser, O.; Kemp, J.A.; Loetscher, H. Role of caspase-3 activation in cerebral ischemia- induced neurodegeneration in adult and neonatal brain. J. Cereb. Blood Flow. Metab. 2002, 22, 420–430. [Google Scholar]
- Northington, F.J.; Zelaya, M.E.; O’Riordan, D.P.; Blomgren, K.; Flock, D.L.; Hagberg, H.; Ferriero, D.M.; Martin, L.J. Failure to complete apoptosis following neonatal hypoxia-ischemia manifests as “continuum” phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain. Neuroscience 2007, 149, 822–833. [Google Scholar] [CrossRef]
- Lay, C.C.; Davis, M.F.; Chen-Bee, C.H.; Frostig, R.D. Mild sensory stimulation completely protects the adult rodent cortex from ischemic stroke. PLoS One 2010, 5, e11270. [Google Scholar]
- Goñi-de-Cerio, F.; Alvarez, A.; Caballero, A.; Mielgo, V.E.; Alvarez, F.J.; Rey-Santano, M.C.; Gastiasoro, E.; Valls-i-Soler, A.; Bilbao, J.; Hilario, E. Early cell death in the brain of fetal preterm lambs after hypoxic-ischemic injury. Brain Res. 2007, 1151, 161–171. [Google Scholar] [CrossRef]
- McLendon, D.; Check, J.; Carteaux, P.; Michael, L.; Moehring, J.; Secrest, J.W.; Clark, S.E.; Cohen, H.; Klein, S.A.; Boyle, D.; et al. Implementation of potentially better practices for the prevention of brain hemorrhage and ischemic brain injury in very low birth weight infants. Pediatrics 2003, 111, 497–503. [Google Scholar]
- Felderhoff-Mueser, U.; Buhrer, C. Clinical measures to preserve cerebral integrity in preterm infants. Early Hum. Dev. 2005, 81, 237–244. [Google Scholar] [CrossRef]
- Badr Zahr, L.K.; Purdy, I. Brain injury in the infant: The old, the new, and the uncertain. J. Perinat. Neonatal Nurs. 2006, 20, 163–175. [Google Scholar]
- Hunt, E.A. Phenytoin in traumatic brain injury. Arch. Dis. Child. 2002, 86, 62–63. [Google Scholar] [CrossRef]
- McGuire, W.; Fowlie, P.W.; Evans, D.J. Naloxone for preventing morbidity and mortality in newborn infants of greater than 34 weeks’ gestation with suspected perinatal asphyxia. Cochrane Database Syst. Rev. 2004. [Google Scholar] [CrossRef]
- Kecskes, Z.; Healy, G.; Jensen, A. Fluid restriction for term infants with hypoxic-ischaemic encephalopathy following perinatal asphyxia. Cochrane Database Syst. Rev. 2005. [Google Scholar] [CrossRef]
- Beveridge, C.J.; Wilkinson, A.R. Sodium bicarbonate infusion during resuscitation of infants at birth. Cochrane Database Syst. Rev. 2006. [Google Scholar] [CrossRef]
- Evans, D.J.; Levene, M.I.; Tsakmakis, M. Anticonvulsants for preventing mortality and morbidity in full term newborns with perinatal asphyxia. Cochrane Database Syst. Rev. 2007. [Google Scholar] [CrossRef]
- Jacobs, S.; Hunt, R.; Tarnow-Mordi, W.; Inder, T.; Davis, P. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Sys. Rev. 2007. [Google Scholar] [CrossRef]
- Vannucci, R.; Towfighi, J.; Bucklacher, R.; Vannucci, S.J. Effect of extreme hypercapnia on hypoxic-ishcemic brain damage in the immature rat. Pediatr. Res. 2001, 49, 799–803. [Google Scholar] [CrossRef]
- Compagnoni, G.; Pogliani, L.; Lista, G.; Castoldi, F.; Fontana, P.; Mosca, F. Hypothermia reduces neurological damage in asphyxiated newborn infants. Biol. Neonate 2002, 82, 222–227. [Google Scholar] [CrossRef]
- Thoresen, M.; Whitelaw, A. Therapeutic hypothermia for hypoxic-ischaemic encephalopathy in the newborn infant. Curr. Opin. Neurol. 2005, 18, 111–116. [Google Scholar] [CrossRef]
- Perlman, J.M.; Wyllie, J.; Kattwinkel, J.; Atkins, D.L.; Chameides, L.; Goldsmith, J.P.; Guinsburg, R.; Hazinski, M.F.; Morley, C.; Richmond, S.; et al. Neonatal resuscitation: 2010 international consensus on cardiopulmonary resuscitation and emergency cardiovascular care science with treatment recommendations. Pediatrics 2010, 126, 1319–1344. [Google Scholar] [CrossRef]
- Shankaran, S.; Pappas, A.; McDonald, S.A.; Vohr, B.R.; Hintz, S.R.; Yolton, K.; Gustafson, K.E.; Leach, T.M.; Green, C.; Bara, R.; et al. Childhood outcomes after hypothermia for neonatal encephalopathy. N. Engl. J. Med. 2012, 366, 2085–2092. [Google Scholar] [CrossRef]
- Varon, J.; Marik, P.E.; Einav, S. Therapeutic hypothermia: A state-of-the-art emergency medicine perspective. Am. J. Emerg. Med. 2012, 30, 800–810. [Google Scholar] [CrossRef]
- Barrett, R.D.; Bennet, L.; Davidson, J.; Dean, J.M.; George, S.; Emerald, B.S.; Gunn, A.J. Destruction and reconstruction: Hypoxia and the developing brain. Birth Defects Res. C Embryo Today 2007, 81, 163–176. [Google Scholar] [CrossRef]
- Wagner, C.L.; Eicher, D.J.; Katikaneni, L.D. The use of hypothermia: A role in the treatment of neonatal asphyxia? Pediatr. Neurol. 1999, 21, 429–443. [Google Scholar] [CrossRef]
- Silverstein, F.S.; Barks, J.D.; Hagan, P.; Liu, X.H.; Ivacko, J.; Szaflarski, J. Cytokines and perinatal brain injury. Neurochem. Int. 1997, 30, 375–383. [Google Scholar] [CrossRef]
- Azzopardi, D.; Edwards, A.D. Hypothermia. Semin. Fetal Neonatal Med. 2007, 12, 303–310. [Google Scholar] [CrossRef]
- Salazar-Reyes, H.; Varon, J. Hypoxic tisue damageand the protective effects of therapeutic hypothermia. Crit. Care Shock 2005, 8, 28–31. [Google Scholar]
- Soto-Ruiz, K.M.; Varon, J. Resuscitation great. George W. Crile: A visionary mind in resuscitation. Resuscitation 2009, 80, 6–8. [Google Scholar] [CrossRef]
- Hua, Y.; Hisano, K.; Morimoto, Y. Effect of mild and moderate hypothermia on hypoxic injury in nearly pure neuronal culture. J. Anesth. 2010, 5, 726–732. [Google Scholar]
- Alzaga, A.G.; Cerdan, M.; Varon, J. Therapeutic hypothermia. Resuscitation 2006, 70, 369–380. [Google Scholar] [CrossRef]
- Varon, J.; Acosta, P. Therapeutic hypothermia: Past, present, and future. Chest 2008, 133, 1267–1274. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Wyatt, J.S.; Azzopardi, D.; Ballard, R.; Edwards, A.D.; Ferriero, D.M.; Polin, R.A.; Robertson, C.M.; Thoresen, M.; Whitelaw, A.; Gunn, A.J. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: Multicentre randomized trial. Lancet 2005, 365, 663–670. [Google Scholar]
- Shankaran, S.; Laptook, A.R.; Ehrenkranz, R.A.; Tyson, J.E.; McDonald, S.A.; Donovan, E.F.; Fanaroff, A.A.; Poole, W.K.; Wright, L.L.; Higgins, R.D.; et al. Whole body hypothermia for neonates with hypoxic-ischemic encephalopathy. N. Engl. J. Med. 2005, 353, 1574–1584. [Google Scholar] [CrossRef]
- Lin, Z.; Yu, H.; Lin, J.; Chen, S.; Liang, Z.; Zhang, Z. Mild hypothermia via selective head cooling as neuroprotective therapy in term neonates with perinatal asphyxia: An experience from a single neonatal intensive care unit. J. Perinatol. 2006, 26, 180–184. [Google Scholar] [CrossRef]
- Eicher, D.J.; Wagner, C.L.; Katikaneni, L.P.; Hulsey, T.C.; Bass, W.T.; Kaufman, D.A.; Horgan, M.J.; Languani, S.; Bhatia, J.J.; Givelichian, L.M.; et al. Moderate hypothermia in neonatal encephalopathy: Efficacy outcomes. Pediatr. Neurol. 2005, 32, 11–17. [Google Scholar] [CrossRef]
- Shah, P.S.; Ohlsson, A.; Perlman, M. Hypothermia to treat neonatal hypoxic ischemic encephalopathy: A systemic review. Arch. Pediatr. Adolesc. Med. 2007, 161, 951–958. [Google Scholar] [CrossRef]
- Pfister, R.H.; Soll, R.F. Hypothermia for the treatment of infants with hypoxic-ischemic encephalopathy. J. Perinatol. 2010, 30, 82–87. [Google Scholar] [CrossRef]
- Marks, K.; Shany, E.; Shelef, I.; Golan, A.; Zmora, E. Hypothermia: A neuroprotective therapy for neonatal hypoxic ischemic encephalopathy. Isr. Med. Assoc. J. 2010, 12, 494–500. [Google Scholar]
- Laptook, A.R. Use of therapeutic hypothermia for term infants with hypoxic-ischemic encephalopathy. Pediatr. Clin. North Am. 2009, 56, 601–616. [Google Scholar] [CrossRef]
- Kelen, D.; Robertson, N.J. Experimental treatments for hypoxic ischaemic encephalopathy. Early Hum. Dev. 2010, 86, 369–377. [Google Scholar] [CrossRef]
- Marro, P.J.; Mishra, O.P.; Delivoria-Papadopoulos, M. Effect of allopurinol on brain adenosine levels during hypoxia in newborn piglets. Brain Res. 2006, 1073–1074, 444–450. [Google Scholar] [CrossRef]
- Palmer, C.; Towfighi, J.; Roberts, R.L.; Heitjan, D.F. Allopurinol administered after inducing hypoxia-ischemia reduces brain injury in 7-day-old rats. Pediatr. Res. 1993, 33, 405–411. [Google Scholar]
- Shadid, M.; Moison, R.; Steendijk, P.; Hiltermann, L.; Berger, H.M.; van Bel, F. The effect of antioxidative combination therapy on post hypoxic-ischemic perfusion, metabolism, and electrical activity of the newborn brain. Pediatr. Res. 1998, 44, 119–124. [Google Scholar] [CrossRef]
- Betz, A.L. Identification of hypoxanthine transport and xanthine oxidase activity in brain capillaries. J. Neurochem. 1985, 44, 574–579. [Google Scholar] [CrossRef]
- Phillis, J.W.; Sen, S. Oxypurinol attenuates hydroxyl radical production during ischemia/reperfusion injury of the rat cerebral cortex: An ESR study. Brain Res. 1993, 628, 309–312. [Google Scholar] [CrossRef]
- Moorhouse, P.C.; Grootveld, M.; Halliwell, B.; Quinlan, J.G.; Gutteridge, J.M. Allopurinol and oxypurinol are hydroxyl radical scavengers. FEBS Lett. 1987, 213, 23–28. [Google Scholar] [CrossRef]
- Hudome, S.; Palmer, C.; Roberts, R.L.; Mauger, D.; Housman, C.; Towfighi, J. The role of neutrophils in the production of hypoxic-ischemic brain injury in the neonatal rat. Pediatr. Res. 1997, 41, 607–616. [Google Scholar] [CrossRef]
- Ko, K.M.; Godin, D.V. Inhibition of transition metal ion-catalysed ascorbate oxidation and lipid peroxidation by allopurinol and oxypurinol. Biochem. Pharmacol. 1990, 40, 803–809. [Google Scholar]
- Peterson, D.A.; Kelly, B.; Gerrard, J.M. Allopurinol can act as an electron transferagent. Is this relevant during reperfusion injury? Biochem. Biophys. Res. Commun. 1986, 137, 76–79. [Google Scholar] [CrossRef]
- Torrance, H.L.; Benders, M.J.; Derks, J.B.; Rademaker, C.M.; Bos, A.F.; van Den Berg, P.; Longini, M.; Buonocore, G.; Venegas, M.; Baquero, H.; et al. Maternal allopurinol during fetal hypoxia lowers cord blood levels of the brain injury marker S-100B. Pediatrics 2009, 124, 350–357. [Google Scholar] [CrossRef]
- Peeters-Scholte, C.; Braun, K.; Koster, J.; Kops, N.; Blomgren, K.; Buonocore, G.; van Buul-Offers, S.; Hagberg, H.; Nicolay, K.; van Bel, F.; Groenendaal, F. Effects of allopurinol and deferoxamine on reperfusion injury of the brain in newborn piglets after neonatal hypoxia-ischemia. Pediatr. Res. 2003, 54, 516–522. [Google Scholar] [CrossRef]
- Rodríguez-Yáñez, M.; Castillo, J. Role of inflammatory markers in brain ischemia. Curr. Opin. Neurol. 2008, 21, 353–357. [Google Scholar] [CrossRef]
- Balduini, W.; Mazzoni, E.; Carloni, S.; De Simoni, M.G.; Perego, C.; Sironi, L.; Cimino, M. Prophylactic but not delayed administration of simvastatin protects against long-lasting cognitive and morphological consequences of neonatal hypoxic-ischemic brain injury, reduces interleukin-1beta and tumor necrosis factor-alpha mRNA induction, and does not affect endothelial nitric oxide synthase expression. Stroke 2003, 34, 2007–2012. [Google Scholar] [CrossRef]
- Balduini, W.; de Angelis, V.; Mazzoni, E.; Cimino, M. Simvastatin protects against long-lasting behavioral and morphological consequences of neonatal hypoxic/ischemic brain injury. Stroke 2001, 32, 2185–2191. [Google Scholar]
- Li, A.; Lv, S.; Yu, Z.; Zhang, Y.; Ma, H.; Zhao, H.; Piao, H.; Li, S.; Zhang, N.; Sun, C. Simvastatin attenuates hypomyelination induced by hypoxia-ischemia in neonatal rats. Neurol. Res. 2010, 32, 945–952. [Google Scholar] [CrossRef]
- Franks, N.; Dickinson, R.; de Sousa, S.; Hall, A.; Lieb, W. How does xenon produce anaesthesia? Nature 1998, 26, 324. [Google Scholar]
- Perrone, S.; Stazzoni, G.; Tataranno, M.L.; Buonocore, G. New pharmacologic and therapeutic approaches for hypoxic-ischemic encephalopathy in the newborn. J. Matern. Fetal Neonatal Med. 2012, 25, 83–88. [Google Scholar]
- Petzelt, C.P.; Kodirov, S.; Taschenberger, G.; Kox, W.J. Participation of the Ca2+-calmodulin-activated kinase in the control of metaphase-anaphase transition in human cells. Cell Biol. Int. 2001, 25, 403–409. [Google Scholar] [CrossRef]
- Ma, D.; Williamson, P.; Januszewski, A.; Nogaro, M.C.; Hossain, M.; Ong, L.; Shu, Y.; Franks, N.P.; Maze, M. Xenon mitigates isoflurane-induced neuronal apoptosis in the developing rodent brain. Anaesthesiology 2007, 106, 746–753. [Google Scholar] [CrossRef]
- Ma, D.; Lim, T.; Xu, J.; Tang, H.; Wan, Y.; Zhao, H.; Hossain, M.; Maxwell, P.H.; Maze, M. Xenon preconditioning protects against renal ischemic-reperfusion injury via HIF-1alpha activation. J. Am. Soc. Nephrol. 2009, 20, 713–720. [Google Scholar] [CrossRef]
- Ma, D.; Hossain, M.; Chow, A.; Arshad, M.; Battson, R.M.; Sanders, R.D.; Mehmet, H.; Edwards, A.D.; Franks, N.P.; Maze, M. Xenon and hypothermia combine to provide neuroprotection from neonatal asphyxia. Ann. Neurol. 2005, 58, 182–193. [Google Scholar] [CrossRef]
- Dingley, J.; Tooley, J.; Porter, H.; Thoresen, M. Xenon provides short-term neuroprotection in neonatal rats when administered after hypoxia-ischemia. Stroke 2006, 37, 501–506. [Google Scholar] [CrossRef]
- Martin, J.L.; Ma, D.; Hossain, M.; Xu, J.; Sanders, R.D.; Franks, N.P.; Maze, M. Asynchronous administration of xenon and hypothermia significantly reduces brain infarction in the neonatal rat. Br. J. Anaesth. 2007, 98, 236–240. [Google Scholar] [CrossRef]
- Thoresen, M.; Hobbs, C.E.; Wood, T.; Chakkarapani, E.; Dingley, J. Cooling combined with immediate or delayed xenon inhalation provides equivalent long-term neuroprotection after neonatal hypoxia-ischemia. J. Cereb. Blood Flow Metab. 2009, 4, 707–714. [Google Scholar]
- Ryang, Y.M.; Fahlenkamp, A.V.; Rossaint, R.; Wesp, D.; Loetscher, P.D.; Beyer, C.; Coburn, M. Neuroprotective effects of argon in an in vivo model of transient middle cerebral artery occlusion in rats. Crit. Care Med. 2011, 39, 1448–1453. [Google Scholar] [CrossRef]
- Ovbiagele, B.; Kidwell, C.S.; Starkman, S.; Saber, J.L. Neuroprotective agents for the treatment of acute ischemic stroke. Curr. Neurol. Neurosci. Rep. 2003, 3, 9–20. [Google Scholar] [CrossRef]
- Spandou, E.; Soubasi, V.; Papoutsopoulou, S.; Augoustides-Savvopoulou, P.; Loizidis, T.; Pazaiti, A.; Karkavelas, G.; Guiba-Tziampiri, O. Neuroprotective effect of long-term MgSO4 administration after cerebral hypoxia-ischemia in newborn rats is related to the severity of brain damage. Reprod. Sci. 2007, 14, 667–677. [Google Scholar] [CrossRef]
- Marret, S.; Gressens, P.; Gadisseux, J.F.; Evrard, P. Prevention by magnesium of excitotoxic neuronal death in the developing brain: An animal model for clinical intervention studies. Dev. Med. Child Neurol. 1995, 37, 473–484. [Google Scholar]
- Imer, M.; Omay, B.; Uzunkol, A.; Erdem, T.; Sabanci, P.A.; Karasu, A.; Albayrak, S.B.; Sencer, A.; Hepgul, K.; Kaya, M. Effect of magnesium, MK-801 and combination of magnesium and MK-801 on blood-brain barrier permeability and brain edema after experimental traumatic diffuse brain injury. Neurol. Res. 2009, 31, 977–981. [Google Scholar] [CrossRef]
- Enomoto, T.; Osugi, T.; Satoh, H.; McIntosh, T.K.; Nabeshima, T. Pre-Injury magnesium treatment prevents traumatic brain injury-induced hippocampal ERK activation, neuronal loss, and cognitive dysfunction in the radial-arm maze test. J. Neurotrauma 2005, 22, 783–792. [Google Scholar] [CrossRef]
- Türkyilmaz, C.; Türkyilmaz, Z.; Atalay, Y.; Söylemezoglu, F.; Celasun, B. Magnesium pre-treatment reduces neuronal apoptosis in newborn rats in hypoxia-ischemia. Brain Res. 2002, 955, 133–137. [Google Scholar] [CrossRef]
- Goñi-de-Cerio, F.; Alvarez, A.; Alvarez, F.J.; Rey-Santano, M.C.; Alonso-Alconada, D.; Mielgo, V.E.; Gastiasoro, E.; Hilario, E. MgSO4 treatment preserves the ischemia-induced reduction in S-100 protein without modification of the expression of endothelial tight junction molecules. Histol. Histopathol. 2009, 24, 1129–1138. [Google Scholar]
- Westermaier, T.; Zausinger, S.; Baethmann, A.; Schmid-Elsaesser, R. Dose finding study of intravenous magnesium sulphate in transient focal cerebral ischemia in rats. Acta Neurochir. 2005, 147, 525–532. [Google Scholar] [CrossRef]
- Rouse, D.J.; Hirtz, D.G.; Thom, E. A randomized, controlled trial of magnesium sulfate for the prevention of cerebral palsy. N. Engl. J. Med. 2008, 359, 895–905. [Google Scholar] [CrossRef]
- Szemraj, J.; Sobolewska, B.; Gulczynska, E.; Wilczynski, J.; Zylinska, L. Magnesium sulfate effect on erythrocyte membranes of asphyxiated newborns. Clin. Biochem. 2005, 38, 457–464. [Google Scholar] [CrossRef]
- Groenendaal, F.; Rademaker, C.M.; Toet, M.C.; de Vries, L.S. Effects of magnesium sulphate on amplitude-integrated continuous EEG in asphyxiated term neonates. Acta Paediatr. 2002, 91, 1073–1077. [Google Scholar] [CrossRef]
- Khashaba, M.T.; Shouman, B.O.; Shaltout, A.A.; Al-Marsafawy, H.M.; Abdel-Aziz, M.M.; Patel, K.; Aly, H. Excitatory amino acids and magnesium sulfate in neonatal asphyxia. Brain Dev. 2006, 28, 375–379. [Google Scholar] [CrossRef]
- Levene, M.; Blennow, M.; Whitelaw, A.; Hankø, E.; Fellman, V.; Hartley, R. Acute effects of two different doses of magnesium sulphate in infants with birth asphyxia. Arch. Dis. Child. Fetal Neonatal Ed. 1995, 73, F174–F177. [Google Scholar] [CrossRef]
- Temkin, N.; Anderson, G.D.; Winn, H.R.; Ellenbogen, R.G.; Britz, G.W.; Schuster, J.; Lucas, T.; Newell, D.W.; Mansfield, P.N.; Machamer, J.E.; et al. Magnesium sulfate for neuroprotection after traumatic brain injury: A randomised controlled trial. Lancet Neurol. 2007, 6, 29–38. [Google Scholar]
- Alison, G.; Cahill, M.D.; Aaron, B.; Caughey, M.D. Magnesium for neuroprophylaxis: Fact or fiction? Am. J. Obstet. Gynecol. 2009, 200, 590–594. [Google Scholar] [CrossRef]
- Elliott, J.P.; Lewis, D.F.; Morrison, J.C.; Garite, T.J. In defense of magnesium sulfate. Obstet. Gynecol. 2009, 113, 341–1348. [Google Scholar]
- Costantine, M.M.; Drever, N. Antenatal exposure to magnesium sulfate and neuroprotection in preterm infants. Obstet. Gynecol. Clin. North Am. 2011, 38, 351–366. [Google Scholar] [CrossRef]
- Zhu, H.; Meloni, B.P.; Bojarski, C.; Knuckey, M.W.; Knuckey, N.W. Post-ischemic modest hypothermia (35 degrees C) combined with intravenous magnesium is more effective at reducing CA1 neuronal death than either treatment used alone following global cerebral ischemia in rats. Exp. Neurol. 2005, 193, 361–368. [Google Scholar] [CrossRef]
- Chacón, A.; Lisott, E.; Eblen-Zajjur, A. Magnesium sulphate reduces cell volume in physiological conditions but not in the cytotoxic oedema during global brain ischemia. Brain Inj. 2006, 20, 1087–1091. [Google Scholar] [CrossRef]
- Pazaiti, A.; Soubasi, V.; Spandou, E.; Karkavelas, G.; Georgiou, T.; Karalis, P.; Guiba-Tziampiri, O. Evaluation of long-lasting sensorimotor consequences following neonatal hypoxic-ischemic brain injury in rats: the neuroprotective role of MgSO4. Neonatology 2009, 95, 33–40. [Google Scholar] [CrossRef]
- Cetinkaya, M.; Alkan, T.; Ozyener, F.; Kafa, I.M.; Kurt, M.A.; Koksal, N. Possible neuroprotective effects of magnesium sulfate and melatonin as both pre- and post-treatment in a neonatal hypoxic-ischemic rat model. Neonatology 2010, 99, 302–310. [Google Scholar]
- Solaroglu, A.; Suat Dede, F.; Gelisen, O.; Secilmis, O.; Dede, H. Neuroprotective effect of magnesium sulfate treatment on fetal brain in experimental intrauterine ischemia reperfusion injury. J. Matern. Fetal Neonatal Med. 2011, 24, 1259–1261. [Google Scholar] [CrossRef]
- Kang, S.W.; Choi, S.K.; Park, E.; Chae, S.J.; Choi, S.; Joo, H.J.; Lee, G.J.; Park, H.K. Neuroprotective effects of magnesium-sulfate on ischemic injury mediated by modulating the release of glutamate and reduced of hyperreperfusion. Brain Res. 2011, 1371, 121–128. [Google Scholar] [CrossRef]
- Greenwood, K.; Cox, P.; Mehmet, H.; Penrice, J.; Amess, P.N.; Cady, E.B.; Wyatt, J.S.; Edwards, A.D. Magnesium sulfate treatment after transient hypoxia-ischemia in newborn piglet does not protect against cerebral damage. Pediatric Res. 2000, 48, 346–350. [Google Scholar] [CrossRef]
- Gee, J.B.; Rorbett, R.J.; Perlman, J.; Laptook, A.R. The effects of systemic magnesium sulfate infusion on brain magnesium concentrations and energy state during hypoxia-ischemia in newborn miniswine. Pediatr. Res. 2004, 55, 93–100. [Google Scholar] [CrossRef]
- Zhu, H.D.; Meloni, B.P.; Moore, S.R.; Majda, B.T.; Knuckey, N.W. Intravenous administration of magnesium is only neuroprotective following transient global ischemia when present with post-ischemic mild hypothermia. Brain Res. 2004, 1014, 53–60. [Google Scholar] [CrossRef]
- Kalincik, T.; Maresova, D. Influence of magnesium sulphate on evoked activity of rat brain after exposure to short-term hypoxia. Physiol. Res. 2005, 54, 229–234. [Google Scholar]
- Dribben, W.H.; Creeley, C.E.; Wang, H.H.; Smith, D.J.; Farber, N.B.; Olney, J.W. High dose magnesium sulfate exposure induces apoptotic cell death in the developing neonatal mouse brain. Neonatology 2009, 96, 23–32. [Google Scholar] [CrossRef]
- Yager, J.Y. Animal models of hypoxic-ischemic brain damage in the newborn. Semin. Pediatr. Neurol. 2004, 11, 31–46. [Google Scholar] [CrossRef]
- Mechoulam, R.; Panikashvili, D.; Shohami, E. Cannabinoids and brain injury: Therapeutic implications. Trends Mol. Med. 2002, 8, 58–61. [Google Scholar] [CrossRef]
- Amar, M.B. Cannabinoids in medicine: A review of their therapeutic potential. J. Ethnopharmacol. 2006, 105, 1–25. [Google Scholar] [CrossRef]
- Maresz, K.; Pryce, G.; Ponomarev, E.D.; Marsicano, G.; Croxford, J.L.; Shriver, L.P.; Ledent, C.; Cheng, X.; Carrier, E.J.; Mann, M.K.; et al. Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB(1) on neurons and CB(2) on autoreactive T cells. Nat. Med. 2007, 13, 492–497. [Google Scholar]
- Baker, D.; Pryce, G.; Croxford, J.L.; Brown, P.; Pertwee, R.G.; Makriyannis, A.; Khanolkar, A.; Layward, L.; Fezza, F.; Bisogno, T.; Di Marzo, V. Endocannabinoids control spasticity in a multiple sclerosis model. FASEB J. 2001, 15, 300–302. [Google Scholar]
- Marsicano, G.; Goodenough, S.; Monory, K.; Hermann, H.; Eder, M.; Cannich, A.; Azad, S.C.; Cascio, M.G.; Gutiérrez, S.O.; van der Stelt, M.; et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 2003, 302, 84–88. [Google Scholar] [CrossRef]
- Chang, Y.H.; Lee, S.T.; Lin, W.W. Effects of cannabinoids on LPS-stimulated inflammatory mediator release from macrophages: Involvement of eicosanoids. J. Cell. Biochem. 2001, 81, 715–723. [Google Scholar]
- Walter, L.; Franklin, A.; Witting, A.; Wade, C.; Xie, Y.; Kunos, G.; Mackie, K.; Stella, N. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J. Neurosci. 2003, 23, 1398–1405. [Google Scholar]
- Parmentier-Batteur, S.; Jin, K.; Mao, X.O.; Xie, L.; Greenberg, D.A. Increased severity of stroke in CB1 cannabinoid receptor knock-out mice. J. Neurosci. 2002, 22, 9771–9775. [Google Scholar]
- Freund, T.F.; Katona, I.; Piomelli, D. Role of endogenous cannabinoids in synaptic signaling. Physiol. Rev. 2003, 83, 1017–1066. [Google Scholar]
- Pacher, P.; Bátkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef]
- Hajos, N.; Ledent, C.; Freund, T.F. Novel cannabinoid-sensitive receptor mediates inhibition of glutamatergic synaptic transmission in the hippocampus. Neuroscience 2001, 106, 1–4. [Google Scholar] [CrossRef]
- Breivogel, C.S.; Walker, J.M.; Huang, S.M.; Roy, M.B.; Childers, S.R. Cannabinoid signaling in rat cerebellar granule cells: G-protein activation, inhibition of glutamate release and endogenous cannabinoids. Neuropharmacology 2004, 47, 81–91. [Google Scholar] [CrossRef]
- Hamrick, S.E.; Ferriero, D.M. The injury response in the term newborn brain. Can we neuroprotect? Curr. Opin. Neurol. 2003, 16, 147–154. [Google Scholar] [CrossRef]
- Kim, S.H.; Won, S.J.; Mao, X.O.; Jin, K.; Greenberg, D.A. Molecular mechanisms of cannabinoid protection from neuronal excitotoxicity. Mol. Pharmacol. 2006, 69, 691–696. [Google Scholar]
- Hampson, A.J.; Grimaldi, M.; Axelrod, J.; Wink, D. Cannabidiol and (−)Δ9-tetrahydrocannabinol are neuroprotective antioxidants. Proc. Natl. Acad. Sci. USA 1998, 95, 8268–8273. [Google Scholar] [CrossRef]
- Marsicano, G.; Moosmann, B.; Hermann, H.; Lutz, B.; Behl, C. Neuroprotective properties of cannabinoids against oxidative stress: Role of the cannabinoid receptor CB1. J. Neurochem. 2002, 80, 448–456. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Fernández-Ruiz, J. An overview of Parkinson’s disease and the cannabinoid system and possible benefits of cannabinoid-based treatments. Curr. Med. Chem. 2006, 13, 3705–3718. [Google Scholar] [CrossRef]
- Hillard, C.J. Endocannabinoids and vascular function. J. Pharmacol. Exp. Ther. 2000, 294, 27–32. [Google Scholar]
- Golech, S.A.; McCarron, R.M.; Chen, Y.; Bembry, J.; Lenz, F.; Mechoulam, R.; Shohami, E.; Spatz, M. Human brain endothelium: Coexpression and function of vanilloid and endocannabinoid receptors. Brain Res. Mol. Brain Res. 2004, 132, 87–92. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, J.; Berrendero, F.; Hernandez, M.L.; Ramos, J.A. The endogenous cannabinoid system and brain development. Trends Neurosci. 2000, 23, 14–20. [Google Scholar] [CrossRef]
- Stella, N. Cannabinoid signaling in glial cells. Glia 2004, 48, 267–277. [Google Scholar] [CrossRef]
- Aguado, T.; Palazuelos, J.; Monory, K.; Stella, N.; Cravatt, B.; Lutz, B.; Marsicano, G.; Kokaia, Z.; Guzmán, M.; Galve-Roperh, I. The endocannabinoid system promotes astroglial differentiation by acting on neural progenitor cells. J. Neurosci. 2006, 26, 1551–1561. [Google Scholar] [CrossRef]
- Sinor, A.D.; Irvin, S.M.; Greenberg, D.A. Endocannabinoids protect cerebral cortical neurons from in vitro ischemia in rats. Neurosci. Lett. 2000, 278, 157–160. [Google Scholar] [CrossRef]
- Van der Stelt, M.; Veldhuis, W.B.; van Haaften, G.W.; Fezza, F.; Bisogno, T.; Bar, P.R.; Veldink, G.A.; Vliegenthart, J.F.; Di Marzo, V.; Nicolay, K. Exogenous anandamide protects rat brain against acute neuronal injury in vivo. J. Neurosci. 2001, 21, 8765–8771. [Google Scholar]
- Panikashvili, D.; Simeonidou, C.; Ben-Shabat, S.; Hanus, L.; Breuer, A.; Mechoulam, R.; Shohami, E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 2001, 413, 527–531. [Google Scholar]
- Lara-Celador, I.; Castro-Ortega, L.; Alvarez, A.; Goñi-de-Cerio, F.; Lacalle, J.; Hilario, E. Endocannabinoids reduce cerebral damage after hypoxic-ischemic injury in perinatal rats. Brain Res. 2012, 1474, 91–99. [Google Scholar] [CrossRef]
- Chen, Y.C.; Tain, Y.L.; Sheen, J.M.; Huang, L.T. Melatonin utility in neonates and children. J. Formos. Med. Assoc. 2012, 111, 57–66. [Google Scholar] [CrossRef]
- Carloni, S.; Buonocore, G.; Balduini, W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol. Dis. 2008, 32, 329–339. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Mayo, J.C.; Sainz, R.M.; Lopez-Burillo, S. Melatonin, longevity and health in the aged: An assessment. Free Radic. Res. 2002, 36, 1323–1329. [Google Scholar] [CrossRef]
- Sola, A.; Wen, T.C.; Hamrick, S.E.; Ferriero, D.M. Potential for protection and repair following injury to the developing brain: A role for erythropoietin. Pediatr. Res. 2005, 57, 110–117. [Google Scholar] [CrossRef]
- Chang, Y.S.; Mu, D.; Wendland, M.; Sheldon, R.A.; Vexler, Z.S.; McQuillen, P.S.; Ferriero, D.M. Eryhropoietin improves functional and histological outcome in neonatal stroke. Pediatr. Res. 2005, 59, 106–111. [Google Scholar]
- Gonzalez, F.F.; McQuillen, P.; Mu, D.; Chang, Y.; Wendland, M.; Vexler, Z.; Ferriero, D.M. Erythropoietin enhaces long term neuropretection and neurogenesis in neonatal stroke. Dev. Neurosci. 2007, 29, 321–330. [Google Scholar] [CrossRef]
- Brion, L.P.; Bell, E.F.; Raghuveer, T.S. Vitamin E supplementation for prevention of morbidity and mortality in preterm infants. Cochrane Database Syst. Rev. 2003. [Google Scholar] [CrossRef]
- Peeters-Scholte, C.; van den Tweel, E.; Ioroi, T.; Post, I.; Braun, K.; Veldhuis, W.; Nicolay, K.; Groenendaal, F.; van Bel, F. Pharmacological interventions in the newborn piglet in the first 24 h after hypoxia-ischemia. A hemodynamic and electrophysiological perspective. Exp. Brain Res. 2002, 147, 200–208. [Google Scholar] [CrossRef]
- deLemos, R.A.; Roberts, R.J.; Coalson, J.J.; deLemos, J.A.; Null, D.M.; Gerstmann, D.R. Toxic effects associated with the administration of deferoxamine in the premature baboon with hyaline membrane disease. Am. J. Dis. Child. 1990, 144, 915–919. [Google Scholar]
- Coopman, K.; Smith, L.D.; Wright, K.L.; Ward, S.G. Temporal variation in CB2R levels following T lymphocyte activation: evidence that cannabinoids modulate CXCL12-induced chemotaxis. Int. Immunopharmacol. 2007, 7, 360–371. [Google Scholar] [CrossRef]
- Romero-Sandoval, E.A.; Horvath, R.; Landry, R.P.; DeLeo, J.A. Cannabinoid receptor type 2 activation induces a microglial anti-inflammatory phenotype and reduces migration via MKP induction and ERK dephosphorylation. Mol. Pain 2009, 5, 25. [Google Scholar] [CrossRef]
- Fernández-López, D.; Pradillo, J.M.; García-Yébenes, I.; Martínez-Orgado, J.A.; Moro, M.A.; Lizasoain, I. The cannabinoid WIN55212-2 promotes neural repair after neonatal hypoxia-ischemia. Stroke 2010, 41, 2956–2964. [Google Scholar] [CrossRef]
- Zhang, M.; Martin, B.R.; Adler, M.W.; Razdan, R.J.; Kong, W.; Ganea, D.; Tuma, R.F. Modulation of cannabinoid receptor activation as a neuroprotective strategy for EAE and stroke. J. Neuroimmun. Pharmacol. 2009, 4, 249–259. [Google Scholar]
- Sekhon, B.; Jatana, M.; Giri, S.; Gilg, A.G.; Sekhon, C.; Singh, I.; Singh, A.K. Administration of N-acetylcysteine after focal cerebral ischemia protects brain and reduces inflammation in a rat model of experimental stroke. J. Neurosci. Res. 2004, 76, 519–527. [Google Scholar] [CrossRef]
- Ahola, T.; Lapatto, R.; Raivio, K.O.; Selander, B.; Stigson, L.; Jonsson, B.; Jonsbo, F.; Esberg, G.; Stövring, S.; Kjartansson, S.; et al. N-acetylcysteine does not prevent bronchopulmonary dysplasia in immature infants: a randomized controlled trial. J. Pediatr. 2003, 143, 713–719. [Google Scholar] [CrossRef]
- Jatana, M.; Singh, I.; Singh, A.; Jenkins, D. Combination of systemic hypothermia and N-acetylcysteine attenuates hypoxic-ischemic brain injury in neonatal rats. Pediatr. Res. 2006, 59, 684–689. [Google Scholar] [CrossRef]
- Esposito, E.; Cuzzocrea, S. Antiinflammatory activity of melatonin in central nervous system. Curr. Neuropharmacol. 2010, 8, 228–242. [Google Scholar] [CrossRef]
- Deng, W.G.; Tang, S.T.; Tseng, H.P.; Wu, K.K. Melatonin suppresses macrophage cyclooxygenase-2 and inducible nitric oxide synthase expression by inhibiting p52 acetylation and binding. Blood 2006, 108, 518–524. [Google Scholar] [CrossRef]
- Welin, A.K.; Svedin, P.; Lapatto, R.; Sultan, B.; Hagberg, H.; Gressens, P.; Kjellmer, I.; Mallard, C. Melatonin reduces inflammation and cell death in white matter in the mid-gestation fetal sheep following umbilical cord occlusion. Pediatr. Res. 2007, 61, 153–158. [Google Scholar] [CrossRef]
- Pandi-Perumal, S.R.; Srinivasan, V.; Maestroni, G.J.; Cardinali, D.P.; Poeggeler, B.; Hardeland, R. Melatonin: Natures most versatile biological signal? FEBS J. 2006, 273, 2813–2838. [Google Scholar] [CrossRef]
- Elmahdy, H.; El-Mashad, A.R.; El-Bahrawy, H.; El-Gohary, T.; El-Barbary, A.; Aly, H. Human recombinant erythropoietin in asphyxia neonatorum: Pilot trial. Pediatrics 2010, 125, 1135–1142. [Google Scholar] [CrossRef]
- Wei, L.; Han, B.H.; Li, Y.; Keogh, C.L.; Holtzman, D.M.; Yu, S.P. Cell death mechanism and protective effect of erythropoietin after focal ischemia in the whisker-barrel cortex of neonatal rats. J. Pharmacol. Exp. Ther. 2006, 317, 109–116. [Google Scholar]
- Sun, Y.; Calvert, J.W.; Zhang, J.H. Neonatal hypoxia/ischemia is associated with decreased inflammatory mediators after erythropoietin administration. Stroke 2005, 36, 1672–1678. [Google Scholar] [CrossRef]
- Juul, S.E.; Beyer, R.P.; Bammler, T.K.; McPherson, R.J.; Wilkerson, J.; Farin, F.M. Microarray analysis of high-dose recombinant erythropoietin treatment of unilateral brain injury in neonatal mouse hippocampus. Pediatr. Res. 2009, 65, 485–492. [Google Scholar] [CrossRef]
- Slusarski, J.D.; McPherson, R.J.; Wallace, G.N.; Juul, S.E. High-dose erythropoietin does not exacerbate retinopathy of prematurity in rats. Pediatr. Res. 2009, 66, 625–630. [Google Scholar] [CrossRef]
- Iwai, M.; Stetler, R.A.; Xing, J.; Hu, X.; Gao, Y.; Zhang, W.; Chen, J.; Cao, G. Enhanced oligodendrogenesis and recovery of neurological function by erythropoietin after neonatal hypoxic/ischemic brain injury. Stroke 2010, 4, 1032–1037. [Google Scholar]
- Nijboer, C.H.; Groenendaal, F.; Kavelaars, A.; Hagberg, H.H.; van Bel, F.; Heijnen, C.J. Gender-specific neuroprotection by 2-iminobiotin after hypoxia-ischemia in the neonatal rat via a nitric oxide independent pathway. J. Cereb. Blood Flow Metab. 2007, 27, 282–292. [Google Scholar]
- Bartley, J.; Soltau, T.; Wimborne, H.; Kim, S.; Martin-Studdard, A.; Hess, D. BRDU-positive cells in the neonatal mouse hippocampus following hypoxic-ischemic brain injury. BMC Neurosci. 2005, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Covey, M.V.; Bitel, C.L.; Ni, L.; Jonakait, G.M.; Levison, S.W. Sustained neocortical neurogenesis after neonatal hypoxic/ischemic injury. Ann. Neurol. 2007, 61, 199–208. [Google Scholar] [CrossRef]
- Ong, J.; Plane, J.M.; Parent, J.M.; Silverstein, F.S. Hypoxic-ischemic injury stimulates subventricular zone proliferation and neurogenesis in the neonatal rat. Pediatr. Res. 2005, 58, 600–606. [Google Scholar] [CrossRef]
- Van Velthoven, C.T.; Kavelaars, A.; van Bel, F. Regeneration of the ischemic brain by engineered stem cells: fuelling endogenous repair processes. Brain Res. Rev. 2009, 61, 1–13. [Google Scholar] [CrossRef]
- Wei, X.; Du, Z.; Zhao, L.; Feng, D.; Wei, G.; He, Y.; Tan, J.; Lee, W.H.; Hampel, H.; Dodel, R.; et al. IFATS collection: The conditioned media of adipose stromal cells protect against hypoxia-ischemia-induced brain damage in neonatal rats. Stem Cells 2009, 27, 478–488. [Google Scholar] [CrossRef]
- Rivera, F.J.; Sierralta, W.D.; Minguell, J.J.; Aigner, L. Adult hippocampus derived soluble factors induce a neuronal-like phenotype in mesenchymal stem cells. Neurosci. Lett. 2006, 406, 49–54. [Google Scholar] [CrossRef]
- Van Velthoven, C.T.; Kavelaars, A.; van Bel, F.; Heijnen, C.J. Mesenchymal stem cell treatment after neonatal hypoxic-ischemic brain injury improves behavioral outcome and induces neuronal and oligodendrocyte regeneration. Brain Behav. Immun. 2010, 24, 387–393. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cerio, F.G.d.; Lara-Celador, I.; Alvarez, A.; Hilario, E. Neuroprotective Therapies after Perinatal Hypoxic-Ischemic Brain Injury. Brain Sci. 2013, 3, 191-214. https://doi.org/10.3390/brainsci3010191
Cerio FGd, Lara-Celador I, Alvarez A, Hilario E. Neuroprotective Therapies after Perinatal Hypoxic-Ischemic Brain Injury. Brain Sciences. 2013; 3(1):191-214. https://doi.org/10.3390/brainsci3010191
Chicago/Turabian StyleCerio, Felipe Goñi de, Idoia Lara-Celador, Antonia Alvarez, and Enrique Hilario. 2013. "Neuroprotective Therapies after Perinatal Hypoxic-Ischemic Brain Injury" Brain Sciences 3, no. 1: 191-214. https://doi.org/10.3390/brainsci3010191