Properties of Thermosets Derived from Chemically Modified Triglycerides and Bio-Based Comonomers

Abstract
:1. Introduction
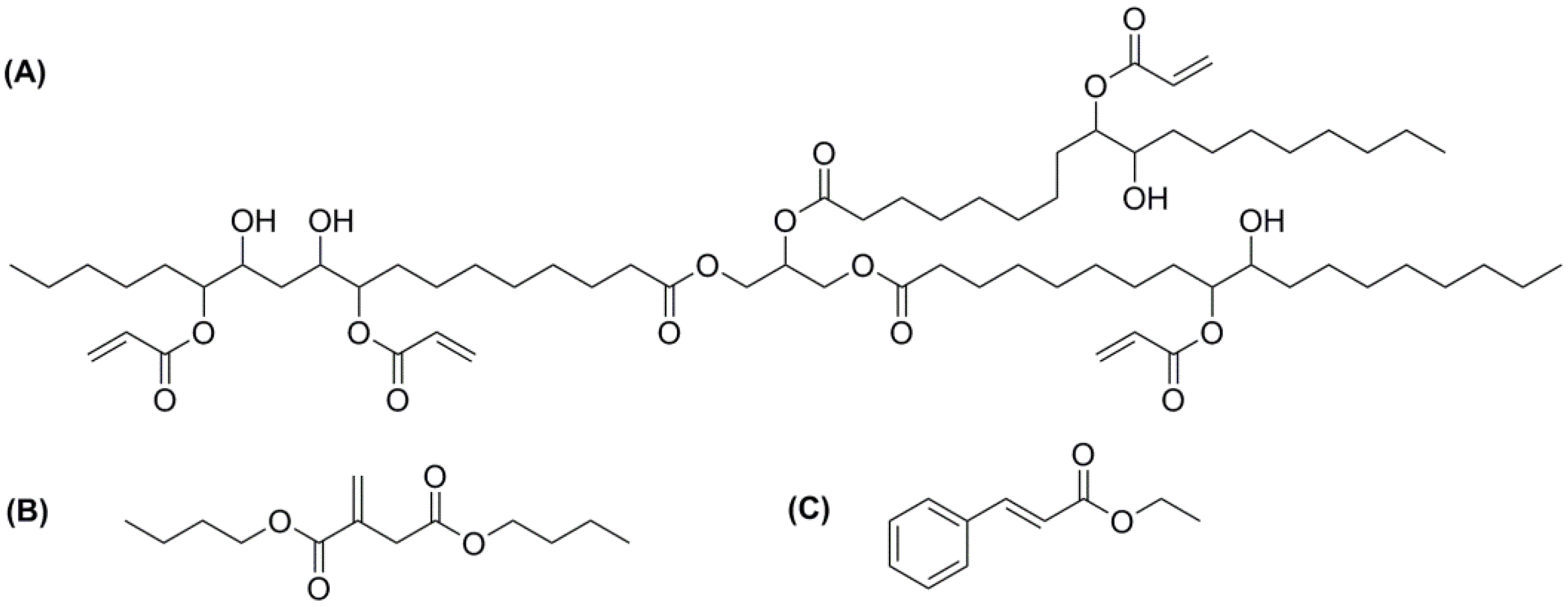
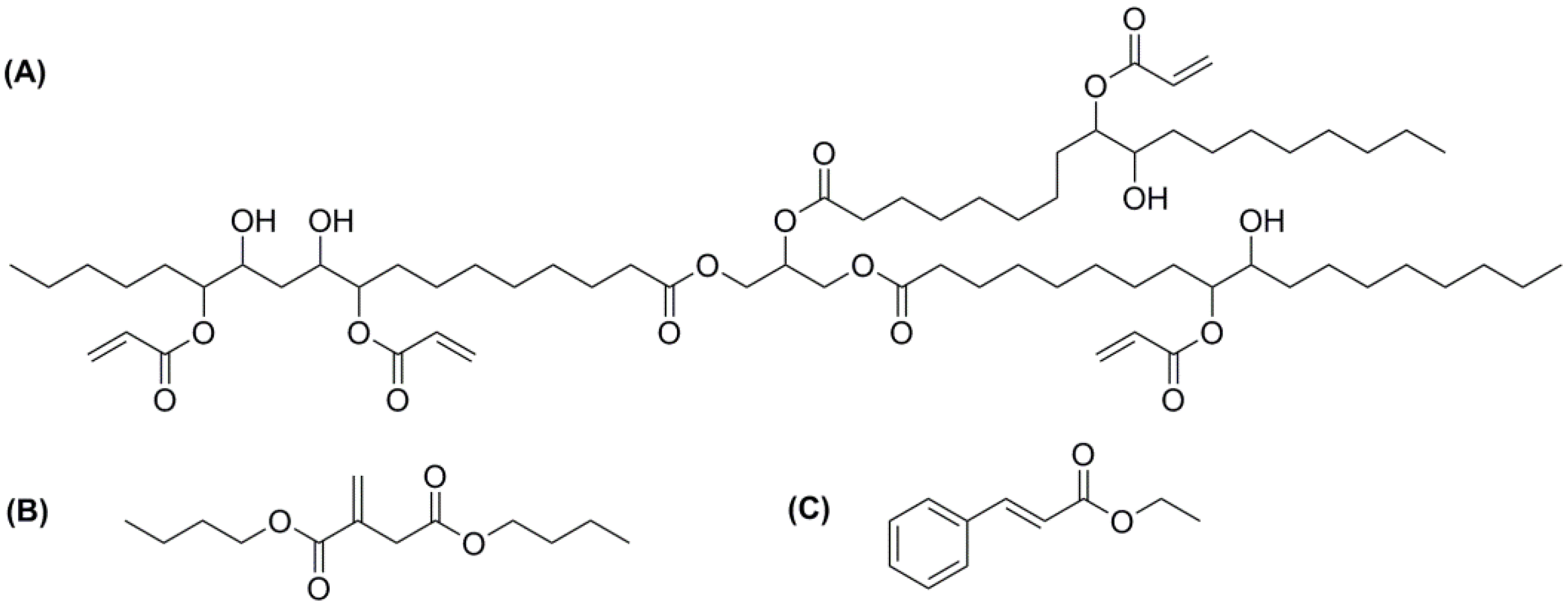
2. Experimental Section
2.1. General
2.2. Synthesis of Homopolymers
2.3. Representative Procedure for Copolymerization of the Bio-Based Resins
2.4. Determination of Extractable Content in the Bio-Based Resins
3. Results and Discussion
3.1. Degree of Incorporation of Bio-Based Monomers
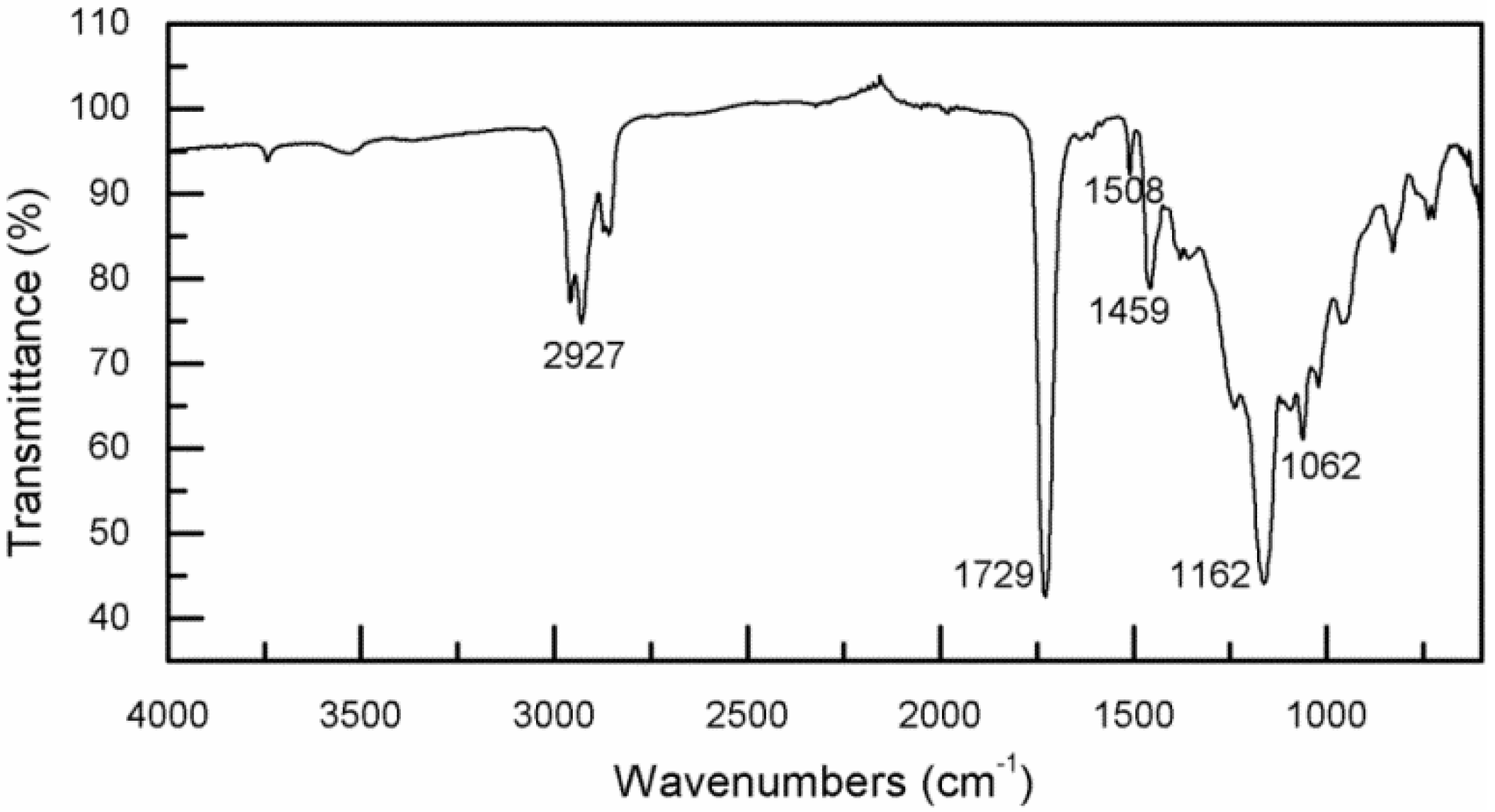
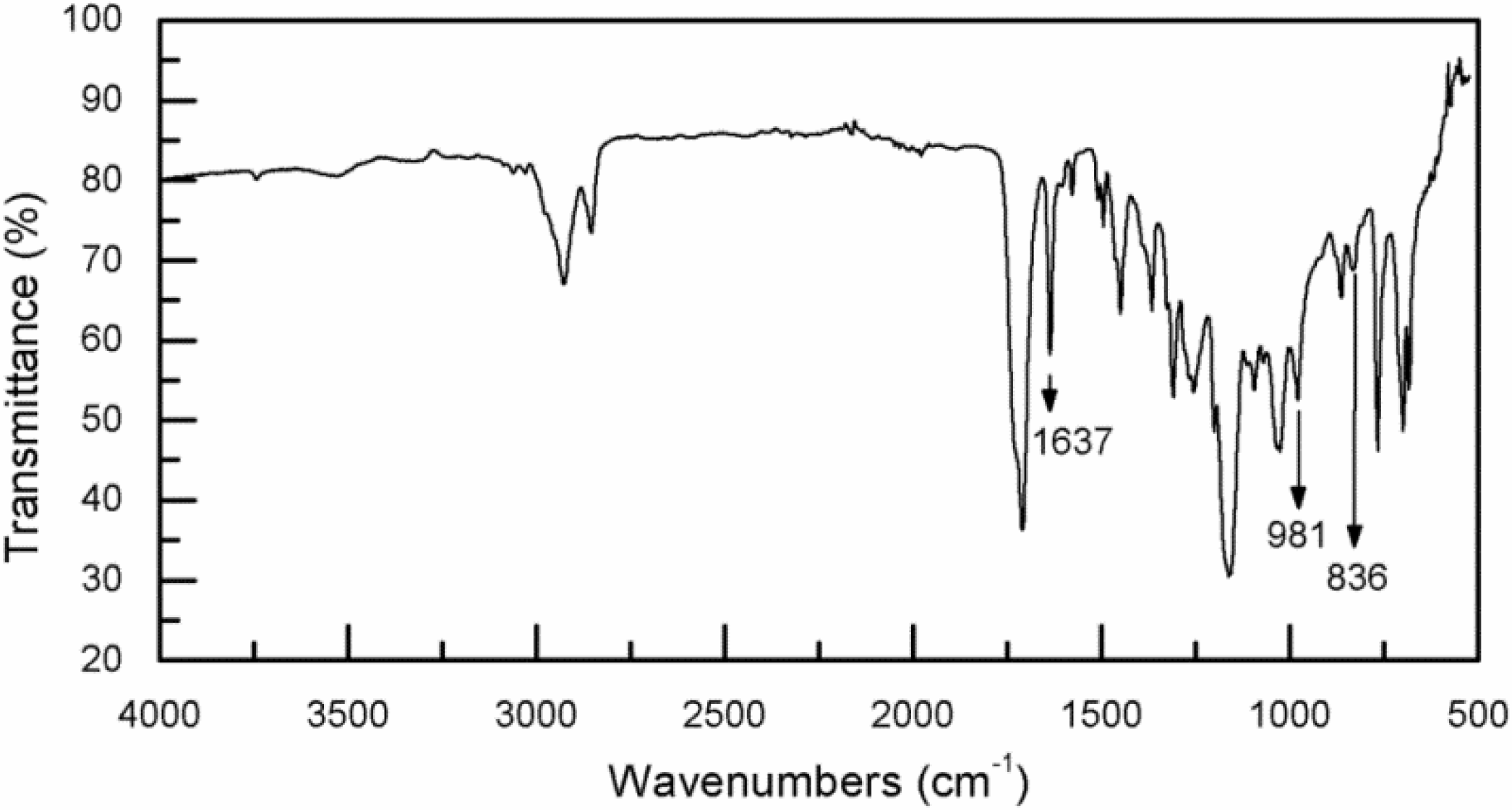
3.2. Infrared Spectroscopy


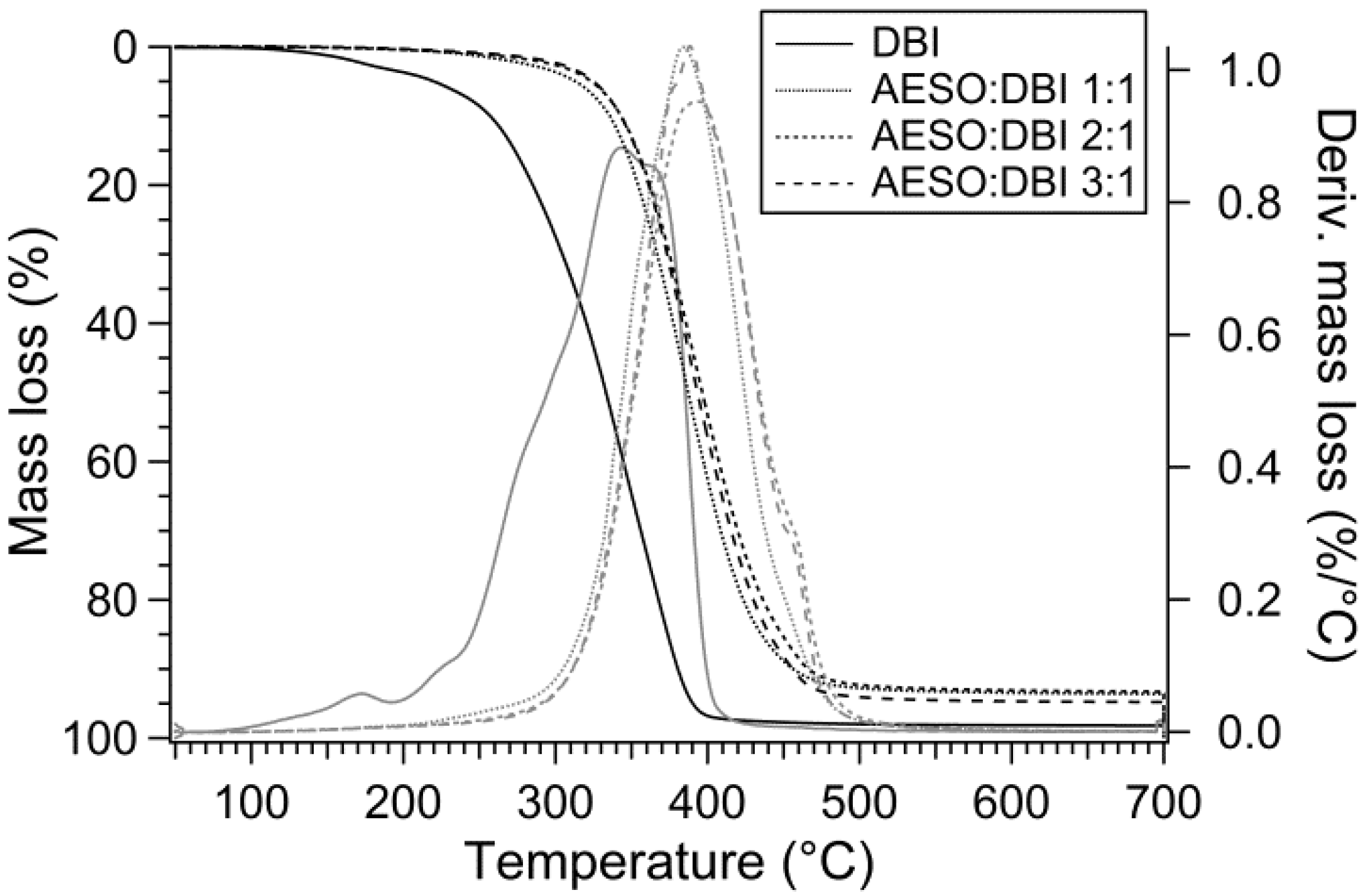
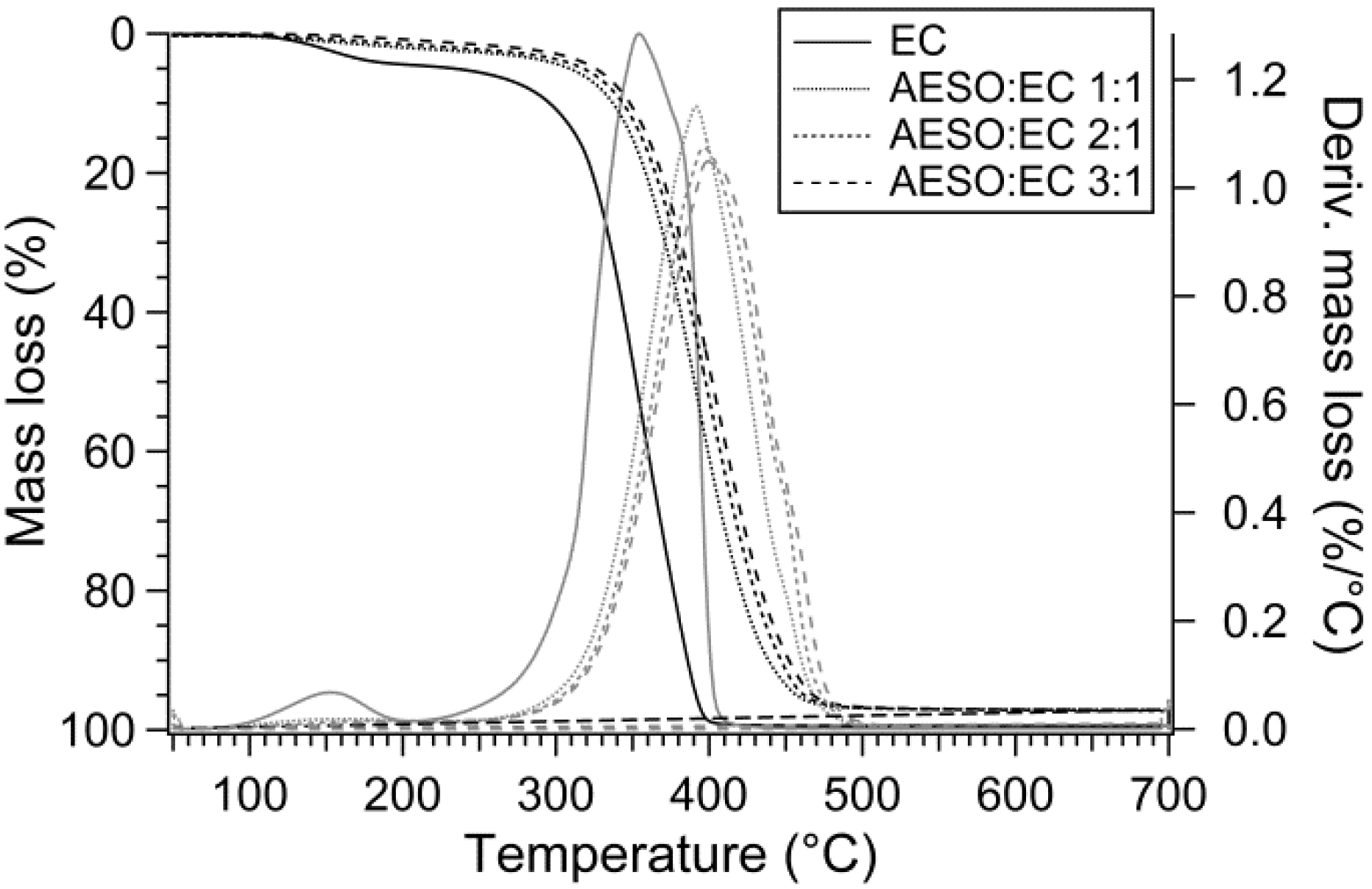
3.3. Thermal Stability


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | T5 (°C) | T10 (°C) | T50 (°C) | Char yield |
|---|---|---|---|---|
| DBI homopolymer | 220 | 257 | 333 | 1.74% |
| AESO:DBI 1:1 | 313 | 336 | 387 | 6.34% |
| AESO:DBI 2:1 | 324 | 343 | 397 | 6.66% |
| AESO:DBI 3:1 | 325 | 343 | 393 | 5.16% |
| EC homopolymer | 233 | 296 | 352 | 0.53% |
| AESO:EC 1:1 | 309 | 337 | 390 | 2.86% |
| AESO:EC 2:1 | 318 | 344 | 398 | 2.42% |
| AESO:EC 3:1 | 326 | 348 | 402 | 2.84% |


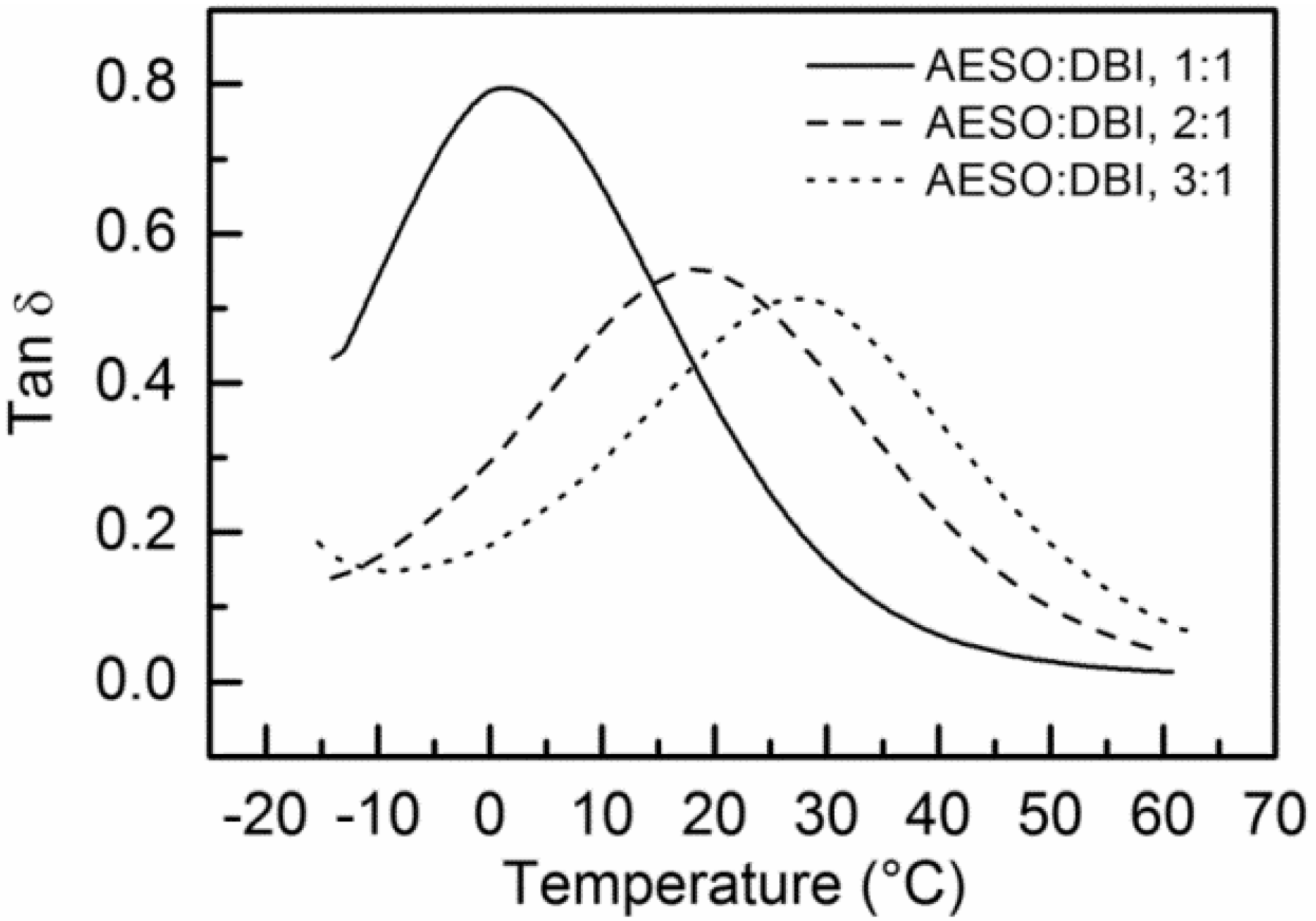

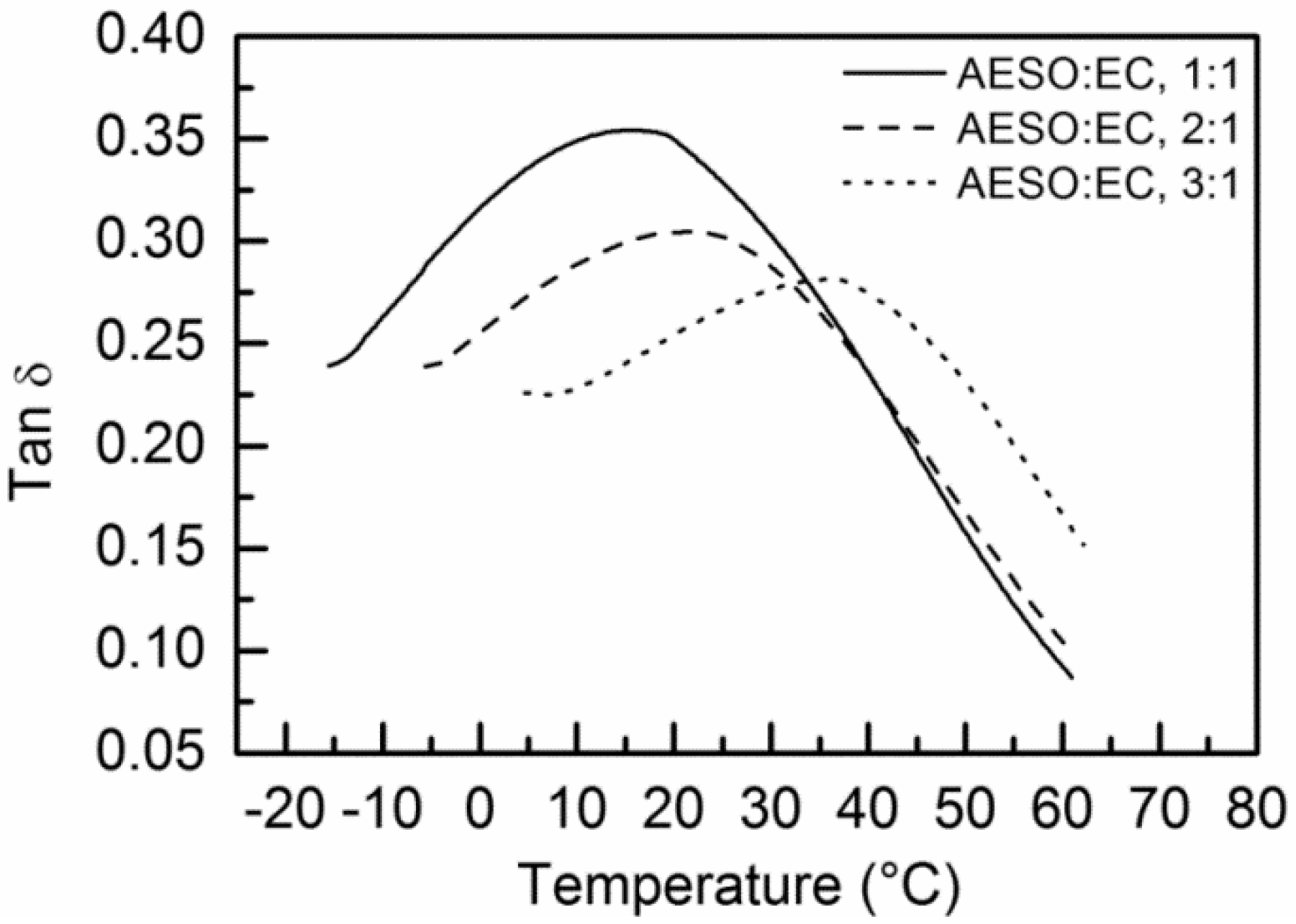
| Sample | AESO: EC, 1:1 | AESO: EC, 2:1 | AESO: EC, 3:1 | AESO: DBI, 1:1 | AESO: DBI, 2:1 | AESO: DBI, 3:1 |
|---|---|---|---|---|---|---|
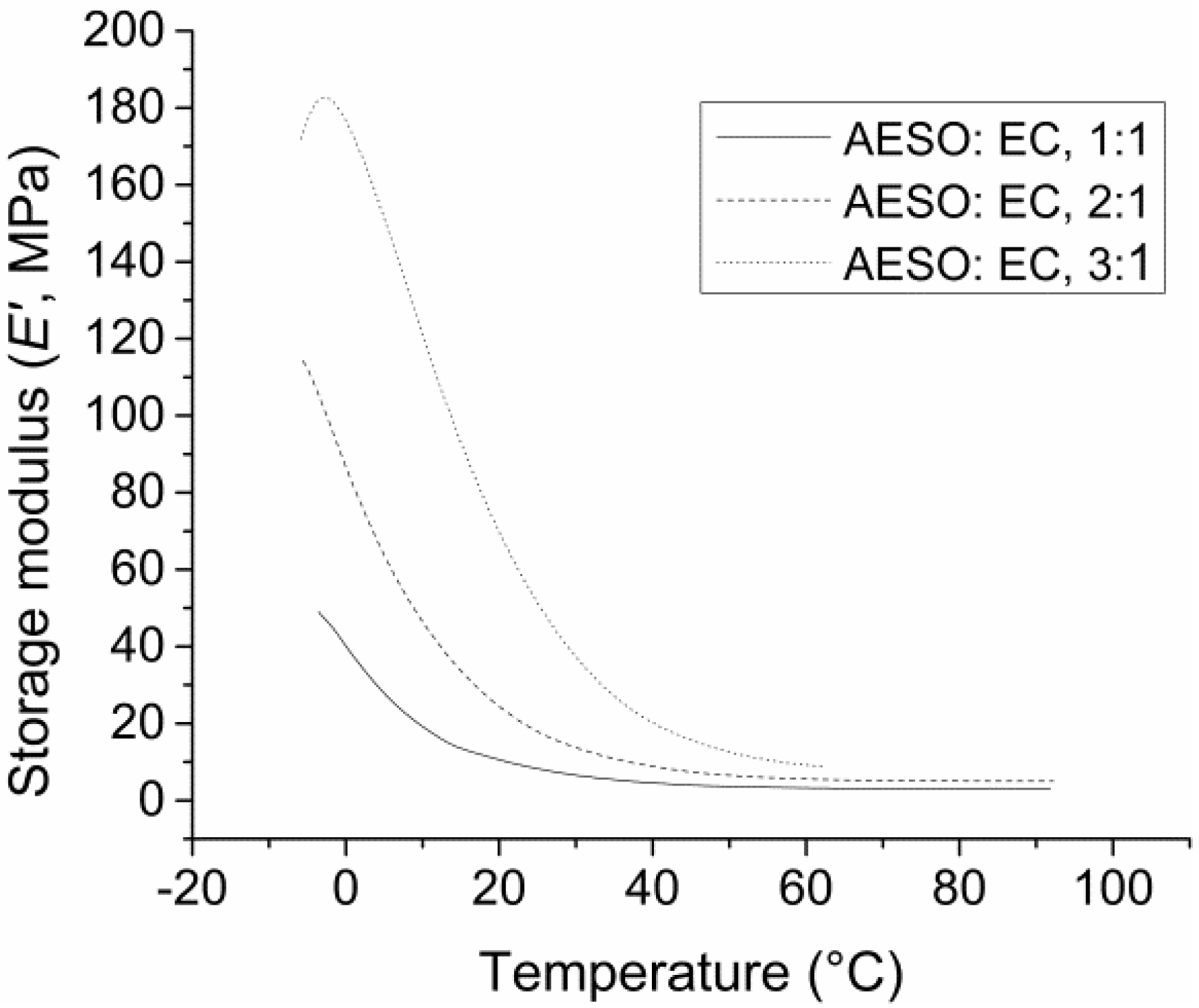
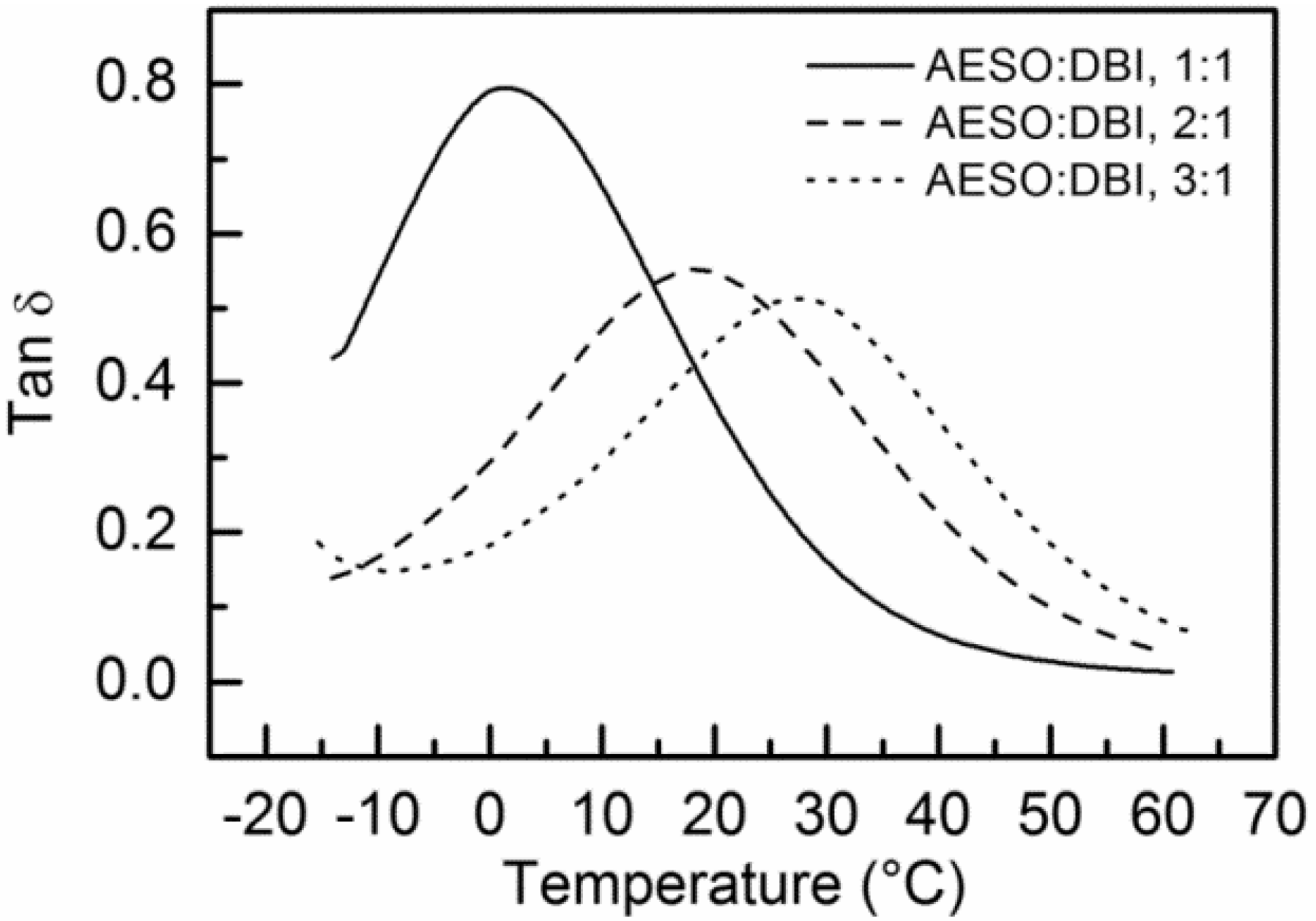
| Tg (as Tan δ peak, °C) | 14.2, 15.6 | 21.0, 21.1 | 41.4, 36.3 | 1.4, 2.5 | 18.4, 19.2 | 27.3, 25.4 |
| Storage modulus E’ (at 30 °C, MPa) | 6.5, 9.1 | 16.6, 13.7 | 35.2, 37.0 | 1.5, 1.6 | 6.6, 7.1 | 17.8, 10.2 |
4. Conclusions
Conflicts of Interest
References
- Zoebelein, H. Dictionary of Renewable Resources, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2001. [Google Scholar]
- Quirino, R.L.; Larock, R.C. Bioplastics, Biocomposites, and Biocoatings from Natural Oils. In Renewable and Sustainable Polymers; American Chemical Society: Washington, DC, USA, 2011; Volume 1063, pp. 37–59. [Google Scholar]
- Xia, Y.; Quirino, R.L.; Larock, R.C. Bio-based thermosetting polymers from vegetable oils. J. Renew. Mat. 2013, 1, 3–27. [Google Scholar]
- Khot, S.N.; La Scala, J.J.; Can, E.; Morye, S.S.; Williams, G.I.; Palmese, G.R.; Kusefoglu, S.H.; Wool, R.P. Development and application of triglyceride-based polymers and composites. J. Appl. Polym. Sci. 2001, 82, 703–723. [Google Scholar] [CrossRef]
- Wool, R.P.; Sun, X.S. Bio-Based Polymers and Composites; Elsevier Academic Press: Boston, MA, USA, 2005. [Google Scholar]
- Anastas, P.T. The transformative innovations needed by green chemistry for sustainability. ChemSusChem 2009, 2, 391–392. [Google Scholar] [CrossRef]
- Beach, E.S.; Cui, Z.; Anastas, P.T. Green chemistry: A design framework for sustainability. Energ. Environ. Sci. 2009, 2, 1038–1049. [Google Scholar] [CrossRef]
- Werpy, T.; Petersen, G. Top Value Added Chemicals from Biomass, Volume I: Results of Screening for Potential Candidates from Sugars and Synthetic Gas; US Department of Energy: Oak Ridge, TN, USA, 2004. [Google Scholar]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US Department of Energy’s “Top 10” revisited. Green Chem. 2010, 12, 539. [Google Scholar]
- Holladay, J.E.; White, J.F.; Bozell, J.J.; Johnson, D. Top Value Added Chemicals from Biomass, Volume II: Results of Screening for Potential Candidates from Biorefinery Lignin; US Department of Energy: Oak Ridge, TN, USA, 2007. [Google Scholar]
- Stanzione, J.F.; Giangiulio, P.A.; Sadler, J.M.; La Scala, J.J.; Wool, R.P. Lignin-based bio-oil mimic as biobased resin for composite applications. ACS Sustain. Chem. Eng. 2013, 1, 419–426. [Google Scholar] [CrossRef]
- Stanzione, J.F.; Sadler, J.M.; La Scala, J.J.; Wool, R.P. Lignin model compounds as bio-based reactive diluents for liquid molding resins. ChemSusChem 2012, 5, 1291–1297. [Google Scholar] [CrossRef]
- Ma, Q.; Liu, X.; Zhang, R.; Zhu, J.; Jiang, Y. Synthesis and properties of full bio-based thermosetting resins from rosin acid and soybean oil: The role of rosin acid derivatives. Green Chem. 2013, 15, 1300. [Google Scholar] [CrossRef]
- Willke, T.; Vorlop, K.D. Biotechnological production of itaconic acid. Appl. Microbiol. Biotechnol. 2001, 56, 289–295. [Google Scholar] [CrossRef]
- Sastry, G.S.R.; Murthy, B.G.K.; Aggarwal, J.S. Diels-Alder adducts of safflower oil fatty acids. Itaconic acid as a dienophile. Farbe und Lack 1972, 78, 927–929. [Google Scholar]
- Sugama, T.; Cook, M. Poly(itaconic acid)-modified chitosan coatings for mitigating corrosion of aluminum substrates. Prog. Org. Coat. 2000, 38, 79–87. [Google Scholar] [CrossRef]
- Ma, S.; Liu, X.; Jiang, Y.; Tang, Z.; Zhang, C.; Zhu, J. Bio-based epoxy resin from itaconic acid and its thermosets cured with anhydride and comonomers. Green Chem. 2013, 15, 245–254. [Google Scholar] [CrossRef]
- Cowie, J.M.G.; Henshall, S.A.E.; McEwen, I.J.; Velickovic, J. Poly(alkyl itaconates). 4. Glass and sub-glass transitions in the di-alkyl ester series, methyl to hexyl. Polymer 1977, 18, 612–616. [Google Scholar] [CrossRef]
- Marvel, C.S.; McCain, G.H. Polymerization of esters of cinnamic acid. J. Am. Chem. Soc. 1953, 75, 3272–3273. [Google Scholar] [CrossRef]
- La Scala, J.; Wool, R.P. Rheology of chemically modified triglycerides. J. Appl. Polym. Sci. 2005, 95, 774–783. [Google Scholar] [CrossRef]
- Campanella, A.; La Scala, J.J.; Wool, R.P. Fatty acid-based comonomers as styrene replacements in soybean and castor oil-based thermosetting polymers. J. Appl. Polym. Sci. 2011, 119, 1000–1010. [Google Scholar] [CrossRef]
- Lu, J.; Wool, R.P. Novel thermosetting resins for smc applications from linseed oil: Synthesis, characterization, and properties. J. Appl. Polym. Sci. 2006, 99, 2481–2488. [Google Scholar] [CrossRef]
- Sung, S.-J.; Cho, K.-Y.; Hah, H.; Lee, S.; Park, J.-K. Effect of plasticization of poly(vinyl cinnamate) on liquid crystal orientation stability. Jpn. J. Appl. Phys. 2005, 44, L412–L415. [Google Scholar] [CrossRef]
- Fernández-García, M.; Madruga, E.L. Glass transitions in dimethyl and di-n-butyl poly(itaconate ester)s and their copolymers with methyl methacrylate. Polymer 1997, 38, 1367–1371. [Google Scholar] [CrossRef]
- La Scala, J. The Effects of Triglyceride Structure on the Properties of Plant Oil-Based Resins, Vol. 1. Ph.D. Thesis, University of Delaware, Newark, DE, USA, 2002. [Google Scholar]
- La Scala, J.; Wool, R.P. Fundamental thermo-mechanical property modeling of triglyceride-based thermosetting resins. J. Appl. Polym. Sci. 2013, 127, 1812–1826. [Google Scholar] [CrossRef]
- Wool, R.P. Twinkling fractal theory of the glass transition. J. Polym. Sci. B 2008, 46, 2765–2778. [Google Scholar] [CrossRef]
- Hong, C.K.; Wool, R.P. Development of a bio-based composite material from soybean oil and keratin fibers. J. Appl. Polym. Sci. 2005, 95, 1524–1538. [Google Scholar] [CrossRef]
- Senoz, E.; Stanzione, J.F.; Reno, K.H.; Wool, R.P.; Miller, M.E.N. Pyrolyzed chicken feather fibers for biobased composite reinforcement. J. Appl. Polym. Sci. 2013, 128, 983–989. [Google Scholar] [CrossRef]
- Lee, K.-Y.; Ho, K.K.C.; Schlufter, K.; Bismarck, A. Hierarchical composites reinforced with robust short sisal fibre preforms utilising bacterial cellulose as binder. Compos. Sci. Technol. 2012, 72, 1479–1486. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Beach, E.S.; Cui, Z.; Anastas, P.T.; Zhan, M.; Wool, R.P. Properties of Thermosets Derived from Chemically Modified Triglycerides and Bio-Based Comonomers. Appl. Sci. 2013, 3, 684-693. https://doi.org/10.3390/app3040684
Beach ES, Cui Z, Anastas PT, Zhan M, Wool RP. Properties of Thermosets Derived from Chemically Modified Triglycerides and Bio-Based Comonomers. Applied Sciences. 2013; 3(4):684-693. https://doi.org/10.3390/app3040684
Chicago/Turabian StyleBeach, Evan S., Zheng Cui, Paul T. Anastas, Mingjiang Zhan, and Richard P. Wool. 2013. "Properties of Thermosets Derived from Chemically Modified Triglycerides and Bio-Based Comonomers" Applied Sciences 3, no. 4: 684-693. https://doi.org/10.3390/app3040684
APA StyleBeach, E. S., Cui, Z., Anastas, P. T., Zhan, M., & Wool, R. P. (2013). Properties of Thermosets Derived from Chemically Modified Triglycerides and Bio-Based Comonomers. Applied Sciences, 3(4), 684-693. https://doi.org/10.3390/app3040684