Evaluation of a Wet Chemistry Method for Isolation of Cyclotron Produced [211At]Astatine

Abstract
:
1. Introduction
2. Experimental Section
2.1. General
2.2. Dose Calibrator Measurements and Calibration
2.3. Preparation and Irradiation of Bismuth Targets
2.4. Procedure for Wet Chemistry Isolation of Na[211At]At
2.5. HPLC Analysis of Isolated Na[211At]At
2.6. Evaluation of Bismuth Attenuation in Quantification of 211At
3. Results
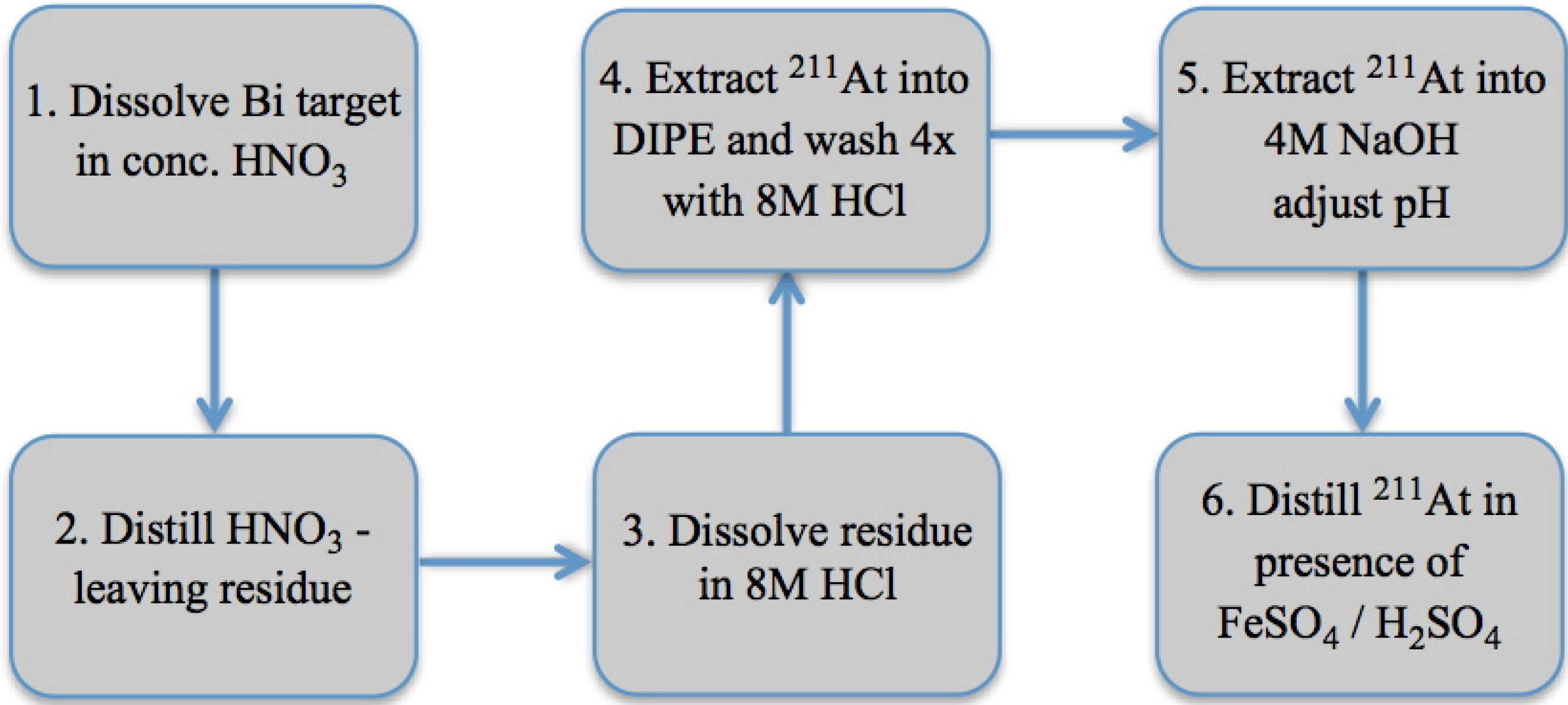
3.1. Wet Chemistry Isolation of 211At
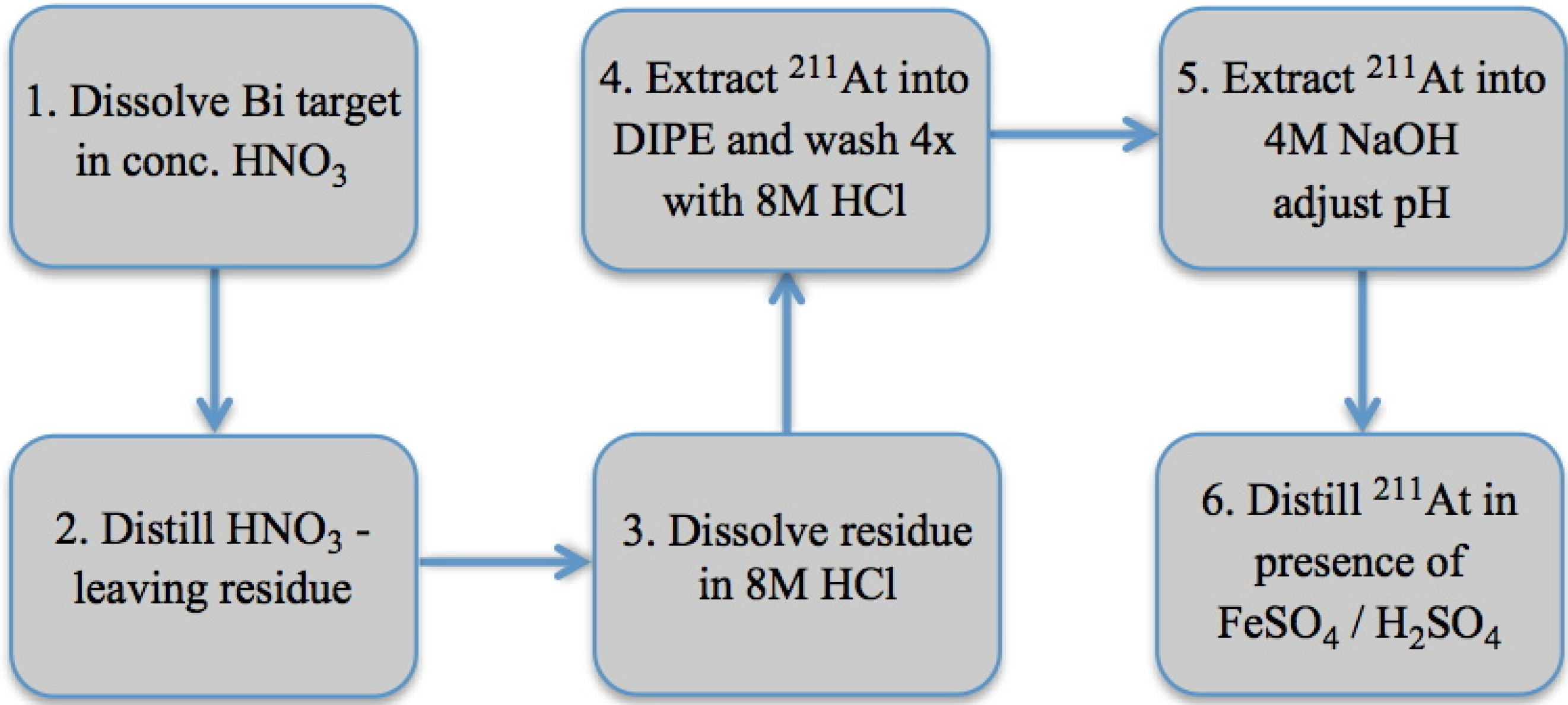

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 211At Isolation Runs (most ~0.75 h irrad.) | Activity Produced (mCi) ** | Time to Isolation (min) | Isolated Activity (mCi) | Decay Corrected Recovery (%) *** |
|---|---|---|---|---|
| Non-optimized (n = 53) | 20.7 ± 1.7 | 171 ± 46 | 13.6 ± 2.3 | 87 ± 15 |
| Optimized (n = 55) | 19.6 ± 2.0 | 155 ± 50 | 15.9 ± 2.7 | 104 ±15 |
| Preparative Run (2 h) | 54.0 | 118 | 43.2 | 97 |
| Preparative Run (4 h) | 100.5 | 114 | 72.0 | 86 |
| Run # | 211At Isolate to Distill (mCi) | 211At Activity Purified (mCi) | Volume Collected (mL) | % 211At Recovered |
|---|---|---|---|---|
| 1 | 14.3 | 12.2 | 3.4 | 85 |
| 2 | 8.0 | 4.8 | 1.5 | 61 |
| 3 | 12.3 | 9.1 | 1.5 | 74 |
| 4 | 7.5 | 5.4 | 1.2 | 72 |
| 5 | 15.1 | 10.8 | 1.8 | 72 |
| 6 | 16.5 | 10.5 | 1.8 | 64 |
| 7 | 34.0 | 23.0 | 2.0 | 67 |
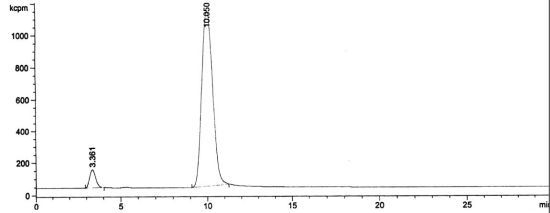
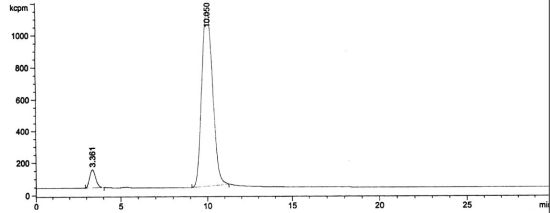
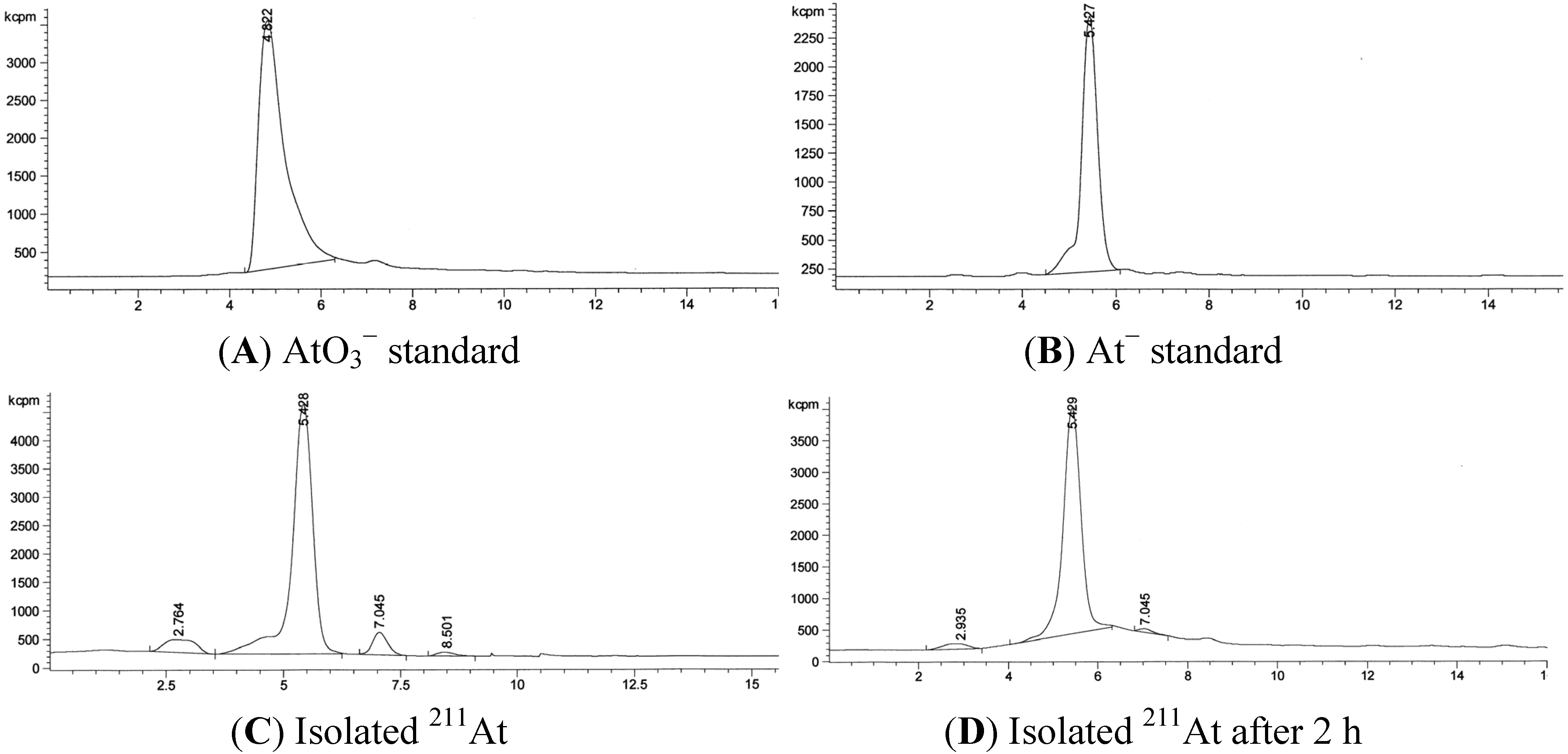
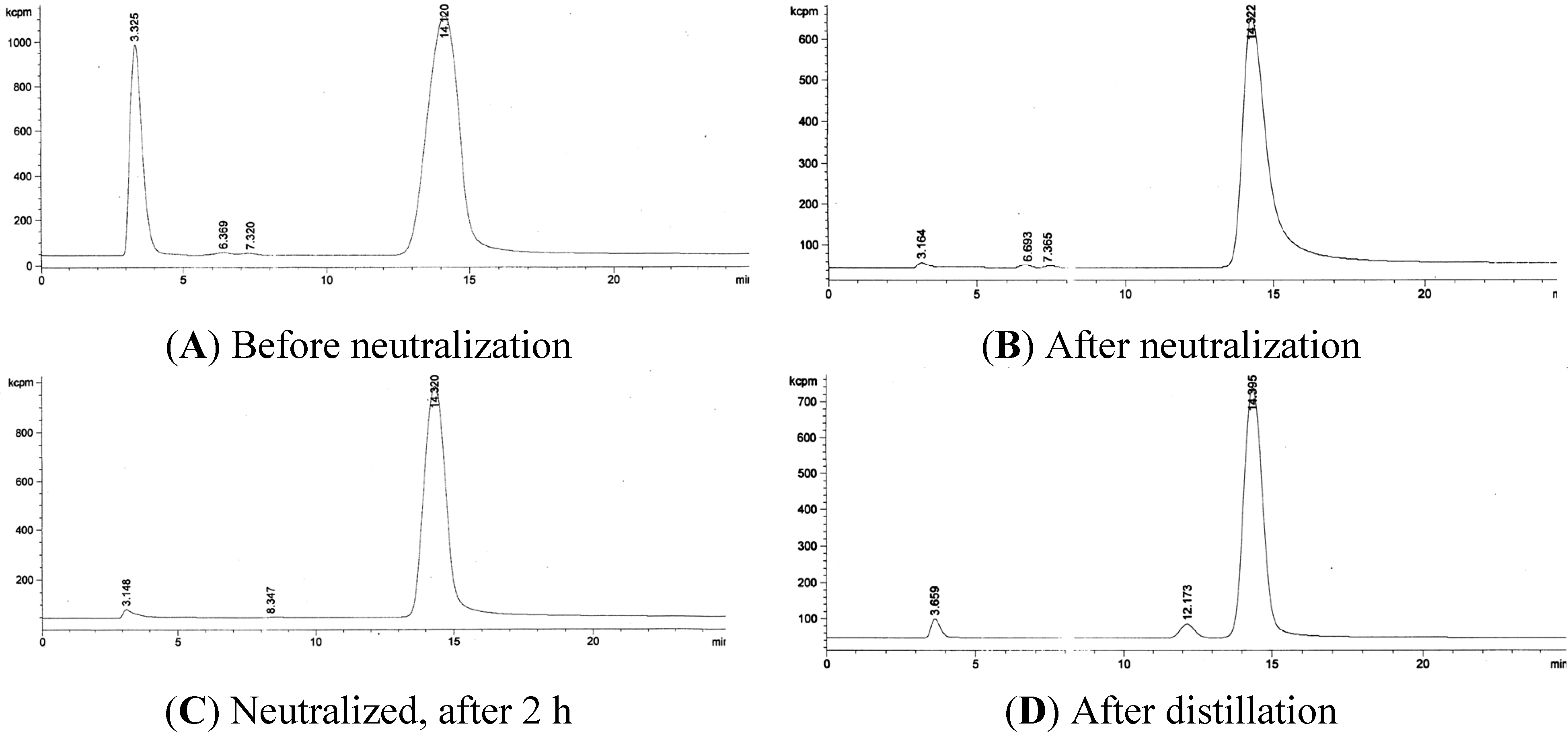
3.2. RadioHPLC of 211At Solutions
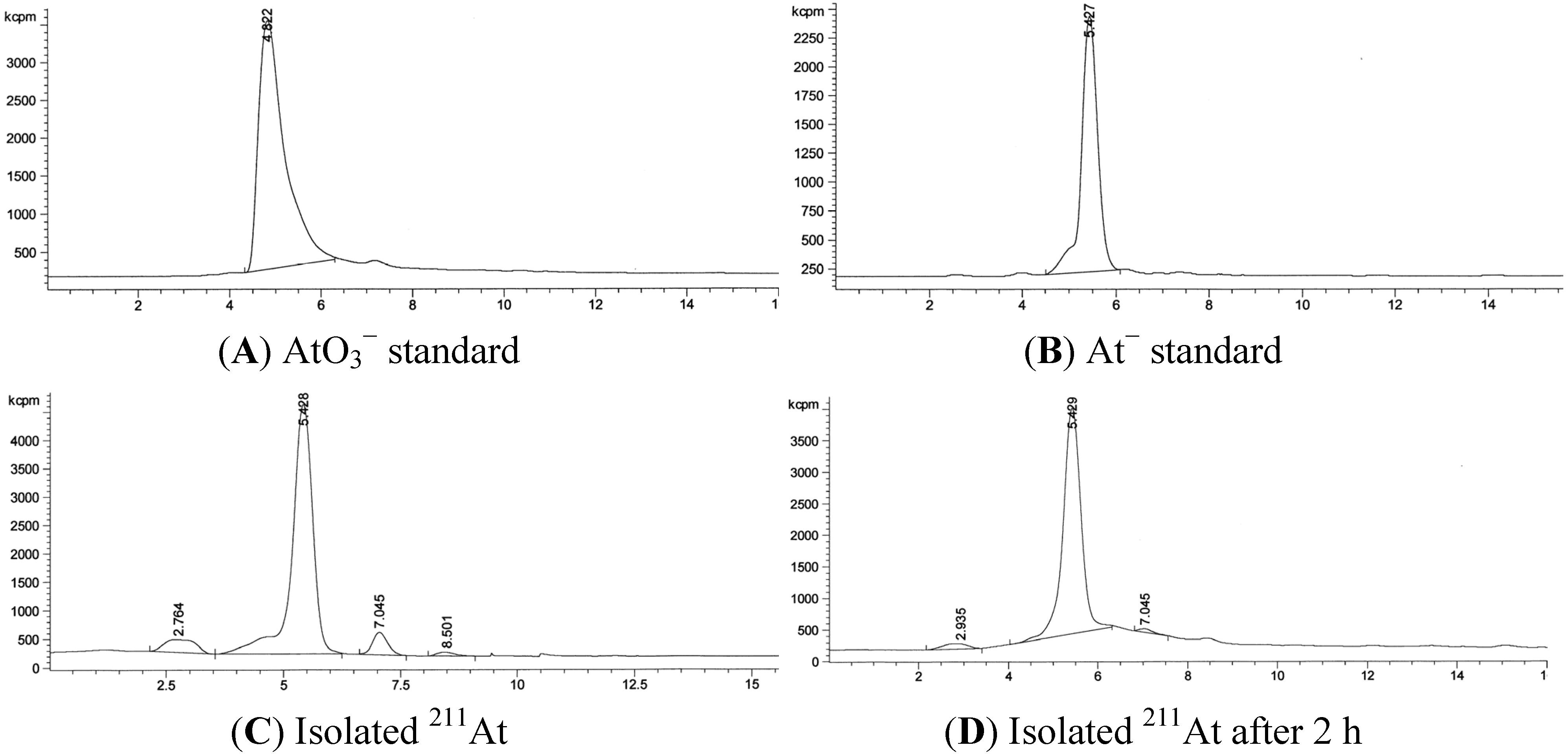
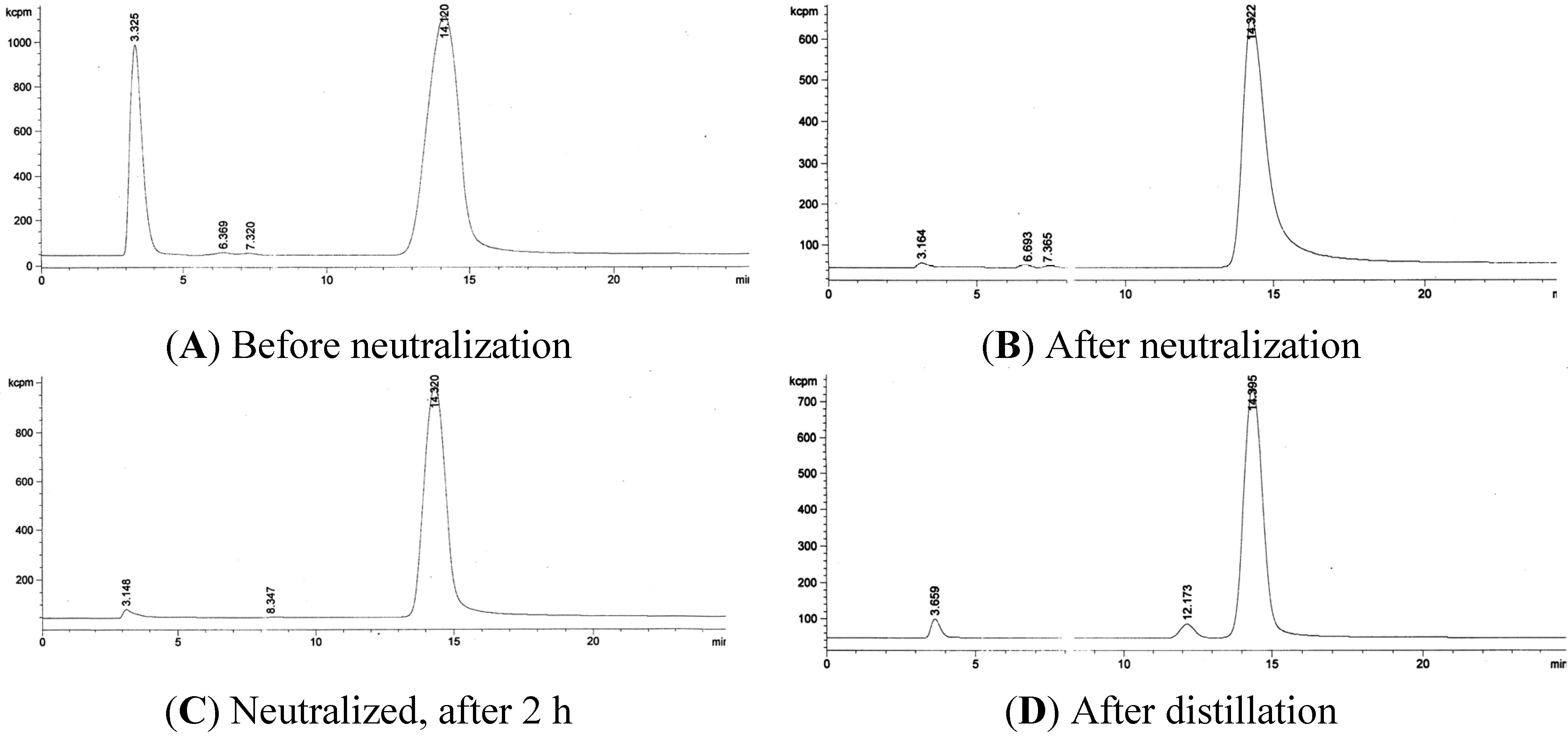
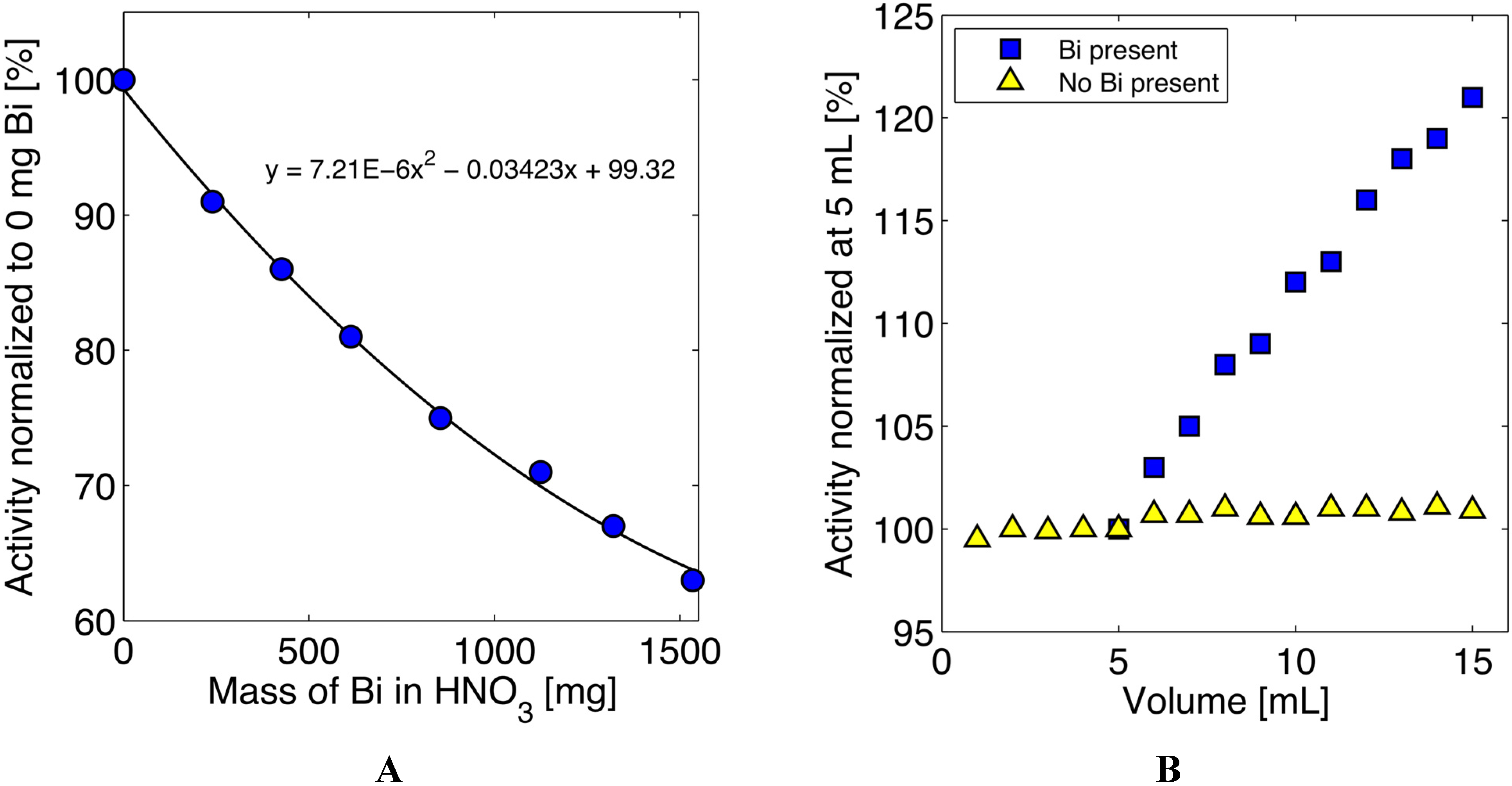
3.3. Quantification of 211At in the Presence of Bismuth
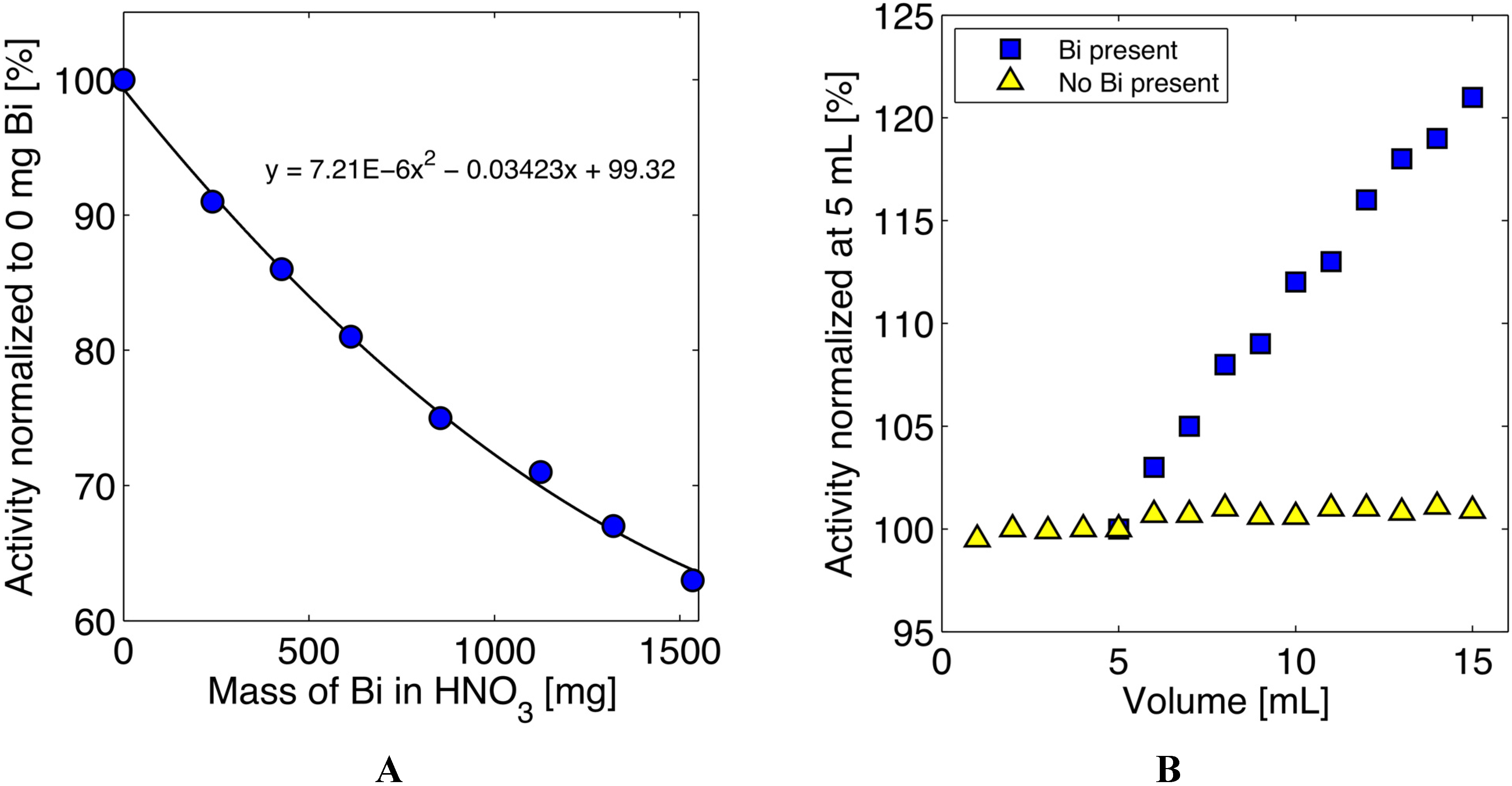
| 211At Isolation Runs (most ~0.75 h irrad.) | Corrected Act. Produced (mCi) ** | Isolated Activity (mCi) | Est. Attenuation* & Decay Corrected Recovery (%) *** |
|---|---|---|---|
| Non-optimized (n = 53) | 27.6 ± 2.3 | 13.6 ± 2.3 | 65 ± 11 |
| Optimized (n = 55) | 26.1 ± 2.7 | 15.9 ± 2.7 | 78 ± 11 |
| Preparative Run (2 h) | 72 | 43.2 | 73 |
| Preparative Run (4 h) | 134 | 72.0 | 65 |
4. Discussion
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Wilbur, D.S. [211At]astatine-labeled compound stability: Issues with released [211At]astatide and development of labeling reagents to increase stability. Curr. Radiopharm. 2008, 1, 144–176. [Google Scholar] [CrossRef]
- Lucignani, G. Alpha-particle radioimmunotherapy with astatine-211 and bismuth-213. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1729–1733. [Google Scholar] [CrossRef]
- Zalutsky, M.R.; Reardon, D.A.; Pozzi, O.R.; Vaidyanathan, G.; Bigner, D.D. Targeted α-particle radiotherapy with 211At-labeled monoclonal antibodies. Nucl. Med. Biol. 2007, 34, 779–785. [Google Scholar] [CrossRef]
- Zalutsky, M.R.; Reardon, D.A.; Akabani, G.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; McLendon, R.E.; Wong, T.Z.; Bigner, D.D. Clinical experience with alpha-particle emitting 211At: Treatment of recurrent brain tumor patients with 211At-labeled chimeric antitenascin monoclonal antibody 81C6. J. Nucl. Med. 2008, 49, 30–38. [Google Scholar]
- Andersson, H.; Cederkrantz, E.; Back, T.; Divgi, C.; Elgqvist, J.; Himmelman, J.; Horvath, G.; Jacobsson, L.; Jensen, H.; Lindegren, S.; et al. Intraperitoneal alpha-particle radioimmunotherapy of ovarian cancer patients: Pharmacokinetics and dosimetry of (211)At-MX35 F(ab')2—A phase I study. J. Nucl. Med. 2009, 50, 1153–1160. [Google Scholar] [CrossRef]
- Andersson, H.; Elgqvist, J.; Horvath, G.; Hultborn, R.; Jacobsson, L.; Jensen, H.; Karlsson, B.; Lindegren, S.; Palm, S. Astatine-211-labeled antibodies for treatment of disseminated ovarian cancer: An overview of results in an ovarian tumor model. Clin. Cancer Res. 2003, 9, 3914S–3921S. [Google Scholar]
- Palm, S.; Back, T.; Claesson, I.; Danielsson, A.; Elgqvist, J.; Frost, S.; Hultborn, R.; Jensen, H.; Lindegren, S.; Jacobsson, L. Therapeutic efficacy of astatine-211-labeled trastuzumab on radioresistant SKOV-3 tumors in nude mice. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 572–579. [Google Scholar] [CrossRef]
- Sundberg, A.L.; Almqvist, Y.; Tolmachev, V.; Carlsson, J. Treatment of cultured glioma cells with the EGFR-TKI gefitinib (\“Iressa\”, ZD1839) increases the uptake of astatinated EGF despite the absence of gefitinib-mediated growth inhibition. Eur. J. Nucl. Med. Mol. Imag. 2003, 30, 727–729. [Google Scholar] [CrossRef]
- Petrich, T.; Korkmaz, Z.; Krull, D.; Fromke, C.; Meyer, G.J.; Knapp, W.H. In vitro experimental 211At-anti-CD33 antibody therapy of leukaemia cells overcomes cellular resistance seen in vivo against gemtuzumab ozogamicin. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 851–861. [Google Scholar] [CrossRef]
- Zhang, M.; Yao, Z.; Zhang, Z.; Garmestani, K.; Talanov, V.S.; Plascjak, P.S.; Yu, S.; Kim, H.S.; Goldman, C.K.; Paik, C.H.; Brechbiel, M.W.; Carrasquillo, J.A.; Waldmann, T.A. The Anti-CD25 monoclonal antibody 7G7/B6, armed with the alpha-emitter 211At, provides effective radioimmunotherapy for a murine model of leukemia. Cancer Res. 2006, 66, 8227–8232. [Google Scholar] [CrossRef]
- Aurlien, E.; Larsen, R.H.; Kvalheim, G.; Bruland, O.S. Demonstration of highly specific toxicity of the alpha-emitting radioimmunoconjugate 211At-rituximab against non-Hodgkin’s lymphoma cells. Br. J. Cancer 2000, 83, 1375–1379. [Google Scholar] [CrossRef]
- Persson, M.I.; Gedda, L.; Jensen, H.J.; Lundqvist, H.; Malmstrom, P.U.; Tolmachev, V. Astatinated trastuzumab, a putative agent for radionuclide immunotherapy of ErbB2-expressing tumours. Oncol. Rep. 2006, 15, 673–680. [Google Scholar]
- Robinson, M.K.; Shaller, C.; Garmestani, K.; Plascjak, P.S.; Hodge, K.M.; Yuan, Q.A.; Marks, J.D.; Waldmann, T.A.; Brechbiel, M.W.; Adams, G.P. Effective treatment of established human breast tumor xenografts in immunodeficient mice with a single dose of the alpha-emitting radioisotope astatine-211 conjugated to anti-HER2/neu diabodies. Clin. Cancer Res. 2008, 14, 875–882. [Google Scholar] [CrossRef]
- Boskovitz, A.; McLendon, R.E.; Okamura, T.; Sampson, J.H.; Bigner, D.D.; Zalutsky, M.R. Treatment of HER2-positive breast carcinomatous meningitis with intrathecal administration of alpha-particle-emitting 211At-labeled trastuzumab. Nucl. Med. Biol. 2009, 36, 659–669. [Google Scholar] [CrossRef]
- Nestor, M.; Persson, M.; van Dongen, G.A.; Jensen, H.J.; Lundqvist, H.; Anniko, M.; Tolmachev, V. In vitro evaluation of the astatinated chimeric monoclonal antibody U36, a potential candidate for treatment of head and neck squamous cell carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 1296–1304. [Google Scholar] [CrossRef]
- Cheng, J.; Ekberg, T.; Engstrom, M.; Nestor, M.; Jensen, H.J.; Tolmachev, V.; Anniko, M. Radioimmunotherapy with astatine-211 using chimeric monoclonal antibody U36 in head and neck squamous cell carcinoma. The Laryngoscope 2007, 117, 1013–1018. [Google Scholar] [CrossRef]
- Willhauck, M.J.; Samani, B.R.; Wolf, I.; Senekowitsch-Schmidtke, R.; Stark, H.J.; Meyer, G.J.; Knapp, W.H.; Goke, B.; Morris, J.C.; Spitzweg, C. The potential of 211Astatine for NIS-mediated radionuclide therapy in prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1272–1281. [Google Scholar] [CrossRef]
- Hall, E.J. LET and RBE. In Radiobiology for the Radiologist, 3rd ed.; J. B. Lippincott Company: Philadelphia, PA, USA, 1988; pp. 161–178. [Google Scholar]
- Hall, E.J. Radiation Damage and Dose Rate Effect. In Radiobiology for the Radiologist, 3rd ed.; J. B. Lippincott Company: Philadelphia, PA, USA, 1988; pp. 107–136. [Google Scholar]
- Hall, E.J. The Oxygen Effect and Reoxygenation. In Radiobiology for the Radiologist, 3rd ed.; J. B. Lippincott Company: Philadelphia, PA, USA, 1988; pp. 137–160. [Google Scholar]
- Cancer Facts and Figures; American Cancer Society: Atlanta, GA, USA, 2013.
- Zalutsky, M.R.; Pruszynski, M. Astatine-211: Production and availability. Curr. Radiopharm. 2011, 4, 177–185. [Google Scholar] [CrossRef]
- Eberle, S.H. Chemical Behavior and Compounds of Astatine. In Gmelin Handbook of Chemistry, Astatine; Kugler, H.K., Keller, C., Eds.; Springer-Verlag: Berlin, Germany, 1985; pp. 183–259. [Google Scholar]
- Meyer, G.-J.; Rössler, K. Preparation of inorganic forms and interhalogen compounds of 211At via distillation techniques. Radiochem. Radioanal. Lett. 1976, 25, 377–390. [Google Scholar]
- Lambrecht, R.M.; Mirzadeh, S. Cyclotron Isotopes and Radiopharmaceuticals-XXXV. Astatine-211. Int. J. Appl. Radiat. Isot. 1985, 36, 443–450. [Google Scholar] [CrossRef]
- Lindegren, S.; Back, T.; Jensen, H.J. Dry-distillation of astatine-211 from irradiated bismuth targets: A time-saving procedure with high recovery yields. Appl. Radiat. Isot. 2001, 55, 157–160. [Google Scholar] [CrossRef]
- Wilbur, D.S.; Hadley, S.W.; Hines, J.J.; Atcher, R.W. Assessment of Dry Distillation Methods for Improving Protein Labeling Yields with Astatine-211. J. Labelled Compd. Radiopharm. 1990, 30, 214–215. [Google Scholar]
- Wilbur, D.S.; Vessella, R.L.; Stray, J.E.; Goffe, D.K.; Blouke, K.A.; Atcher, R.W. Preparation and evaluation of para-[211At]astatobenzoyl labeled anti-renal cell carcinoma antibody A6H F(ab')2. In vivo distribution comparison with para-[125I]iodobenzoyl labeled A6H F(ab')2. Nucl. Med. Biol. 1993, 20, 917–927. [Google Scholar] [CrossRef]
- Gagnon, K.; Risler, R.; Pal, S.; Hamlin, D.; Orzechowski, J.; Pavan, R.; Zeisler, S.; Wilbur, D.S. Design and evaluation of an external high-current target for production of 211At. J. Labelled Compd. Radiopharm. 2012, 55, 436–440. [Google Scholar] [CrossRef]
- Neumann, H.M. Solvent distribution studies of the chemistry of astatine. J. Inorg. Nucl. Chem. 1957, 4, 349–353. [Google Scholar] [CrossRef]
- Neirinckx, R.D.; Smit, J.A. Separation of astatine-211 from bismuth metal. Anal. Chem. Acta 1973, 63, 201–204. [Google Scholar] [CrossRef]
- Yordanov, A.T.; Pozzi, O.; Carlin, S.; Akabani, G.; Wieland, B.; Zalutsky, M. Wet harvesting of no-carrier-added 211At from an irradiated 209Bi target for radiopharmaceutical applications. J. Radioanal. Nucl. Chem. 2004, 262, 593–599. [Google Scholar] [CrossRef]
- Zona, C.; Bonardi, M.L.; Groppi, F.; Morzenti, S.; Canella, L.; Persico, E.; Menapace, E.; Alfassi, Z.B.; Abbas, K.; Holzwarth, U.; Gibson, N. Wet-chemistry method for the separation of no-carrier-added 211At/211gPo from 209Bi target irradiated by alpha-beam cyclotron. J. Radioanal. Nucl. Chem. 2008, 276, 819–824. [Google Scholar] [CrossRef]
- Sajonz, P.; Bookalam, J.; Miller, R.A. Separation of periodate, iodate and iodide on a C-18 stationary phase. Dependence of the retention on the temperature and solvent composition. monitoring of an oxidative clevage reaction. Chromatographia 2006, 64, 635–640. [Google Scholar] [CrossRef]
- Alliot, C.; Cherel, M.; Barbet, J.; Sauvage, T.; Montavon, G. Extraction of astatine-211 in diisopropylether (DIPE). Radiochim. Acta 2009, 97, 161–165. [Google Scholar]
- Ruth, T.J.; Dombsky, M.; D’Auria, J.M.; Ward, T.E. Radiochemistry of Astatine; U.S. Department of Energy (DOE): Springfield, VA, USA, 1988. [Google Scholar]
- Johnson, G.L.; Leininger, R.F.; Segre, E. Chemical properties of astatine. I. J. Chem. Phys. 1949, 17, 1–10. [Google Scholar] [CrossRef]
- Norseyev, Y.V.; Khalkin, V.A. Discovery and study of new inorganic and organic astatine compounds. Radiochemistry 1999, 41, 318–321. [Google Scholar]
- Champion, J.; Alliot, C.; Renault, E.; Mokili, B.M.; Cherel, M.; Galland, N.; Montavon, G. Astatine standard redox potentials and speciation in acidic medium. J. Phys. Chem. A 2010, 114, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Rössler, K.; Tornau, W.; Stöcklin, G. Rapid separation of carrier-free inorganic and organic compounds of radioiodine and astatine by high-pressure liquid chromatography. J. Radioanal. Chem. 1974, 21, 199–209. [Google Scholar] [CrossRef]
- Sabatié-Gogova, A.; Champion, J.; Huclier, S.; Michel, N.; Pottier, F.; Galland, N.; Asfari, Z.; Chérel, M.; Montavon, G. Characterization of At−species in simple and biological media by high performance anion exchange chromatography coupled to gamma detector. Anal. Chim. Acta 2012, 721, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Champion, J.; Sabatie-Gogova, A.; Bassal, F.; Ayed, T.; Alliot, C.; Galland, N.; Montavon, G. Investigation of astatine(III) hydrolyzed species: Experiments and relativistic calculations. J. Phys. Chem. A 2013, 117, 1983–1990. [Google Scholar] [CrossRef]
- Larsen, R.H.; Wieland, B.W.; Zalutsky, M.R. Evaluation of an internal cyclotron target for the production of 211At via the 209Bi (alpha,2n)211At reaction. Appl. Radiat. Isot. 1996, 47, 135–143. [Google Scholar] [CrossRef]
- Lebeda, O.; Jiran, R.; Ralis, J.; Stursa, J. A new internal target system for production of 211At on the cyclotron U-120M. Appl. Radiat. Isot. 2005, 63, 49–53. [Google Scholar] [CrossRef]
- Schwarz, U.P.; Plascjak, P.; Beitzel, M.P.; Gansow, O.A.; Eckelman, W.C.; Waldmann, T.A. Preparation of 211At-labeled humanized anti-Tac using 211At produced in disposable internal and external bismuth targets. Nucl. Med. Biol. 1998, 25, 89–93. [Google Scholar] [CrossRef]
- Bourgeois, M.; Guerard, F.; Alliot, C.; Mougin-Degraef, M.; Rajerison, H.; Remaud-Le Saec, P.; Gestin, J.-F.; Davodeau, F.; Cherel, M.; Barbet, J.; Faivre-Chauvet, A. Feasibility of the radioastatination of a monoclonal antibody with astatine-211 purified by wet extraction. J. Labelled Compd. Radiopharm. 2008, 51, 379–383. [Google Scholar] [CrossRef]
- Wilbur, D.S. Chemical and radiochemical considerations in radiolabeling with α-emitting radionuclides. Curr. Radiopharm. 2011, 4, 214–247. [Google Scholar] [CrossRef]
- Wilbur, D.S. Enigmatic astatine. Nat. Chem. 2013, 5. [Google Scholar] [CrossRef]
- Berry, F.J.; Collins, R.D.; Parish, R.V.; Moore, L.S. Studies of iodine in nitric acid and of iodine adsorbed onto silver-impregnated silica. Inorg. Chim. Acta 1987, 126, 119–124. [Google Scholar] [CrossRef]
- Norseyev, Y.V.; Khalkin, V.A. The stability constants of chloride complexes of mono-valent astatine in nitric acid solution. J. Inorg. Nucl. Chem. 1968, 30, 3239–3243. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Balkin, E.R.; Hamlin, D.K.; Gagnon, K.; Chyan, M.-K.; Pal, S.; Watanabe, S.; Wilbur, D.S. Evaluation of a Wet Chemistry Method for Isolation of Cyclotron Produced [211At]Astatine. Appl. Sci. 2013, 3, 636-655. https://doi.org/10.3390/app3030636
Balkin ER, Hamlin DK, Gagnon K, Chyan M-K, Pal S, Watanabe S, Wilbur DS. Evaluation of a Wet Chemistry Method for Isolation of Cyclotron Produced [211At]Astatine. Applied Sciences. 2013; 3(3):636-655. https://doi.org/10.3390/app3030636
Chicago/Turabian StyleBalkin, Ethan R., Donald K. Hamlin, Katherine Gagnon, Ming-Kuan Chyan, Sujit Pal, Shigeki Watanabe, and D. Scott Wilbur. 2013. "Evaluation of a Wet Chemistry Method for Isolation of Cyclotron Produced [211At]Astatine" Applied Sciences 3, no. 3: 636-655. https://doi.org/10.3390/app3030636
APA StyleBalkin, E. R., Hamlin, D. K., Gagnon, K., Chyan, M.-K., Pal, S., Watanabe, S., & Wilbur, D. S. (2013). Evaluation of a Wet Chemistry Method for Isolation of Cyclotron Produced [211At]Astatine. Applied Sciences, 3(3), 636-655. https://doi.org/10.3390/app3030636