
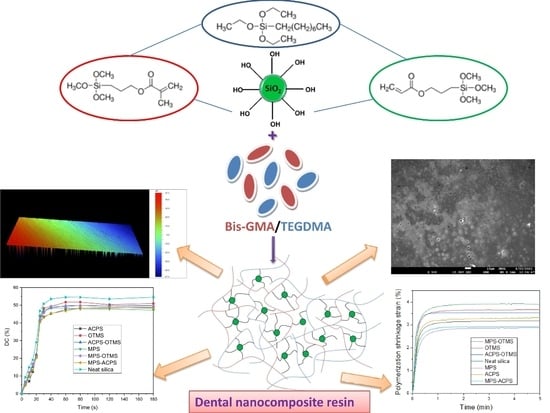
Effect of Silica Nanoparticles Silanized by Functional/Functional or Functional/Non-Functional Silanes on the Physicochemical and Mechanical Properties of Dental Nanocomposite Resins


 , and
, and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Surface Modification of Nanosilica with Quaternary Ammonium Silane Coupling Agents
2.3. Preparation of the Uncured Dental Composite Pastes
2.4. Measuremens
2.4.1. Characterization of the Organically Modified Nanosilica
2.4.2. Surface Morphology Measurements of Nanocomposite Resins
2.4.3. Physicochemical Properties
2.4.4. Mechanical Properties
3. Results and Discussion
3.1. Confirmation of Silica Silanization Reaction
3.2. Surface Morphology Evaluation of the Produced Dental Nanocomposite Resins
3.3. Polymerization Kinetics Evaluation
3.4. Polymerization Shrinkage Evaluation
3.5. Water Sorption and Solubility Parameters
3.6. Flexural Properties
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Li, Z.; Zhang, H.; Xiong, G.; Zhang, J.; Guo, R.; Li, L.; Zhou, H.; Chen, G.; Zhou, Z.; Li, Q. A low-shrinkage dental composite with epoxy-polyhedral oligomeric silsesquioxane. J. Mech. Behav. Biomed. Mater. 2020, 103, 103515. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.D.; Bishara, S.E.; Quinn, J.K. Biodegradation of orthodontic appliances. Part I. Biodegradation of nickel and chromium in vitro. Am. J. Orthod. Dentofac. Orthop. 1993, 103, 8–14. [Google Scholar] [CrossRef]
- Eliades, T.; Athanasiou, A.E. In vivo aging of orthodontic alloys: Implications for corrosion potential, nickel release, and biocompatibility. Angle Orthod. 2002, 72, 222–237. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Garg, P.K.; Dubey, A.K. Insights into the human oral microbiome. Arch. Microbiol. 2018, 200, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Gerdolle, D.A.; Mortier, E.; Droz, D. Microleakage and polymerization shrinkage of various polymer restorative materials. J. Dent. Child. 2008, 75, 125–133. [Google Scholar]
- Bento, A.P.; Bickelhaupt, F.M. Nucleophilic Substitution at Silicon (SN2@Si) via a Central Reaction Barrier. J. Org. Chem. 2007, 72, 2201–2207. [Google Scholar] [CrossRef] [PubMed]
- Sideridou, I.D.; Karabela, M.M. Effect of the amount of 3-methacyloxypropyltrimethoxysilane coupling agent on physical properties of dental resin nanocomposites. Dent. Mater. 2009, 25, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, J.M.; Dickens, S.H.; Fowler, B.O.; Xu, H.H.K. Chemistry of silanes: Interfaces in dental polymers and composites. J. Res. Natl. Inst. Stand. Technol. 2005, 110, 541–558. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, Y.; Nishiwaki, T.; Nemoto, K.; Ben, G. Effect of filler content on bending properties of dental composites: Numerical simulation with the use of the finite-element method. J. Biomed. Mater. Res. 2004, 71B, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.K. Mechanisms of fracture in filled thermosetting resins. Br. Polym. J. 1980, 12, 24–30. [Google Scholar] [CrossRef]
- Mohsen, N.M.; Craig, R.G. Effect of silanation of fillers on their dispersability by monomer systems. J. Oral Rehabil. 1995, 22, 183–189. [Google Scholar] [CrossRef]
- Söderholm, K.-J. Degradation of Glass Filler in Experimental Composites. J. Dent. Res. 1981, 60, 1867–1875. [Google Scholar] [CrossRef]
- Matinlinna, J.; Özcan, M.; Lassila, L.; Kalk, W.; Vallittu, P. Effect of the cross-linking silane concentration in a novel silane system on bonding resin-composite cement. Acta Odontol. Scand. 2008, 66, 250–255. [Google Scholar] [CrossRef]
- Habib, E.; Wang, R.; Zhu, X.X. Monodisperse silica-filled composite restoratives mechanical and light transmission properties. Dent. Mater. 2017, 33, 280–287. [Google Scholar] [CrossRef]
- Hosseinalipour, M.; Javadpour, J.; Rezaie, H.; Dadras, T.; Hayati, A.N. Investigation of mechanical properties of experimental Bis-GMA/TEGDMA dental composite resins containing various mass fractions of silica nanoparticles. J. Prosthodont. 2010, 19, 112–117. [Google Scholar] [CrossRef]
- Kim, J.W.; Kim, L.U.; Kim, C.K. Size control of silica nanoparticles and their surface treatment for fabrication of dental nanocomposites. Biomacromolecules 2007, 8, 215–222. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Z.; Zhao, C.; Bu, W.; Na, H. Preparation and characterization of silane-modified SiO 2 particles reinforced resin composites with fluorinated acrylate polymer. J. Mech. Behav. Biomed. Mater. 2018, 80, 11–19. [Google Scholar] [CrossRef]
- Miao, X.; Li, Y.; Zhang, Q.; Zhu, M.; Wang, H. Low shrinkage light curable dental nanocomposites using SiO2 microspheres as fillers. Mater. Sci. Eng. C 2012, 32, 2115–2121. [Google Scholar] [CrossRef]
- Rahim, T.N.A.T.; Mohamad, D.; Ismail, A.R.; Akil, H.M. Synthesis of nanosilica fillers for experimental dental nanocomposites and their characterisations. J. Phys. Sci. 2011, 22, 93–105. [Google Scholar]
- Rodríguez-Quirós, H.A.; Casanova-Yepes, H.F. Effect of the functionalization of silica nanoparticles as a reinforcing agent on dental composite materials. Rev. Fac. Ing. Univ. Antioq. 2015, 75, 36–44. [Google Scholar] [CrossRef]
- Rodríguez, H.A.; Kriven, W.M.; Casanova, H. Development of mechanical properties in dental resin composite: Effect of filler size and filler aggregation state. Mater. Sci. Eng. C 2019, 101, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Zanchi, C.H.; Ogliari, F.A.; Marques e Silva, R.; Lund, R.G.; Machado, H.H.; Prati, C.; Carreño, N.L.V.; Piva, E. Effect of the silane concentration on the selected properties of an experimental microfilled composite resin. Appl. Adhes. Sci. 2015, 3, 27. [Google Scholar] [CrossRef] [Green Version]
- Karabela, M.M.; Sideridou, I.D. Synthesis and study of properties of dental resin composites with different nanosilica particles size. Dent. Mater. 2011, 27, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Matinlinna, J.P.; Vallittu, P.K.; Lassila, L.V. Effects of Different Silane Coupling Agent Monomers on Flexural Strength of an Experimental Filled Resin Composite. J. Adhes. Sci. Technol. 2011, 25, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Fronza, B.M.; Lewis, S.; Shah, P.K.; Barros, M.D.; Giannini, M.; Stansbury, J.W. Modification of filler surface treatment of composite resins using alternative silanes and functional nanogels. Dent. Mater. 2019, 35, 928–936. [Google Scholar] [CrossRef]
- Nikolaidis, A.K.; Koulaouzidou, E.A.; Gogos, C.; Achilias, D.S. Synthesis of Novel Dental Nanocomposite Resins by Incorporating Polymerizable, Quaternary Ammonium Silane-Modified Silica Nanoparticles. Polymers 2021, 13, 1682. [Google Scholar] [CrossRef]
- Karabela, M.; Sideridou, I. Effect of the structure of silane coupling agent on sorption characteristics of solvents by dental resin-nanocomposites. Dent. Mater. 2008, 24, 1631–1639. [Google Scholar] [CrossRef]
- Karabela, M.M.; Sideridou, I.D. Synthesis and study of physical properties of dental light-cured nanocomposites using different amounts of a urethane dimethacrylate trialkoxysilane coupling agent. Dent. Mater. 2011, 27, 1144–1152. [Google Scholar] [CrossRef]
- Wang, R.; Bao, S.; Liu, F.; Jiang, X.; Zhang, Q.; Sun, B.; Zhu, M. Wear behavior of light-cured resin composites with bimodal silica nanostructures as fillers. Mater. Sci. Eng. C 2013, 33, 4759–4766. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, M.; Liu, F.; Bao, S.; Wu, T.; Jiang, X.; Zhang, Q.; Zhu, M. Investigation on the physical–mechanical properties of dental resin composites reinforced with novel bimodal silica nanostructures. Mater. Sci. Eng. C 2015, 50, 266–273. [Google Scholar] [CrossRef]
- Wilson, K.S.; Antonucci, J.M. Interphase structure–property relationships in thermoset dimethacrylate nanocomposites. Dent. Mater. 2006, 22, 995–1001. [Google Scholar] [CrossRef]
- Wilson, K.S.; Zhang, K.; Antonucci, J.M. Systematic variation of interfacial phase reactivity in dental nanocomposites. Biomaterials 2005, 26, 5095–5103. [Google Scholar] [CrossRef]
- Wilson, K.; Allen, A.; Washburn, N.; Antonucci, J. Interphase Effects in Dental Nanocomposites Investigated by Small-Angle Neutron Scattering. J. Biomed. Mater. Res. 2007, 81A, 113–123. [Google Scholar] [CrossRef]
- Matinlinna, J. Silane Chemistry Aspects in Some conventional and Novel Dental Biomaterials. Ph.D. Thesis, University of Turku, Turku, Finland, 2004. [Google Scholar]
- Khan, A.A.; Al Kheraif, A.A.; Syed, J.; Divakar, D.D.; Matinlinna, J.P. Enhanced resin titanium adhesion with silane primers using tribochemical silica-coating. Dent. Mater. J. 2017, 36, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Matinlinna, J.P.; Lassila, L.V.J.; Vallittu, P.K. The effect of three silane coupling agents and their blends with a cross-linker silane on bonding a bis-GMA resin to silicatized titanium (a novel silane system). J. Dent. 2006, 34, 740–746. [Google Scholar] [CrossRef]
- Lung, C.Y.K.; Kukk, E.; Matinlinna, J.P. Shear bond strength between resin and zirconia with two different silane blends. Acta Odontol. Scand. 2012, 70, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Durgesh, B.H.; Alhijji, S.; Hashem, M.I.; Kheraif, A.A.A.; Malash, A.M.; Divakar, D.D.; Shahrani, O.A.A.; Asmari, M.A.; Matinlinna, J.P. Evaluation of an Experimental Silane Primer System in Promoting Adhesion Between Orthodontic Bracket and Ceramic. J. Biomater. Tissue Eng. 2016, 6, 239–245. [Google Scholar] [CrossRef]
- Khan, A.A.; Al Kheraif, A.A.; Syed, J.; Divakar, D.D.; Matinlinna, J.P. Effect of experimental primers on hydrolytic stability of resin zirconia bonding. J. Adhes. Sci. Technol. 2017, 31, 1094–1104. [Google Scholar] [CrossRef]
- Kurata, S.; Yamazaki, N. Effect of Silane Coupling Agents with a Bisfunctional Hydrolyzable Group. Dent. Mater. J. 1993, 12, 127–135, 272. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Ding, J.; Chambers, D.E.; Debnath, S.; Wunder, S.L.; Baran, G.R. Filler-coupling agent-matrix interactions in silica/polymethylmethacrylate composites. J. Biomed. Mater. Res. 2001, 57, 384–393. [Google Scholar] [CrossRef]
- Söderholm, K.-J.M.; Shang, S.-W. Molecular Orientation of Silane at the Surface of Colloidal Silica. J. Dent. Res. 1993, 72, 1050–1054. [Google Scholar] [CrossRef]
- Chen, T.M.; Brauer, G.M. Solvent Effects on Bonding Organo-silane to Silica Surfaces. J. Dent. Res. 1982, 61, 1439–1443. [Google Scholar] [CrossRef]
- Watts, D.; Marouf, A. Optimal specimen geometry in bonded-disk shrinkage-strain measurements on light-cured biomaterials. Dent. Mater. 2000, 16, 447–451. [Google Scholar] [CrossRef]
- Watts, D. Photo-polymerization shrinkage-stress kinetics in resin-composites: Methods development. Dent. Mater. 2003, 19, 1–11. [Google Scholar] [CrossRef]
- Al Sunbul, H.; Silikas, N.; Watts, D.C. Polymerization shrinkage kinetics and shrinkage-stress in dental resin-composites. Dent. Mater. 2016, 32, 998–1006. [Google Scholar] [CrossRef] [Green Version]
- Rueggeberg, F.A.; Hashinger, D.T.; Fairhurst, C.W. Calibration of FTIR conversion analysis of contemporary dental resin composites. Dent. Mater. 1990, 6, 241–249. [Google Scholar] [CrossRef]
- International Organization for Standardization. ISO 4049:2000 (E) Dentistry—Polymer-Based Filling, Restorative and Luting Materials; ISO: Geneva, Switzerland, 2000. [Google Scholar]
- Nikolaidis, A.K.; Koulaouzidou, E.A.; Achilias, D.S. Synthesis and Characterization of Novel Organomodified Nanoclays for Application in Dental Materials. Curr. Nanosci. 2019, 15, 512–524. [Google Scholar] [CrossRef]
- Halvorson, R.H.; Erickson, R.L.; Davidson, C.L. The effect of filler and silane content on conversion of resin-based composite. Dent. Mater. 2003, 19, 327–333. [Google Scholar] [CrossRef]
- Gonçalves, F.; Kawano, Y.; Pfeifer, C.; Stansbury, J.W.; Braga, R.R. Influence of BisGMA, TEGDMA, and BisEMA contents on viscosity, conversion, and flexural strength of experimental resins and composites. Eur. J. Oral Sci. 2009, 117, 442–446. [Google Scholar] [CrossRef]
- Sideridou, I.D.; Karabela, M.M. Effect of the structure of silane-coupling agent on dynamic mechanical properties of dental resin-nanocomposites. J. Appl. Polym. Sci. 2008, 110, 507–516. [Google Scholar] [CrossRef]
- Mourouzis, P.; Koulaouzidou, E.A.; Vassiliadis, L.; Helvatjoglu-Antoniades, M. Effects of sonic scaling on the surface roughness of restorative materials. J. Oral Sci. 2009, 51, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griggs, J.A.; Thompson, J.Y.; Anusavice, K.J. Effects of flaw size and auto-glaze treatment on porcelain strength. J. Dent. Res. 1996, 75, 1414–1417. [Google Scholar] [CrossRef] [PubMed]
- De Jager, N.; Feilzer, A.J.; Davidson, C.L. The influence of surface roughness on porcelain strength. Dent. Mater. 2000, 16, 381–388. [Google Scholar] [CrossRef]
- Yilmaz, C.; Korkmaz, T.; Demirköprülü, H.; Ergün, G.; Ozkan, Y. Color stability of glazed and polished dental porcelains. J. Prosthodont. 2008, 17, 20–24. [Google Scholar] [CrossRef]
- Ruyter, I.E.; Øysaed, H. Composites for use in posterior teeth: Composition and conversion. J. Biomed. Mater. Res. 1987, 21, 11–23. [Google Scholar] [CrossRef]
- Achilias, D.S. A Review of Modeling of Diffusion Controlled Polymerization Reactions. Macromol. Theory Simul. 2007, 16, 319–347. [Google Scholar] [CrossRef]
- Achilias, D.S.; Verros, G.D. Modeling of diffusion-controlled reactions in free radical solution and bulk polymerization: Model validation by DSC experiments. J. Appl. Polym. Sci. 2010, 116, 1842–1856. [Google Scholar] [CrossRef]
- Verros, G.D.; Achilias, D.S. Modeling gel effect in branched polymer systems: Free-radical solution homopolymerization of vinyl acetate. J. Appl. Polym. Sci. 2009, 111, 2171–2185. [Google Scholar] [CrossRef]
- Verros, G.D.; Latsos, T.; Achilias, D.S. Development of a unified framework for calculating molecular weight distribution in diffusion controlled free radical bulk homo-polymerization. Polymer 2005, 46, 539–552. [Google Scholar] [CrossRef]
- Peutzfeldt, A. Resin composites in dentistry: The monomer systems. Eur. J. Oral Sci. 1997, 105, 97–116. [Google Scholar] [CrossRef]
- Luo, S.; Liu, F.; He, J. Preparation of low shrinkage stress dental composite with synthesized dimethacrylate oligomers. J. Mech. Behav. Biomed. Mater. 2019, 94, 222–228. [Google Scholar] [CrossRef]
- Ferracane, J.L.; Mitchem, J.C. Relationship between composite contraction stress and leakage in Class V cavities. Am. J. Dent. 2003, 16, 239–243. [Google Scholar]
- Par, M.; Mohn, D.; Attin, T.; Tarle, Z.; Tauböck, T.T. Polymerization shrinkage behaviour of resin composites functionalized with unsilanized bioactive glass fillers. Sci. Rep. 2020, 10, 15237. [Google Scholar] [CrossRef]
- Wang, X.; Cai, Q.; Zhang, X.; Wei, Y.; Xu, M.; Yang, X.; Ma, Q.; Cheng, Y.; Deng, X. Improved performance of Bis-GMA/TEGDMA dental composites by net-like structures formed from SiO2 nanofiber fillers. Mater. Sci. Eng. C 2016, 59, 464–470. [Google Scholar] [CrossRef]
- Kleverlaan, C.J.; Feilzer, A.J. Polymerization shrinkage and contraction stress of dental resin composites. Dent. Mater. 2005, 21, 1150–1157. [Google Scholar] [CrossRef]
- Nikolaidis, A.K.; Koulaouzidou, E.A.; Gogos, C.; Achilias, D.S. Synthesis and Characterization of Dental Nanocomposite Resins Filled with Different Clay Nanoparticles. Polymers 2019, 11, 730. [Google Scholar] [CrossRef] [Green Version]
- Ferracane, J.L. Hygroscopic and hydrolytic effects in dental polymer networks. Dent. Mater. 2006, 22, 211–222. [Google Scholar] [CrossRef]
- Aliabadian, E.; Sadeghi, S.; Kamkar, M.; Chen, Z.; Sundararaj, U. Rheology of fumed silica nanoparticles/partially hydrolyzed polyacrylamide aqueous solutions under small and large amplitude oscillatory shear deformations. J. Rheol. 2018, 62, 1197–1216. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | ||||
|---|---|---|---|---|
| Code | Fumed Silica Nanopowder (AEROSIL OX 50) | 3-(Trimethoxysilyl)propyl methacrylate (γ-MPS) (Amount wt% Relative to Silica) | Octyltrimethoxysilane (OTMS) (Amount wt% Relative to Silica) | 3-(Trimethoxysilyl)propyl acrylate (ACPS) (Amount wt% Relative to Silica) |
| Neat silica | 100% | - | - | - |
| MPS | √ | 10 | - | - |
| OTMS | √ | - | 10 | - |
| ACPS | √ | - | - | 10 |
| MPS-OTMS | √ | 5 | 5 | - |
| ACPS-OTMS | √ | - | 5 | 5 |
| MPS-ACPS | √ | 5 | - | 5 |
| Nanocomposite | Surface Roughness, Sα (nm) | Final DC (%) | Total Strain (%) |
|---|---|---|---|
| Neat silica | 308.00 ± 336.08 | 54.5 | 3.68 ± 0.34 |
| MPS | 227.23 ± 204.84 | 47.2 | 2.87 ± 0.35 |
| OTMS | 254.26 ± 261.40 | 50.9 | 3.65 ± 0.15 |
| ACPS | 127.41 ± 49.39 | 48.6 | 3.35 ± 0.53 |
| MPS-OTMS | 140.50 ± 19.00 | 49.4 | 3.18 ± 0.39 |
| ACPS-OTMS | 169 ± 101.00 | 49.7 | 3.51 ± 0.56 |
| MPS-ACPS | 171.5 ± 74.00 | 48.3 | 2.91 ± 0.60 |
| Nanocomposite | Sorption, Wsp (μg/mm3) | Solubility, Wsl (μg/mm3) | Flexural Modulus (GPa) | Flexural Strength (MPa) |
|---|---|---|---|---|
| Neat silica | 2.58 ± 0.51 | 0.08 ± 0.09 | 1.59 ± 0.81 | 15.31 ± 3.66 |
| MPS | 2.48 ± 0.43 | 0.32 ± 0.17 | 3.66 ± 0.67 | 44.30 ± 21.23 |
| OTMS | 2.29 ± 0.15 | 0.56 ± 0.13 | 1.75 ± 0.71 | 15.38 ± 23.45 |
| ACPS | 2.68 ± 0.84 | 0.16 ± 0.10 | 3.08 ± 0.40 | 40.18 ± 7.48 |
| MPS-OTMS | 2.34 ± 0.26 | 0.49 ± 0.15 | 2.04 ± 0.62 | 22.21 ± 12.63 |
| ACPS-OTMS | 2.52 ± 0.25 | 0.39 ± 0.16 | 2.60 ± 0.92 | 30.65 ± 10.53 |
| MPS-ACPS | 2.72 ± 0.29 | 0.25 ± 0.11 | 3.30 ± 0.93 | 42.83 ± 9.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karkanis, S.; Nikolaidis, A.K.; Koulaouzidou, E.A.; Achilias, D.S. Effect of Silica Nanoparticles Silanized by Functional/Functional or Functional/Non-Functional Silanes on the Physicochemical and Mechanical Properties of Dental Nanocomposite Resins. Appl. Sci. 2022, 12, 159. https://doi.org/10.3390/app12010159
Karkanis S, Nikolaidis AK, Koulaouzidou EA, Achilias DS. Effect of Silica Nanoparticles Silanized by Functional/Functional or Functional/Non-Functional Silanes on the Physicochemical and Mechanical Properties of Dental Nanocomposite Resins. Applied Sciences. 2022; 12(1):159. https://doi.org/10.3390/app12010159
Chicago/Turabian StyleKarkanis, Stefanos, Alexandros K. Nikolaidis, Elisabeth A. Koulaouzidou, and Dimitris S. Achilias. 2022. "Effect of Silica Nanoparticles Silanized by Functional/Functional or Functional/Non-Functional Silanes on the Physicochemical and Mechanical Properties of Dental Nanocomposite Resins" Applied Sciences 12, no. 1: 159. https://doi.org/10.3390/app12010159