Crosstalk between Fibroblast Growth Factor (FGF) Receptor and Integrin through Direct Integrin Binding to FGF and Resulting Integrin-FGF-FGFR Ternary Complex Formation


Abstract
:1. Introduction
2. Fibroblast Growth Factors (FGFs) are Multi-Functional Growth Factors
3. Signal Transduction of FGF and FGFR
4. Role of FGF and FGFR in Tumorigenesis, Tumor Angiogenesis, and Inflammation
5. Integrin-Growth Factor Receptor Crosstalk

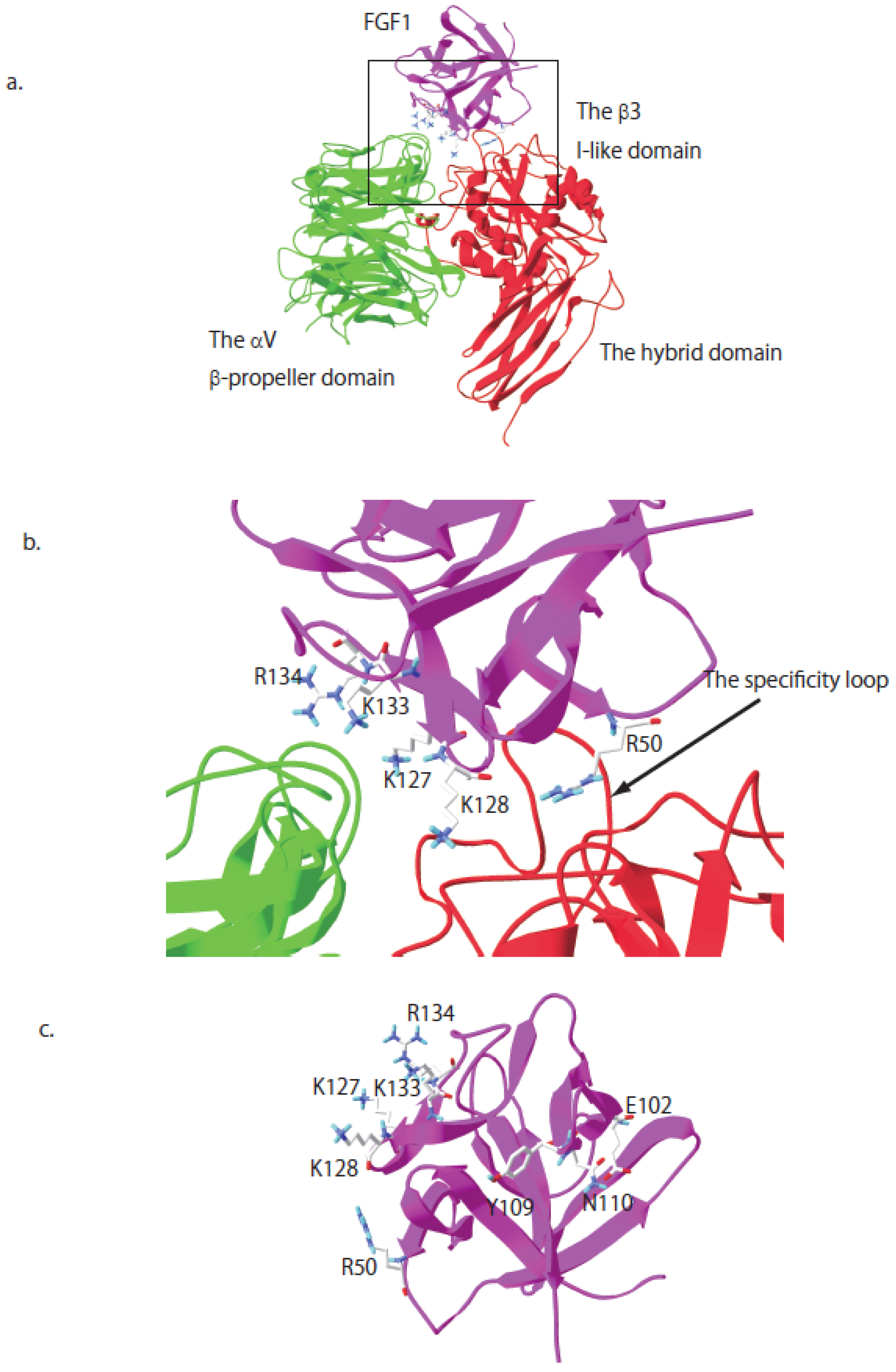
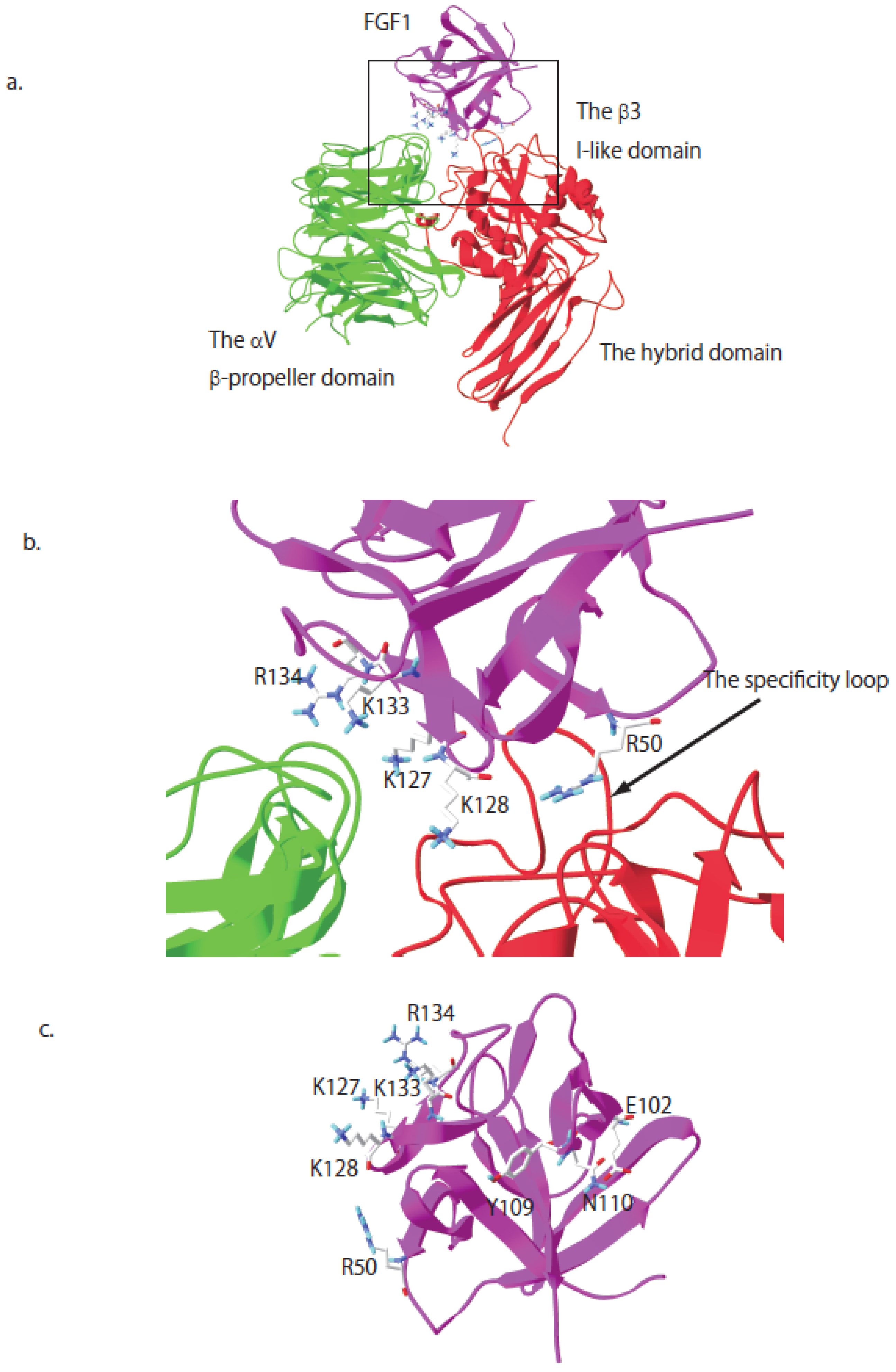{kind=link}
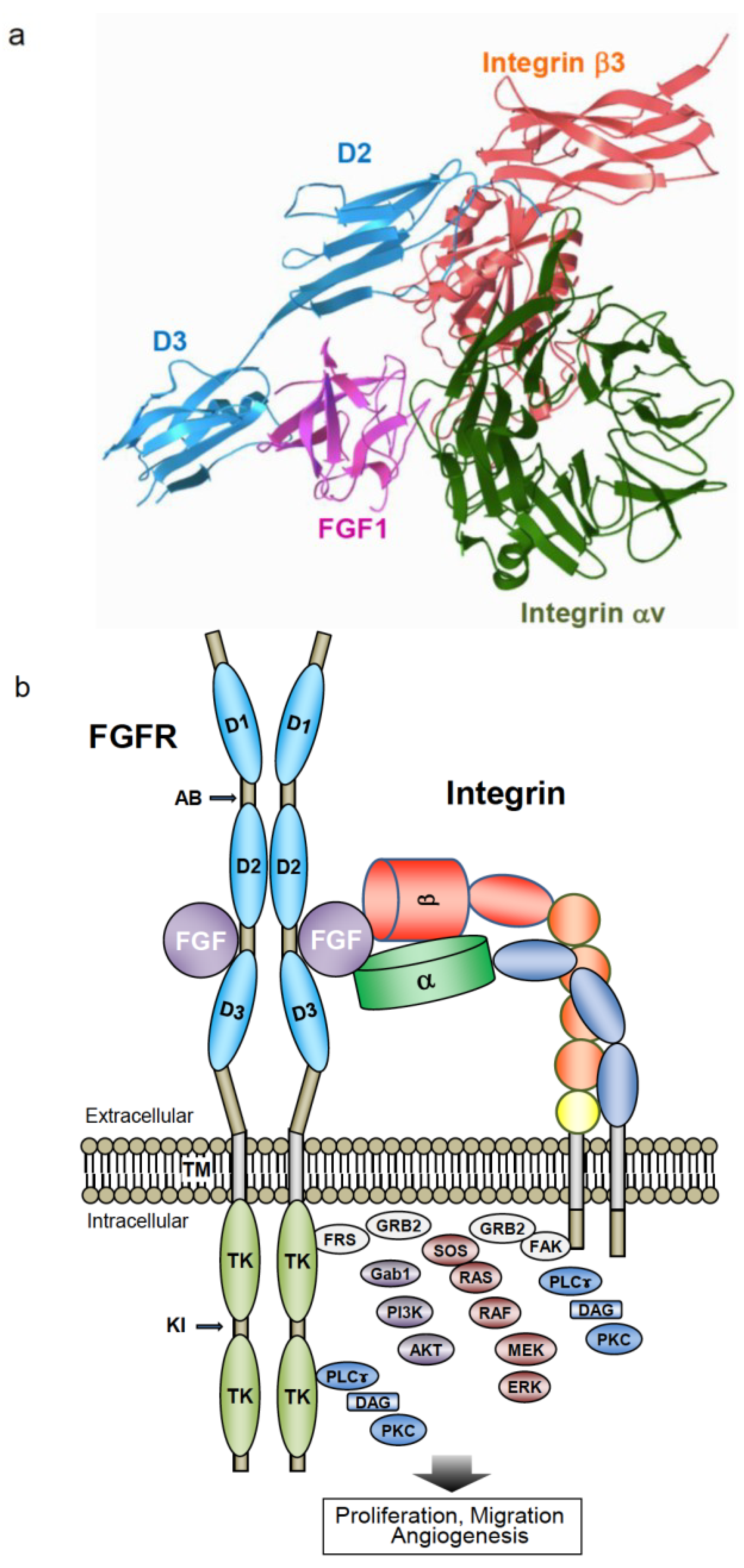{kind=link}
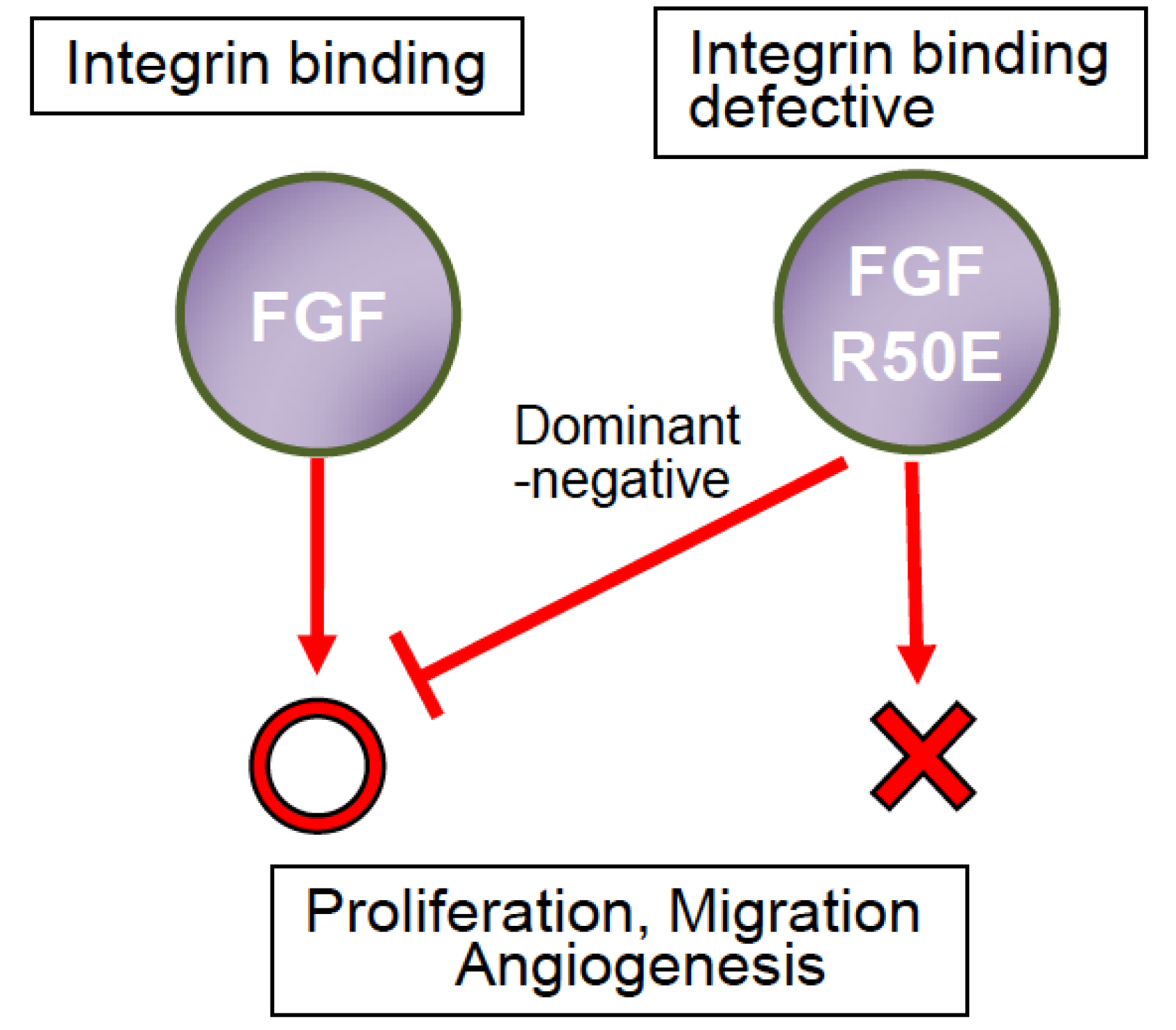{kind=link}
| Growth factor | Integrins | References |
|---|---|---|
| Angiopoietin-1 | α5β1 | [68] |
| Angiopoietin-2 | α5β1, β2 | [68,69] |
| CD40 ligand | αIIbβ3, α5β1, αMβ2 | [70,71,72] |
| FGF-1 | αvβ3 | [10] |
| FGF-2 | αvβ3 | [73] |
| Fractalkine | αvβ3, α4β1 | [74] |
| Pro HB-EGF | α3β1 | [75] |
| IGF-1 | αvβ3, α6β4 | [60,61] |
| LTGF-β | α6β4, αvβ8 | [66,67] |
| Neuregulin-1 | αvβ3, α6β4 | [62] |
| NGF | α9β1 | [76] |
| NT3 | α9β1 | [76] |
| Semaphorin 7A | α1β1 | [77] |
| VEGF-A | α3β1 | [64] |
| VEGF-C | α9β1 | [63] |
| VEGF-D | α9β1 | [63] |
6. Crosstalk between Integrin αvβ3 and FGFR through Direct Binding to FGF

7. R50E Is a Dominant-Negative Antagonist of FGFR
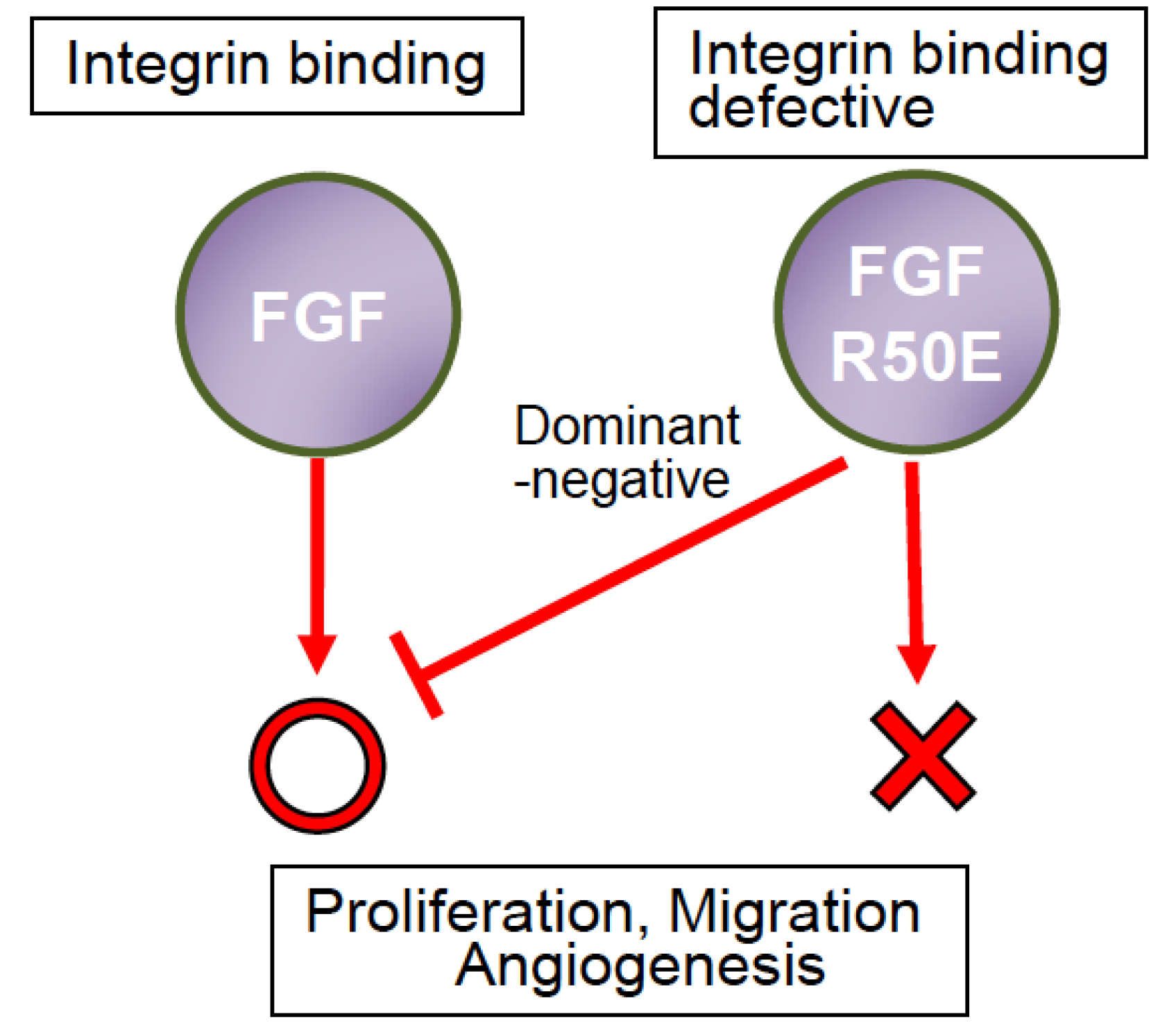
8. Potential Advantage of R50E over Antibodies and Kinase Inhibitors
9. Can Mutant Human Protein Be Used As a Therapeutic Agent?
10. Targeting and Delivery of R50E to Cancer
11. Conclusions
Acknowledgments
Conflict of Interest
References
- Schwartz, M.A.; Ginsberg, M.H. Networks and crosstalk: Integrin signalling spreads. Nat. Cell Biol. 2002, 4, E65–E68. [Google Scholar] [CrossRef]
- Itoh, N.; Ornitz, D.M. Evolution of the FGF and FGFR gene families. Trends Genet. 2004, 20, 563–569. [Google Scholar] [CrossRef]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef]
- Guillemot, F.; Zimmer, C. From cradle to grave: The multiple roles of fibroblast growth factors in neural development. Neuron 2011, 71, 574–588. [Google Scholar] [CrossRef]
- Polanska, U.M.; Fernig, D.G.; Kinnunen, T. Extracellular interactome of the FGF receptor-ligand system: Complexities and the relative simplicity of the worm. Dev. Dyn. 2009, 238, 277–293. [Google Scholar] [CrossRef]
- Murakami, M.; Elfenbein, A.; Simons, M. Non-canonical fibroblast growth factor signalling in angiogenesis. Cardiovasc. Res. 2008, 78, 223–231. [Google Scholar] [CrossRef]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007. [Google Scholar] [CrossRef]
- Legate, K.R.; Wickstrom, S.A.; Fassler, R. Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev. 2009, 23, 397–418. [Google Scholar] [CrossRef]
- Ivaska, J.; Heino, J. Interplay between cell adhesion and growth factor receptors: From the plasma membrane to the endosomes. Cell Tissue Res. 2010, 339, 111–120. [Google Scholar] [CrossRef]
- Mori, S.; Wu, C.Y.; Yamaji, S.; Saegusa, J.; Shi, B.; Ma, Z.; Kuwabara, Y.; Lam, K.S.; Isseroff, R.R.; Takada, Y.K.; et al. Direct binding of integrin αvβ3 to FGF1 plays a role in FGF1 signaling. J. Biol. Chem. 2008, 283, 18066–18075. [Google Scholar] [CrossRef]
- Lanner, F.; Rossant, J. The role of FGF/ERK signaling in pluripotent cells. Development 2010, 137, 3351–3360. [Google Scholar] [CrossRef]
- Jackson, A.; Tarantini, F.; Gamble, S.; Friedman, S.; Maciag, T. The release of fibroblast growth factor-1 from NIH 3T3 cells in response to temperature involves the function of cysteine residues. J. Biol. Chem. 1995, 270, 33–36. [Google Scholar] [CrossRef]
- Shi, J.; Friedman, S.; Maciag, T. A carboxyl-terminal domain in fibroblast growth factor (FGF)-2 inhibits FGF-1 release in response to heat shock in vitro. J. Biol. Chem. 1997, 272, 1142–1147. [Google Scholar] [CrossRef]
- Mouta Carreira, C.; Landriscina, M.; Bellum, S.; Prudovsky, I.; Maciag, T. The comparative release of FGF1 by hypoxia and temperature stress. Growth Factors 2001, 18, 277–285. [Google Scholar] [CrossRef]
- Schafer, T.; Zentgraf, H.; Zehe, C.; Brugger, B.; Bernhagen, J.; Nickel, W. Unconventional secretion of fibroblast growth factor 2 is mediated by direct translocation across the plasma membrane of mammalian cells. J. Biol. Chem. 2004, 279, 6244–6251. [Google Scholar]
- Mohan, S.K.; Rani, S.G.; Yu, C. The heterohexameric complex structure, a component in the non-classical pathway for fibroblast growth factor 1 (FGF1) secretion. J. Biol. Chem. 2010, 285, 15464–15475. [Google Scholar] [CrossRef]
- Goldfarb, M. Fibroblast growth factor homologous factors: Evolution, structure, and function. Cytokine Growth Factor Rev. 2005, 16, 215–220. [Google Scholar] [CrossRef]
- Schoorlemmer, J.; Goldfarb, M. Fibroblast growth factor homologous factors are intracellular signaling proteins. Curr. Biol. 2001, 11, 793–797. [Google Scholar] [CrossRef]
- Liu, C.; Dib-Hajj, S.D.; Waxman, S.G. Fibroblast growth factor homologous factor 1β binds to the c terminus of the tetrodotoxin-resistant sodium channel rnav1.9a (nan). J. Biol. Chem. 2001, 276, 18925–18933. [Google Scholar] [CrossRef]
- Tomlinson, E.; Fu, L.; John, L.; Hultgren, B.; Huang, X.; Renz, M.; Stephan, J.P.; Tsai, S.P.; Powell-Braxton, L.; French, D.; et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 2002, 143, 1741–1747. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Invest. 2005, 115, 1627–1635. [Google Scholar] [CrossRef]
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of FGF23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin d metabolism. J. Clin. Invest. 2004, 113, 561–568. [Google Scholar]
- Gospodarowicz, D. Purification of a fibroblast growth factor from bovine pituitary. J. Biol. Chem. 1975, 250, 2515–2520. [Google Scholar]
- Itoh, N.; Ornitz, D.M. Fibroblast growth factors: From molecular evolution to roles in development, metabolism and disease. J. Biochem. 2011, 149, 121–130. [Google Scholar] [CrossRef]
- Miller, D.L.; Ortega, S.; Bashayan, O.; Basch, R.; Basilico, C. Compensation by fibroblast growth factor 1 (FGF1) does not account for the mild phenotypic defects observed in FGF2 null mice. Mol. Cell Biol. 2000, 20, 2260–2268. [Google Scholar] [CrossRef]
- Zhou, M.; Sutliff, R.L.; Paul, R.J.; Lorenz, J.N.; Hoying, J.B.; Haudenschild, C.C.; Yin, M.; Coffin, J.D.; Kong, L.; Kranias, E.G.; et al. Fibroblast growth factor 2 control of vascular tone. Nat. Med. 1998, 4, 201–207. [Google Scholar] [CrossRef]
- Dono, R.; Texido, G.; Dussel, R.; Ehmke, H.; Zeller, R. Impaired cerebral cortex development and blood pressure regulation in FGF-2-deficient mice. EMBO J. 1998, 17, 4213–4225. [Google Scholar] [CrossRef]
- Powers, C.J.; McLeskey, S.W.; Wellstein, A. Fibroblast growth factors, their receptors and signaling. Endocr. Relat. Cancer 2000, 7, 165–197. [Google Scholar] [CrossRef]
- Groth, C.; Lardelli, M. The structure and function of vertebrate fibroblast growth factor receptor 1. Int. J. Dev. Biol. 2002, 46, 393–400. [Google Scholar]
- Mohammadi, M.; Olsen, S.K.; Ibrahimi, O.A. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005, 16, 107–137. [Google Scholar] [CrossRef]
- Zhang, X.; Ibrahimi, O.A.; Olsen, S.K.; Umemori, H.; Mohammadi, M.; Ornitz, D.M. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J. Biol. Chem. 2006, 281, 15694–15700. [Google Scholar]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef]
- Dorkin, T.J.; Robinson, M.C.; Marsh, C.; Bjartell, A.; Neal, D.E.; Leung, H.Y. FGF8 over-expression in prostate cancer is associated with decreased patient survival and persists in androgen independent disease. Oncogene 1999, 18, 2755–2761. [Google Scholar] [CrossRef]
- Feng, S.; Dakhova, O.; Creighton, C.J.; Ittmann, M. Endocrine fibroblast growth factor FGF19 promotes prostate cancer progression. Cancer Res. 2013, 73, 2551–2562. [Google Scholar] [CrossRef]
- Okunieff, P.; Fenton, B.M.; Zhang, L.; Kern, F.G.; Wu, T.; Greg, J.R.; Ding, I. Fibroblast growth factors (FGFs) increase breast tumor growth rate, metastases, blood flow, and oxygenation without significant change in vascular density. Adv. Exp. Med. Biol. 2003, 530, 593–601. [Google Scholar] [CrossRef]
- Gruel, N.; Lucchesi, C.; Raynal, V.; Rodrigues, M.J.; Pierron, G.; Goudefroye, R.; Cottu, P.; Reyal, F.; Sastre-Garau, X.; Fourquet, A.; et al. Lobular invasive carcinoma of the breast is a molecular entity distinct from luminal invasive ductal carcinoma. Eur. J. Cancer 2010, 46, 2399–2407. [Google Scholar] [CrossRef]
- Naidu, R.; Wahab, N.A.; Yadav, M.; Kutty, M.K.; Nair, S. Detection of amplified int-2/FGF-3 gene in primary breast carcinomas using differential polymerase chain reaction. Int. J. Mol. Med. 2001, 8, 193–198. [Google Scholar]
- Birrer, M.J.; Johnson, M.E.; Hao, K.; Wong, K.K.; Park, D.C.; Bell, A.; Welch, W.R.; Berkowitz, R.S.; Mok, S.C. Whole genome oligonucleotide-based array comparative genomic hybridization analysis identified fibroblast growth factor 1 as a prognostic marker for advanced-stage serous ovarian adenocarcinomas. J. Clin. Oncol. 2007, 25, 2281–2287. [Google Scholar] [CrossRef]
- Giri, D.; Ropiquet, F.; Ittmann, M. Alterations in expression of basic fibroblast growth factor (FGF) 2 and its receptor FGFR-1 in human prostate cancer. Clin. Cancer Res. 1999, 5, 1063–1071. [Google Scholar]
- Arora, N.; Scognamiglio, T.; Lubitz, C.C.; Moo, T.A.; Kato, M.A.; Zhu, B.; Zarnegar, R.; Chen, Y.T.; Fahey, T.J., 3rd. Identification of borderline thyroid tumors by gene expression array analysis. Cancer 2009, 115, 5421–5431. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Toratani, S.; Sato, J.D.; Kan, M.; McKeehan, W.L.; Okamoto, T. Growth inhibition by keratinocyte growth factor receptor of human salivary adenocarcinoma cells through induction of differentiation and apoptosis. Proc. Natl. Acad. Sci. USA 2001, 98, 11336–11340. [Google Scholar]
- Amann, T.; Bataille, F.; Spruss, T.; Dettmer, K.; Wild, P.; Liedtke, C.; Muhlbauer, M.; Kiefer, P.; Oefner, P.J.; Trautwein, C.; et al. Reduced expression of fibroblast growth factor receptor 2iiib in hepatocellular carcinoma induces a more aggressive growth. Am. J. Pathol. 2010, 176, 1433–1442. [Google Scholar] [CrossRef]
- Nakamura, N.; Iijima, T.; Mase, K.; Furuya, S.; Kano, J.; Morishita, Y.; Noguchi, M. Phenotypic differences of proliferating fibroblasts in the stroma of lung adenocarcinoma and normal bronchus tissue. Cancer Sci. 2004, 95, 226–232. [Google Scholar] [CrossRef]
- Presta, M.; Dell’Era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Mahabeleshwar, G.H.; Feng, W.; Reddy, K.; Plow, E.F.; Byzova, T.V. Mechanisms of integrin-vascular endothelial growth factor receptor cross-activation in angiogenesis. Circ. Res. 2007, 101, 570–580. [Google Scholar] [CrossRef]
- Borges, E.; Jan, Y.; Ruoslahti, E. Platelet-derived growth factor receptor β and vascular endothelial growth factor receptor 2 bind to the β3 integrin through its extracellular domain. J. Biol. Chem. 2000, 275, 39867–39873. [Google Scholar] [CrossRef]
- Cybulsky, A.V.; McTavish, A.J.; Cyr, M.D. Extracellular matrix modulates epidermal growth factor receptor activation in rat glomerular epithelial cells. J. Clin. Invest. 1994, 94, 68–78. [Google Scholar] [CrossRef]
- Soldi, R.; Mitola, S.; Strasly, M.; Defilippi, P.; Tarone, G.; Bussolino, F. Role of αvβ3 integrin in the activation of vascular endothelial growth factor receptor-2. EMBO J. 1999, 18, 882–892. [Google Scholar] [CrossRef]
- Clemmons, D.R.; Horvitz, G.; Engleman, W.; Nichols, T.; Moralez, A.; Nickols, G.A. Synthetic αvβ3 antagonists inhibit insulin-like growth factor-i-stimulated smooth muscle cell migration and replication. Endocrinology 1999, 140, 4616–4621. [Google Scholar] [CrossRef]
- Jones, P.L.; Crack, J.; Rabinovitch, M. Regulation of tenascin-c, a vascular smooth muscle cell survival factor that interacts with the αvβ3 integrin to promote epidermal growth factor receptor phosphorylation and growth. J. Cell Biol. 1997, 139, 279–293. [Google Scholar] [CrossRef]
- Brooks, P.C.; Clark, R.A.; Cheresh, D.A. Requirement of vascular integrin αvβ3 for angiogenesis. Science 1994, 264, 569–571. [Google Scholar]
- Friedlander, M.; Brooks, P.C.; Shaffer, R.W.; Kincaid, C.M.; Varner, J.A.; Cheresh, D.A. Definition of two angiogenic pathways by distinct αv integrins. Science 1995, 270, 1500–1502. [Google Scholar]
- Eliceiri, B.P.; Klemke, R.; Stromblad, S.; Cheresh, D.A. Integrin αvβ3 requirement for sustained mitogen-activated protein kinase activity during angiogenesis. J. Cell Biol. 1998, 140, 1255–1263. [Google Scholar] [CrossRef]
- Kim, S.; Bell, K.; Mousa, S.A.; Varner, J.A. Regulation of angiogenesis in vivo by ligation of integrin α5β1 with the central cell-binding domain of fibronectin. Am. J. Pathol. 2000, 156, 1345–1362. [Google Scholar] [CrossRef]
- Zou, L.; Cao, S.; Kang, N.; Huebert, R.C.; Shah, V.H. Fibronectin induces endothelial cell migration through β1 integrin and src-dependent phosphorylation of fibroblast growth factor receptor-1 at tyrosines 653/654 and 766. J. Biol. Chem. 2012, 287, 7190–7202. [Google Scholar]
- Schneller, M.; Vuori, K.; Ruoslahti, E. αvβ3 integrin associates with activated insulin and PDGFβ receptors and potentiates the biological activity of PDGF. EMBO J. 1997, 16, 5600–5607. [Google Scholar] [CrossRef]
- Mahabeleshwar, G.H.; Chen, J.; Feng, W.; Somanath, P.R.; Razorenova, O.V.; Byzova, T.V. Integrin affinity modulation in angiogenesis. Cell Cycle 2008, 7, 335–347. [Google Scholar] [CrossRef]
- Saegusa, J.; Yamaji, S.; Ieguchi, K.; Wu, C.Y.; Lam, K.S.; Liu, F.T.; Takada, Y.K.; Takada, Y. The direct binding of insulin-like growth factor-1 (IGF-1) to integrin αvβ3 is involved in IGF-1 signaling. J. Biol. Chem. 2009, 284, 24106–24114. [Google Scholar]
- Fujita, M.; Ieguchi, K.; Davari, P.; Yamaji, S.; Taniguchi, Y.; Sekiguchi, K.; Takada, Y.K.; Takada, Y. Cross-talk between integrin α6β4 and insulin-like growth factor-1 receptor (igf1r) through direct α6β4 binding to IGF1 and subsequent α6β4-IGF1-IGF1R ternary complex formation in anchorage-independent conditions. J. Biol. Chem. 2012, 287, 12491–12500. [Google Scholar] [CrossRef]
- Ieguchi, K.; Fujita, M.; Ma, Z.; Davari, P.; Taniguchi, Y.; Sekiguchi, K.; Wang, B.; Takada, Y.K.; Takada, Y. Direct binding of the EGF-like domain of neuregulin-1 to integrins (αvβ3 and α6β4) is involved in neuregulin-1/ErbB signaling. J. Biol. Chem. 2010, 285, 31388–31398. [Google Scholar] [CrossRef]
- Vlahakis, N.E.; Young, B.A.; Atakilit, A.; Sheppard, D. The lymphangiogenic vascular endothelial growth factors VEGF-C and -D are ligands for the integrin α9β1. J. Biol. Chem. 2005, 280, 4544–4552. [Google Scholar]
- Hutchings, H.; Ortega, N.; Plouet, J. Extracellular matrix-bound vascular endothelial growth factor promotes endothelial cell adhesion, migration, and survival through integrin ligation. FASEB J. 2003, 17, 1520–1522. [Google Scholar]
- Vlahakis, N.E.; Young, B.A.; Atakilit, A.; Hawkridge, A.E.; Issaka, R.B.; Boudreau, N.; Sheppard, D. Integrin α9β1 directly binds to vascular endothelial growth factor (VEGF)-A and contributes to VEGF-A-induced angiogenesis. J. Biol. Chem. 2007, 282, 15187–15196. [Google Scholar]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin αvβ6 binds and activates latent TGF β 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef]
- Cambier, S.; Gline, S.; Mu, D.; Collins, R.; Araya, J.; Dolganov, G.; Einheber, S.; Boudreau, N.; Nishimura, S.L. Integrin αvβ8-mediated activation of transforming growth factor-β by perivascular astrocytes: An angiogenic control switch. Am. J. Pathol. 2005, 166, 1883–1894. [Google Scholar] [CrossRef]
- Carlson, T.R.; Feng, Y.; Maisonpierre, P.C.; Mrksich, M.; Morla, A.O. Direct cell adhesion to the angiopoietins mediated by integrins. J. Biol. Chem. 2001, 276, 26516–26525. [Google Scholar]
- Bezuidenhout, L.; Zilla, P.; Davies, N. Association of ang-2 with integrin β2 controls ang-2/PDGF-BB-dependent upregulation of human peripheral blood monocyte fibrinolysis. Inflammation 2009, 32, 393–401. [Google Scholar] [CrossRef]
- Andre, P.; Prasad, K.S.; Denis, C.V.; He, M.; Papalia, J.M.; Hynes, R.O.; Phillips, D.R.; Wagner, D.D. CD40L stabilizes arterial thrombi by a β3 integrin--dependent mechanism. Nat. Med. 2002, 8, 247–252. [Google Scholar] [CrossRef]
- Leveille, C.; Bouillon, M.; Guo, W.; Bolduc, J.; Sharif-Askari, E.; El-Fakhry, Y.; Reyes-Moreno, C.; Lapointe, R.; Merhi, Y.; Wilkins, J.A.; et al. CD40 ligand binds to α5β1 integrin and triggers cell signaling. J. Biol. Chem. 2007, 282, 5143–5151. [Google Scholar]
- Zirlik, A.; Maier, C.; Gerdes, N.; MacFarlane, L.; Soosairajah, J.; Bavendiek, U.; Ahrens, I.; Ernst, S.; Bassler, N.; Missiou, A.; et al. CD40 ligand mediates inflammation independently of CD40 by interaction with mac-1. Circulation 2007, 115, 1571–1580. [Google Scholar] [CrossRef]
- Rusnati, M.; Tanghetti, E.; Dell’Era, P.; Gualandris, A.; Presta, M. αvβ3 integrin mediates the cell-adhesive capacity and biological activity of basic fibroblast growth factor (FGF-2) in cultured endothelial cells. Mol. Biol. Cell 1997, 8, 2449–2461. [Google Scholar] [CrossRef]
- Fujita, M.; Takada, Y.K.; Takada, Y. Integrins αvβ3 and α4β1 act as coreceptors for fractalkine, and the integrin-binding defective mutant of fractalkine is an antagonist of CX3CR1. J. Immunol. 2012, 189, 5809–5819. [Google Scholar] [CrossRef]
- Nakamura, K.; Iwamoto, R.; Mekada, E. Membrane-anchored heparin-binding EGF-like growth factor (HB-EGF) and diphtheria toxin receptor-associated protein (drap27)/CD9 form a complex with integrin α3β1 at cell-cell contact sites. J. Cell Biol. 1995, 129, 1691–1705. [Google Scholar] [CrossRef]
- Staniszewska, I.; Sariyer, I.K.; Lecht, S.; Brown, M.C.; Walsh, E.M.; Tuszynski, G.P.; Safak, M.; Lazarovici, P.; Marcinkiewicz, C. Integrin α9β1 is a receptor for nerve growth factor and other neurotrophins. J. Cell Sci. 2008, 121, 504–513. [Google Scholar] [CrossRef]
- Suzuki, K.; Okuno, T.; Yamamoto, M.; Pasterkamp, R.J.; Takegahara, N.; Takamatsu, H.; Kitao, T.; Takagi, J.; Rennert, P.D.; Kolodkin, A.L.; et al. Semaphorin 7a initiates t-cell-mediated inflammatory responses through α1β1 integrin. Nature 2007, 446, 680–684. [Google Scholar] [CrossRef]
- Tanghetti, E.; Ria, R.; Dell'Era, P.; Urbinati, C.; Rusnati, M.; Ennas, M.G.; Presta, M. Biological activity of substrate-bound basic fibroblast growth factor (FGF2): Recruitment of FGF receptor-1 in endothelial cell adhesion contacts. Oncogene 2002, 21, 3889–3897. [Google Scholar] [CrossRef]
- Sahni, A.; Francis, C.W. Stimulation of endothelial cell proliferation by FGF-2 in the presence of fibrinogen requires αvβ3. Blood 2004, 104, 3635–3641. [Google Scholar] [CrossRef]
- Sahni, A.; Khorana, A.A.; Baggs, R.B.; Peng, H.; Francis, C.W. FGF-2 binding to fibrin(ogen) is required for augmented angiogenesis. Blood 2006, 107, 126–131. [Google Scholar] [CrossRef]
- Sahni, A.; Altland, O.D.; Francis, C.W. FGF-2 but not FGF-1 binds fibrin and supports prolonged endothelial cell growth. J. Thromb. Haemost. 2003, 1, 1304–1310. [Google Scholar] [CrossRef]
- Yamaji, S.; Saegusa, J.; Ieguchi, K.; Fujita, M.; Mori, S.; Takada, Y.K.; Takada, Y. A novel fibroblast growth factor-1 (FGF1) mutant that acts as an FGF antagonist. PLoS One 2010, 5, e10273. [Google Scholar]
- Takagi, J.; Petre, B.M.; Walz, T.; Springer, T.A. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 2002, 110, 599–611. [Google Scholar] [CrossRef]
- Xiong, J.P.; Stehle, T.; Zhang, R.; Joachimiak, A.; Frech, M.; Goodman, S.L.; Arnaout, M.A. Crystal structure of the extracellular segment of integrin αvβ3 in complex with an Arg-Gly-Asp ligand. Science 2002, 296, 151–155. [Google Scholar] [CrossRef]
- Beglova, N.; Blacklow, S.C.; Takagi, J.; Springer, T.A. Cysteine-rich module structure reveals a fulcrum for integrin rearrangement upon activation. Nat. Struct. Biol. 2002, 9, 282–287. [Google Scholar] [CrossRef]
- Adair, B.D.; Xiong, J.P.; Maddock, C.; Goodman, S.L.; Arnaout, M.A.; Yeager, M. Three-dimensional em structure of the ectodomain of integrin αvβ3 in a complex with fibronectin. J. Cell Biol. 2005, 168, 1109–1118. [Google Scholar] [CrossRef]
- Sharrocks, A.D. Cell cycle: Sustained ERK signalling represses the inhibitors. Curr. Biol. 2006, 16, R540–R542. [Google Scholar] [CrossRef]
- Schlessinger, J.; Plotnikov, A.N.; Ibrahimi, O.A.; Eliseenkova, A.V.; Yeh, B.K.; Yayon, A.; Linhardt, R.J.; Mohammadi, M. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 2000, 6, 743–750. [Google Scholar] [CrossRef]
- Mori, S.; Tran, V.; Nishikawa, K.; Kaneda, T.; Hamada, Y.; Kawaguchi, N.; Fujita, M.; Takada, Y.K.; Matsuura, N.; Zhao, M.; et al. A dominant-negative FGF1 mutant (the R50E mutant) suppresses tumorigenesis and angiogenesis. PLoS One 2013, 8, e57927. [Google Scholar] [CrossRef]
- Kopchick, J.J.; Parkinson, C.; Stevens, E.C.; Trainer, P.J. Growth hormone receptor antagonists: Discovery, development, and use in patients with acromegaly. Endocr. Rev. 2002, 23, 623–646. [Google Scholar] [CrossRef]
- Schreiber, I.; Buchfelder, M.; Droste, M.; Forssmann, K.; Mann, K.; Saller, B.; Strasburger, C.J. Treatment of acromegaly with the gh receptor antagonist pegvisomant in clinical practice: Safety and efficacy evaluation from the german pegvisomant observational study. Eur. J. Endocrinol. 2007, 156, 75–82. [Google Scholar] [CrossRef]
- Wang, M.; Thanou, M. Targeting nanoparticles to cancer. Pharmacol. Res. 2010, 62, 90–99. [Google Scholar] [CrossRef]
- Moya, M.L.; Morley, M.; Khanna, O.; Opara, E.C.; Brey, E.M. Stability of alginate microbead properties in vitro. J. Mater. Sci. Mater. Med. 2012, 23, 903–912. [Google Scholar] [CrossRef]
- Khanna, O.; Moya, M.L.; Opara, E.C.; Brey, E.M. Synthesis of multilayered alginate microcapsules for the sustained release of fibroblast growth factor-1. J. Biomed. Mater. Res. A 2010, 95, 632–640. [Google Scholar]
- Witzenbichler, B.; Mahfoudi, A.; Soubrier, F.; Le Roux, A.; Branellec, D.; Schultheiss, H.P.; Isner, J.M. Intramuscular gene transfer of fibroblast growth factor-1 using improved pcor plasmid design stimulates collateral formation in a rabbit ischemic hindlimb model. J. Mol. Med. (Berl) 2006, 84, 491–502. [Google Scholar] [CrossRef]
- Baumgartner, I.; Chronos, N.; Comerota, A.; Henry, T.; Pasquet, J.P.; Finiels, F.; Caron, A.; Dedieu, J.F.; Pilsudski, R.; Delaere, P. Local gene transfer and expression following intramuscular administration of FGF-1 plasmid DNA in patients with critical limb ischemia. Mol. Ther. 2009, 17, 914–921. [Google Scholar] [CrossRef]
- Eliceiri, B.P. Integrin and growth factor receptor crosstalk. Circ. Res. 2001, 89, 1104–1110. [Google Scholar] [CrossRef]
- Somanath, P.R.; Ciocea, A.; Byzova, T.V. Integrin and growth factor receptor alliance in angiogenesis. Cell Biochem. Biophys. 2009, 53, 53–64. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mori, S.; Takada, Y. Crosstalk between Fibroblast Growth Factor (FGF) Receptor and Integrin through Direct Integrin Binding to FGF and Resulting Integrin-FGF-FGFR Ternary Complex Formation. Med. Sci. 2013, 1, 20-36. https://doi.org/10.3390/medsci1010020
Mori S, Takada Y. Crosstalk between Fibroblast Growth Factor (FGF) Receptor and Integrin through Direct Integrin Binding to FGF and Resulting Integrin-FGF-FGFR Ternary Complex Formation. Medical Sciences. 2013; 1(1):20-36. https://doi.org/10.3390/medsci1010020
Chicago/Turabian StyleMori, Seiji, and Yoshikazu Takada. 2013. "Crosstalk between Fibroblast Growth Factor (FGF) Receptor and Integrin through Direct Integrin Binding to FGF and Resulting Integrin-FGF-FGFR Ternary Complex Formation" Medical Sciences 1, no. 1: 20-36. https://doi.org/10.3390/medsci1010020