Regenerative Inflammation: Lessons from Drosophila Intestinal Epithelium in Health and Disease

Abstract
:
1. Introduction
2. Early Lessons from Drosophila Systemic Immune Response
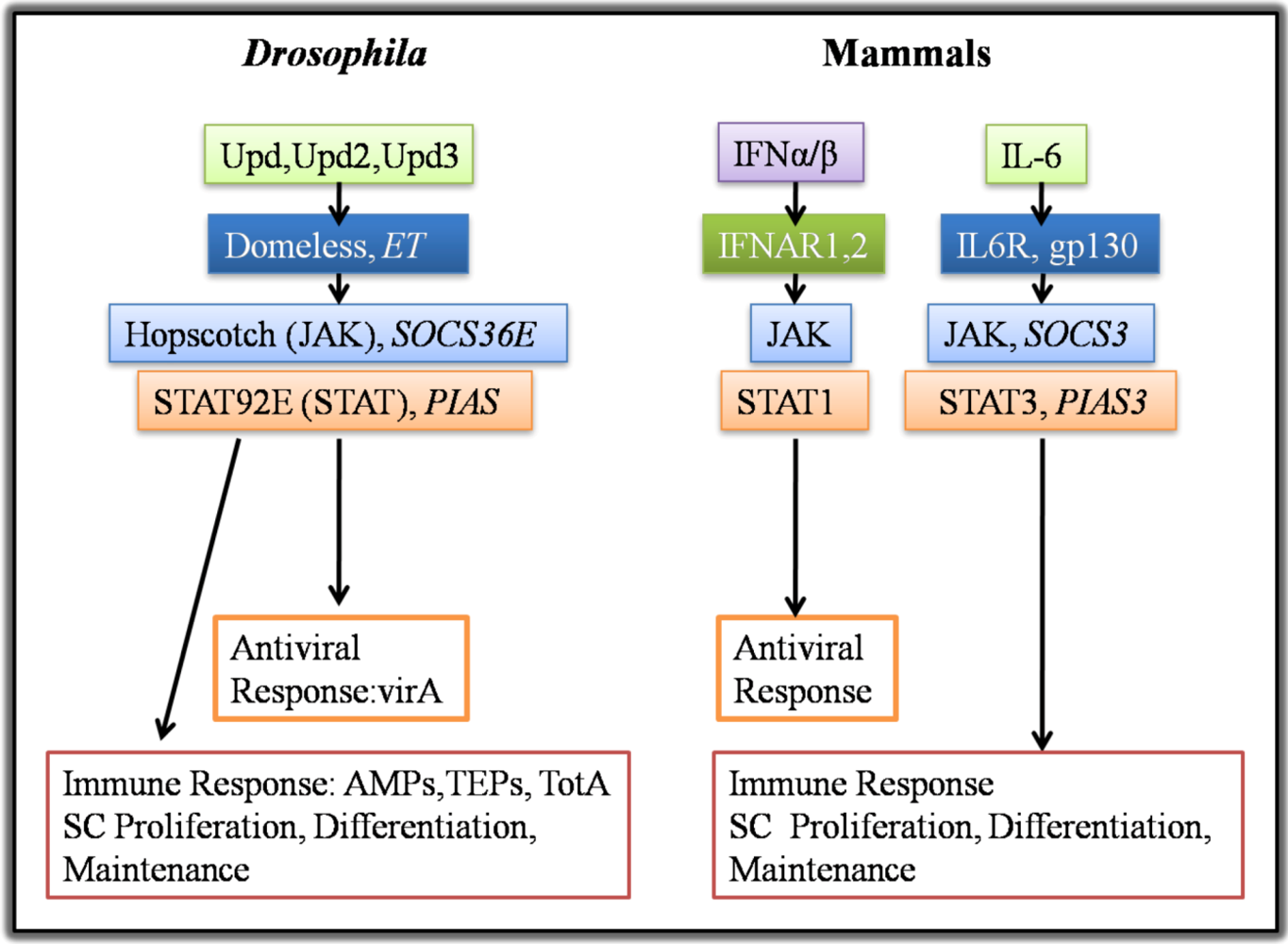
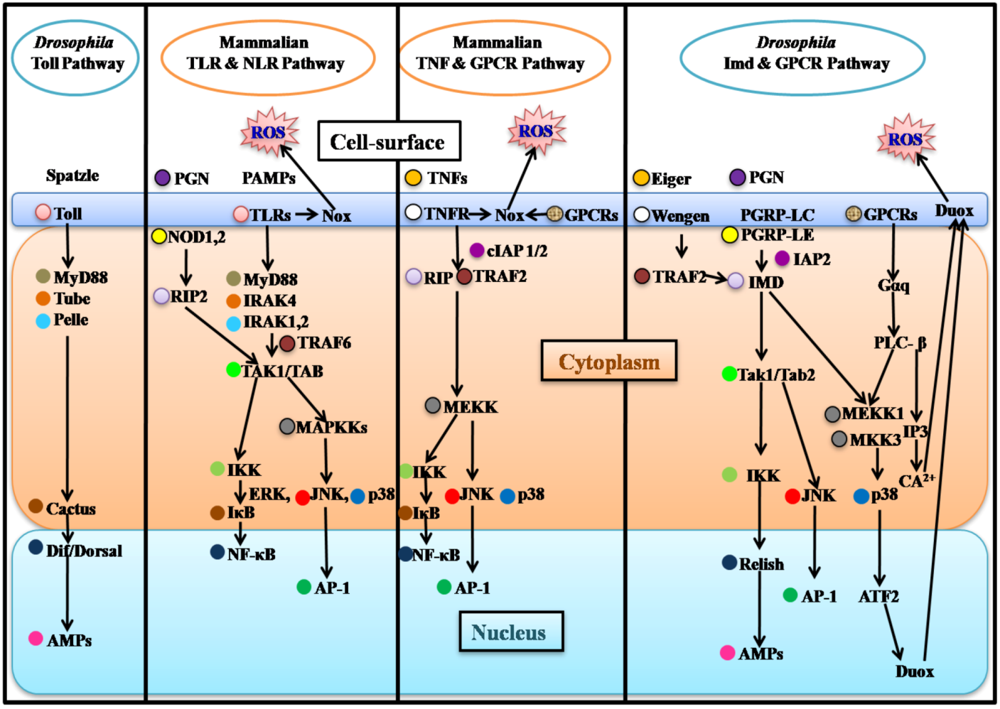
2.1. Drosophila Systemic Immune Response
2.2. Mammalian Systemic Immune Response and Parallels with Drosophila
3. Epithelial Immune Responses of Flies and Mammals
3.1. Drosophila Epithelial Immune Responses
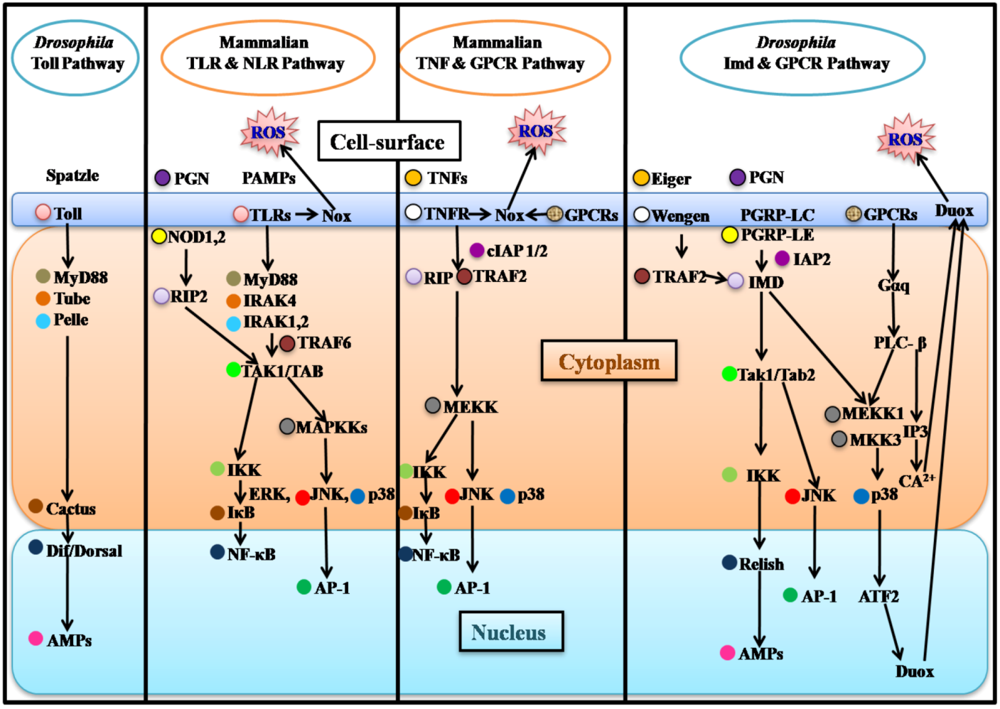
3.2. Mammalian Epithelial Immune Responses and Parallels with Drosophila
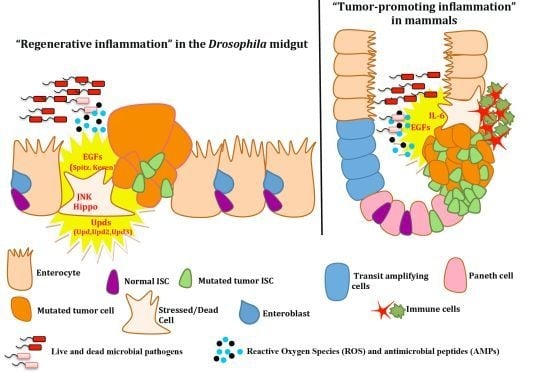
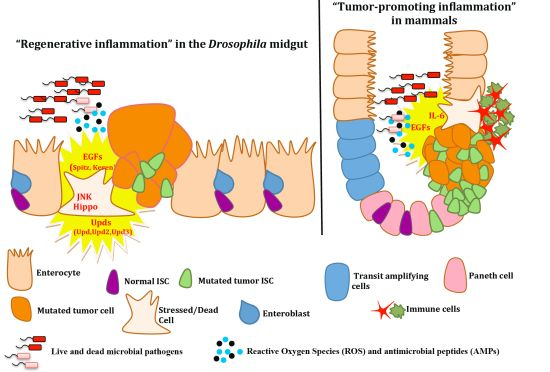
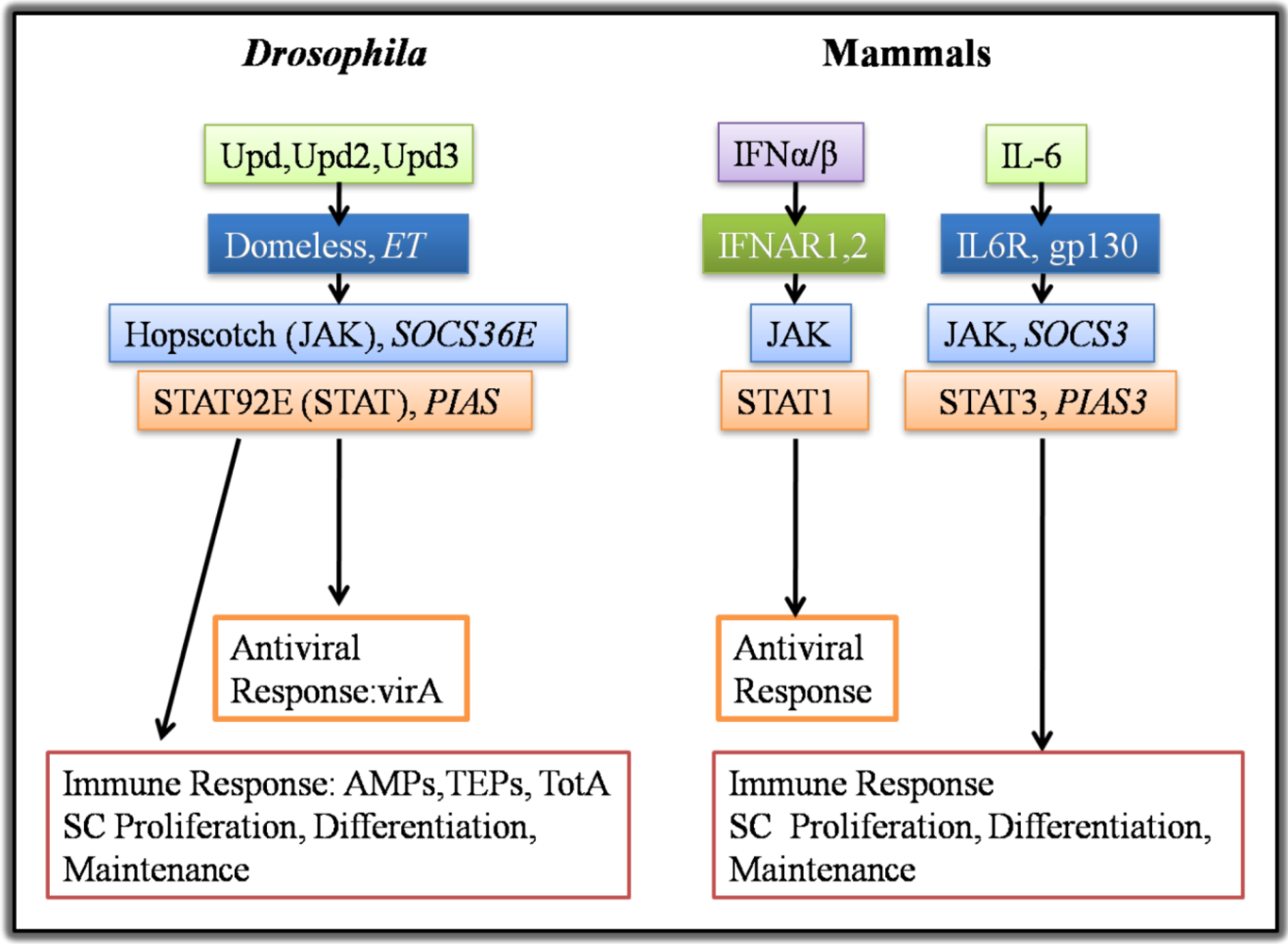
4. Epithelial ISC Responses: Regenerative Inflammation
4.1. Growth Factors and Cytokines in Intestinal Stem Cell Maintenance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mammals | Drosophila | |
|---|---|---|
| ISC Proliferation/ Maintenance | Wnt | Wingless |
| IL-6 (Stat3) | Upds (JAK/STAT) | |
| EGF (EGFR-Ras) | Spitz,Keren,Vein (EGFR-Ras1) | |
| ISC Differentiation | Ihh | Hh |
| BMP | Dpp? | |
| Wnt | Wingless | |
| IL-6 (Stat3) | Upds (JAK/STAT) | |
| EGF (EGFR-Ras) | Spitz,Keren,Vein (EGFR-Ras1) | |
| EC Apoptosis | TNF (PAMPs/NF-κB/JNK) | Eiger (PAMPs/NF-κB/JNK) |
| EC Immune Response | TNF (PAMPs/NF-κB/JNK) | Eiger (PAMPs/NF-κB/JNK) |
| IL-6 (Stat3) | Upds (JAK/STAT) |
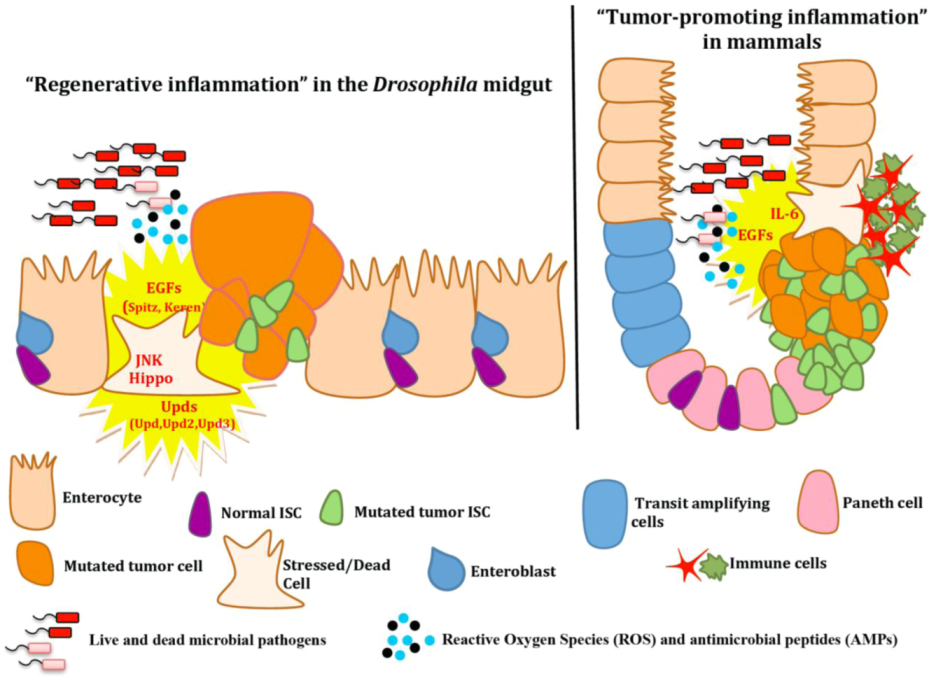
4.2. Intestinal Epithelium Regeneration and Cancer-Promoting Inflammation
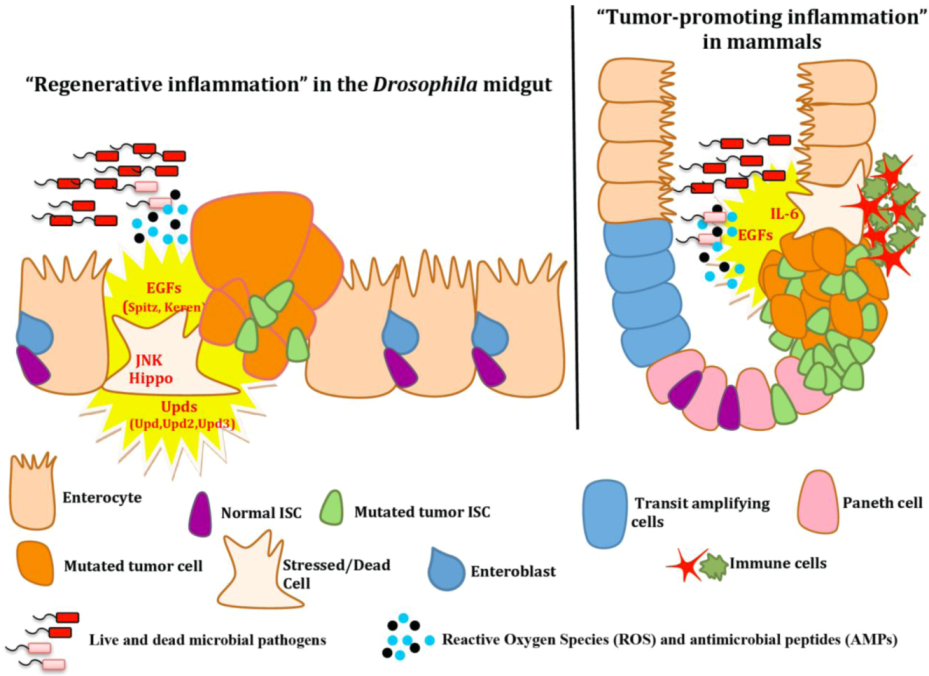
5. Other Frontiers in Drosophila Inflammation
5.1. Organ Communication: Inflammatory Signal Cross-Talk between Different Organs
5.2. Intestinal Microbiota and Inflammation
6. Conclusions
Conflict of Interest
References and Notes
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer. 2006, 6, 24–37. [Google Scholar] [CrossRef]
- Trinchieri, G. Cancer and Inflammation: An old intuition with rapidly evolving new concepts. Annu. Rev. Immunol. 2012, 30, 677–706. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and Cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar]
- Mladenova, D.; Kohonen-Corish, M.R. Review: Mouse Models of Inflammatory Bowel Disease--Insights into the Mechanisms of Inflammation-Associated Colorectal Cancer. In Vivo 2012, 26, 627–646. [Google Scholar]
- Apidianakis, Y.; Rahme, L.G. Drosophila Melanogaster as a Model for Human Intestinal Infection and Pathology. Dis. Model. Mech. 2011, 4, 21–30. [Google Scholar] [CrossRef]
- Limmer, S.; Quintin, J.; Hetru, C.; Ferrandon, D. Virulence on the Fly: Drosophila Melanogaster as a Model Genetic Organism to Decipher Host-Pathogen Interactions. Curr. Drug Targets 2011, 12, 978–999. [Google Scholar] [CrossRef]
- Valanne, S.; Wang, J.H.; Ramet, M. The Drosophila Toll Signaling Pathway. J. Immunol. 2011, 186, 649–656. [Google Scholar]
- Kounatidis, I.; Ligoxygakis, P. Drosophila as a Model System to Unravel the Layers of Innate Immunity to Infection. Open Biol. 2012, 2, 120075. [Google Scholar] [CrossRef]
- Bier, E.; Guichard, A. Deconstructing Host-Pathogen Interactions in Drosophila. Dis. Model. Mech. 2012, 5, 48–61. [Google Scholar] [CrossRef]
- Agaisse, H.; Petersen, U.M.; Boutros, M.; Mathey-Prevot, B.; Perrimon, N. Signaling Role of Hemocytes in Drosophila JAK/STAT-Dependent Response to Septic Injury. Dev. Cell 2003, 5, 441–450. [Google Scholar] [CrossRef]
- Towb, P.; Sun, H.; Wasserman, S.A. Tube is an IRAK-4 Homolog in a Toll Pathway Adapted for Development and Immunity. J. Innate Immun. 2009, 1, 309–321. [Google Scholar] [CrossRef]
- Huang, G.; Shi, L.Z.; Chi, H. Regulation of JNK and p38 MAPK in the Immune System: Signal Integration, Propagation and Termination. Cytokine 2009, 48, 161–169. [Google Scholar] [CrossRef]
- Ashwell, J.D. The Many Paths to p38 Mitogen-Activated Protein Kinase Activation in the Immune System. Nat. Rev. Immunol. 2006, 6, 532–540. [Google Scholar] [CrossRef]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern Recognition Receptors and the Innate Immune Response to Viral Infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen Recognition by the Innate Immune System. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef]
- Shin, O.S.; Harris, J.B. Innate Immunity and Transplantation Tolerance: The Potential Role of TLRs/NLRs in GVHD. Korean J. Hematol. 2011, 46, 69–79. [Google Scholar] [CrossRef]
- Mahoney, D.J.; Cheung, H.H.; Mrad, R.L.; Plenchette, S.; Simard, C.; Enwere, E.; Arora, V.; Mak, T.W.; Lacasse, E.C.; Waring, J.; et al. Both cIAP1 and cIAP2 Regulate TNFalpha-Mediated NF-kappaB Activation. Proc. Natl. Acad. Sci. USA 2008, 105, 11778–11783. [Google Scholar] [CrossRef]
- Valanne, S.; Kleino, A.; Myllymaki, H.; Vuoristo, J.; Ramet, M. Iap2 is Required for a Sustained Response in the Drosophila Imd Pathway. Dev. Comp. Immunol. 2007, 31, 991–1001. [Google Scholar] [CrossRef]
- Wang, W.B.; Levy, D.E.; Lee, C.K. STAT3 Negatively Regulates Type I IFN-Mediated Antiviral Response. J. Immunol. 2011, 187, 2578–2585. [Google Scholar] [CrossRef]
- Naugler, W.E.; Karin, M. The Wolf in Sheep's Clothing: The Role of Interleukin-6 in Immunity, Inflammation and Cancer. Trends Mol. Med. 2008, 14, 109–119. [Google Scholar] [CrossRef]
- Camporeale, A.; Poli, V. IL-6, IL-17 and STAT3: A Holy Trinity in Auto-Immunity? Front. Biosci. 2012, 17, 2306–2326. [Google Scholar] [CrossRef]
- Nishimura, T.; Andoh, A.; Inatomi, O.; Shioya, M.; Yagi, Y.; Tsujikawa, T.; Fujiyama, Y. Amphiregulin and Epiregulin Expression in Neoplastic and Inflammatory Lesions in the Colon. Oncol. Rep. 2008, 19, 105–110. [Google Scholar]
- Kallio, J.; Myllymaki, H.; Gronholm, J.; Armstrong, M.; Vanha-aho, L.M.; Makinen, L.; Silvennoinen, O.; Valanne, S.; Ramet, M. Eye Transformer is a Negative Regulator of Drosophila JAK/STAT Signaling. FASEB J. 2010, 24, 4467–4479. [Google Scholar] [CrossRef]
- Callus, B.A.; Mathey-Prevot, B. SOCS36E, a Novel Drosophila SOCS Protein, Suppresses JAK/STAT and EGF-R Signalling in the Imaginal Wing Disc. Oncogene 2002, 21, 4812–4821. [Google Scholar] [CrossRef]
- Betz, A.; Lampen, N.; Martinek, S.; Young, M.W.; Darnell, J.E., Jr. A Drosophila PIAS Homologue Negatively Regulates stat92E. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 9563–9568. [Google Scholar]
- Barclay, J.L.; Anderson, S.T.; Waters, M.J.; Curlewis, J.D. Regulation of Suppressor of Cytokine Signaling 3 (SOC3) by Growth Hormone in Pro-B Cells. Mol. Endocrinol. 2007, 21, 2503–2515. [Google Scholar] [CrossRef]
- Duechting, A.; Tschope, C.; Kaiser, H.; Lamkemeyer, T.; Tanaka, N.; Aberle, S.; Lang, F.; Torresi, J.; Kandolf, R.; Bock, C.T. Human Parvovirus B19 NS1 Protein Modulates Inflammatory Signaling by Activation of STAT3/PIAS3 in Human Endothelial Cells. J. Virol. 2008, 82, 7942–7952. [Google Scholar] [CrossRef]
- Ha, E.M.; Oh, C.T.; Ryu, J.H.; Bae, Y.S.; Kang, S.W.; Jang, I.H.; Brey, P.T.; Lee, W.J. An Antioxidant System Required for Host Protection Against Gut Infection in Drosophila. Dev. Cell 2005, 8, 125–132. [Google Scholar]
- Charroux, B.; Royet, J. Drosophila Immune Response: From Systemic Antimicrobial Peptide Production in Fat Body Cells to Local Defense in the Intestinal Tract. Fly (Austin) 2010, 4, 40–47. [Google Scholar]
- Vallet-Gely, I.; Lemaitre, B.; Boccard, F. Bacterial Strategies to Overcome Insect Defences. Nat. Rev. Microbiol. 2008, 6, 302–313. [Google Scholar]
- Pitsouli, C.; Apidianakis, Y.; Perrimon, N. Homeostasis in Infected Epithelia: Stem Cells Take the Lead. Cell Host Microbe 2009, 6, 301–307. [Google Scholar]
- Ahn, H.M.; Lee, K.S.; Lee, D.S.; Yu, K. JNK/FOXO Mediated PeroxiredoxinV Expression Regulates Redox Homeostasis during Drosophila Melanogaster Gut Infection. Dev. Comp. Immunol. 2012, 38, 466–473. [Google Scholar]
- Biteau, B.; Hochmuth, C.E.; Jasper, H. JNK Activity in Somatic Stem Cells Causes Loss of Tissue Homeostasis in the Aging Drosophila Gut. Cell Stem Cell 2008, 3, 442–455. [Google Scholar]
- Wang, M.C.; Bohmann, D.; Jasper, H. JNK Signaling Confers Tolerance to Oxidative Stress and Extends Lifespan in Drosophila. Dev. Cell 2003, 5, 811–816. [Google Scholar]
- Bae, Y.S.; Choi, M.K.; Lee, W.J. Dual Oxidase in Mucosal Immunity and Host-Microbe Homeostasis. Trends Immunol. 2010, 31, 278–287. [Google Scholar]
- Wu, H.; Wang, M.C.; Bohmann, D. JNK Protects Drosophila from Oxidative Stress by Trancriptionally Activating Autophagy. Mech. Dev. 2009, 126, 624–637. [Google Scholar]
- Schneider, D.S.; Ayres, J.S.; Brandt, S.M.; Costa, A.; Dionne, M.S.; Gordon, M.D.; Mabery, E.M.; Moule, M.G.; Pham, L.N.; Shirasu-Hiza, M.M. Drosophila Eiger Mutants are Sensitive to Extracellular Pathogens. PLoS Pathog. 2007, 3, e41. [Google Scholar]
- Berkey, C.D.; Blow, N.; Watnick, P.I. Genetic Analysis of Drosophila Melanogaster Susceptibility to Intestinal Vibrio Cholerae Infection. Cell Microbiol. 2009, 11, 461–474. [Google Scholar]
- Bangi, E.; Pitsouli, C.; Rahme, L.G.; Cagan, R.; Apidianakis, Y. Immune Response to Bacteria Induces Dissemination of Ras-Activated Drosophila Hindgut Cells. EMBO Rep. 2012, 13, 569–576. [Google Scholar]
- Christofi, T.; Apidianakis, Y. Ras-Oncogenic Drosophila Hindgut but Not Midgut Cells use an Inflammation-Like Program to Disseminate to Distant Sites. Gut Microbes 2013, 4, 54–59. [Google Scholar]
- Latz, E. NOX-Free Inflammasome Activation. Blood 2010, 116, 1393–1394. [Google Scholar]
- Swanson, P.A., 2nd; Kumar, A.; Samarin, S.; Vijay-Kumar, M.; Kundu, K.; Murthy, N.; Hansen, J.; Nusrat, A.; Neish, A.S. Enteric Commensal Bacteria Potentiate Epithelial Restitution Via Reactive Oxygen Species-Mediated Inactivation of Focal Adhesion Kinase Phosphatases. Proc. Natl. Acad. Sci. USA 2011, 108, 8803–8808. [Google Scholar]
- Geiszt, M.; Witta, J.; Baffi, J.; Lekstrom, K.; Leto, T.L. Dual Oxidases Represent Novel Hydrogen Peroxide Sources Supporting Mucosal Surface Host Defense. FASEB J. 2003, 17, 1502–1504. [Google Scholar]
- El Hassani, R.A.; Benfares, N.; Caillou, B.; Talbot, M.; Sabourin, J.C.; Belotte, V.; Morand, S.; Gnidehou, S.; Agnandji, D.; Ohayon, R.; et al. Dual oxidase2 is Expressed all Along the Digestive Tract. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G933–G942. [Google Scholar]
- Rokutan, K.; Kawahara, T.; Kuwano, Y.; Tominaga, K.; Nishida, K.; Teshima-Kondo, S. Nox Enzymes and Oxidative Stress in the Immunopathology of the Gastrointestinal Tract. Semin. Immunopathol. 2008, 30, 315–327. [Google Scholar]
- Matsuzawa, A.; Ichijo, H. Redox Control of Cell Fate by MAP Kinase: Physiological Roles of ASK1-MAP Kinase Pathway in Stress Signaling. Biochim. Biophys. Acta 2008, 1780, 1325–1336. [Google Scholar]
- Schramm, A.; Matusik, P.; Osmenda, G.; Guzik, T.J. Targeting NADPH Oxidases in Vascular Pharmacology. Vascul Pharmacol. 2012, 56, 216–231. [Google Scholar]
- Tae Lim, Y.; Sup Song, D.; Joon Won, T.; Lee, Y.J.; Yoo, J.S.; Eun Hyung, K.; Won Yoon, J.; Park, S.Y.; Woo Hwang, K. Peroxiredoxin-1, a Possible Target in Modulating Inflammatory Cytokine Production in Macrophage Like Cell Line RAW264.7. Microbiol. Immunol. 2012, 56, 411–419. [Google Scholar]
- Bast, A.; Erttmann, S.F.; Walther, R.; Steinmetz, I. Influence of iNOS and COX on Peroxiredoxin Gene Expression in Primary Macrophages. Free Radic. Biol. Med. 2010, 49, 1881–1891. [Google Scholar]
- Yano, T.; Kurata, S. Intracellular Recognition of Pathogens and Autophagy as an Innate Immune Host Defence. J. Biochem. 2011, 150, 143–149. [Google Scholar]
- Cadwell, K.; Patel, K.K.; Maloney, N.S.; Liu, T.C.; Ng, A.C.; Storer, C.E.; Head, R.D.; Xavier, R.; Stappenbeck, T.S.; Virgin, H.W. Virus-Plus-Susceptibility Gene Interaction Determines Crohn's Disease Gene Atg16L1 Phenotypes in Intestine. Cell 2010, 141, 1135–1145. [Google Scholar]
- Kabi, A.; Nickerson, K.P.; Homer, C.R.; McDonald, C. Digesting the Genetics of Inflammatory Bowel Disease: Insights from Studies of Autophagy Risk Genes. Inflamm. Bowel Dis. 2012, 18, 782–792. [Google Scholar]
- Hisamatsu, T.; Kanai, T.; Mikami, Y.; Yoneno, K.; Matsuoka, K.; Hibi, T. Immune Aspects of the Pathogenesis of Inflammatory Bowel Disease. Pharmacol. Ther. 2013, 137, 283–297. [Google Scholar]
- Brest, P.; Lapaquette, P.; Souidi, M.; Lebrigand, K.; Cesaro, A.; Vouret-Craviari, V.; Mari, B.; Barbry, P.; Mosnier, J.F.; Hebuterne, X.; et al. A Synonymous Variant in IRGM Alters a Binding Site for miR-196 and Causes Deregulation of IRGM-Dependent Xenophagy in Crohn's Disease. Nat. Genet. 2011, 43, 242–245. [Google Scholar]
- Ioannidou, E. Therapeutic Modulation of Growth Factors and Cytokines in Regenerative Medicine. Curr. Pharm. Des. 2006, 12, 2397–2408. [Google Scholar]
- Broughton, S.E.; Hercus, T.R.; Lopez, A.F.; Parker, M.W. Cytokine Receptor Activation at the Cell Surface. Curr. Opin. Struct. Biol. 2012, 22, 350–359. [Google Scholar]
- Cutler, A.; Brombacher, F. Cytokine Therapy. Ann. N. Y. Acad. Sci. 2005, 1056, 16–29. [Google Scholar]
- Feliciani, C.; Gupta, A.K.; Sauder, D.N. Keratinocytes and cytokine/growth Factors. Crit. Rev. Oral Biol. Med. 1996, 7, 300–318. [Google Scholar]
- Broughton, S.E.; Dhagat, U.; Hercus, T.R.; Nero, T.L.; Grimbaldeston, M.A.; Bonder, C.S.; Lopez, A.F.; Parker, M.W. The GM-CSF/IL-3/IL-5 Cytokine Receptor Family: From Ligand Recognition to Initiation of Signaling. Immunol. Rev. 2012, 250, 277–302. [Google Scholar]
- Kuraishy, A.; Karin, M.; Grivennikov, S.I. Tumor Promotion Via Injury- and Death-Induced Inflammation. Immunity 2011, 35, 467–477. [Google Scholar]
- Takashima, S.; Hartenstein, V. Genetic Control of Intestinal Stem Cell Specification and Development: A Comparative View. Stem Cell Rev. 2012, 8, 597–608. [Google Scholar] [CrossRef]
- Singh, S.R.; Mishra, M.K.; Kango-Singh, M.; Hou, S.X. Generation and Staining of Intestinal Stem Cell Lineage in Adult Midgut. Methods Mol. Biol. 2012, 879, 47–69. [Google Scholar]
- Takashima, S.; Hartenstein, V. Genetic Control of Intestinal Stem Cell Specification and Development: A Comparative View. Stem Cell Rev. 2012, 8, 597–608. [Google Scholar] [CrossRef]
- Lucchetta, E.M.; Ohlstein, B. The Drosophila Midgut: A Model for Stem Cell Driven Tissue Regeneration. Wiley Interdisciplinary Reviews: Develop. Biol. 2012, 1, 781–788. [Google Scholar]
- Perdigoto, C.N.; Bardin, A.J. Sending the Right Signal: Notch and Stem Cells. Biochim. Biophys. Acta 2013, 1830, 2307–2322. [Google Scholar] [CrossRef]
- Fre, S.; Pallavi, S.K.; Huyghe, M.; Lae, M.; Janssen, K.P.; Robine, S.; Artavanis-Tsakonas, S.; Louvard, D. Notch and Wnt Signals Cooperatively Control Cell Proliferation and Tumorigenesis in the Intestine. Proc. Natl. Acad. Sci. USA 2009, 106, 6309–6314. [Google Scholar] [CrossRef]
- Fre, S.; Bardin, A.; Robine, S.; Louvard, D. Notch Signaling in Intestinal Homeostasis Across Species: The Cases of Drosophila, Zebrafish and the Mouse. Exp. Cell Res. 2011, 317, 2740–2747. [Google Scholar] [CrossRef]
- Takashima, S.; Mkrtchyan, M.; Younossi-Hartenstein, A.; Merriam, J.R.; Hartenstein, V. The Behaviour of Drosophila Adult Hindgut Stem Cells is Controlled by Wnt and Hh Signalling. Nature 2008, 454, 651–655. [Google Scholar]
- Kosinski, C.; Stange, D.E.; Xu, C.; Chan, A.S.; Ho, C.; Yuen, S.T.; Mifflin, R.C.; Powell, D.W.; Clevers, H.; Leung, S.Y.; et al. Indian Hedgehog Regulates Intestinal Stem Cell Fate through Epithelial-Mesenchymal Interactions during Development. Gastroenterology 2010, 139, 893–903. [Google Scholar] [CrossRef] [Green Version]
- Fevr, T.; Robine, S.; Louvard, D.; Huelsken, J. Wnt/beta-Catenin is Essential for Intestinal Homeostasis and Maintenance of Intestinal Stem Cells. Mol. Cell Biol. 2007, 27, 7551–7559. [Google Scholar] [CrossRef]
- Yeung, T.M.; Chia, L.A.; Kosinski, C.M.; Kuo, C.J. Regulation of Self-Renewal and Differentiation by the Intestinal Stem Cell Niche. Cell Mol. Life Sci. 2011, 68, 2513–2523. [Google Scholar] [CrossRef]
- Jiang, H.; Edgar, B.A. Intestinal Stem Cell Function in Drosophila and Mice. Curr. Opin. Genet. Dev. 2012, 22, 354–360. [Google Scholar] [CrossRef]
- Xu, N.; Wang, S.Q.; Tan, D.; Gao, Y.; Lin, G.; Xi, R. EGFR, Wingless and JAK/STAT Signaling Cooperatively Maintain Drosophila Intestinal Stem Cells. Dev. Biol. 2011, 354, 31–43. [Google Scholar] [CrossRef]
- Jiang, H.; Grenley, M.O.; Bravo, M.J.; Blumhagen, R.Z.; Edgar, B.A. EGFR/Ras/MAPK Signaling Mediates Adult Midgut Epithelial Homeostasis and Regeneration in Drosophila. Cell Stem Cell 2011, 8, 84–95. [Google Scholar] [CrossRef]
- Osman, D.; Buchon, N.; Chakrabarti, S.; Huang, Y.T.; Su, W.C.; Poidevin, M.; Tsai, Y.C.; Lemaitre, B. Autocrine and Paracrine Unpaired Signalling Regulate Intestinal Stem Cell Maintenance and Division. J. Cell Sci. 2012, 125, 5944–5949. [Google Scholar] [CrossRef]
- Liu, W.; Singh, S.R.; Hou, S.X. JAK-STAT is Restrained by Notch to Control Cell Proliferation of the Drosophila Intestinal Stem Cells. J. Cell Biochem. 2010, 109, 992–999. [Google Scholar]
- Lin, G.; Xu, N.; Xi, R. Paracrine Unpaired Signaling through the JAK/STAT Pathway Controls Self-Renewal and Lineage Differentiation of Drosophila Intestinal Stem Cells. J. Mol. Cell Biol. 2010, 2, 37–49. [Google Scholar] [CrossRef]
- Sato, T.; van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; van de Wetering, M.; Clevers, H. Paneth Cells Constitute the Niche for Lgr5 Stem Cells in Intestinal Crypts. Nature 2011, 469, 415–418. [Google Scholar]
- Yeung, T.M.; Chia, L.A.; Kosinski, C.M.; Kuo, C.J. Regulation of Self-Renewal and Differentiation by the Intestinal Stem Cell Niche. Cell Mol. Life Sci. 2011, 68, 2513–2523. [Google Scholar] [CrossRef]
- O'Brien, L.E.; Soliman, S.S.; Li, X.; Bilder, D. Altered Modes of Stem Cell Division Drive Adaptive Intestinal Growth. Cell 2011, 147, 603–614. [Google Scholar] [CrossRef]
- De Navascues, J.; Perdigoto, C.N.; Bian, Y.; Schneider, M.H.; Bardin, A.J.; Martinez-Arias, A.; Simons, B.D. Drosophila Midgut Homeostasis Involves Neutral Competition between Symmetrically Dividing Intestinal Stem Cells. EMBO J. 2012, 31, 2473–2485. [Google Scholar] [CrossRef]
- Snippert, H.J.; van der Flier, L.G.; Sato, T.; van Es, J.H.; van den Born, M.; Kroon-Veenboer, C.; Barker, N.; Klein, A.M.; van Rheenen, J.; Simons, B.D.; et al. Intestinal Crypt Homeostasis Results from Neutral Competition between Symmetrically Dividing Lgr5 Stem Cells. Cell 2010, 143, 134–144. [Google Scholar] [CrossRef]
- Kocks, C.; Cho, J.H.; Nehme, N.; Ulvila, J.; Pearson, A.M.; Meister, M.; Strom, C.; Conto, S.L.; Hetru, C.; Stuart, L.M.; et al. Eater, a Transmembrane Protein Mediating Phagocytosis of Bacterial Pathogens in Drosophila. Cell 2005, 123, 335–346. [Google Scholar] [CrossRef]
- Nehme, N.T.; Liegeois, S.; Kele, B.; Giammarinaro, P.; Pradel, E.; Hoffmann, J.A.; Ewbank, J.J.; Ferrandon, D. A Model of Bacterial Intestinal Infections in Drosophila Melanogaster. PLoS Pathog. 2007, 3, e173. [Google Scholar] [CrossRef]
- Jiang, H.; Edgar, B.A. Intestinal Stem Cells in the Adult Drosophila Midgut. Exp. Cell Res. 2011, 317, 2780–2788. [Google Scholar] [CrossRef]
- Buchon, N.; Broderick, N.A.; Kuraishi, T.; Lemaitre, B. Drosophila EGFR Pathway Coordinates Stem Cell Proliferation and Gut Remodeling Following Infection. BMC Biol. 2010, 8, 152. [Google Scholar] [CrossRef]
- Ren, F.; Wang, B.; Yue, T.; Yun, E.Y.; Ip, Y.T.; Jiang, J. Hippo Signaling Regulates Drosophila Intestine Stem Cell Proliferation through Multiple Pathways. Proc. Natl. Acad. Sci. USA 2010, 107, 21064–21069. [Google Scholar]
- Zhou, F.; Rasmussen, A.; Lee, S.; Agaisse, H. The upd3 Cytokine Couples Environmental Challenge and Intestinal Stem Cell Division through Modulation of JAK/STAT Signaling in the Stem Cell Microenvironment. Dev. Biol. 2012, 373, 383–393. [Google Scholar]
- Cordero, J.B.; Stefanatos, R.K.; Scopelliti, A.; Vidal, M.; Sansom, O.J. Inducible Progenitor-Derived Wingless Regulates Adult Midgut Regeneration in Drosophila. EMBO J. 2012, 31, 3901–3917. [Google Scholar] [CrossRef]
- Buchon, N.; Broderick, N.A.; Chakrabarti, S.; Lemaitre, B. Invasive and Indigenous Microbiota Impact Intestinal Stem Cell Activity through Multiple Pathways in Drosophila. Genes Dev. 2009, 23, 2333–2344. [Google Scholar] [CrossRef]
- Apidianakis, Y.; Pitsouli, C.; Perrimon, N.; Rahme, L. Synergy between Bacterial Infection and Genetic Predisposition in Intestinal Dysplasia. Proc. Natl. Acad. Sci. USA 2009, 106, 20883–20888. [Google Scholar]
- Arthur, J.C.; Perez-Chanona, E.; Muhlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef]
- Ben-Neriah, Y.; Karin, M. Inflammation Meets Cancer, with NF-kappaB as the Matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are Required for Survival of Intestinal Epithelial Cells and Development of Colitis-Associated Cancer. Cancer. Cell 2009, 15, 103–113. [Google Scholar] [CrossRef]
- Vermeulen, L.; De Sousa E Melo, F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt Activity Defines Colon Cancer Stem Cells and is Regulated by the Microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Terzic, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and Colon Cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar]
- Buchon, N.; Broderick, N.A.; Poidevin, M.; Pradervand, S.; Lemaitre, B. Drosophila Intestinal Response to Bacterial Infection: Activation of Host Defense and Stem Cell Proliferation. Cell Host Microbe 2009, 5, 200–211. [Google Scholar] [CrossRef]
- Ryu, J.H.; Ha, E.M.; Lee, W.J. Innate Immunity and Gut-Microbe Mutualism in Drosophila. Dev. Comp. Immunol. 2010, 34, 369–376. [Google Scholar] [CrossRef]
- Shia, A.K.; Glittenberg, M.; Thompson, G.; Weber, A.N.; Reichhart, J.M.; Ligoxygakis, P. Toll-Dependent Antimicrobial Responses in Drosophila Larval Fat Body Require Spatzle Secreted by Haemocytes. J. Cell Sci. 2009, 122, 4505–4515. [Google Scholar] [CrossRef]
- Brennan, C.A.; Delaney, J.R.; Schneider, D.S.; Anderson, K.V. Psidin is Required in Drosophila Blood Cells for both Phagocytic Degradation and Immune Activation of the Fat Body. Curr. Biol. 2007, 17, 67–72. [Google Scholar] [CrossRef]
- Agaisse, H.; Perrimon, N. The Roles of JAK/STAT Signaling in Drosophila Immune Responses. Immunol. Rev. 2004, 198, 72–82. [Google Scholar] [CrossRef]
- Pastor-Pareja, J.C.; Wu, M.; Xu, T. An Innate Immune Response of Blood Cells to Tumors and Tissue Damage in Drosophila. Dis. Model. Mech. 2008, 1, 144–154; discussion 153. [Google Scholar]
- Wu, S.C.; Liao, C.W.; Pan, R.L.; Juang, J.L. Infection-Induced Intestinal Oxidative Stress Triggers Organ-to-Organ Immunological Communication in Drosophila. Cell Host Microbe 2012, 11, 410–417. [Google Scholar] [CrossRef]
- Foley, E.; O'Farrell, P.H. Nitric Oxide Contributes to Induction of Innate Immune Responses to Gram-Negative Bacteria in Drosophila. Genes Dev. 2003, 17, 115–125. [Google Scholar] [CrossRef]
- Charroux, B.; Royet, J. Gut-Microbiota Interactions in Non-Mammals: What can we Learn from Drosophila? Semin. Immunol. 2012, 24, 17–24. [Google Scholar] [CrossRef]
- Broderick, N.A.; Lemaitre, B. Gut-Associated Microbes of Drosophila Melanogaster. Gut Microbes 2012, 3, 307–321. [Google Scholar] [CrossRef]
- Storelli, G.; Defaye, A.; Erkosar, B.; Hols, P.; Royet, J.; Leulier, F. Lactobacillus Plantarum Promotes Drosophila Systemic Growth by Modulating Hormonal Signals through TOR-Dependent Nutrient Sensing. Cell Metab. 2011, 14, 403–414. [Google Scholar]
- Cox, C.R.; Gilmore, M.S. Native Microbial Colonization of Drosophila Melanogaster and its use as a Model of Enterococcus Faecalis Pathogenesis. Infect. Immun. 2007, 75, 1565–1576. [Google Scholar] [CrossRef]
- An, D.; Apidianakis, Y.; Boechat, A.L.; Baldini, R.L.; Goumnerov, B.C.; Rahme, L.G. The Pathogenic Properties of a Novel and Conserved Gene Product, KerV, in Proteobacteria. PLoS One 2009, 4, e7167. [Google Scholar]
- Apidianakis, Y.; Mindrinos, M.N.; Xiao, W.; Lau, G.W.; Baldini, R.L.; Davis, R.W.; Rahme, L.G. Profiling Early Infection Responses: Pseudomonas Aeruginosa Eludes Host Defenses by Suppressing Antimicrobial Peptide Gene Expression. Proc. Natl. Acad. Sci. USA 2005, 102, 2573–2578. [Google Scholar]
- Shin, S.C.; Kim, S.H.; You, H.; Kim, B.; Kim, A.C.; Lee, K.A.; Yoon, J.H.; Ryu, J.H.; Lee, W.J. Drosophila Microbiome Modulates Host Developmental and Metabolic Homeostasis Via Insulin Signaling. Science 2011, 334, 670–674. [Google Scholar] [CrossRef]
- Ryu, J.H.; Kim, S.H.; Lee, H.Y.; Bai, J.Y.; Nam, Y.D.; Bae, J.W.; Lee, D.G.; Shin, S.C.; Ha, E.M.; Lee, W.J. Innate Immune Homeostasis by the Homeobox Gene Caudal and Commensal-Gut Mutualism in Drosophila. Science 2008, 319, 777–782. [Google Scholar] [CrossRef]
- Ha, E.M.; Lee, K.A.; Seo, Y.Y.; Kim, S.H.; Lim, J.H.; Oh, B.H.; Kim, J.; Lee, W.J. Coordination of Multiple Dual Oxidase-Regulatory Pathways in Responses to Commensal and Infectious Microbes in Drosophila Gut. Nat. Immunol. 2009, 10, 949–957. [Google Scholar] [CrossRef]
- Shanahan, F. The Colonic Microbiota in Health and Disease. Curr. Opin. Gastroenterol. 2013, 29, 49–54. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Panayidou, S.; Apidianakis, Y. Regenerative Inflammation: Lessons from Drosophila Intestinal Epithelium in Health and Disease. Pathogens 2013, 2, 209-231. https://doi.org/10.3390/pathogens2020209
Panayidou S, Apidianakis Y. Regenerative Inflammation: Lessons from Drosophila Intestinal Epithelium in Health and Disease. Pathogens. 2013; 2(2):209-231. https://doi.org/10.3390/pathogens2020209
Chicago/Turabian StylePanayidou, Stavria, and Yiorgos Apidianakis. 2013. "Regenerative Inflammation: Lessons from Drosophila Intestinal Epithelium in Health and Disease" Pathogens 2, no. 2: 209-231. https://doi.org/10.3390/pathogens2020209