Rapid Collection of Biospecimens by Automated Identification of Patients Eligible for Pharmacoepigenetic Studies

Abstract
:1. Introduction
1.1. Epigenetics and Human Diseases
1.2. Pharmacoepigenetic Studies
1.3. Epigenetic Modification by Intervention
1.4. Pharmacogenetic (PGx) and PEGx Research Using Electronic Medical Record (EMR)
2. A Novel Design of PEGx Using EMR Database
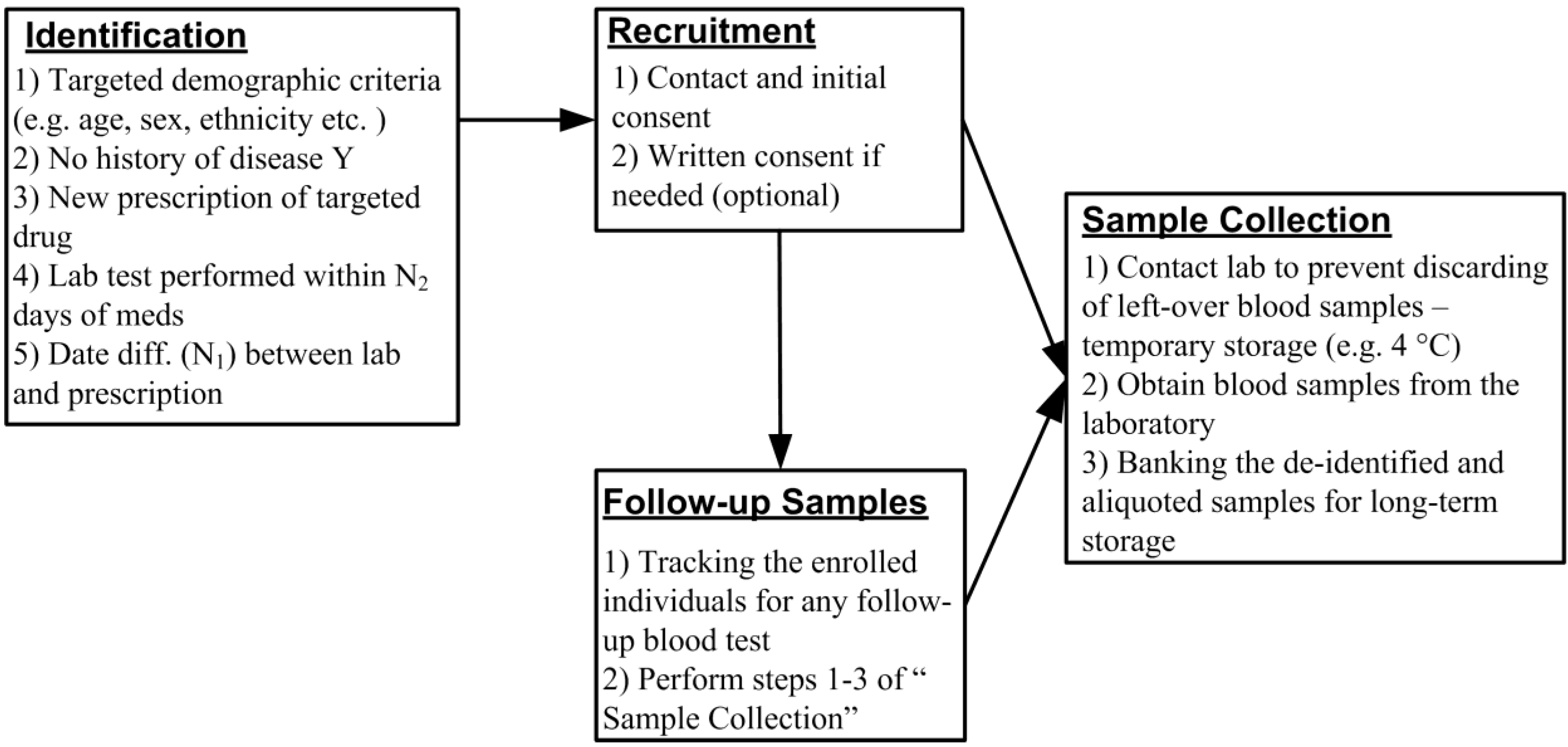
2.1. Rapid Identification and Recruitment of Treatment-Naïve Participants
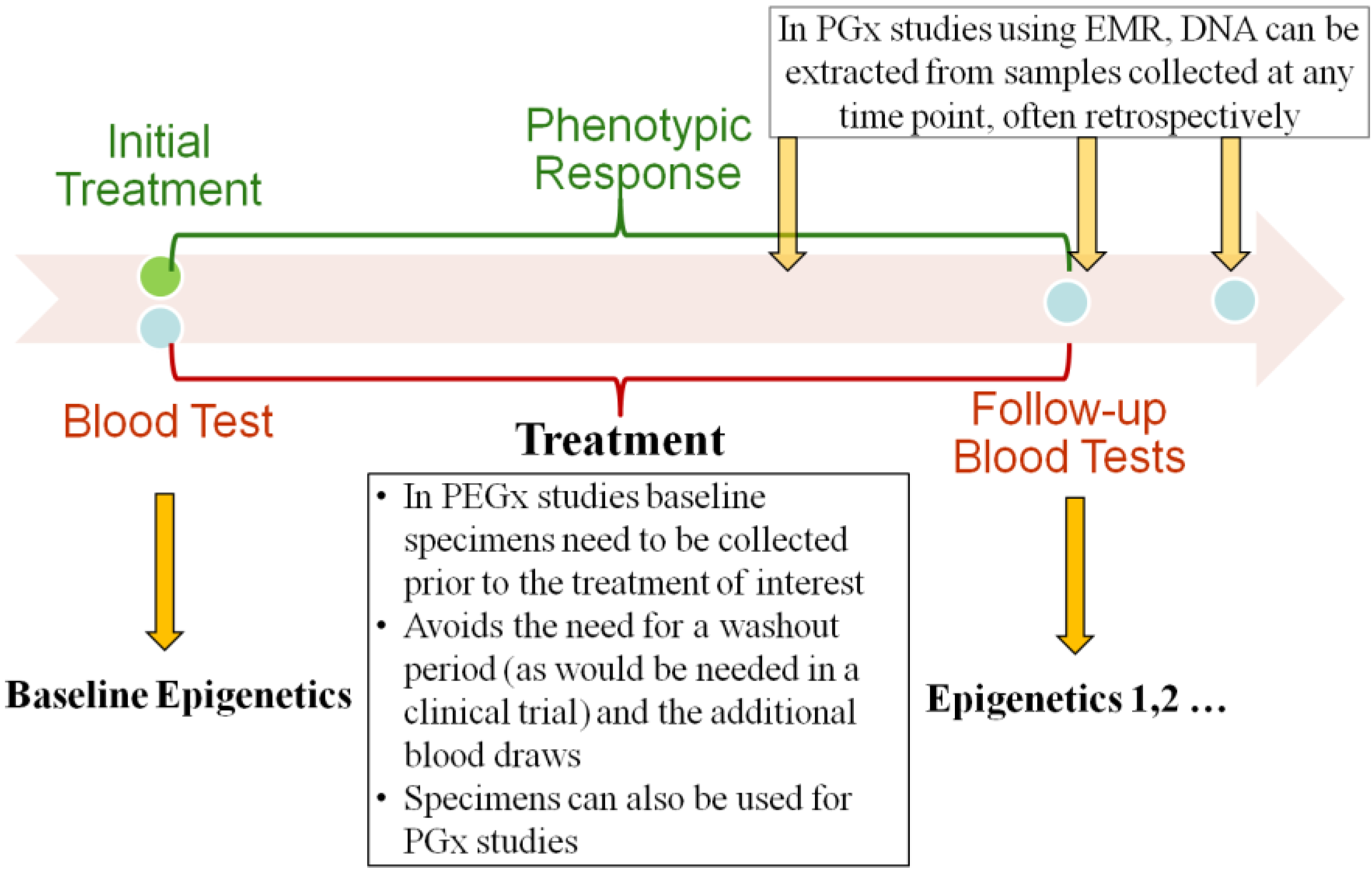
2.2. Baseline and Longitudinal Collection of Specimen

{kind=link}
{kind=link}
| CPT Code | Description (from AMA coding online) |
|---|---|
| 80061 | Lipid panel. This panel must include the following: Cholesterol, serum, total (82465), Lipoprotein, direct measurement, high density cholesterol (HDL cholesterol) (83718), Triglycerides (84478) |
| 82040 | Albumin; serum, plasma or whole blood |
| 82465 | Cholesterol, serum or whole blood, total |
| 80048 | Basic metabolic panel (Calcium, total) This panel must include the following: Calcium, total (82310), Carbon dioxide (82374), Chloride (82435), Creatinine (82565), Glucose (82947), Potassium (84132), Sodium (84295), Urea nitrogen (BUN) (84520) |
| 80053 | Comprehensive metabolic panel. This panel must include the following: Albumin (82040), Bilirubin, total (82247), Calcium, total (82310), Carbon dioxide (bicarbonate) (82374), Chloride (82435), Creatinine (82565), Glucose (82947), Phosphatase, alkaline (84075), Potassium (84132), Protein, total (84155), Sodium (84295), Transferase, alanine amino (ALT) (SGPT) (84460), Transferase, aspartate amino (AST) (SGOT) (84450), Urea nitrogen (BUN) (84520) |
| 83036 | Hemoglobin; glycosylated (A1C) |
| 85025 | Blood count; complete (CBC), automated (Hgb, Hct, RBC, WBC and platelet count) and automated differential WBC count |
3. Discussion
Acknowledgements
Conflicts of Interest
References
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef]
- Bell, J.T.; Pai, A.A.; Pickrell, J.K.; Gaffney, D.J.; Pique-Regi, R.; Degner, J.F.; Gilad, Y.; Pritchard, J.K. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011, 12, R10. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev.Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Van der Maarel, S.M. Epigenetic mechanisms in health and disease. Ann. Rheum. Dis. 2008, 67, ii97–ii100. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Karimi, M.; Johansson, S.; Axelsson, J.; Suliman, M.; Lindholm, B.; Heimburger, O.; Barany, P.; Alvestrand, A.; Nordfors, L.; et al. Impact of inflammation on epigenetic DNA methylation—A novel risk factor for cardiovascular disease? J. Intern. Med. 2007, 261, 488–499. [Google Scholar] [CrossRef]
- Sun, Y.V.; Lazarus, A.; Smith, J.A.; Chuang, Y.H.; Zhao, W.; Turner, S.T.; Kardia, S.L. Gene-specific DNA methylation association with serum levels of C-reactive protein in african americans. PLoS One 2013, 8, e73480. [Google Scholar]
- Teschendorff, A.E.; Menon, U.; Gentry-Maharaj, A.; Ramus, S.J.; Weisenberger, D.J.; Shen, H.; Campan, M.; Noushmehr, H.; Bell, C.G.; Maxwell, A.P.; et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010, 20, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sanchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic predictor of age. PLoS One 2011, 6, e14821. [Google Scholar]
- Waterland, R.A.; Jirtle, R.L. Transposable elements: Targets for early nutritional effects on epigenetic gene regulation. Mol. Cell. Biol. 2003, 23, 5293–5300. [Google Scholar] [CrossRef]
- Ivanov, M.; Kacevska, M.; Ingelman-Sundberg, M. Epigenomics and interindividual differences in drug response. Clin. Pharmacol. Ther. 2012, 92, 727–736. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suner, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef]
- Breitling, L.P.; Yang, R.; Korn, B.; Burwinkel, B.; Brenner, H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am. J. Hum. Genet. 2011, 88, 450–457. [Google Scholar] [CrossRef]
- Shenker, N.S.; Polidoro, S.; van Veldhoven, K.; Sacerdote, C.; Ricceri, F.; Birrell, M.A.; Belvisi, M.G.; Brown, R.; Vineis, P.; Flanagan, J.M. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum. Mol. Genet. 2013, 22, 843–851. [Google Scholar] [CrossRef]
- Sun, Y.V.; Smith, A.K.; Conneely, K.N.; Chang, Q.; Li, W.; Lazarus, A.; Smith, J.A.; Almli, L.M.; Binder, E.B.; Klengel, T.; et al. Epigenomic association analysis identifies smoking-related DNA methylation sites in African Americans. Hum. Genet. 2013, 132, 1027–1037. [Google Scholar] [CrossRef]
- Collotta, M.; Bertazzi, P.A.; Bollati, V. Epigenetics and pesticides. Toxicology 2013, 307, 35–41. [Google Scholar] [CrossRef]
- Pogribny, I.P.; Rusyn, I. Environmental toxicants, epigenetics, and cancer. Adv. Exp. Med. Biol. 2013, 754, 215–232. [Google Scholar] [CrossRef]
- Kohane, I.S. Using electronic health records to drive discovery in disease genomics. Nat. Rev.Genet. 2011, 12, 417–428. [Google Scholar] [CrossRef]
- Roden, D.M.; Pulley, J.M.; Basford, M.A.; Bernard, G.R.; Clayton, E.W.; Balser, J.R.; Masys, D.R. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin. Pharmacol. Ther. 2008, 84, 362–369. [Google Scholar] [CrossRef]
- Denny, J.C.; Ritchie, M.D.; Basford, M.A.; Pulley, J.M.; Bastarache, L.; Brown-Gentry, K.; Wang, D.; Masys, D.R.; Roden, D.M.; Crawford, D.C. PheWAS: Demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 2010, 26, 1205–1210. [Google Scholar] [CrossRef]
- Ritchie, M.D.; Denny, J.C.; Crawford, D.C.; Ramirez, A.H.; Weiner, J.B.; Pulley, J.M.; Basford, M.A.; Brown-Gentry, K.; Balser, J.R.; Masys, D.R.; et al. Robust replication of genotype-phenotype associations across multiple diseases in an electronic medical record. Am. J. Hum. Genet. 2010, 86, 560–572. [Google Scholar] [CrossRef]
- Zhou, K.; Pearson, E.R. Insights from genome-wide association studies of drug response. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 299–310. [Google Scholar] [CrossRef]
- Foley, D.L.; Craig, J.M.; Morley, R.; Olsson, C.A.; Dwyer, T.; Smith, K.; Saffery, R. Prospects for epigenetic epidemiology. Am. J. Epidemiol. 2009, 169, 389–400. [Google Scholar]
- Cortessis, V.K.; Thomas, D.C.; Levine, A.J.; Breton, C.V.; Mack, T.M.; Siegmund, K.D.; Haile, R.W.; Laird, P.W. Environmental epigenetics: Prospects for studying epigenetic mediation of exposure-response relationships. Hum. Genet. 2012, 131, 1565–1589. [Google Scholar] [CrossRef]
- Qiu, W.; Baccarelli, A.; Carey, V.J.; Boutaoui, N.; Bacherman, H.; Klanderman, B.; Rennard, S.; Agusti, A.; Anderson, W.; Lomas, D.A.; et al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am. J. Respir. Crit. Care Med. 2012, 185, 373–381. [Google Scholar] [CrossRef]
- Liu, Y.; Aryee, M.J.; Padyukov, L.; Fallin, M.D.; Hesselberg, E.; Runarsson, A.; Reinius, L.; Acevedo, N.; Taub, M.; Ronninger, M.; et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat. Biotechnol. 2013, 31, 142–147. [Google Scholar] [CrossRef]
- Lemaire, M.; Chabot, G.G.; Raynal, N.J.; Momparler, L.F.; Hurtubise, A.; Bernstein, M.L.; Momparler, R.L. Importance of dose-schedule of 5-aza-2'-deoxycytidine for epigenetic therapy of cancer. BMC Cancer 2008, 8, e128. [Google Scholar] [CrossRef]
- Eadon, M.T.; Wheeler, H.E.; Stark, A.L.; Zhang, X.; Moen, E.L.; Delaney, S.M.; Im, H.K.; Cunningham, P.N.; Zhang, W.; Dolan, M.E. Genetic and epigenetic variants contributing to clofarabine cytotoxicity. Hum. Mol. Genet. 2013, 22, 4007–4020. [Google Scholar] [CrossRef]
- Pai, A.A.; Bell, J.T.; Marioni, J.C.; Pritchard, J.K.; Gilad, Y. A genome-wide study of DNA methylation patterns and gene expression levels in multiple human and chimpanzee tissues. PLoS Genet. 2011, 7, e1001316. [Google Scholar] [CrossRef]
- Sun, Y.V.; Turner, S.T.; Smith, J.A.; Hammond, P.I.; Lazarus, A.; van de Rostyne, J.L.; Cunningham, J.M.; Kardia, S.L. Comparison of the DNA methylation profiles of human peripheral blood cells and transformed B-lymphocytes. Hum. Genet. 2010, 127, 651–658. [Google Scholar] [CrossRef]
- Koestler, D.C.; Marsit, C.J.; Christensen, B.C.; Accomando, W.; Langevin, S.M.; Houseman, E.A.; Nelson, H.H.; Karagas, M.R.; Wiencke, J.K.; Kelsey, K.T. Peripheral blood immune cell methylation profiles are associated with nonhematopoietic cancers. Cancer Epidemiol. Biomarkers Prev. 2012, 21, 1293–1302. [Google Scholar] [CrossRef]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, e86. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sun, Y.V.; Davis, R.L. Rapid Collection of Biospecimens by Automated Identification of Patients Eligible for Pharmacoepigenetic Studies. J. Pers. Med. 2013, 3, 263-274. https://doi.org/10.3390/jpm3040263
Sun YV, Davis RL. Rapid Collection of Biospecimens by Automated Identification of Patients Eligible for Pharmacoepigenetic Studies. Journal of Personalized Medicine. 2013; 3(4):263-274. https://doi.org/10.3390/jpm3040263
Chicago/Turabian StyleSun, Yan V., and Robert L. Davis. 2013. "Rapid Collection of Biospecimens by Automated Identification of Patients Eligible for Pharmacoepigenetic Studies" Journal of Personalized Medicine 3, no. 4: 263-274. https://doi.org/10.3390/jpm3040263