
Support Values for Genome Phylogenies

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods and Data
2.1. Classical Bootstrap
http://evolbioinf.github.io/life2015
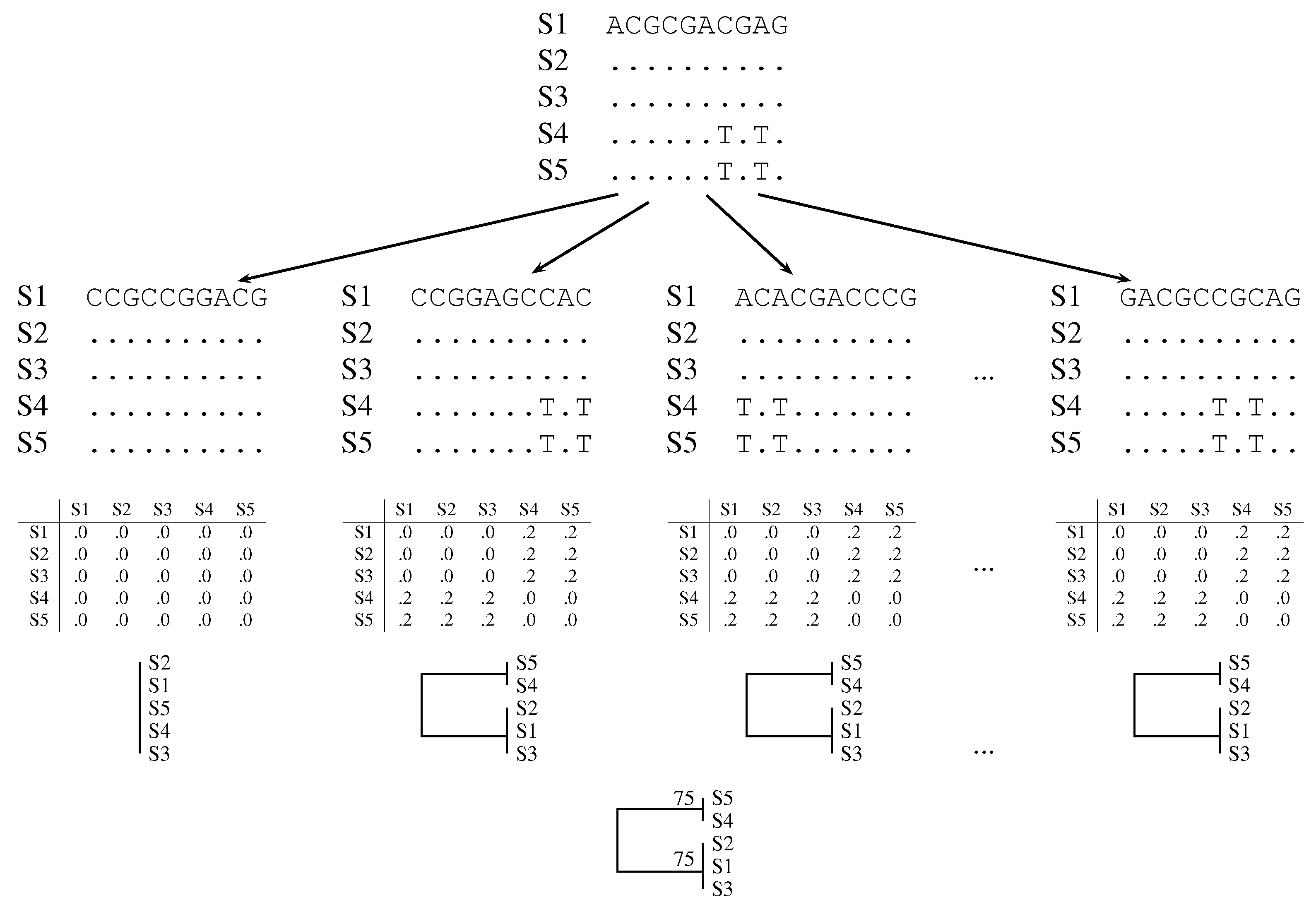
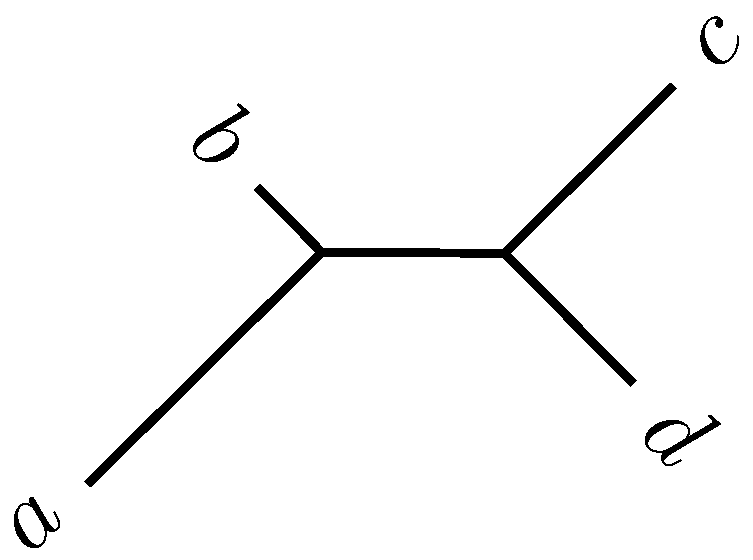
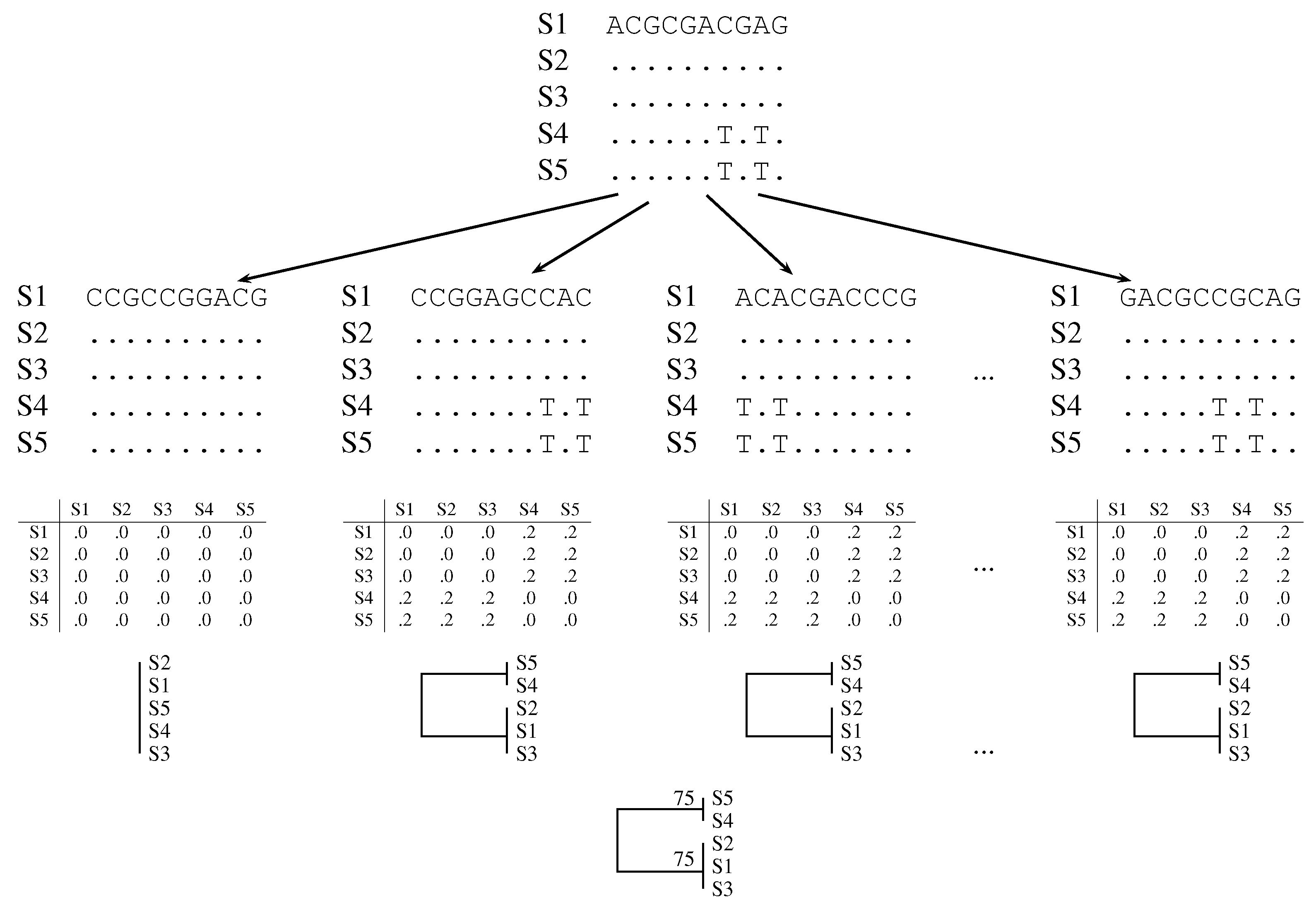
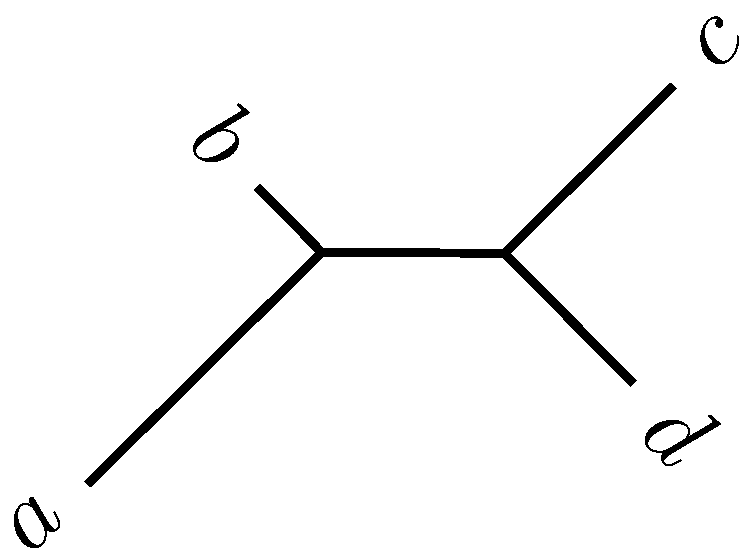
2.2. Quartet Analysis
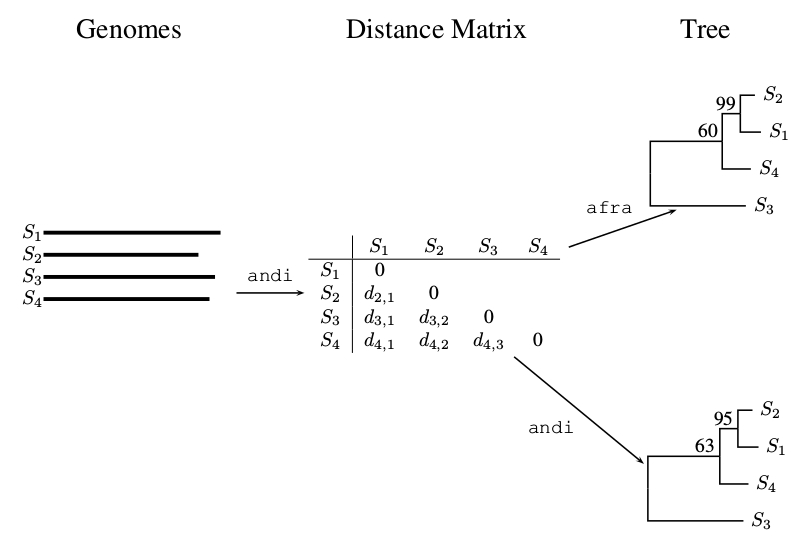
2.3. Pairwise Bootstrap
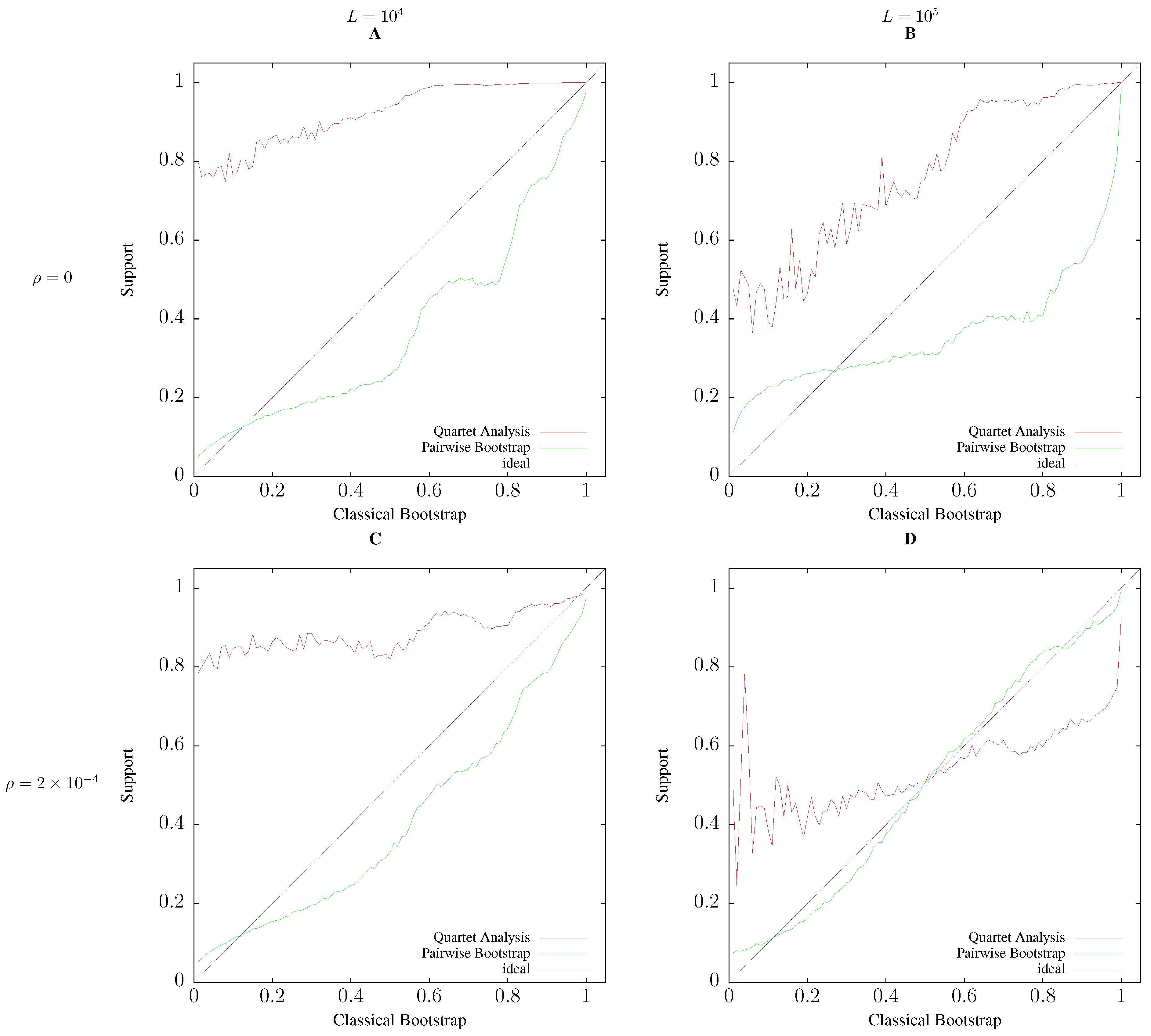
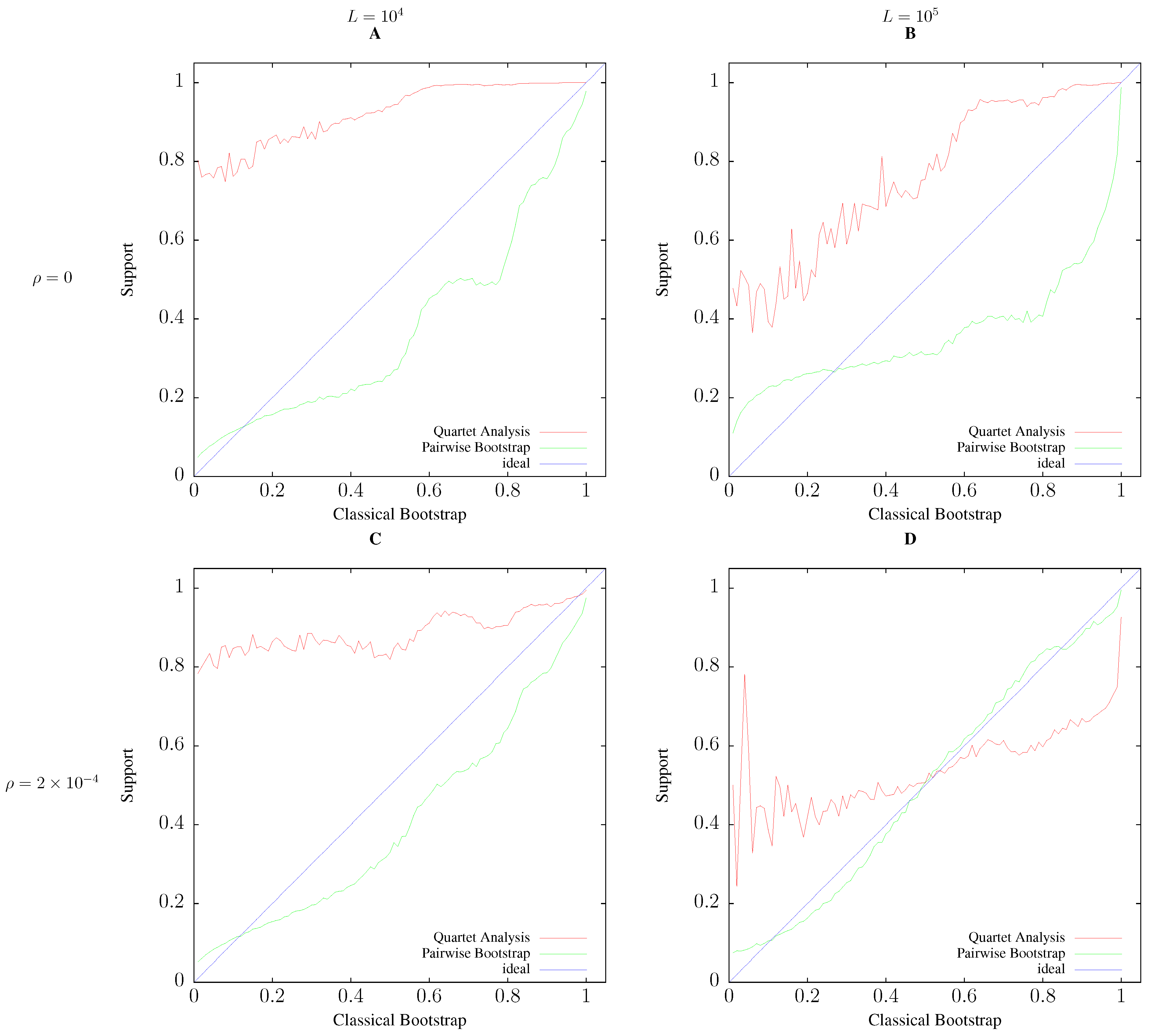
2.4. Simulation
- Simulate n related sequences using the coalescent simulator ms [12].
- Convert the output of ms to an alignment of DNA sequences, A, using ms2dna.
- Subject A to bootstrap analysis using dnaDist.
- Compute the consensus tree and support values from the output of dnaDist using the program consense, which is part of the PHYLIP package [13].
- Subject A to pairwise bootstrap analysis as implemented in the latest version of andi [8] and also calculate the consensus tree using consense.
- Use afra to carry out quartet analysis on andi-distances computed from A.
- For each cluster in the consensus tree, extract the three support values classical, pairwise and quartet using the program correlation.js.
- Repeat.
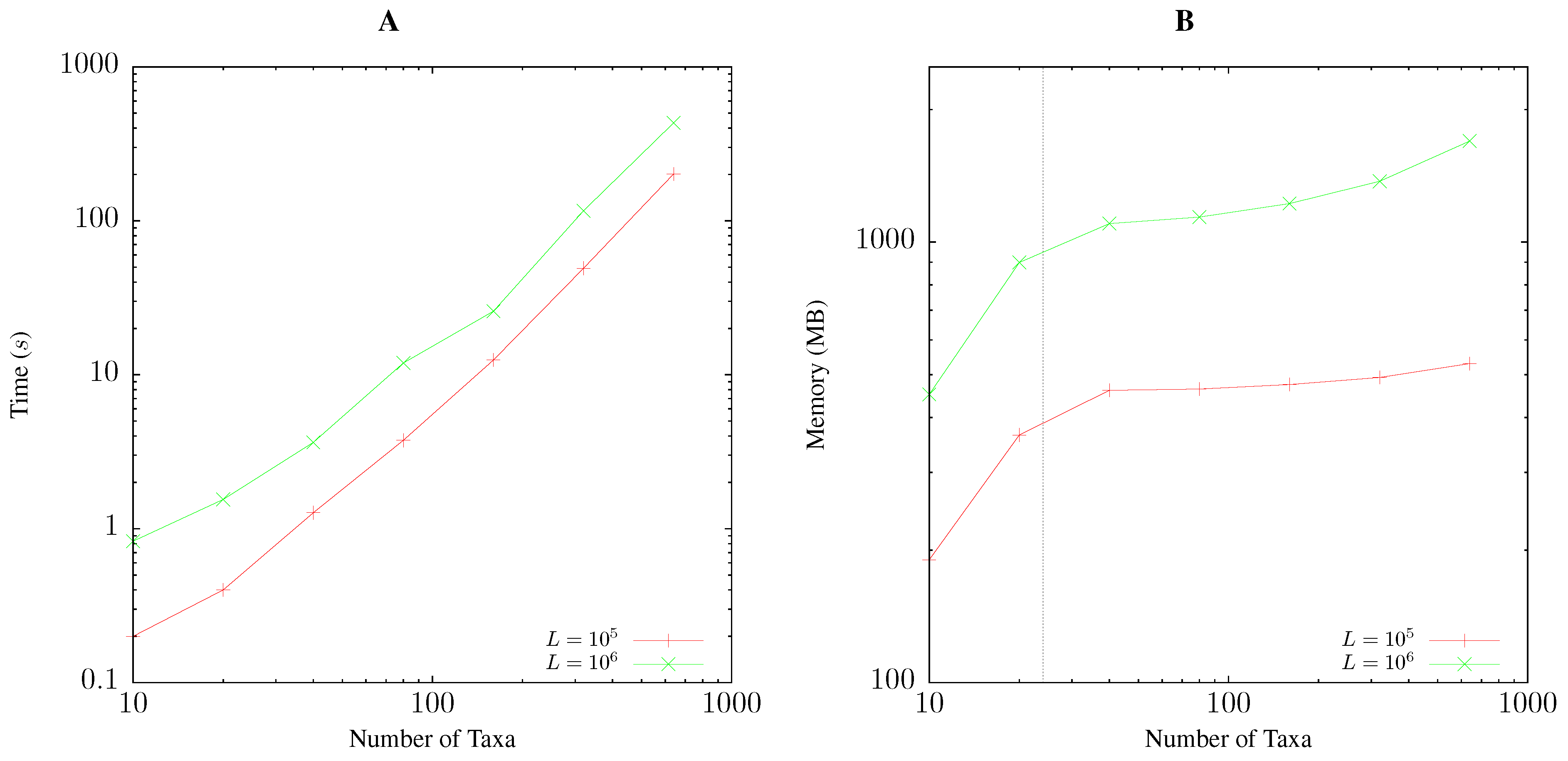
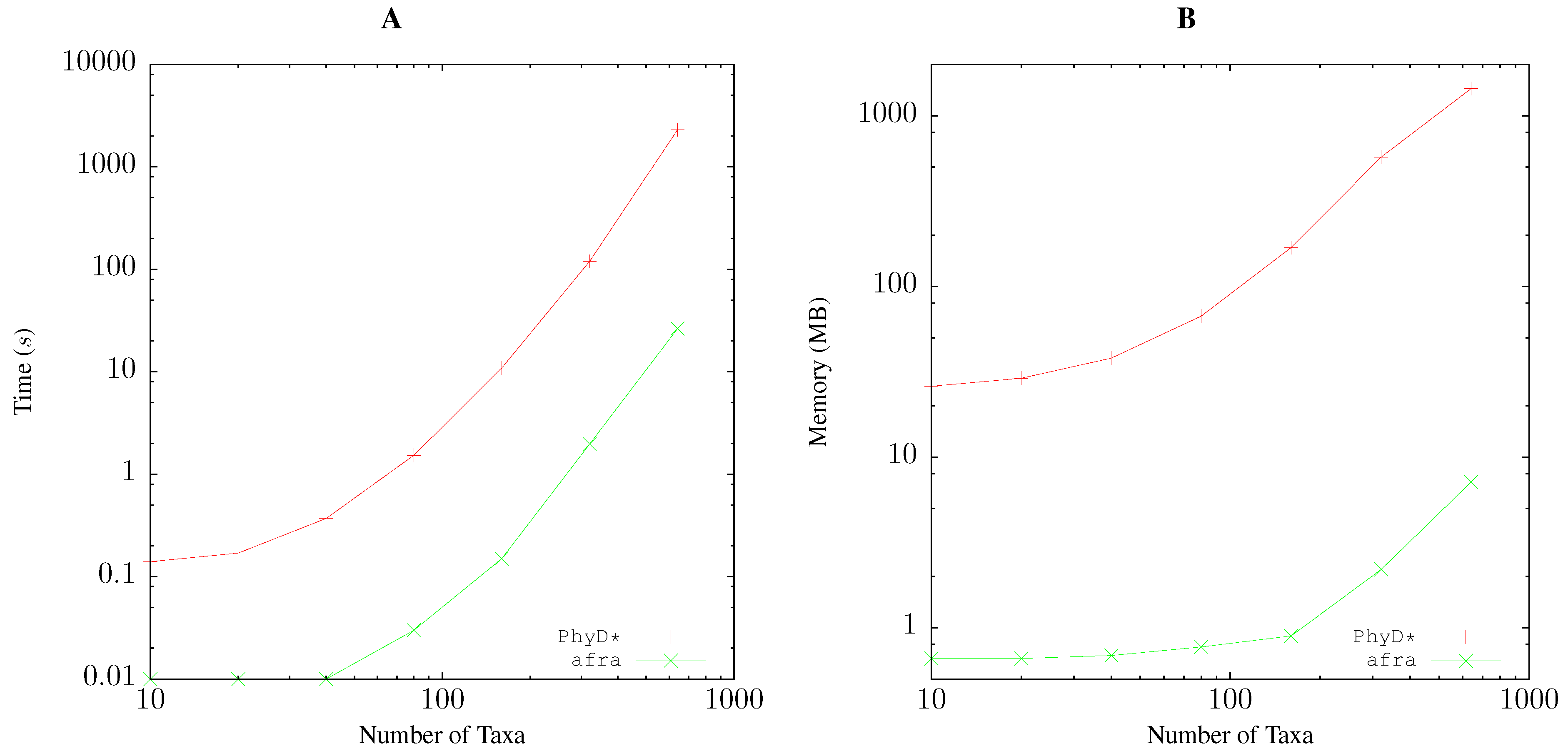
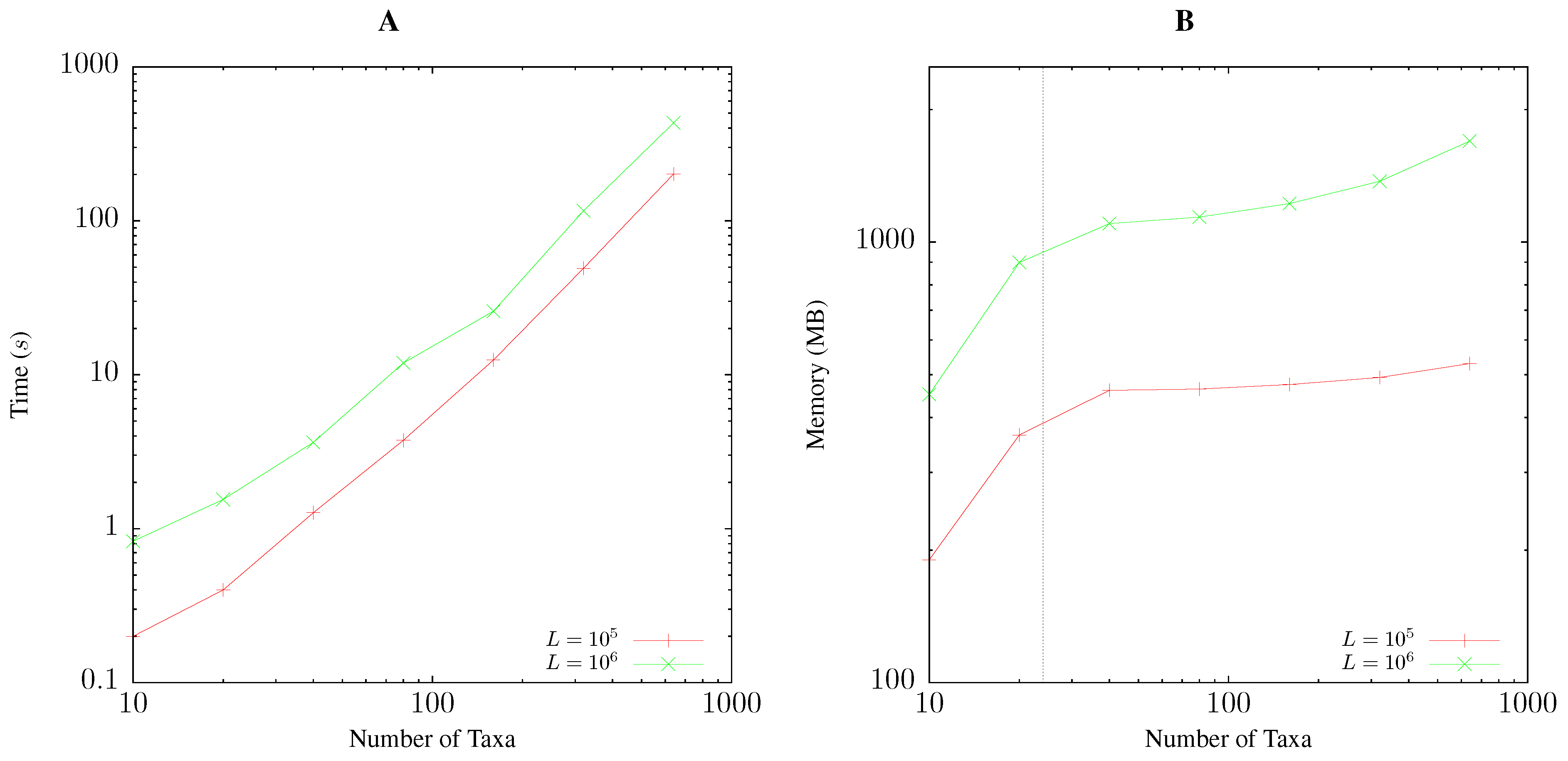
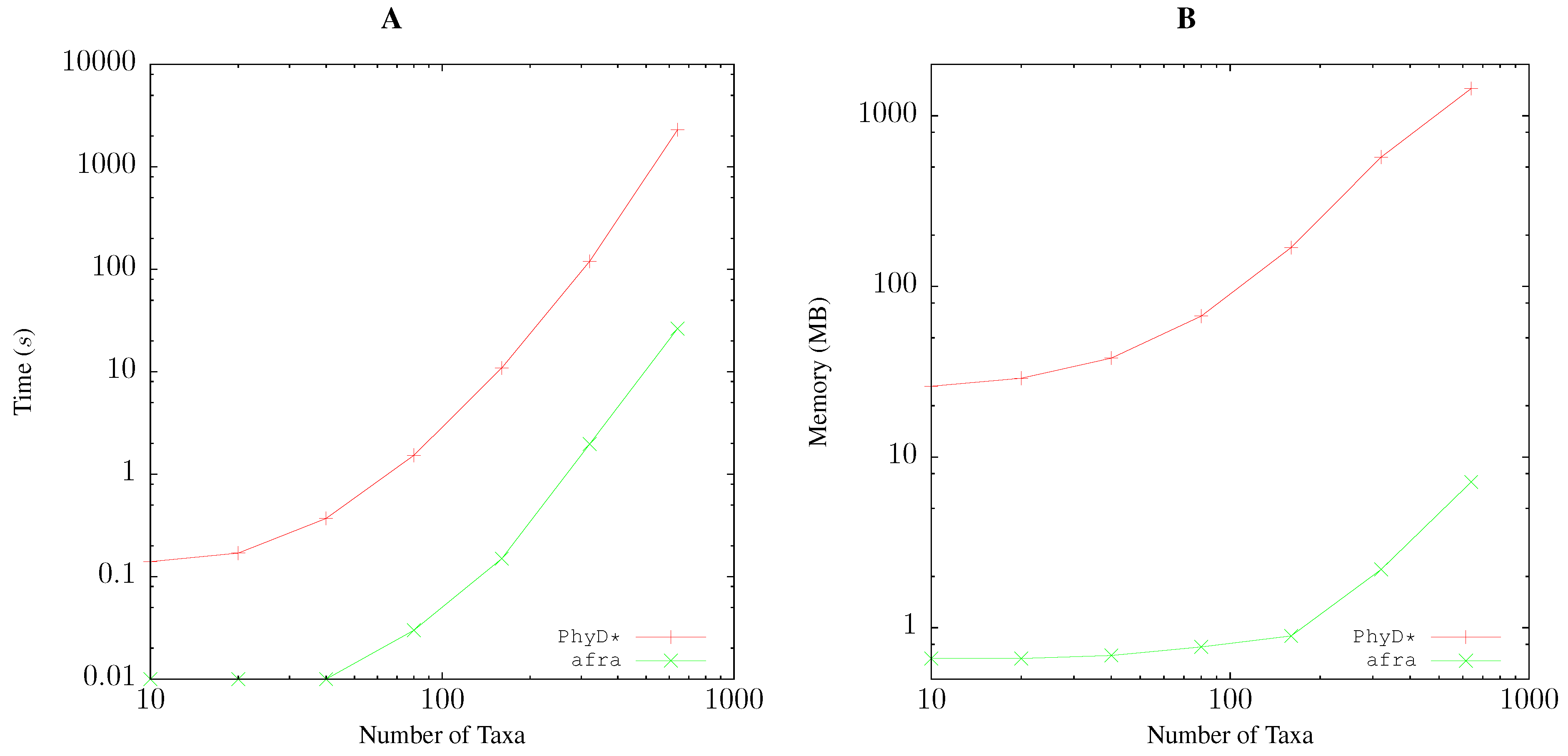
2.5. Resource Consumption
/usr/bin/time -f “elapsed\t%Es\nuser\t%Us\nmem\t%MkB\n” \ andi -b 1000 foo.fasta > foo.distfor pairwise bootstrap analysis, and
/usr/bin/time -f “elapsed\t%Es\nuser\t%Us\nmem\t%MkB\n“ \ java -Xmx4096m -jar PhyDstar.jar -c -i foo.distfor PhyD* [10].
2.6. Data
2.7. Alignment and Phylogeny Computation
3. Results and Discussion
3.1. Resource Consumption
Pairwise Bootstrap
Quartet Analysis
3.2. Accuracy
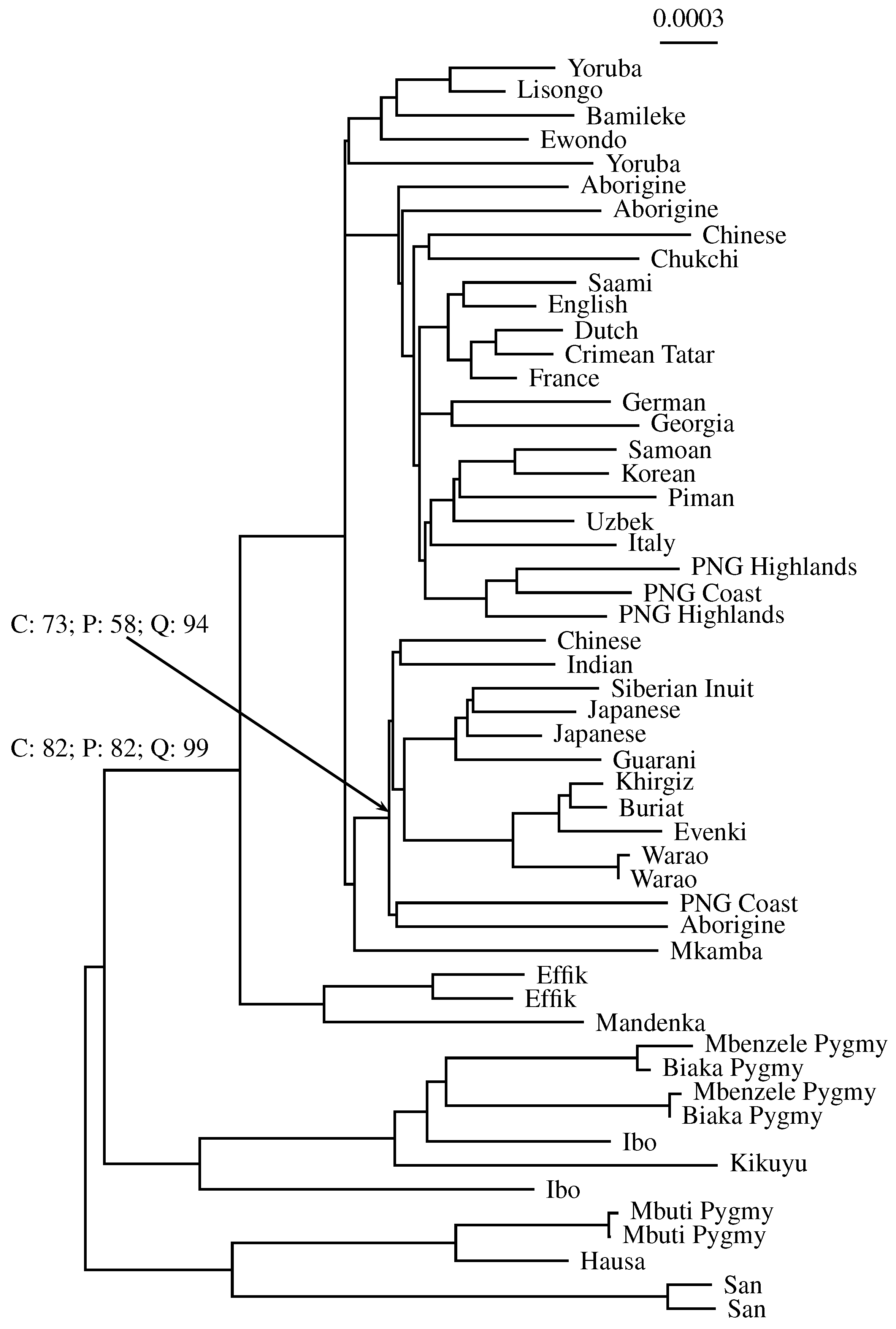
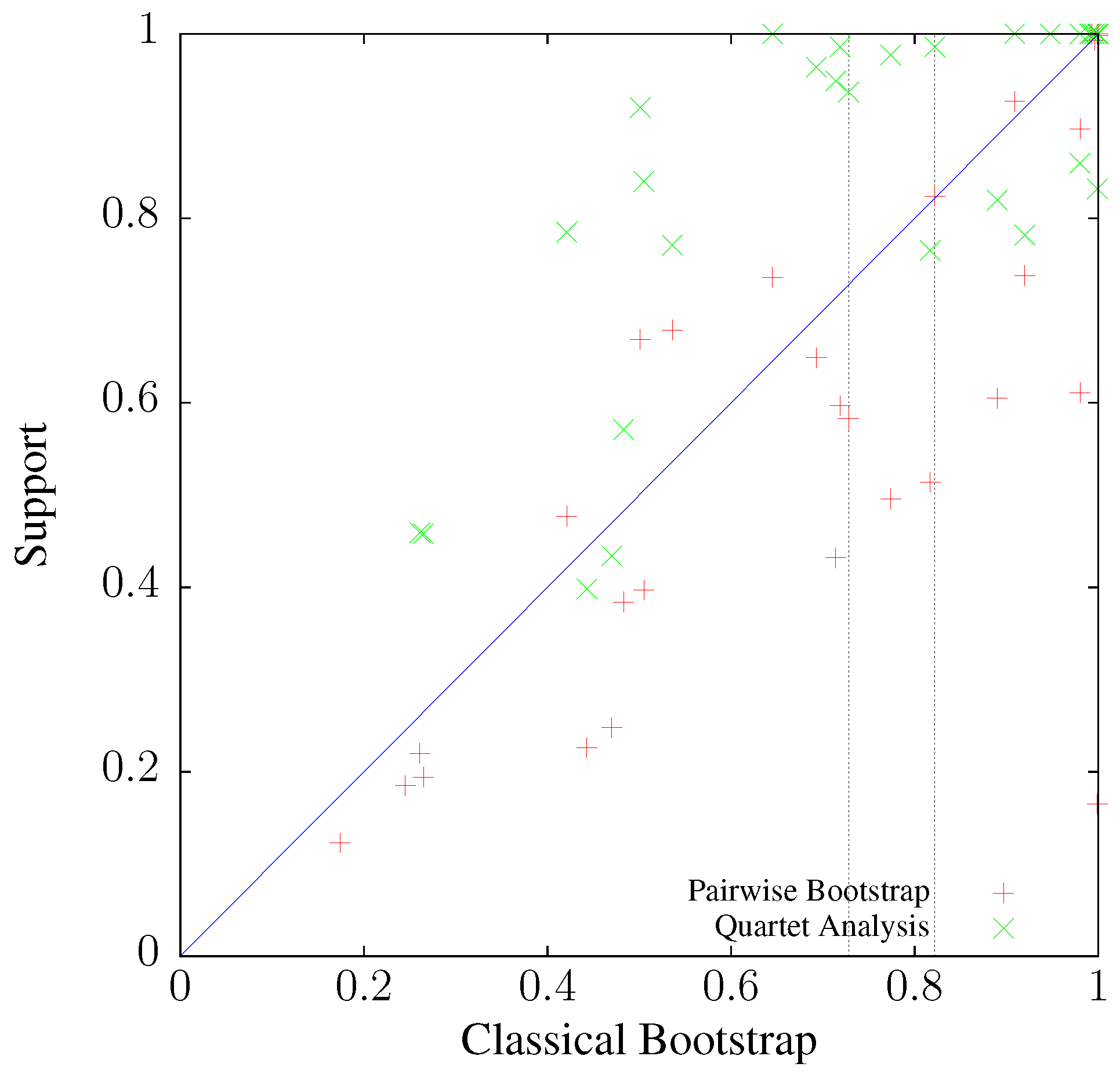
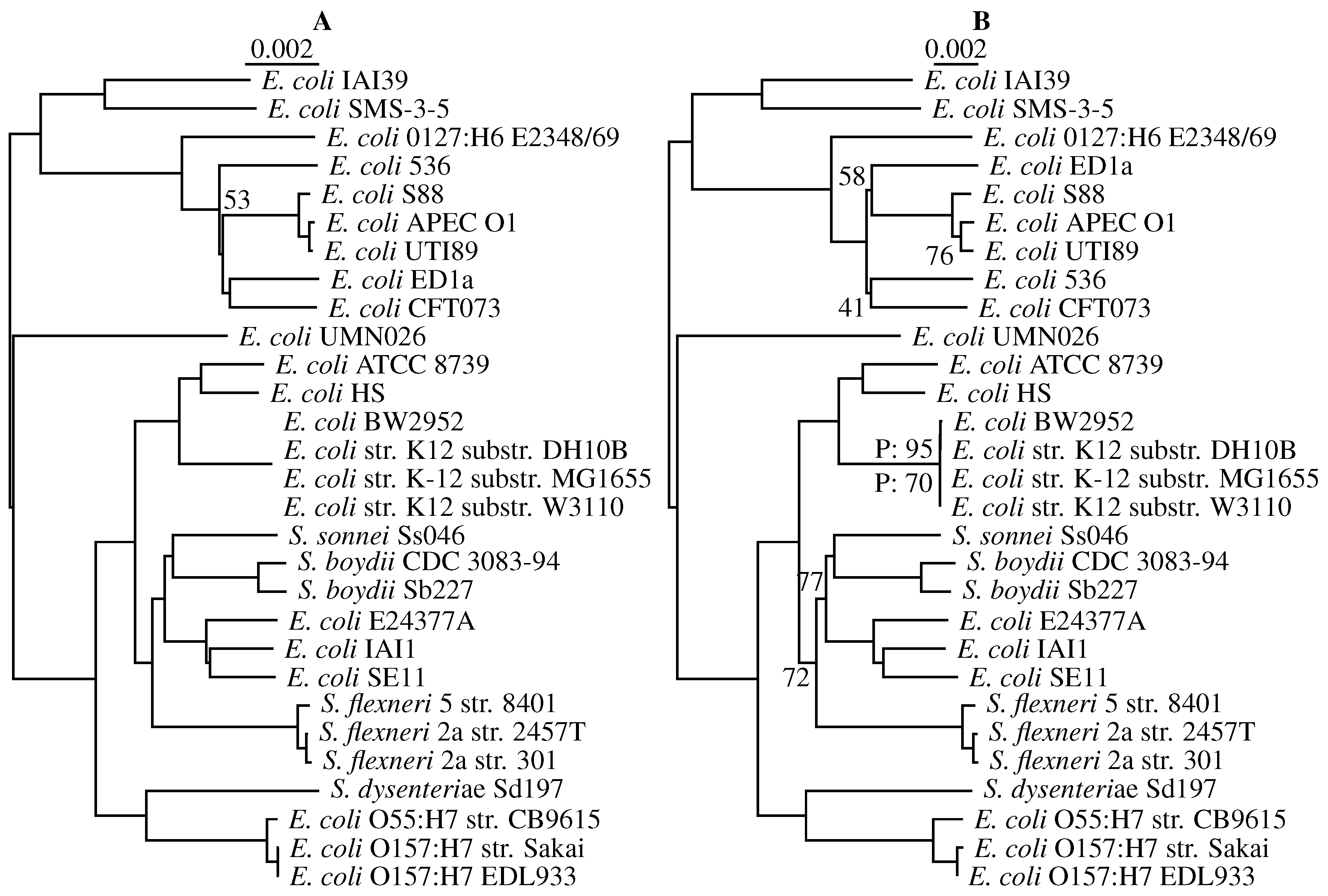
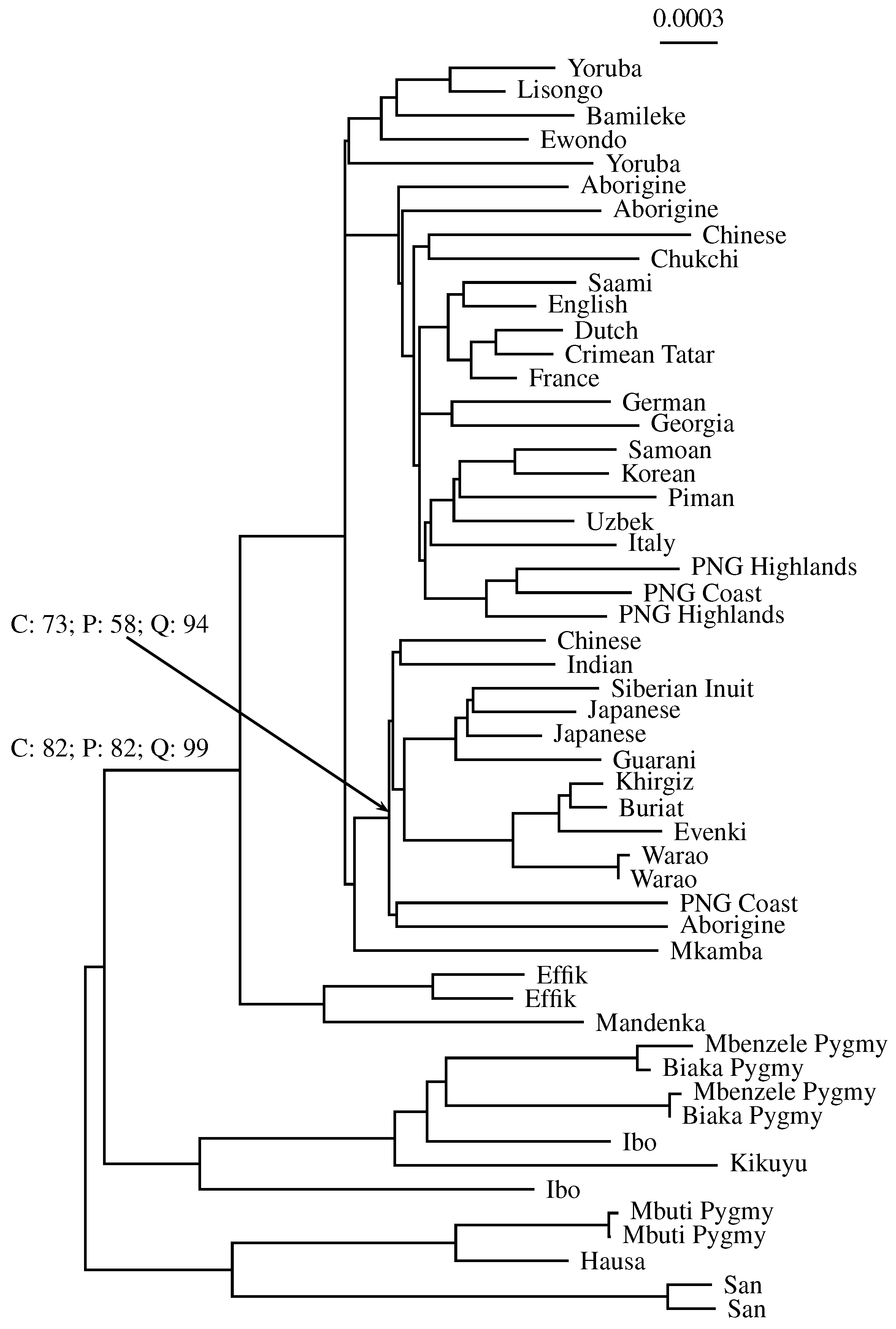
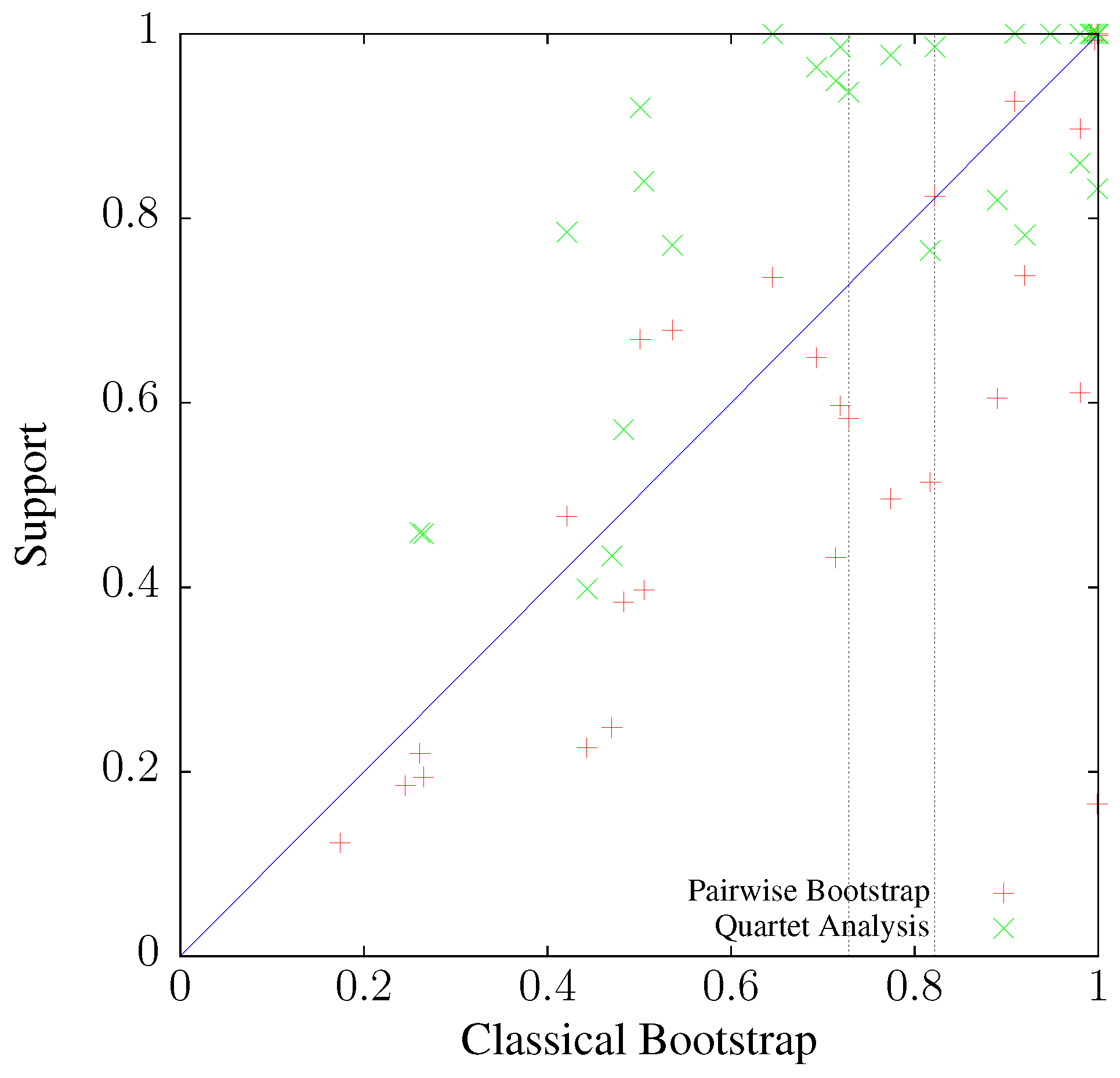
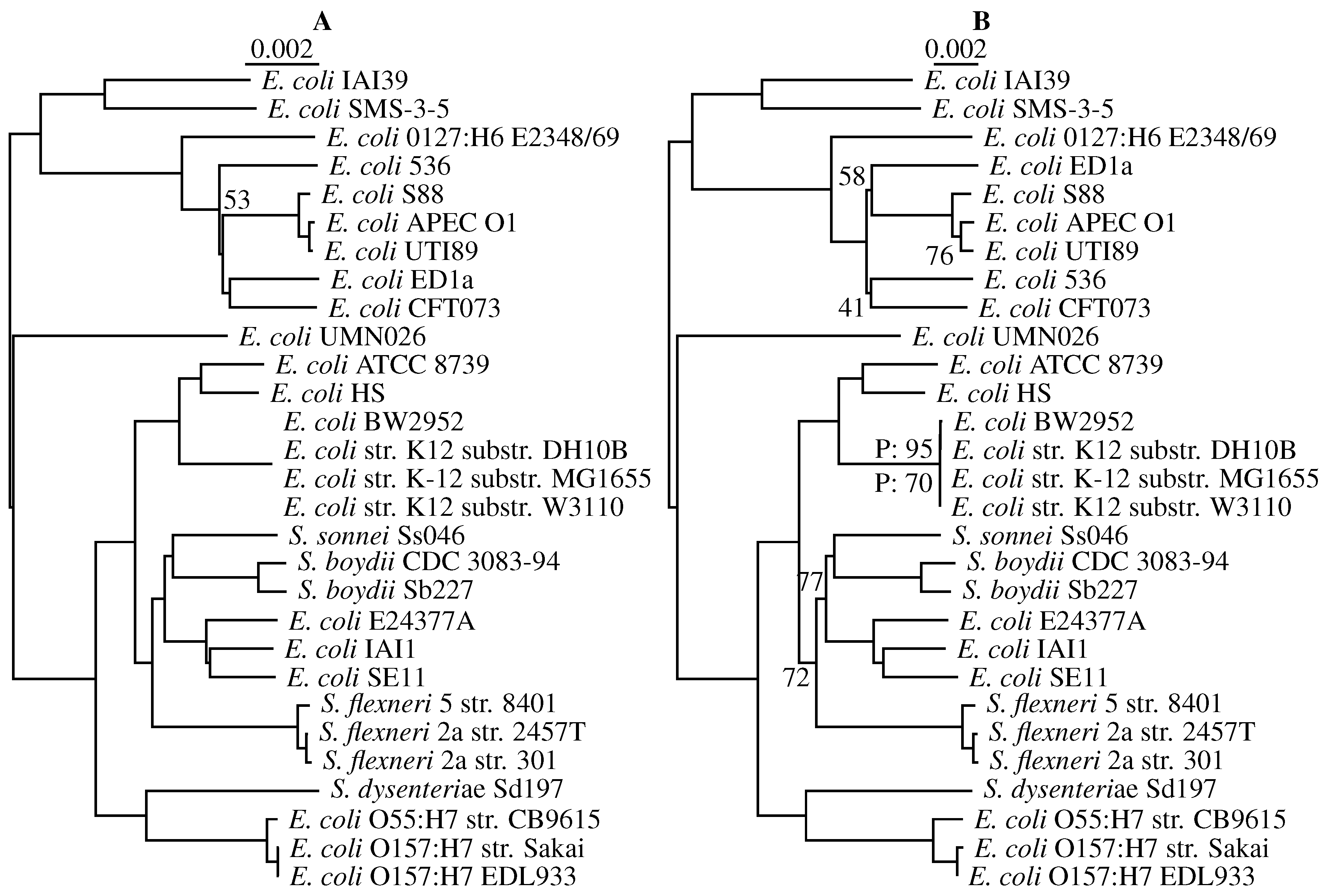
3.3. Application to Real Sequence Data
Human Mitochondrial Genomes
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Soltis, P.S.; Soltis, D.E. Applying the bootstrap in phylogeny reconstruction. Stat. Sci. 2003, 18, 256–267. [Google Scholar] [CrossRef]
- Efron, B. Bootstrap methods: Another look at the Jackknife. Ann. Stat. 1979, 7, 1–26. [Google Scholar] [CrossRef]
- Diaconis, P.; Efron, B. Computer-intensive methods in statistics. Sci. Am. 1983, 248, 116–130. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Chewapreecha, C.; Harris, S.R.; Croucher, N.J.; Turner, C.; Marttinen, P.; Cheng, L.; Pessia, A.; Aanensen, D.M.; Mather, A.E.; Page, A.J.; et al. Dense genomic sampling identifies highways of pneumococcal recombination. Nat. Genet. 2014, 46, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Haubold, B. Alignment-free phylogenetics and population genetics. Brief. Bioinform. 2014, 15, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Vinga, S.; Almeida, J. Alignment-free sequence comparison—A review. Bioinformatics 2003, 19, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Haubold, B.; Klötzl, F.; Pfaffelhuber, P. Andi: Fast and accurate estimation of evolutionary distances between closely related genomes. Bioinformatics 2015, 31, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Guénoche, A.; Garreta, H. Can we have confidence in a tree representation? In JOBIM; Gascuel, O., Sagot, M., Eds.; Lecture Notes in Computer Science; Springer: Berlin, Germany; Heidelberg, Germany, 2000; Volume 2066, pp. 45–56. [Google Scholar]
- Criscuolo, A.; Gascuel, O. Fast NJ-like algorithms to deal with incomplete distance matrices. BMC Bioinform. 2008, 9, 166. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Inferring Phylogenies; Sinauer: Sunderland, MA, USA, 2004. [Google Scholar]
- Hudson, R.R. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 2002, 18, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. PHYLIP (phylogeny interference package) version 3.6, 2005. Available online: http://evolution.genetics.washington.edu/phylip.html (accessed on 25 February 2016).
- Ingman, M.; Kaessmann, H.; Pääbo, S.; Gyllensten, U. Mitochondrial genome variation and the origin of modern humans. Nature 2000, 408, 708–713. [Google Scholar] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Clustal w and clustal x version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Angiuoli, S.V.; Salzberg, S.L. Mugsy: Fast multiple alignment of closely related whole genomes. Bioinformatics 2011, 27, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Domazet-Lošo, M.; Haubold, B. Alignment-free detection of local similarity among viral and bacterial genomes. Bioinformatics 2011, 27, 1466–1472. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klötzl, F.; Haubold, B. Support Values for Genome Phylogenies. Life 2016, 6, 11. https://doi.org/10.3390/life6010011
Klötzl F, Haubold B. Support Values for Genome Phylogenies. Life. 2016; 6(1):11. https://doi.org/10.3390/life6010011
Chicago/Turabian StyleKlötzl, Fabian, and Bernhard Haubold. 2016. "Support Values for Genome Phylogenies" Life 6, no. 1: 11. https://doi.org/10.3390/life6010011
APA StyleKlötzl, F., & Haubold, B. (2016). Support Values for Genome Phylogenies. Life, 6(1), 11. https://doi.org/10.3390/life6010011