3.2. Adsorption Studies and Polymer Cationic Nature
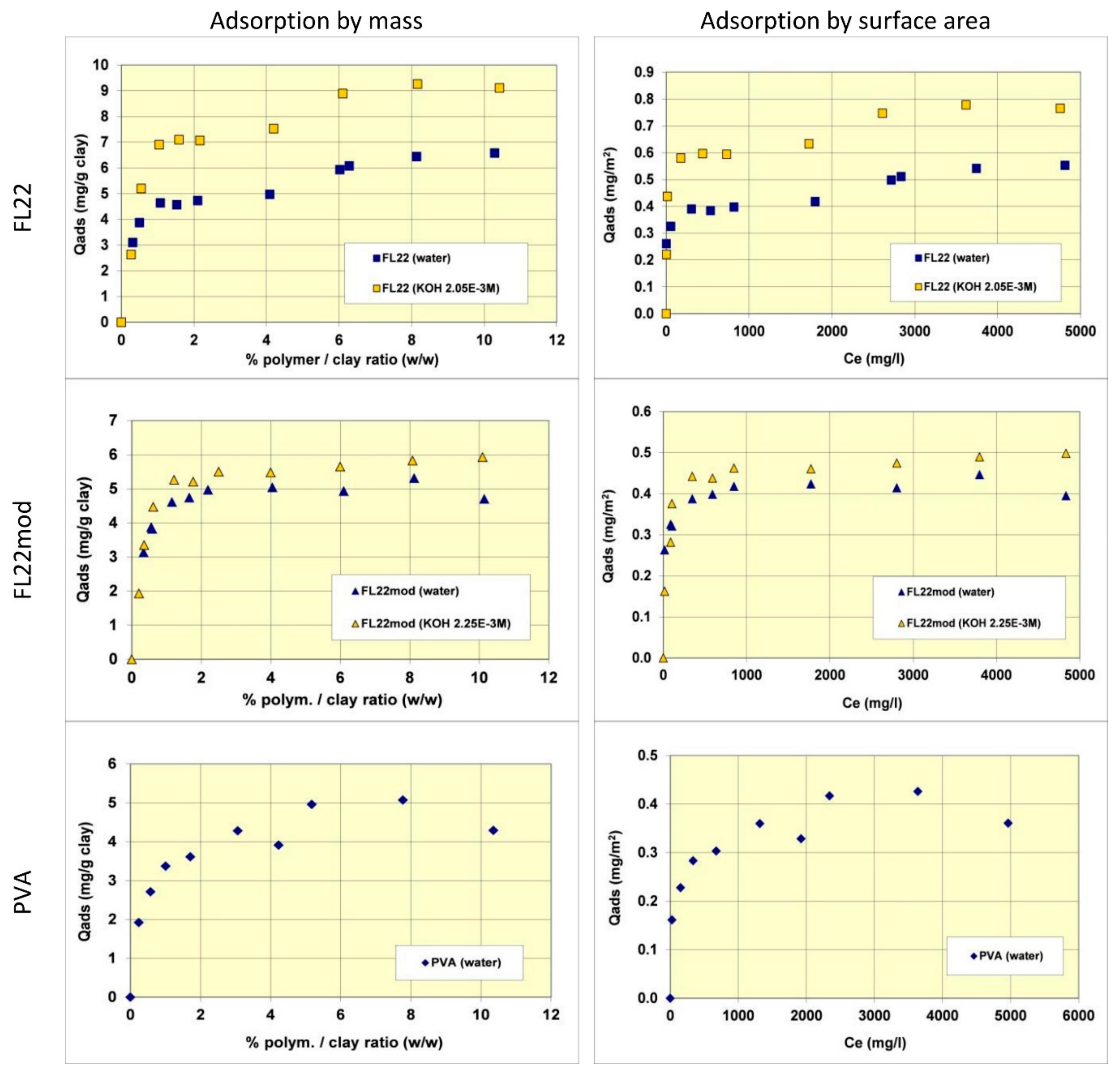
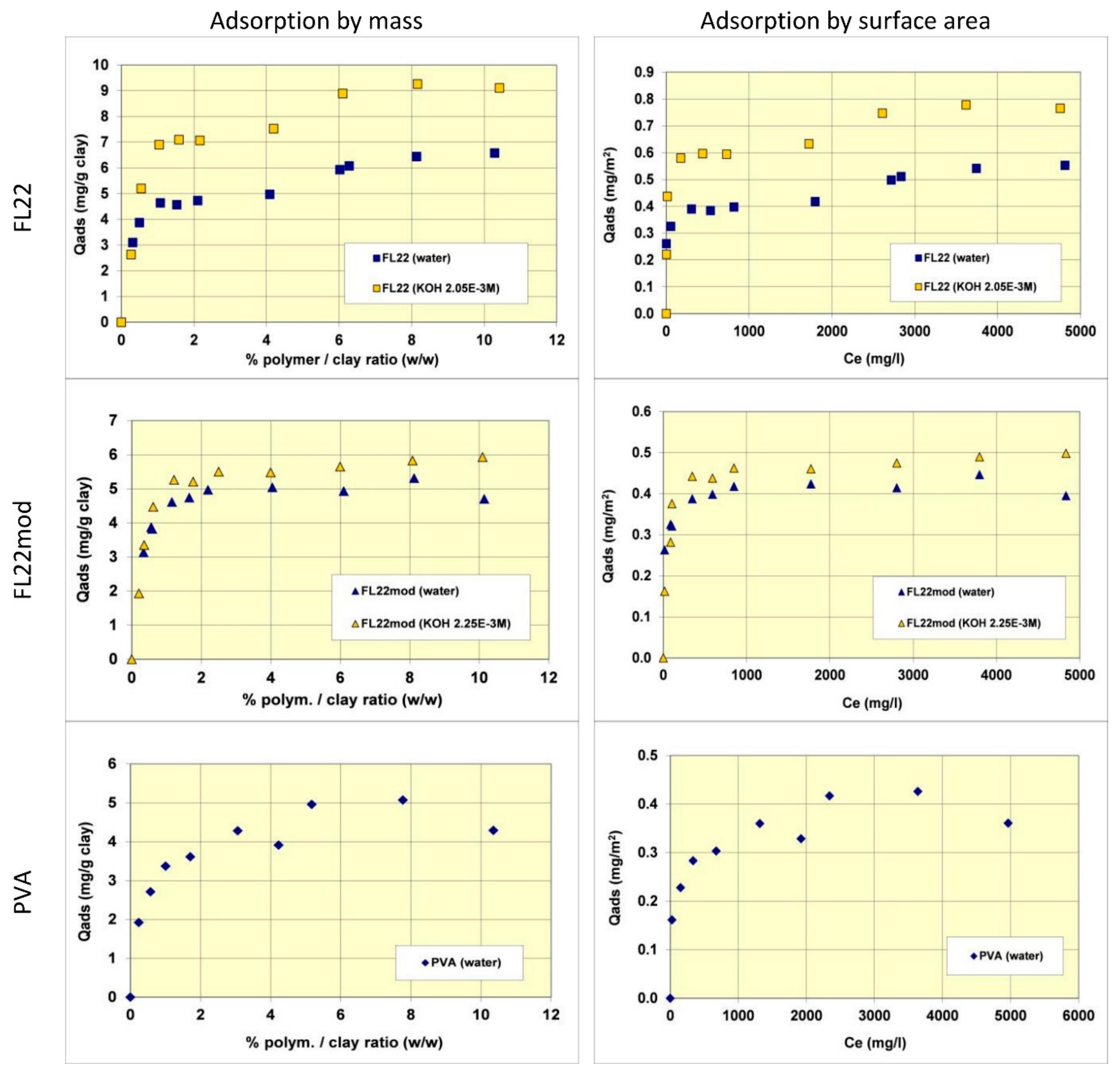
The adsorption isotherms of the three polymers were obtained in suspensions of kaolinite in water. The pH of the mixtures was therefore the natural pH (the pH each polymer solution formed), changing as a function of the ionic exchanges between the medium and the kaolinite surfaces. Subsequently, new isotherms were measured at a basic pH with a known and set quantity of KOH (0.1 M) to study the adsorption at pH values higher than pH 10. As illustrated in
Figure 5, the quantities of adsorbed polymers on this kaolinite, having a relatively low specific surface area (11.9 m
2/g), were not very high. The maximum adsorbed quantities were indeed lower than 1% by mass of the clay. This finding made it necessary to reproduce certain points of the isotherms several times to confirm the obtained experimental values.
Comparisons of isotherms obtained with different polymers at a natural pH (initial pH of the suspension close to 6) showed that the adsorbed quantities (given by mass) were not very different and did not greatly depend on the nature of the polymer. The obtained quantities were close to 5 mg/g of clay, i.e., 0.4 to 0.5 mg/m2 of clay. It should nevertheless be noted that the affinity of the PVA for kaolinite appeared to be lower than that of the polycationic polymers because the adsorption plateau was reached for higher residual polymer concentrations in solution. Furthermore, it should also be noted that the isotherm of the FL22 showed a second adsorption plateau at 7 mg/g of clay, not present in the other polymers. The obtained value does not correspond to the formation of a second layer of polymer on top of the surface of the first and it is therefore difficult to explain this phenomenon unless interactions between polymeric molecules occurred for the highest dosages of FL22. When considering the impact of the pH on the adsorption isotherms, it was evident that the increase of pH had little impact on the adsorption of the polycationic polymer without an OH group (FL22mod), which reached approximately 5.5 mg/g of clay. However, in the case of the FL22 polymer, adsorption greatly increased at a higher pH to approximately 7 mg/g of clay for the first plateau. A second plateau was always visible for this polymer (9 mg/g).
The stability of the OH group of the FL22 polymer at high pH explains these adsorption differences, hence the sensitivity to pH. Cationic charge measurements were carried out on the polymers according to the pH using the method described in
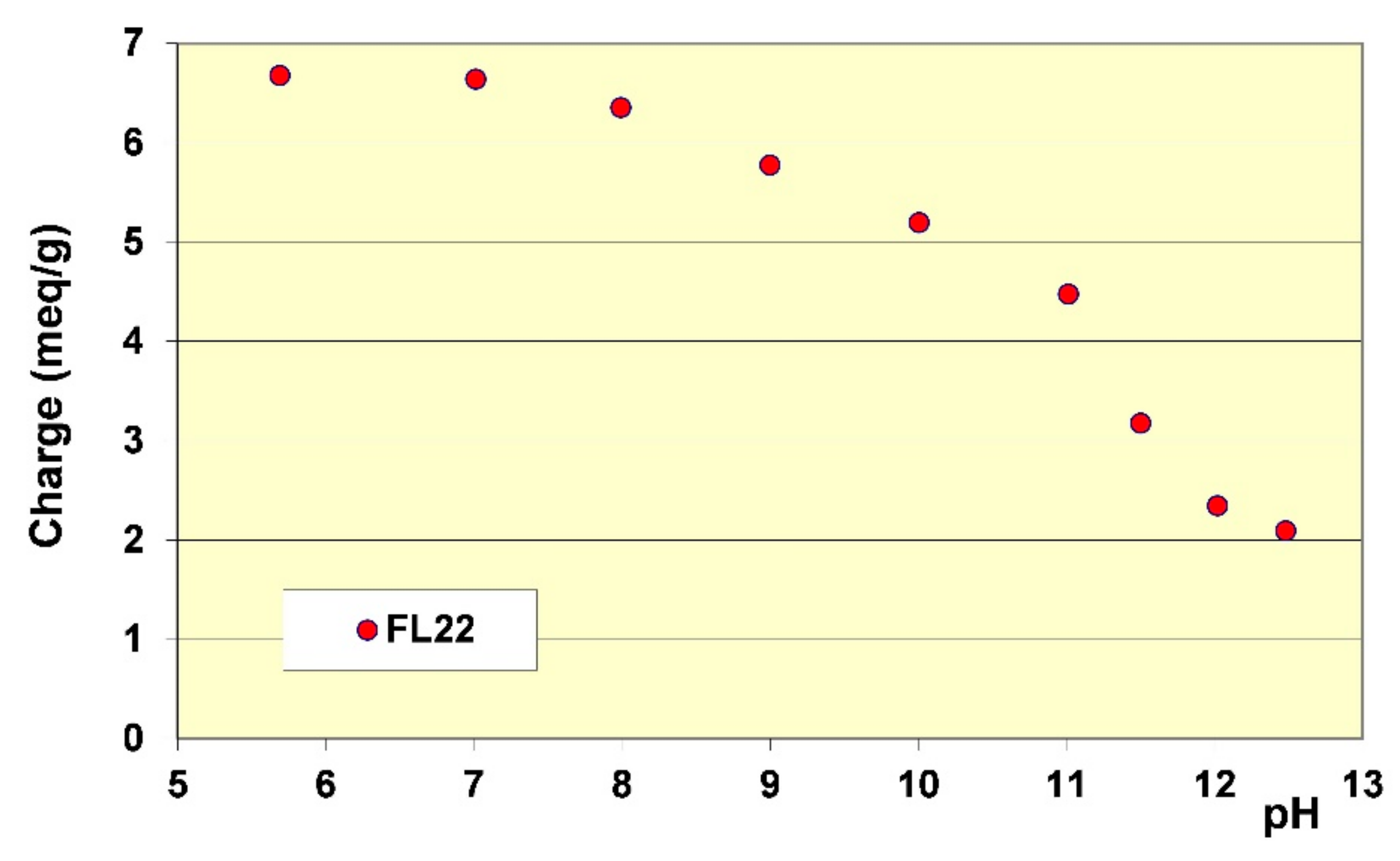
Section 2.2. The results showed that while the cationic charge of FL22mod was not influenced by the pH, this was not the case for the FL22 containing the OH group (see
Figure 6). It appeared that FL22 lost approximately 30% of cationic charge between pH 6 (6.7 meq/g clay) and pH 10.5 (4.8 meq/g clay). This polymer therefore had a fraction of negative sites on its chain at high pH. It would then be possible to infer that the electrostatic interactions created on this zwitterionic polymer induced an increased difficulty for this polymer to adopt certain configurations in solution, and hence also on the solid surface with which it would interact. When comparing the increase in adsorption of the FL22 with the pH and the increase of ionisation, it can be noted that the two were similar: 30% less charges correlating with some 40% more adsorption. The major part of this adsorption increase at high pH could therefore be explained by the variation of charges of the polymer; this variation would result in poorer adsorption efficiency on the clay mineral and aggregate surfaces.
The additional residual adsorption difference of 10%, which cannot be explained by the variation of the cationic charge, could probably be related to variation of the ionisation on the surface of the kaolinite when the pH increased. It is known, for example, that between pH 6 and pH 10.5, sites of the basal aluminous surface (AlOH) can ionize to form AlO−. When the adsorption variation of the FL22mod polymer was considered, which did not contain an OH group, and where the cationic charge was not related to the pH, it was found that between pH 6 and pH 10.5, the increase was equal to circa 10%.
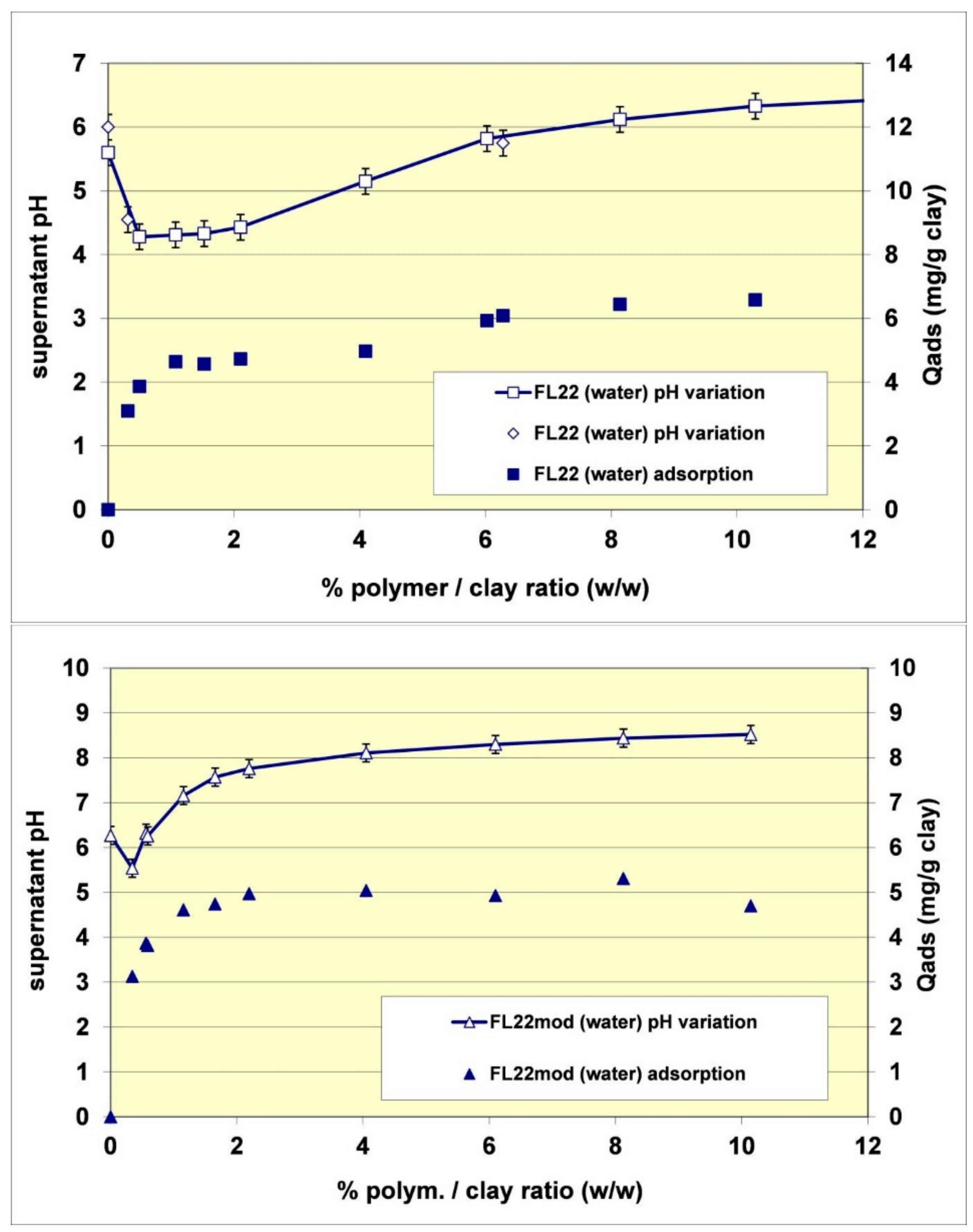
Finally, the adsorption increase of the FL22 on kaolinite when the pH increased from pH 6 to pH 10.5 was 75%, accounted for by the variation of the polymer’s cationic charge and the remaining 25% accounted for by an increase of ionization on the surface of the clay at high pH. The fact that the adsorption of FL22 or FL22mod on kaolinite induced an increase of surface ionization was furthermore confirmed by measurement of the pH during adsorption (see
Figure 7). In the case of the FL22, for example, the addition of a solution of polymer at 19.9% of active matter and at pH 7.4 in a suspension of kaolinite at pH 6 was accompanied during the first additions (complete adsorption of the polymer on the material) by a drop in pH (varying from pH 6 to pH 4.3). Subsequently, and when the supplementary adsorption greatly decreased, further additions of FL22 induced a rise of pH. The FL22mod behaved similarly (see
Figure 7). The pH of the solution of polymer at 21.9% was equal to pH 9.6. Its addition in a suspension of kaolinite at pH 6.3 induced a drop of pH, which reached pH 5.5 before increasing when the additions of polymer were more frequent. We conclude from these results that the affinity of these polycationic molecules for kaolinite is extremely high.
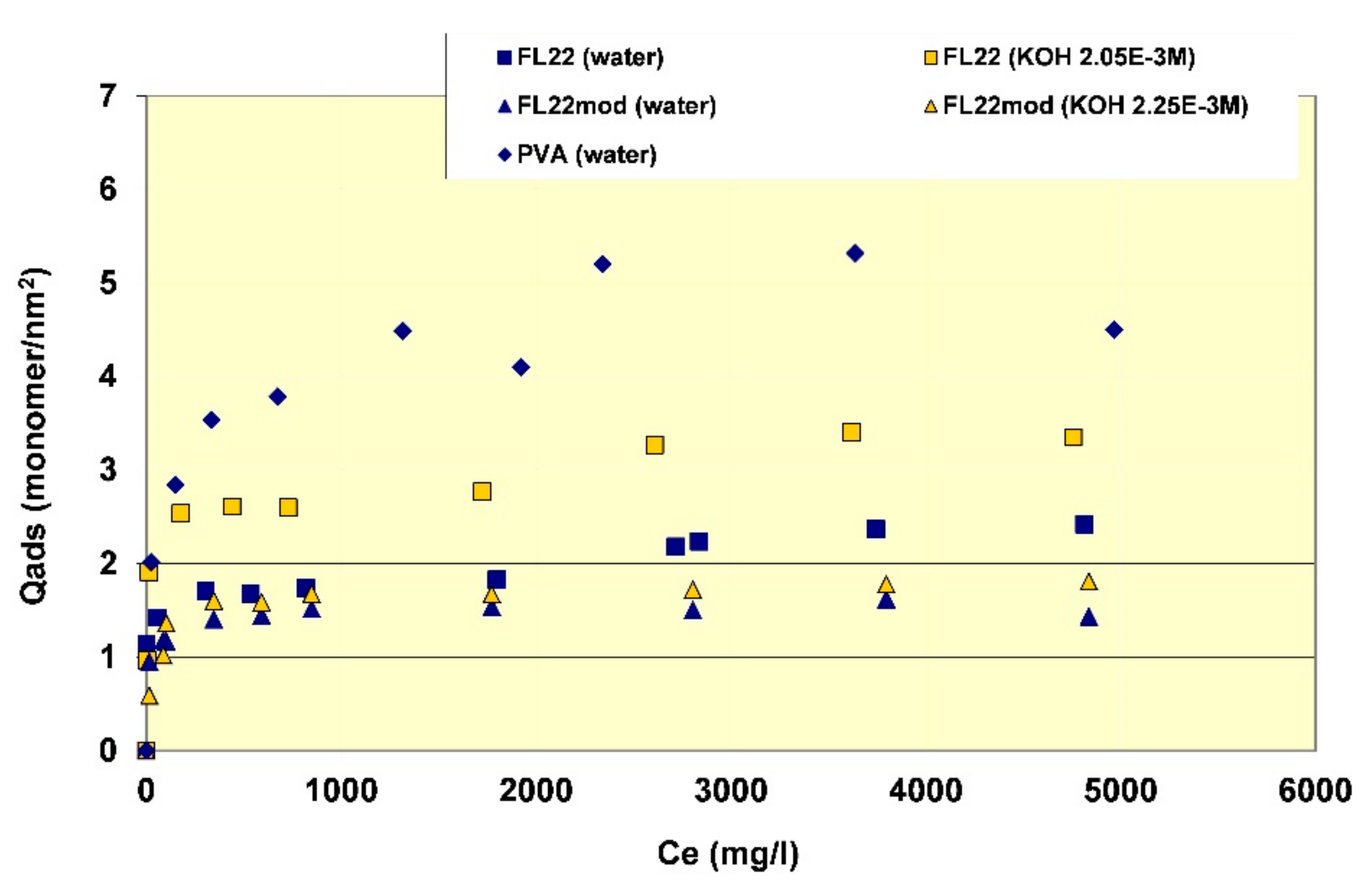
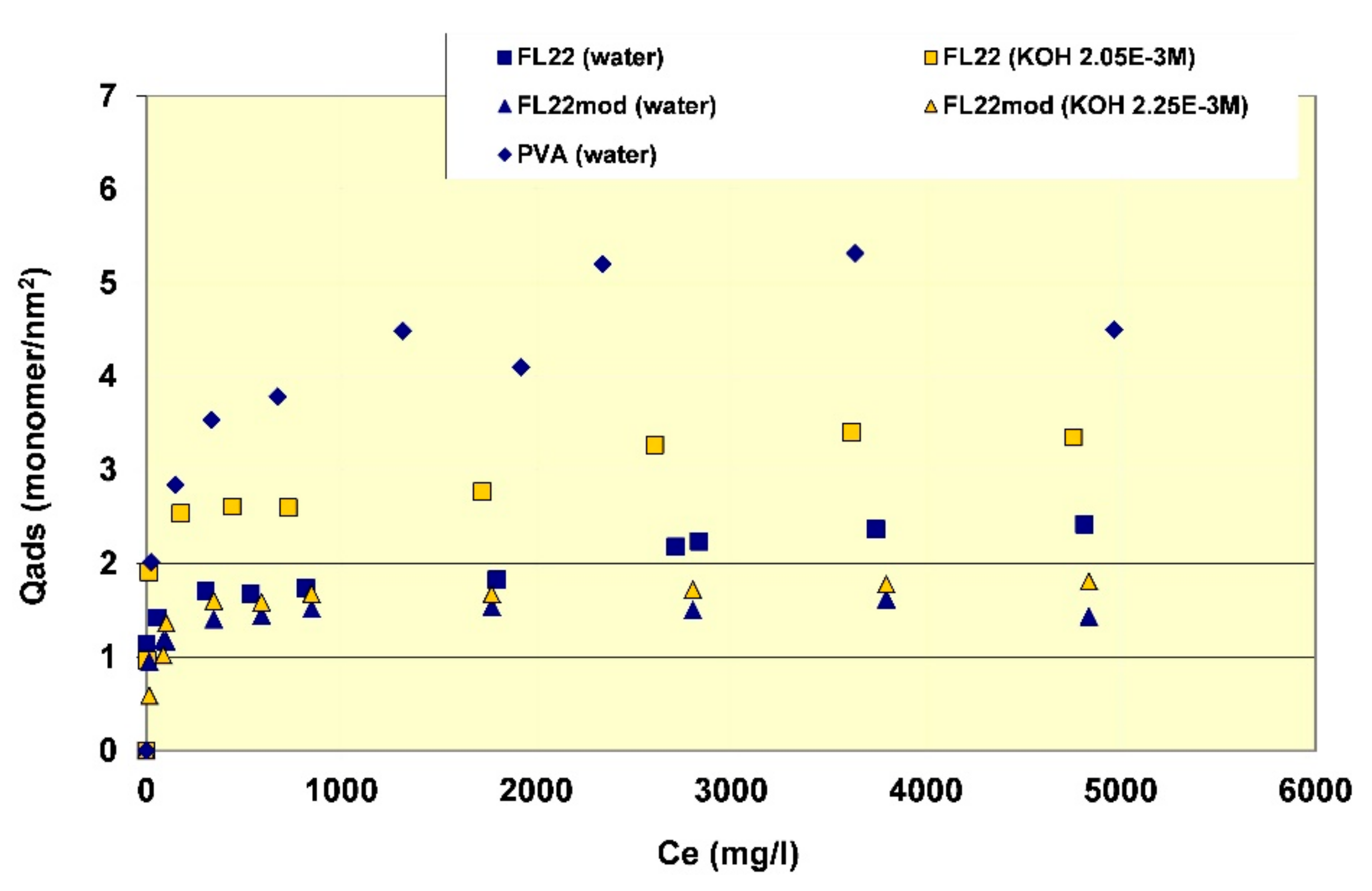
In order to better evaluate the surface coverage of the cationic polymers on kaolinite, the adsorption results are given in number of monomers per unit surface area (see
Figure 8). It clearly appears that FL22mod is adsorbed a little less than FL22 (with the OH group) at the natural pH of the suspension. These differences between adsorption by mass and adsorption by number of monomers can be explained by the fact that FL22 is a polycationic chloride while FL22mod is a bromide, the molar mass of which is higher (79.9 g/mol for the bromide and 35.5 g/mol for the chloride ions, respectively). At the natural pH of the suspension, the surface coverage of the kaolinite was equal to 1.5 monomers/nm
2 for FL22mod and 1.8 monomers/nm
2 for FL22.
For high pH, the average coating level increased from 1.7 monomers/nm2 for FL22mod to 2.6 monomers/nm2 for FL22. It should be noted that only the adsorption values obtained on the first adsorption plateau were taken into account here.
In the second part of this study, the aim was to compare the experimental surface coverages obtained at natural pH with those resulting from molecular modelling. Given the differences between the adsorbed quantities of the two polycationic polymers at natural pH (20% by number of monomers), this study was completed by measuring the adsorption isotherm of polyvinyl alcohol (see
Figure 8) wherein the monomer is not charged and is much smaller in size (length is approximately 3.8 Å for PVA sub-unit; 5.6 Å for FL22). The surface coverage of the PVA was equal to 5.5 monomers/nm
2, i.e., approximately two to three times higher than the level obtained for the polycationic polymers.
Given that the length per monomer of PVA is approximately two fold shorter than the FL22 system monomers, the lower level of coverage for FL22 is perhaps not unexpected, however the conformation space available to each polymer and the polymer specific functional group interactions also need to be accounted for, and we discuss this further when comparing the computational modelling with experiments in
Section 3.4.1.
3.3. Flocculation
Finally, the question was raised whether these differences of affinity between FL22 and FL22mod for kaolinite could have an impact on the stability of the kaolinite suspensions (state of flocculation). Observations were therefore made after 24 h of stirring with and without the polymer (see
Section 2.4). These observations are recorded in
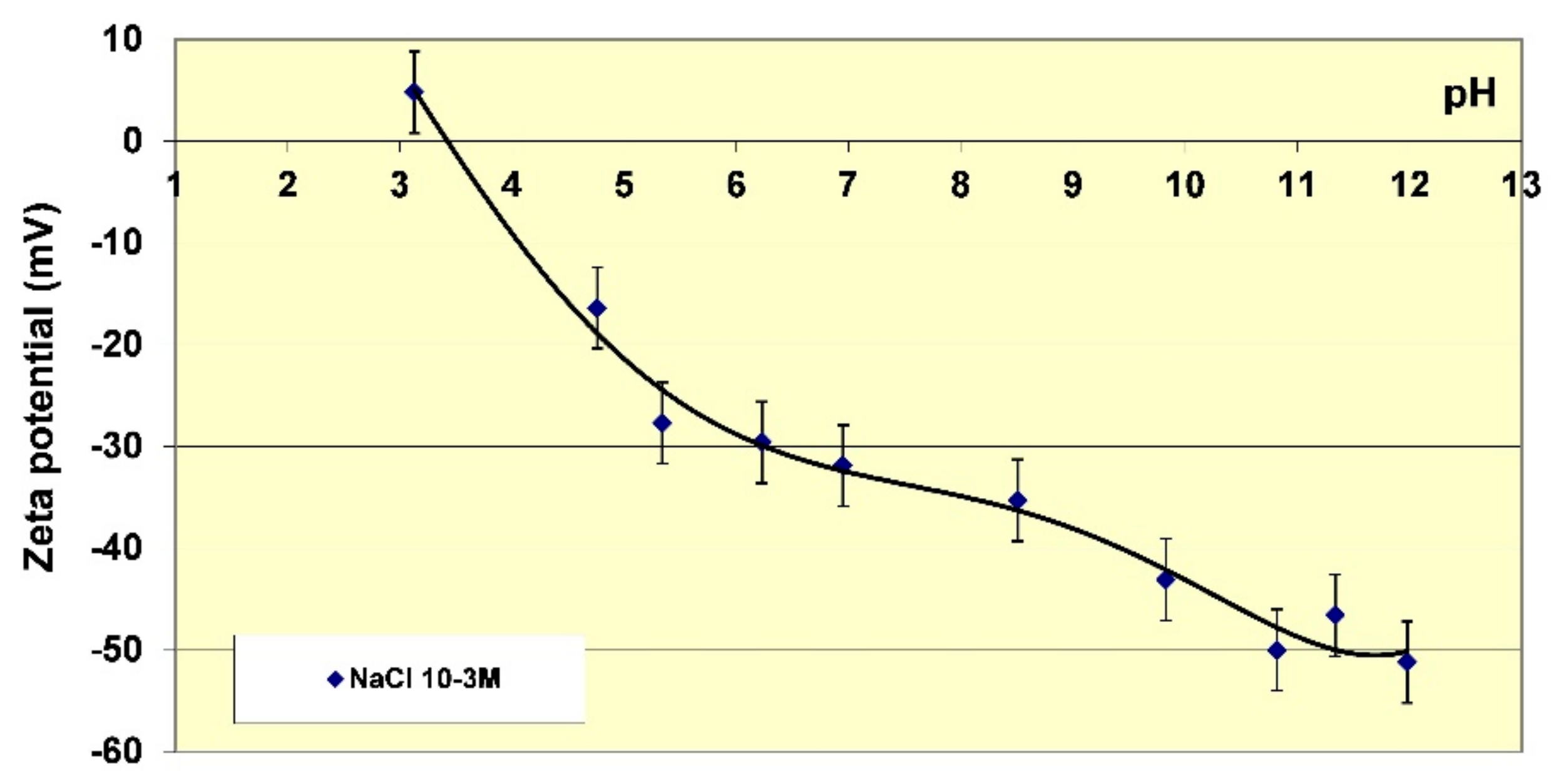
Table 3. At pH 6, the suspension of kaolinite was not stable and all the particles flocculated. If this observation is compared to values of the zeta potential (
Figure 4), this result is coherent because the value obtained is within the range of −25 mV and −30 mV, a value usually considered to be too low to stabilize a colloidal suspension. When adding increasing dosages of the three polymers, differences of behaviour were observed. In the case of the polycationic polymer FL22, several phases of deflocculation/flocculation/deflocculation were observed as increasing dosages of the polymer were added. Fines often tended to be present in the supernatant.
This phenomenon could be explained by an inversion of charges of the particles of kaolinite for the highest dosages of FL22. This finding was already noted for this type of polymer at natural pH [
44]. In the case of FL22mod, flocculation was maintained after the additions. This polycationic polymer therefore does not allow for a re-dispersion of the clay at these pH values. In the case of PVA, however, the suspension did not change very much in relative terms by the increase of adsorption; flocculation was maintained whatever the dosage. At a high pH (pH of approximately 10.5), and as mentioned in
Section 4, the apparent charge of the particles was high and the suspension of kaolinite, when polymer was absent, dispersed. The addition of FL22 induced flocculation, followed by re-dispersion for the higher dosages of polymer. There is every reason to consider that, as in the case of the measurements at natural pH, there was an inversion of the negative charge of the particles of kaolinite. This therefore tends to confirm that surface adsorption is arrested due to the polymer’s N
+ charge being more than the sum of the negative charges of the kaolinite, hence the non-optimum surface coverage efficiency. There will then remain residual positive charges on the polycationic polymer after adsorption. In the case of the additions of FL22mod (without an OH group) in the suspension at high pH, flocculation of kaolinite was observed. No re-dispersion was observed and all the particles flocculated at the highest dosages. It is therefore possible to consider that there was no or very little inversion of charge, making it possible to presume better adsorption efficiency on the surfaces with better neutralization of the negative charges on the surface.
These results, even though they originate from relatively simple observations, are therefore coherent with the hypotheses already advanced on the role of the OH group during the adsorption of polycationic polymers on kaolinite.
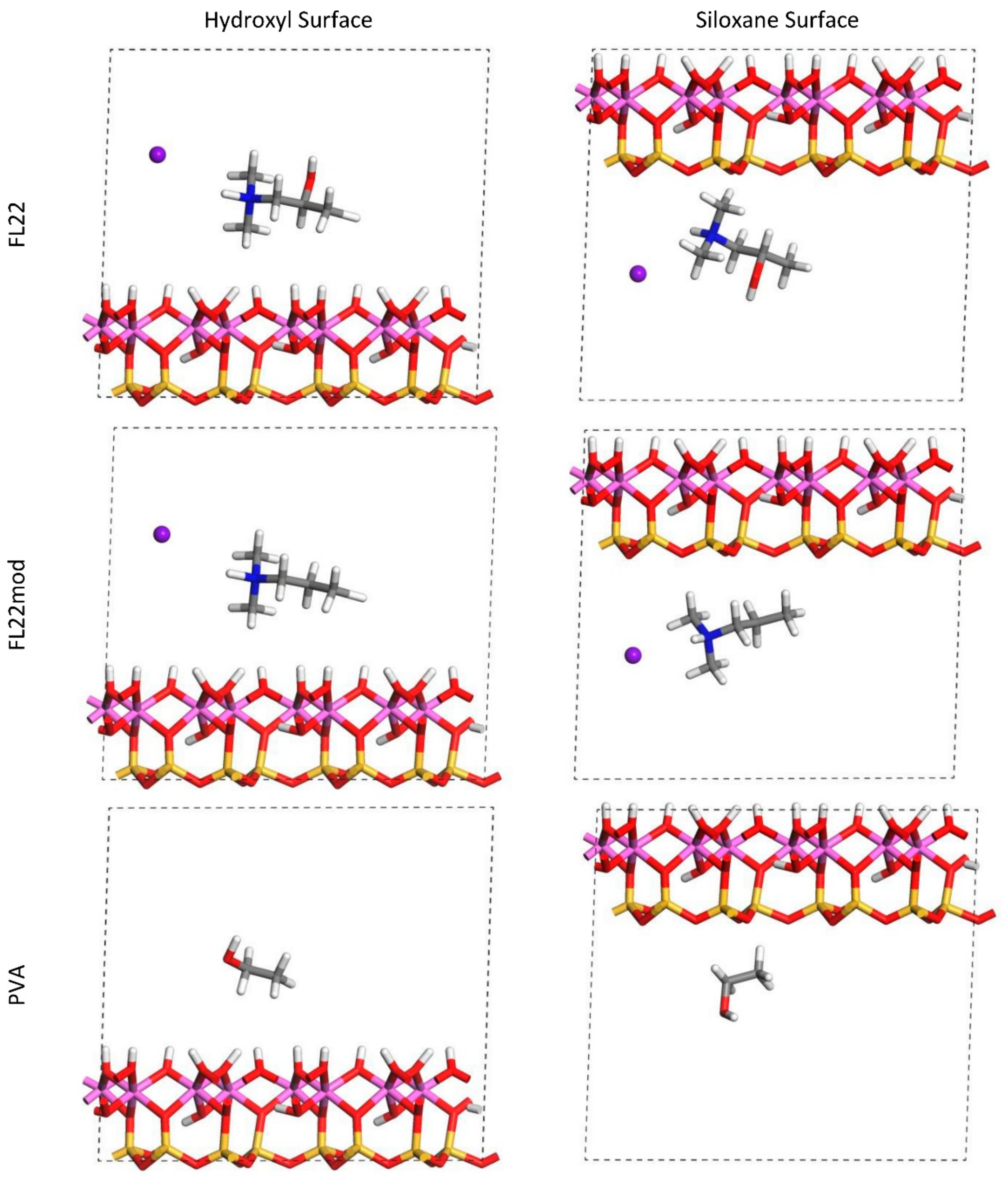
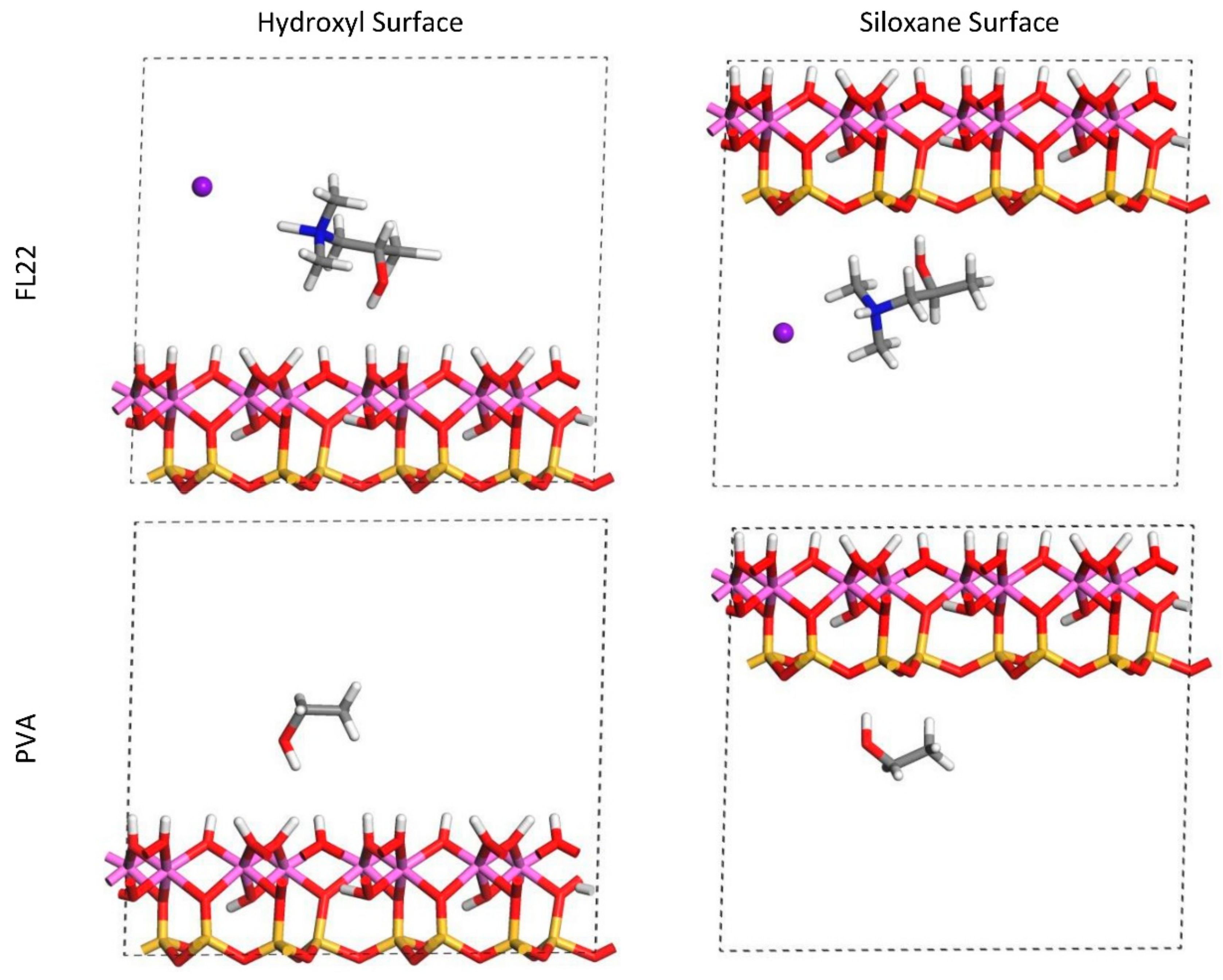
3.4. Geometry and Surface Coverage from Simulations
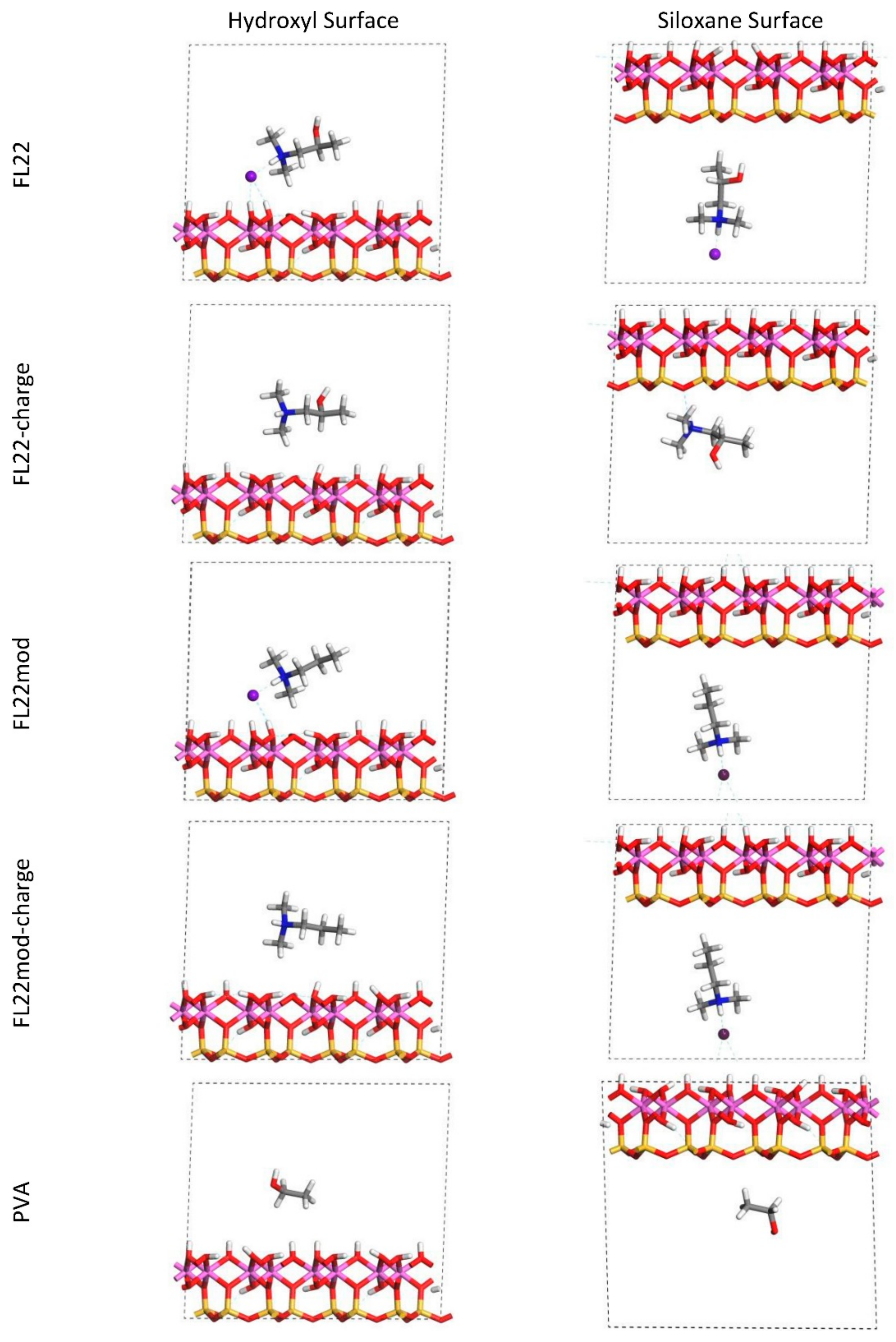
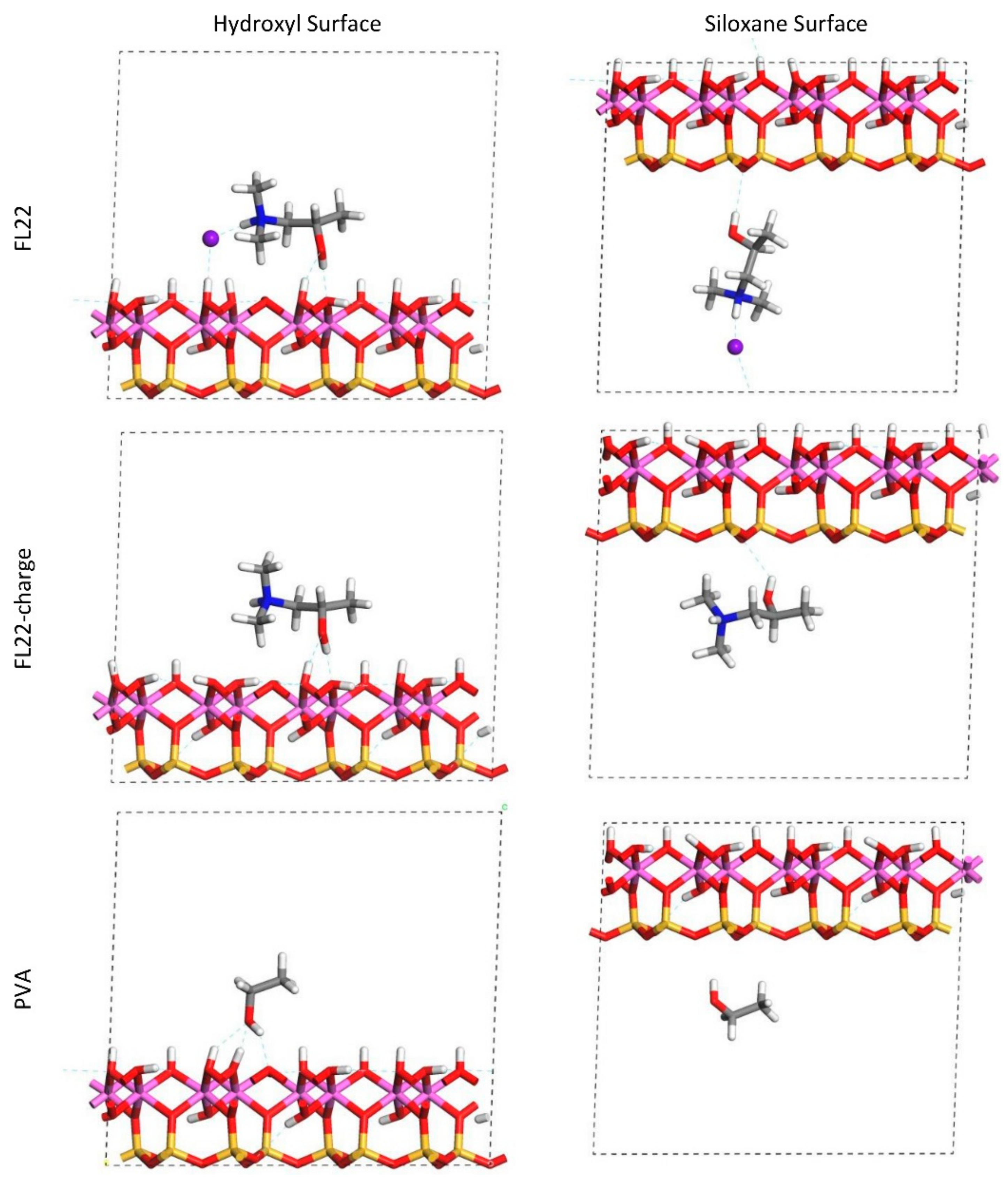
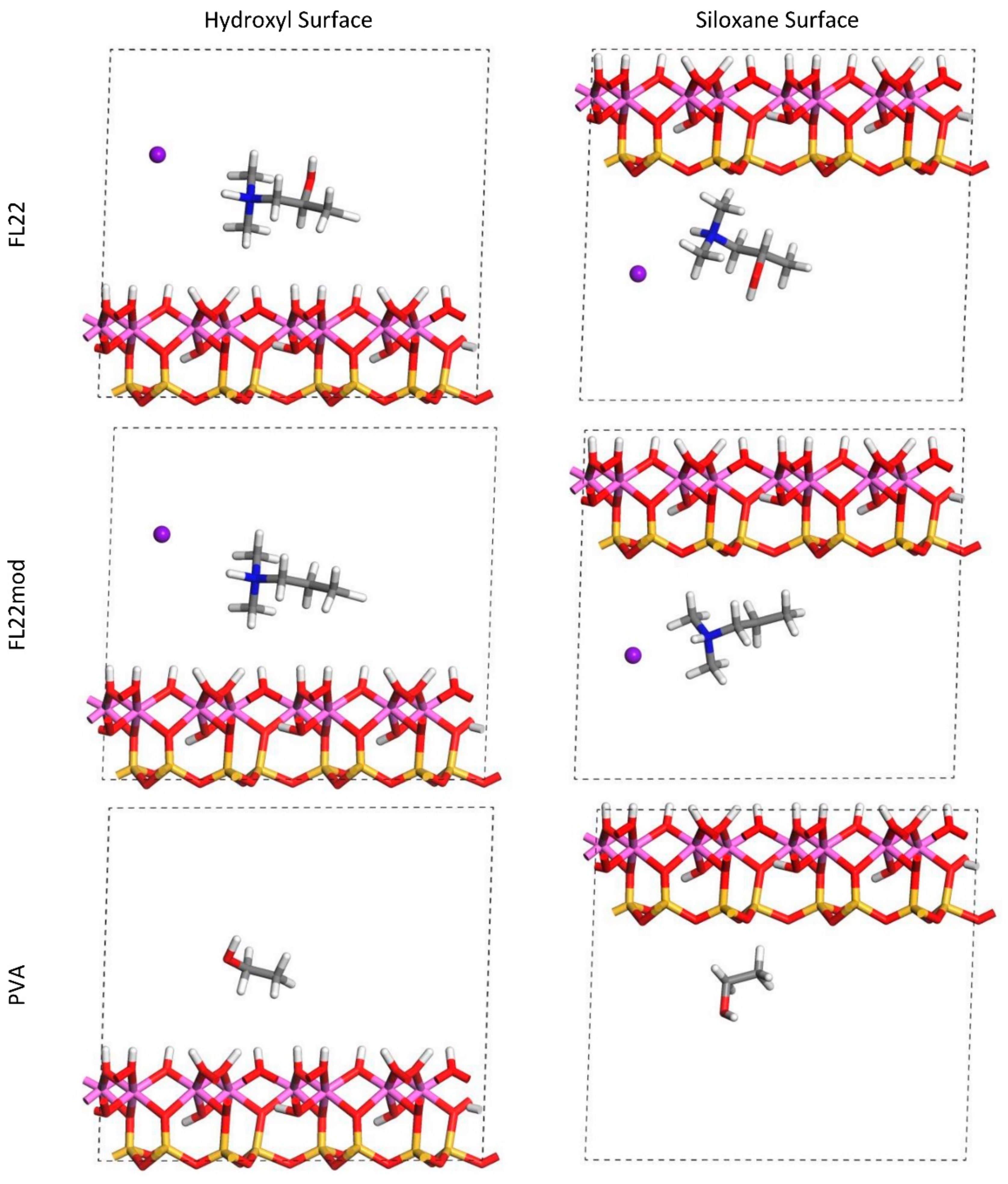
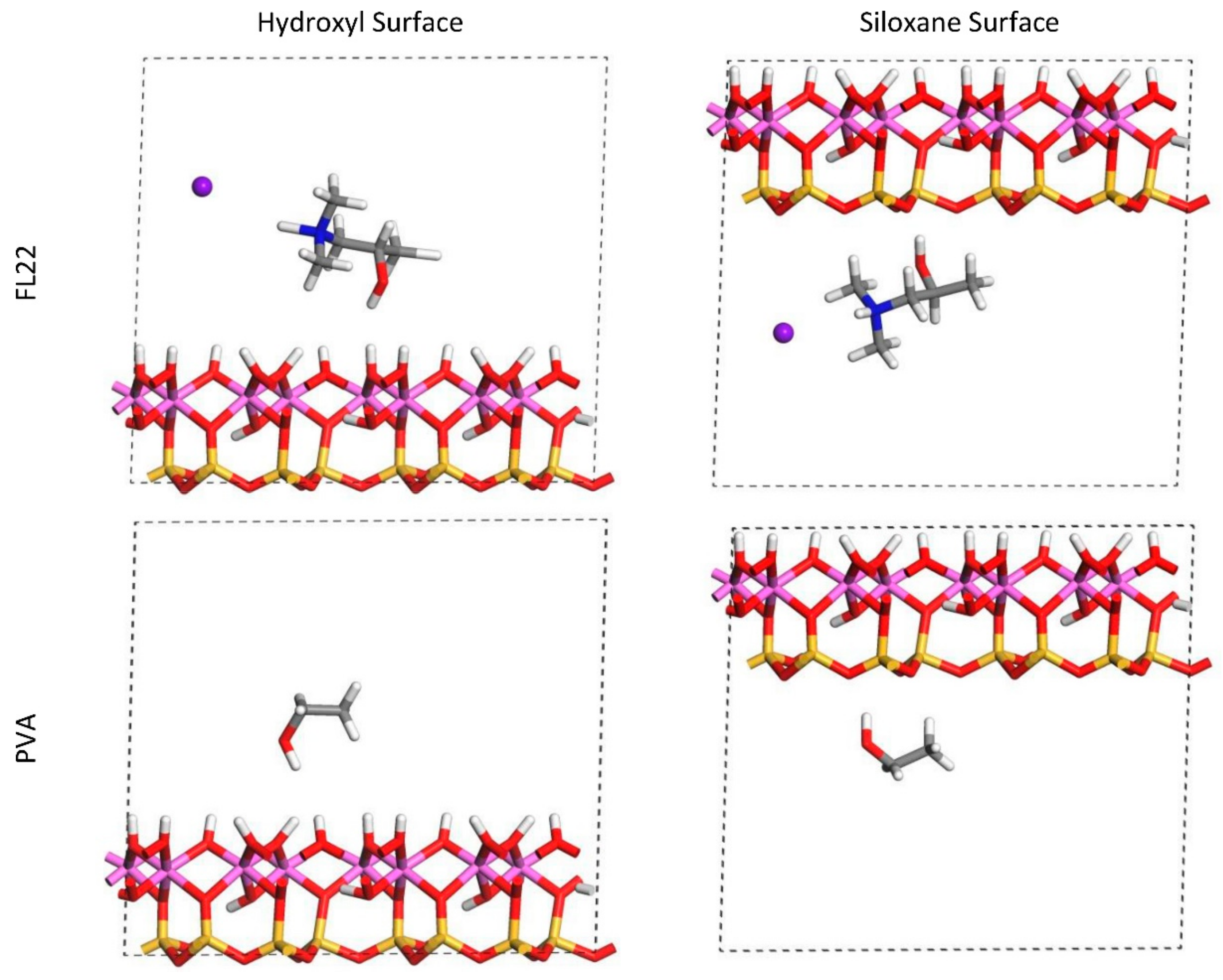
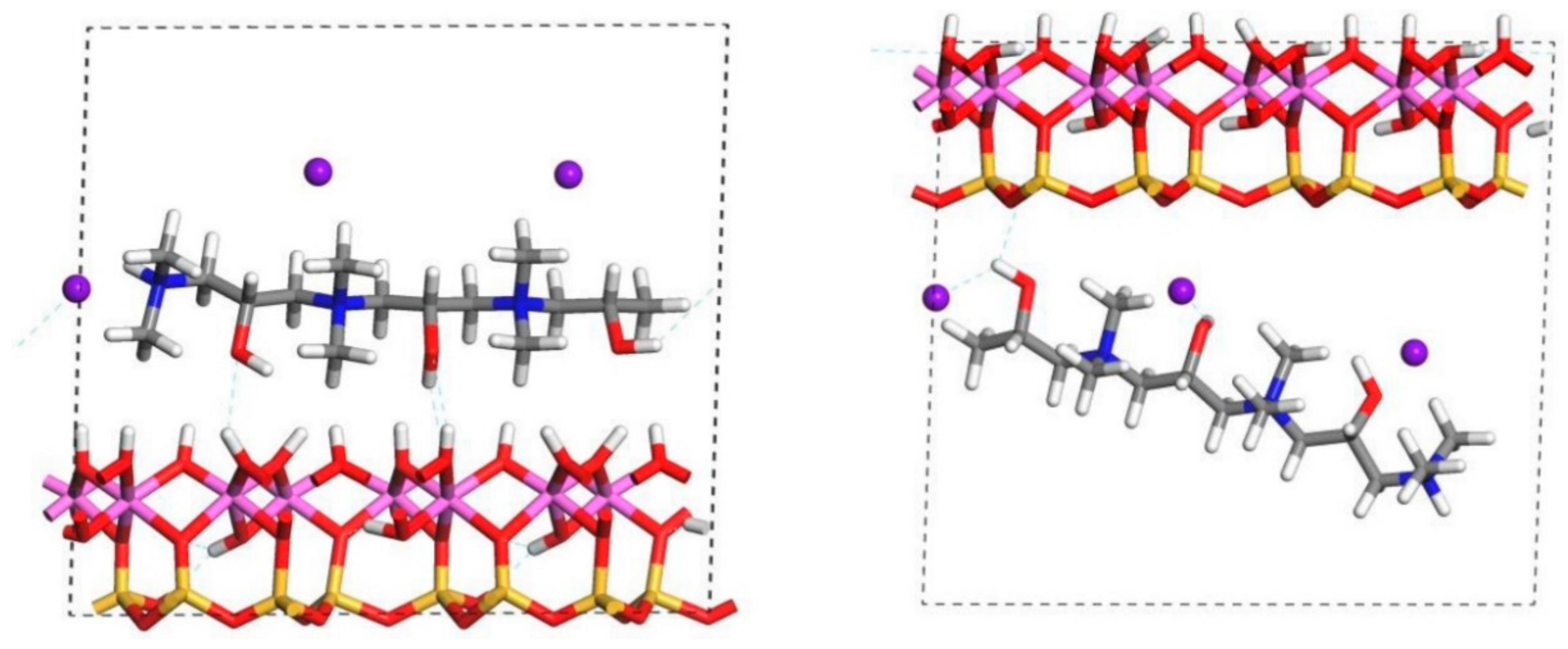
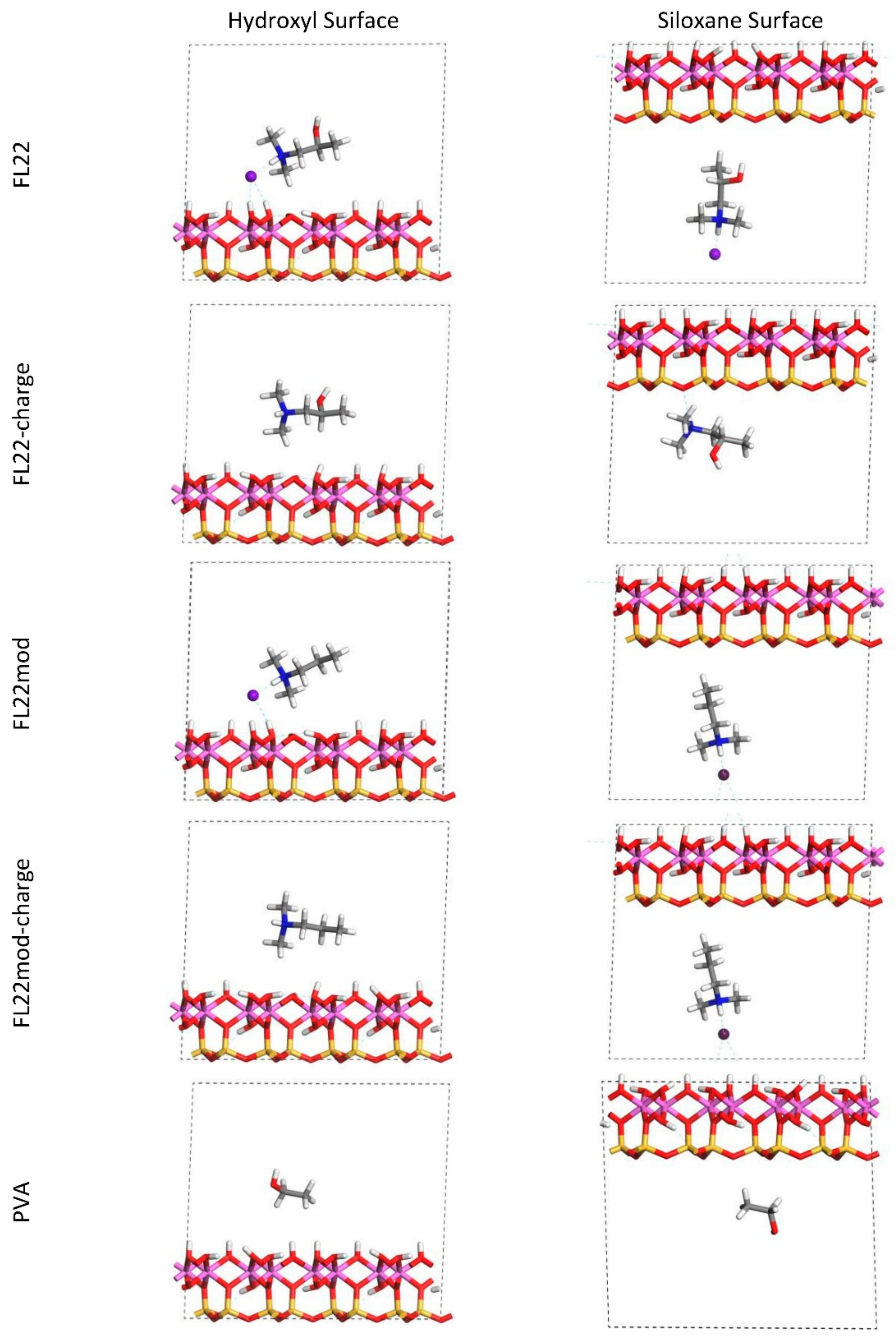
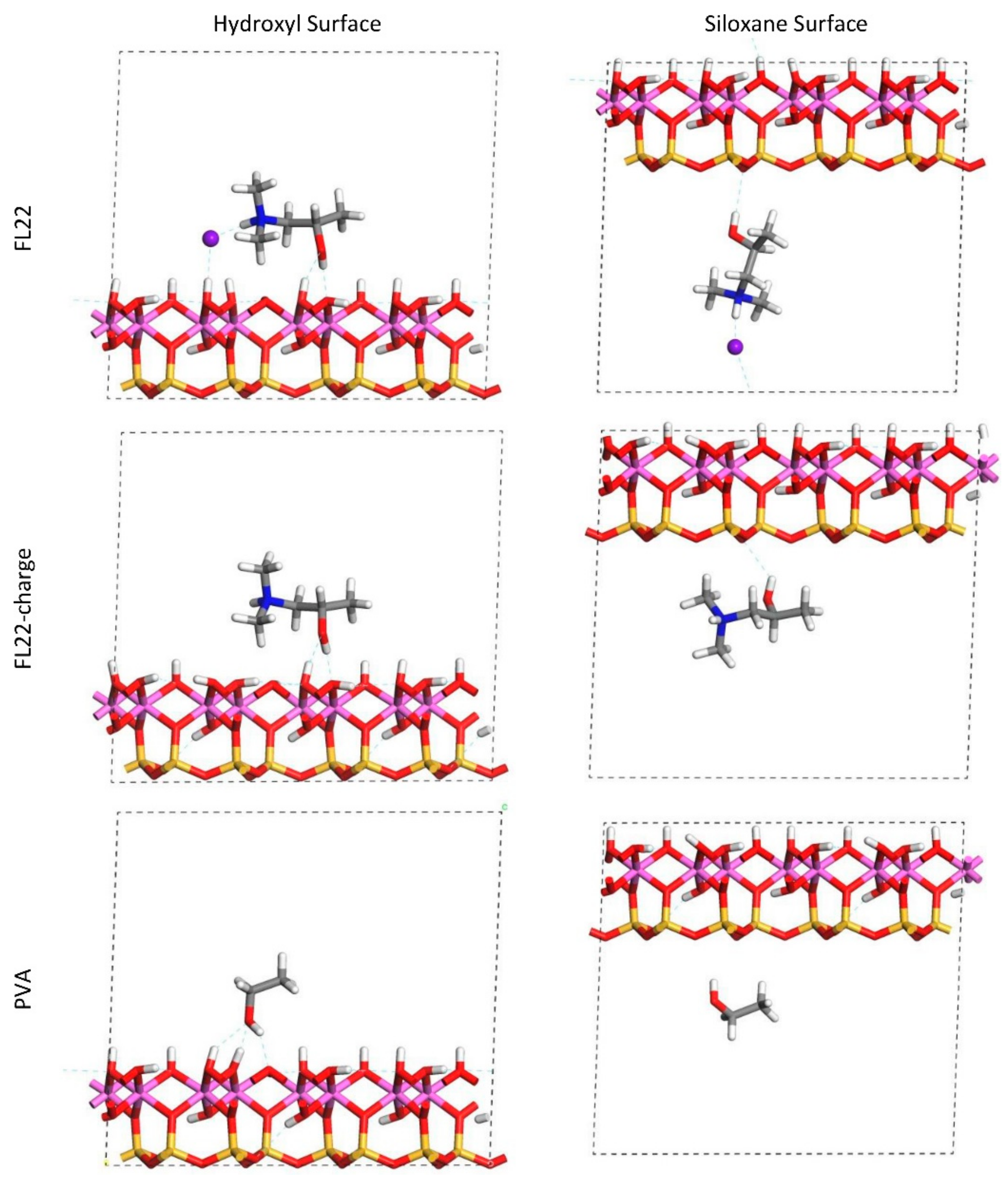
The relaxed configurations of the monomer/kaolinite models can be seen in
Figure 9 and
Figure 10 and comparing these shows that the initial orientation of the hydroxyl group of the monomer affects formation of hydrogen bonds with the basal surfaces. If the OH-group of the monomer is sufficiently close to the hydroxyl surface, the relaxed configuration exhibits hydrogen bonding between the monomer and this surface. This also occurs at the siloxane surface for FL22 but not for PVA. There is also some hydrogen-bonding between the H of the N–H group and the chloride or basal surface, although this is not as significant as the hydroxyl hydrogen-bonding because there is only one N–H group per polymer, compared to one per monomer, hence this contribution to the interactions is relatively insignificant. The presence of water in the system has been studied for a simple ammonium monomer system [
12], and given the hygroscopic nature of the quaternary ammonium group, especially with an adjacent OH group, there may be further hydrogen bonding. However, the previous study showed that addition of explicit water molecules has negligible effect on the orientation of the monomer both with and without chloride, but an analysis of frontier orbitals showed it completely changed the reactivity of the monomer [
12].
The presence of chloride compared to charge has the effect of orienting the monomer towards the hydroxyl surface, mostly due to the formation of hydrogen bonds between the chloride and the monomer and the chloride and the hydroxyl surface. This effect is seen for both orientations 1 and 2 for FL22 and FL22mod and is a consequence of using a monomer [
12]. The same degree of orientation is not expected to occur for a polymer in the presence of chloride. This difference in orientation created a difference in surface coverage as can be seen in
Table 4, where for most models without chloride, the surface coverage is higher.
Surface coverage was calculated by projecting the aspect of the monomer (and chloride where appropriate) facing the basal surface, including the van der Waals radii. Consequently, where a chloride ion lies outside the monomer projection, the total surface coverage is lower, and where it falls within the monomer projection it is higher. This, together with differences in orientation, produces two scenarios where coverage without chloride is lower than with chloride. In the trimer scenarios the positioning of the chlorides was not obvious, while one might lie outside the monomer/surface projection, two of them cannot. As the atomic positions of these models were not relaxed these results serve to indicate what the surface coverage might be, if trimers were oriented as shown in
Figure 3.
Comparison with Experiment
If the computational chloride models are excluded due to their exaggerated effect on the monomers, then the surface coverage for FL22 and FL22mod lies between 2.6–2.9 monomers/nm
2 and there is no significant difference between the values for the two types of monomers. The experimental values for FL22 in water are 1.84 and 2.39 monomers/nm
2 and for FL22mod in water, 1.52 monomers/nm
2 (See
Figure 8). Not only are these different to each other, they are also different to the theoretical results. This suggests that the polymers do not lie completely flat to the basal surfaces. For PVA the computational value lies between 5.1–7.6 monomers/nm
2. This range is wide due to the small size of the monomer, hence slight differences in orientation have greater effects on total surface coverage, than in the cases of FL22 and FL22mod. The experimental value for the adsorption of PVA (in water) is 5.24 monomers/nm
2 and is within 5% of the lower theoretical value, suggesting that PVA polymers lie flat to the basal surface, possibly without forming hydrogen bonds with the surface.
3.5. Calculated Formation Energies
In this study, “formation energy” is defined as the difference between the relaxed, full system and the sum of the energies comprising the parts of this relaxed model, e.g., for FL22 with chloride, the “parts” constitute separate models of the clay layer alone, and a monomer of FL22 plus chloride. Formation energies indicate the relative strength of the interactions between the monomers and the surfaces and hence describe one aspect of their relative adsorption (they have not been calculated for the trimers as the atomic positions of the trimers were not allowed to relax). The higher formation energies of the chloride models (see
Table 5) indicates the strength of the Coulombic repulsion between periodic images of the chlorides in the chloride/monomer models, which is ameliorated by the presence of the clay in the clay/monomer/chloride models. The columns of
Table 5 cannot be compared directly, i.e., the results for the chloride and no chloride models cannot be compared with each other, but the values within each of the columns can be compared. The trends within these two chloride and charge scenarios, however, are comparable. The formation energies of both orientations of FL22 for both surfaces in the chloride models and separately, the charge models, lie within 10% of each other, suggesting that the hydrogen bonding between FL22 and kaolinite surfaces, which is more prevalent in the o2 configurations, does not contribute significantly to the strength of the adsorption of the monomers, as the formation energy does not follow a consistent pattern aligned with the number of hydrogen bonds.
The formation energy of FL22mod is approximately 10% lower than FL22-o1 per comparable scenario, which is not significant, suggesting that the mode of interaction of these two types of monomer and the kaolinite basal surfaces is the same. In all cases the formation energy is lower on the siloxane surface than on the hydroxyl surface. The formation energies of PVA are, in three of the four scenarios, two orders of magnitude higher than those of FL22 and FL22mod. The more negative value of −11.37 kCal/mol for PVA-o2 on the hydroxyl surface, is due to the increased number of hydrogen bonds compared to the numbers in the remaining PVA configurations. Even so, this is still an order of magnitude higher than the formation energies for FL22 and FL22mod, although it does suggest that in the absence of other discernible interactions, hydrogen bonding plays a more significant role. These results say that, in terms of the strength of adsorption, FL22 and FL22mod are comparable and compared to PVA are strongly bound to the kaolinite surface.
3.6. Mulliken Charges
A comparison of the Mulliken charges of the monomers indicates the extent of interaction of the monomer with its environment. Mulliken population analysis is particularly suitable for analysing the results of computations performed using well-converged planewave basis sets, which are the basis set of choice in the CASTEP code [
28]. Furthermore, Mulliken charge analysis is a very quick, post-processing step and, although the resulting charges are not valid as absolute charges for the determination of, for example, the quantitative magnitude of chemical bonds [
45], they do yield qualitative information when making relative comparisons between like-systems [
46].
Table 6 shows that the monomers of FL22-o1 and FL22mod in the chloride models have charges between 0.72 e and 0.76 e and in the models without chloride, this varies from 0.91 e to 0.98 e. Therefore, we can say that the FL22 and FL22mod are positively charged and are interacting with the surfaces electrostatically. The configurations of FL22-o2 contain more hydrogen bonds than those of FL22-o1, which is reflected in the lower charges of FL22-o2 except at the siloxane surface, no chloride models, where FL22-o1 has the lower charge. This is explained by the dissipation of charge occurring on formation of hydrogen bonds and perhaps the NH-group hydrogen bond dissipates more electron density than OH-group hydrogen bonds, although this difference is relatively small (approximately 5% of the total monomer charge).
The Mulliken charges of the PVA monomers are approximately 1% of those of the quaternary ammonium monomers, indicating that PVA and kaolinite do not interact electrostatically. These results mirror the initial charges of these monomers where FL22 and FL22mod are both positively charged and PVA is uncharged, hence there has been relatively little change in electron density in the monomers and kaolinite on their combination.
The relative charges of the trimers show the same trends as for the monomers, i.e., the charges of the FL22 and FL22mod trimers in the no chloride models are positive, ranging from 2.38 e to 2.98 e. It would be reasonable to expect trimer charges to be three times those of the monomers, and would probably be realized if the atomic positions of the trimers were allowed to relax. The differences in relative charges seen in the chloride trimer models are an indication of the difficulty of positioning the chloride ions, especially when their coordinates are constrained, therefore it is probable that the trimer chloride results are not as relevant as those of the no-chloride models. The PVA trimers at the hydroxyl surface have relatively negligible charge, as would be expected but those at the siloxane surface have gained a relatively considerable charge of 1 e for both orientations. Further investigation of the Mulliken charges of both surfaces of kaolinite show that in the siloxane surface models, the charges of the hydrogens of the hydroxyl surface are approximately half the values of the hydrogens of the hydroxyl surface in the hydroxyl surface models. It would be interesting to test whether this positive charge on PVA would decrease and dissipate to the hydrogens on the hydroxyl surface if all atomic positions were allowed to relax, although in this study this is constrained by the nature of the models.
These results show that, with the exception of PVA trimers at the siloxane surface, the trimer models show the same charge trends as the monomer models, and therefore we can state that the interactions of the polymers with the basal surfaces of kaolinite are primarily electrostatic.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}