Molecular Engineering of Therapeutic Cytokines

Garvan Institute of Medical Research, 384 Victoria Street, Darlinghurst, Sydney, New South Wales 2010, Australia
*
Author to whom correspondence should be addressed.
Antibodies 2013, 2(3), 426-451; https://doi.org/10.3390/antib2030426
Submission received: 9 May 2013
/
Revised: 13 June 2013
/
Accepted: 13 June 2013
/
Published: 3 July 2013
(This article belongs to the Special Issue Cytokine Growth Factor Antibodies in Immunotherapy)
Abstract
:Over the past three decades, a large body of work has been directed at the development of therapeutic cytokines. Despite their central role in immune modulation, only a handful of cytokine therapeutics has achieved regulatory approval. One of the major challenges associated with the therapeutic use of cytokines relates to their short serum half-life and low bioavailability. High doses are required to overcome these problems, which often result in dose-limiting toxicities. Consequently, most cytokines require protein engineering approaches to reduce toxicity and increase half-life. For this purpose, PEGylation, fusion proteins, antibody complexes and mutagenesis have been utilized. Here, we summarize past, recent and emerging strategies in this area.
1. Introduction
Cytokines are a large family of mainly soluble proteins and glycoproteins that function as key modulators of the immune system. They encompass interleukins (ILs), interferons (IFNs), growth factors, colony stimulating factors (CSFs), the tumour necrosis factor (TNF) family and chemokines (or chemotactic cytokines). These small signalling molecules (<30 kDa) are mostly secreted by leukocytes, but can also be produced by other cell types (e.g., endothelial cells, epithelial cells and fibroblasts). Cytokines can function in an autocrine, paracrine or endocrine manner to stimulate or suppress the activity of target cell populations. Signals conveyed by cytokines are essential for generation, survival and homeostasis of immune cells, as well as for the generation of immune responses upon external stimuli. Due to their natural role as immune modulators, many cytokines have been identified as suitable therapeutic candidates for the treatment of a number of infectious, inflammatory, autoimmune and malignant diseases.
The discovery of cytokines was guided by initial observations that soluble biological factors could mediate inflammation, cellular stimulation and anti-viral activities. For instance, the pro-inflammatory cytokine, interleukin-1 (IL-1), was initially described as a pyrogenic factor isolated from cultured rabbit leukocyte supernatants [1,2]. Similarly, reports of a T-cell growth factor present in mixed leukocyte cultures led to the identification of IL-2, a master regulator of T-cell function and development [3,4,5]. Furthermore, IFNs were first reported as soluble secreted factors mediating viral interference in chick chorioallantoic membranes [6,7]. In fact, pioneering cytokine immunotherapy in humans was performed with IFN-containing (<1%) protein fractions extracted from growth medium of virus-stimulated human leukocytes [7,8].
The emergence of recombinant DNA technology in the 1970s represented a turning point for exploiting the therapeutic potential of cytokines. Similarly important was the development of high performance liquid chromatography, which eased the laborious task of obtaining cytokine preparations pure enough for characterization [7]. Using these techniques, the first cytokines to be cloned and synthesized in E. coli were IFN-α and IFN-β in 1980 [7,9,10] and IL-2 in 1983 [11,12]. Initial regulatory approval of recombinant IFN-α for the treatment of hairy cell leukaemia was obtained in 1986, while recombinant IL-2 was approved for treating metastatic melanoma in 1992. Additional therapeutic applications for IFN-α, IL-2 and a number of other cytokines have since been approved or are currently under trial, as summarized in Table 1.
2. Cytokine Toxicity
Cytokine immunotherapy often results in the development of severe dose-limiting side effects. Two properties shared by most cytokines are thought to play a crucial role in the development of treatment-associated adverse effects. Firstly, cytokines are pleiotropic, meaning they are able to influence more than a single cell type. In fact, some cytokines are able to stimulate cell types that mediate opposing biological effects. Furthermore, cytokines have a short serum half-life and, thus, need to be administered at high doses to achieve their therapeutic effects [13]. While effectively enhancing therapeutic efficacy, high doses exacerbate pleiotropic activities that manifest as adverse effects in patients. Molecular engineering of cytokines with prolonged half-life, enhanced specificity or localized activity is therefore required to enhance the pharmacological properties of these proteins. Engineered cytokine therapeutics that have been approved for clinical use or are undergoing clinical trials are summarized in Table 2.
A prominent example of cytokine-associated toxicity is the adverse effects observed in patients receiving high-dose (HD) IL-2 cancer immunotherapy. IL-2 stimulates the proliferation of cytotoxic CD8+ T-cells and NK cells, which both act to promote tumour regression [14]. Due to its anti-tumorigenic activity, HD IL-2 therapy has been approved for the treatment of metastatic melanoma and renal cancer, with overall objective responses observed in approximately 16–17% of patients [14,15,16]. However, HD IL-2 therapy can cause excessive vascular permeability, which leads to an adverse state known as vascular leak syndrome (VLS). Symptoms arising from VLS can be life-threatening, making intensive patient management a requirement for the use of IL-2 immunotherapy [16].

{kind=link}
| Cytokine | Main biological functions | Indications | Status | Brand name, ClinicalTrials.gov ID or EudraCT No. |
|---|---|---|---|---|
| IL-1Ra | Binds IL-1 receptor without inducing signalling, natural IL-1 antagonist | Rheumatoid arthritis, neonatal-onset multisystem inflammatory disease | Approved | Kineret (Anakinra) |
| IL-2 | T-cell generation, homeostasis and proliferation; stimulates NK cells. | Metastatic melanoma, renal cell carcinoma | Approved | Proleukin (Aldesleukin) |
| IL-7 | Development of pre-B cells and pre-T-cells | Solid tumours, HIV-induced CD4 lymphopenia, HIV-related PML, T-cell lymphopenia after stem cell transplant | Phase II | NCT00062049, NCT01190111, NCT01190111, NCT00684008, 2010-019773-15, 2012-000725-41 |
| IL-10 | Inhibition of pro-inflammatory cytokine production by macrophages | Psoriasis, scar reduction, Wegener’s granulomatosis | Phase I, II | NCT00001761, NCT00001761, NCT00001761, 2004-001455-11 |
| IL-11 | Promotes haematopoiesis, synergises with IL-3 and IL-4 | Prevention of severe thrombocytopenia after myelosuppressive chemotherapy | Approved | Neumega (Oprelvekin) |
| IL-12 | NK cell activation, polarisation towards development of TH1 CD4+ cells | Ovarian cancer, metastatic melanoma, cutaneous T-cell non-Hodgkin lymphoma and other cancers; HIV-induced CD4 lymphopenia | Phase I, II | NCT00016289, NCT00026143 NCT00052377, NCT00000857 |
| IL-15 | Similar to IL-2, stimulates T-cells and NK cells | Metastatic melanoma, renal cell carcinoma and other cancers | Phase I, I/II | NCT00001761, NCT01021059, NCT01572493, NCT01369888 |
| IL-21 | Induction of B-cells, T-cells and NK cells | Metastatic melanoma, metastatic renal cancer, ovarian cancer, solid tumours | Phase I, II | NCT00336986, NCT00095108, NCT00523380, NCT01629758, 2006-005350-79 |
| IFN-α | Leukocyte interferon, anti-proliferative and anti-viral cytotoxic activity, increased expression of MHC class I antigen | Hairy cell leukaemia, AIDS-related Kaposi's sarcoma, hepatitis B/C, follicular lymphoma, malignant melanoma | Approved | Infergen, Intron-A, Roferon A |
| IFN-β | Fibroblast interferon, similar activity to IFN-α | Relapsing multiple sclerosis | Approved | Avonex, Betaseron, Extavia, Rebif |
| IFN-γ | Modulates T-cell growth and differentiation; promotes development of TH1 CD4+ cells | Chronic granulomatous disease, malignant osteoporosis | Approved | Actinmmune, Imukin |
| G-CSF | Stimulates neutrophil development and differentiation | Chemotherapy-induced neutropenia | Approved | Granocyte (Lenograstim), Neupogen (Filgrastim) |
| GM-CSF | Growth and development of macrophage and granulocyte precursors | Neutropenia under chemotherapy and bone-marrow transplant | Approved | Leucomax (Molgramostim), Leukine (Sargramostim) |
| TNF-α | Promotes inflammation, inhibition of tumorigenesis | Soft tissue sarcoma of the limb—administered via isolated limb perfusion | Approved | Beromun (Tasonermin) |
| TRAP | Induction of T-cell dependent B-cell activation, promotes antibody class-switching, T-cell priming | X-linked hyper IgM syndrome, metastatic melanoma, metastatic kidney cancer | Phase I, II | NCT00001145, NCT00053391, NCT00020540, 2010-023103-94 |
| TRAIL | Apoptosis of tumour cells and activated T-cells | Metastatic colorectal cancer, non-Hodgkin lymphoma | Phase I | NCT00873756, NCT00400764 |
IL-1Ra = IL-1 receptor antagonist, TRAP = TNF-related activation protein (CD40L), TRAIL = TNF-related apoptosis inducing ligand, PML = progressive multifocal leukoencephalopathy.
| Modification /cytokine | Property | Indications | Status | Drug name(s), ClinicaTrials.gov or EudraCT ID | |
|---|---|---|---|---|---|
| PEGylation | |||||
| IFN-α2a | Chronic hepatitis B/C | Approved | Pegasys (peginterferon alfa-2a) | ||
| IFN-α2b | Extended half-life | Chronic hepatitis C | Approved | Pegintron, Sylatron (peginterferon alfa-2b) | |
| G-CSF | Chemotherapy-induced neutropenia | Approved | Neulasta (pegfilgrastim) | ||
| IL-29 (IFN-λ1) | Chronic hepatitis C | Phase I | NCT00565539 | ||
| Fusion to albumin | |||||
| IFN-α2b | Extended half-life | Chronic hepatitis C | Phase III | Albinterferon Alfa-2b | |
| Fusion to toxins | |||||
| IL-2-DT | Targeted cytotoxicity | Cutaneous T-cell lymphoma | Approved | Ontak (denileukin diftitox) | |
| IL-13-PE | Glioblastoma multiform, malignant glioma | Phase I, III | Cintredekin besudotox—NCT00880061, NCT00076986 | ||
| Fusion to Abs (Immunocytokines) | |||||
| F16-IL2 | Targets A1 domain of tenascin C | Breast cancer, solid tumours | Phase II | NCT01134250 | |
| L19-12 | Targets fibronectin EDB splice variant | Metastatic melanoma, renal cell carcinoma, pancreatic cancer and solid tumours | Phase I, II | NCT01058538, NCT01198522, NCT01253096, NCT01055522 | |
| hu14.18-IL2 | Targets GD2 disialoganglioside | Metastatic melanoma, neuroblastoma | Phase II | NCT00590824, NCT00082758, NCT00109863 | |
| KS-IL2 | Targets EpCAM | Various cancers | Phase I | Tucotuzumab celmoleukin - NCT00132522 | |
| NHS-IL2LT | Targets DNA/ histones in necroses | Lung cancer, non-Hodgkin lymphoma | Phase I | Selectikine - NCT00879866, NCT01032681 | |
| NHS-IL12 | Targets DNA/ histones in necroses | Epithelial and mesenchymal solid tumours | Phase I | NCT01417546 | |
| BC1-IL12 | Targets fibronectin EDB splice variant | Metastatic melanoma and renal cell carcinoma | Phase I | NCT00625768 | |
| L19-TNFα | Targets fibronectin EDB splice variant | Solid tumours, colorectal cancer, melanoma of the lower limb | Phase I, II | NCT01253837, NCT01213732 | |
| F8-IL10 | Targets fibronectin EDA splice variant | Rheumatoid arthritis | Phase I | 2008-008729-31 (PH-F8IL10-02/08) | |
| Mutagenesis | |||||
| IL-2 | Mutation of free-cysteine to reduce aggregation | Metastatic melanoma and renal cell carcinoma | Approved | Proleukin (aldesleukin) | |
| IL-4 | Disruption of receptor signalling-subunit binding to generate antagonist | Asthma and atopic eczema | Phase II | Aerovant (pitrakinra) - NCT00801853, NCT00535431, NCT00676884 | |
DT = diphtheria toxin, PE = pseudomonas exotoxin, EpCAM = epithelial cell adhesion molecule.
3. Cytokine PEGylation
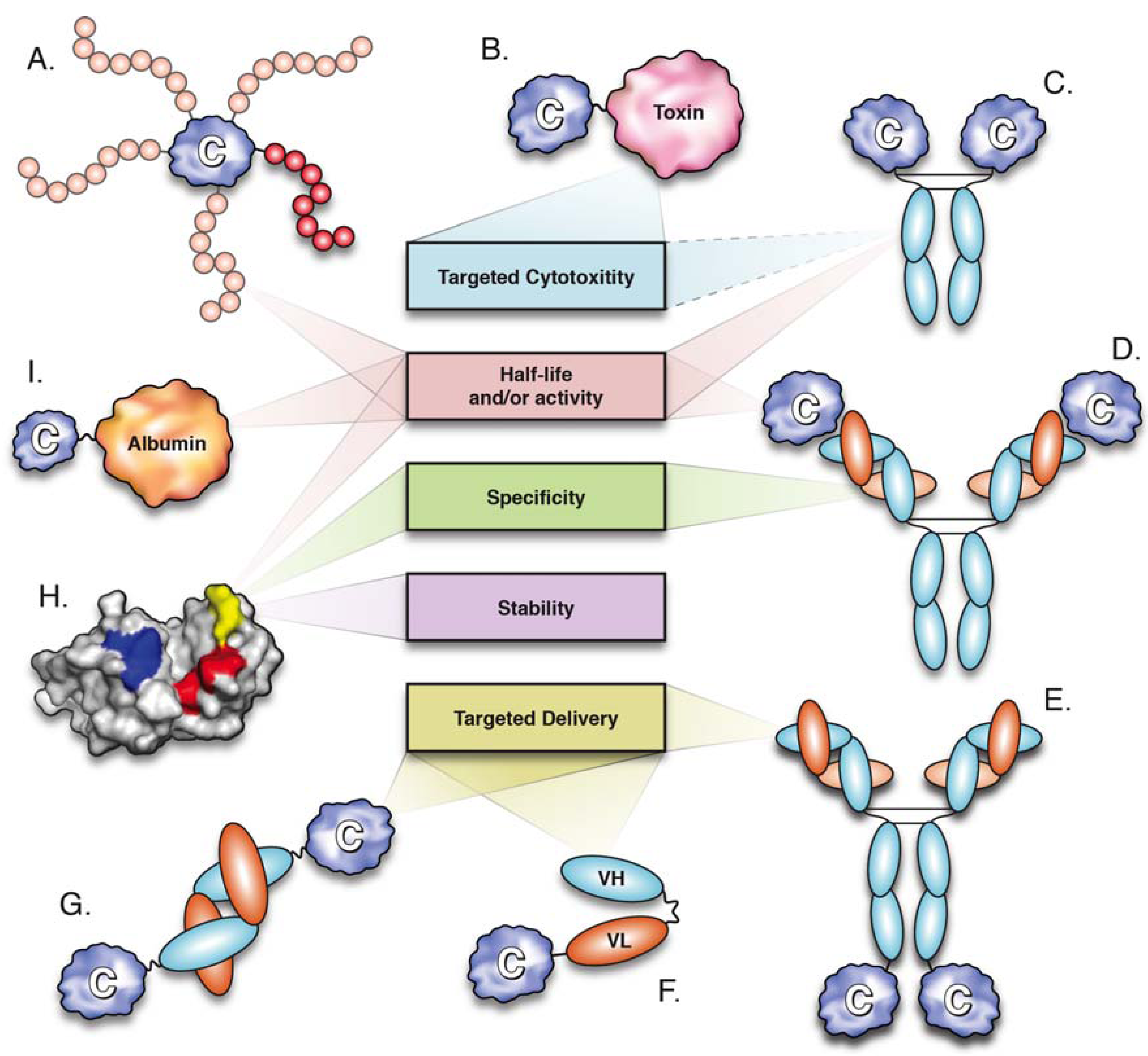
One of the earliest strategies used for half-life extension of therapeutic cytokines was PEGylation, in which the cytokine is conjugated to polyethylene glycol (PEG) chains (5–40 kDa, Figure 1). PEGylation can be achieved chemically or enzymatically, with biophysical and biochemical properties of PEGylated proteins depending on structure, size, number and location of the PEG chains [17]. PEGylation prolongs the circulation half-life of proteins by masking proteolytic cleavage sites and, more importantly, by increasing their hydrodynamic radii, thereby effectively reducing renal clearance [18,19].
Figure 1.
Molecular engineering strategies utilized for cytokine optimization and their major effects. (A) PEGylation (positional isomers displayed); (B) cytokine-toxin fusion; (C) cytokine-Fc fusion; (D) antibody-cytokine immune complex; (E–G) immunocytokines; (E) cytokine-IgG; (F) cytokine-scFv; cytokine-diabody; (H) cytokine mutagenesis; (I) cytokine-albumin fusion.
Figure 1.
Molecular engineering strategies utilized for cytokine optimization and their major effects. (A) PEGylation (positional isomers displayed); (B) cytokine-toxin fusion; (C) cytokine-Fc fusion; (D) antibody-cytokine immune complex; (E–G) immunocytokines; (E) cytokine-IgG; (F) cytokine-scFv; cytokine-diabody; (H) cytokine mutagenesis; (I) cytokine-albumin fusion.
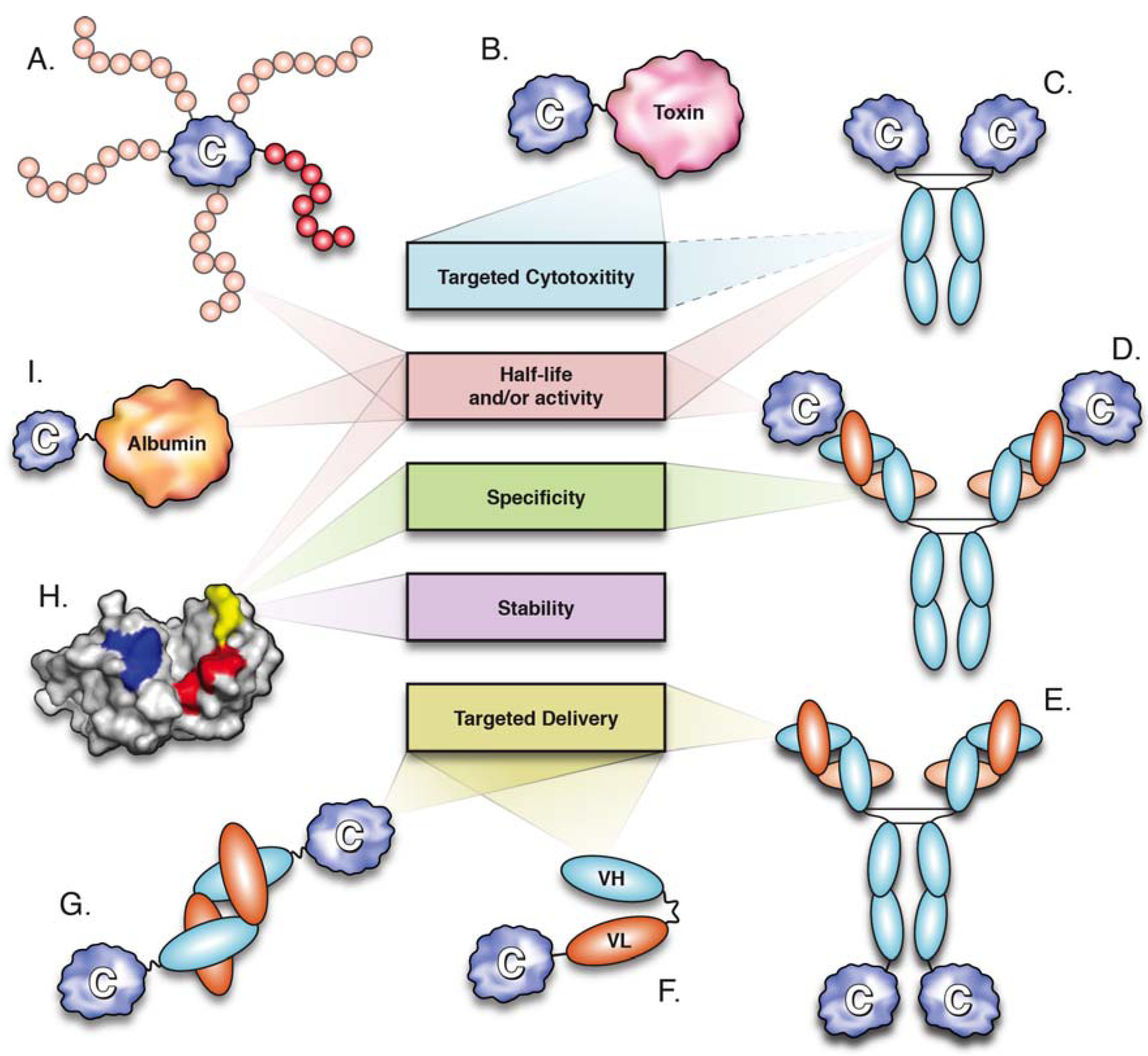
Three PEGylated cytokines have been approved for clinical use: PEG-G-CSF, PEG-IFN-α2a and PEG-IFN-α2b (Table 2). Granulocyte colony stimulating factor (G-CSF) is a 19.6 kDa monomeric cytokine produced by monocytes and fibroblasts that functions to stimulate the growth and differentiation of neutrophils. Recombinant human G-CSF produced in E. coli (filgrastim) is used for the treatment of neutropenia, a pathological reduction in neutrophil numbers often observed in patients receiving myelosuppressive chemotherapy. Daily doses of filgrastim over the course of a chemotherapy cycle are required to prevent neutropenia, mainly because its short serum-half life (t1/2 = 3.5 h) only allows for transient therapeutic action. By significantly increasing serum half-life (t1/2 = 42 h), a PEGylated form of G-CSF (pegfilgrastim) has been used to mediate an equivalent therapeutic effect with only a single dose per chemotherapy cycle [20]. Similarly to pegfilgrastim, PEGylated forms of IFN-α-2a (Pegasys®, branched 40 kDa PEG) and IFN-α-2b (Pegintron®, linear 12 kDa PEG) show approximately 10-fold increases in circulation half-life, correlated with improved therapeutic efficacy for the treatment of chronic hepatitis C [21,22].
Although PEGylation is an effective method for half-life extension, toxicity of PEGylated cytokines remains largely unchanged [20,22,23]. While an overall benefit is observed in therapies that use cytokines at relatively low doses (e.g., G-CSF, IFN-α), PEGylation may be detrimental when high doses are required (e.g., HD IL-2), as increased half-life could prevent rapid reversal of toxicity [24,25].
4. Antibody-IL-2 Immune Complexes
Neutralizing monoclonal antibodies (mAbs) directed against cytokines are commonly used for blocking interactions between cytokines and their receptors. However, several studies in the early 1990s showed dramatic increases in the biological activity of a number of cytokines when mice were administered with a mixture containing cytokine/anti-cytokine mAb at a 2:1 molar ratio [14,26,27,28,29]. These observations suggest that some anti-cytokine mAbs act as cytokine carriers in vivo, identifying a potential use of antibody-cytokine immune complexes in therapeutic applications (Figure 1). Subsequent studies have extensively characterized IL-2/anti-IL-2 mAb complexes, demonstrating that these complexes not only display enhanced biological activity, but also modulate the pleiotropic effects of IL-2 [30].
IL-2 is a member of the common γ chain (γc) family, which comprises cytokines that share the IL-2 receptor γc for signalling (IL-2, -4, -7, -9, -15 and -21). Structurally, IL-2 is classified as a four α-helix bundle cytokine together with several other interleukins, interferons and colony stimulating factors [31].
IL-2 is a major modulator of the immune system capable of stimulating cells bearing either dimeric or trimeric IL-2 receptors (IL-2Rs). The low-affinity dimeric IL-2R (KD = 1 nM), composed of IL-2Rβ (CD122) and γc subunits, is found in CD122high populations, such as memory phenotype (MP) CD8+ T-cells and NK cells. In contrast, the high-affinity (KD = 10 pM) trimeric IL-2R consisting of IL-2Rα (CD25), CD122 and γc subunits is found in CD25high populations, such as activated T-cells, activated B-cells and regulatory T-cells (Tregs). Notably, signal transduction in both types of IL-2Rs is mediated by CD122 and γc, while the CD25 chain acts to confer high-affinity binding of IL-2 to trimeric IL-2Rs [32].
IL-2/mAb complexes formed using different mAbs have been shown to selectively stimulate cells depending on the type of IL-2R displayed. IL-2/mAbCD122 complexes mediate preferential expansion of CD122high cytotoxic CD8+ T-cells and NK cells, resulting in the potent inhibition of tumour growth in various mouse models of cancer [33,34,35]. By contrast, IL-2/mAbCD25 complexes selectively expand immunosuppressive CD25high Tregs [14], resulting in prevention of allogeneic pancreatic islet rejection [36], autoimmune disease [36,37,38] and the reduction of inflammation [39,40].
Further to modulating IL-2 activity, IL-2/mAb complexes can also prolong the biological effects of IL-2. While reduced renal clearance is suggested to play a major role in the increased half-life observed in IL-2 complexes, additional mechanisms are thought to contribute to this feature [14]. These mechanisms include reduced receptor-mediated endocytosis (for IL-2/mAbCD122 complexes) and indirect recycling of IL-2 by the neonatal Fc receptor (for IL-2/mAbCD25 complexes), which rescues IgG antibodies from cellular metabolism [41,42].
Based on the enhanced properties of IL-2/mAb complexes in animal models, the use of mAbs as cytokine carriers has emerged as a potential strategy for improving pharmacodynamics and pharmacokinetics of therapeutic cytokines. However, the therapeutic value of this approach is yet to be tested in clinical trials.
5. Cytokine Fusion Proteins
Genetic fusion to other proteins is a useful strategy for improving or modifying the biophysical properties of cytokine therapeutics (Figure 1). A large body of work has been directed towards the development of cytokine fusion proteins that display extended half-life (e.g., fusion to Fc, albumin or transferrin), increased activity (e.g., fusion to cytokine agonists), enhanced cytotoxicity (e.g., fusion to bacterial toxins or lytic Fc) and localized delivery (e.g., fusion to antibodies or antibody fragments), with several of them currently undergoing clinical trials (Table 2).
5.1. Fusion of Cytokines to the Fc Domain
Several advantages are introduced by fusing the Fc domain of IgG antibodies to therapeutic proteins (Figure 1). These include increased avidity of the fused partner, cost-effective purification via protein G/A affinity chromatography and, more importantly, prolonged circulation half-life [43,44,45,46,47]. Notably, half-life extension of Fc fusion proteins results from a combination of reduced renal clearance (due to increased molecular size) and FcRn-mediated recycling, where Fc binds FcRn at acidic pH in endosomes and is then released at physiological pH to the plasma [41]. Moreover, expression of FcRn in lung epithelium has been shown to mediate pulmonary uptake of Fc fusion proteins, thereby providing an alternative route of administration particularly relevant to the treatment of respiratory conditions [45,46].
In addition, effector functions mediated by Fc can significantly alter the biological activity of Fc fusion proteins. These functions include antibody dependent cell-mediated cytotoxicity (ADCC), which occurs through binding of the Fc domain to Fc receptors (FcR) on immune cells, as well as complement-mediated cytotoxicity (CDC), induced by binding to C1q [48]. Notably, Fc effector functions can be modulated through mutagenesis, modification of glycosylation status and choice of Fc subclass [44,48,49]. For instance, etanercept (Enbrel®), a TNFRII-Fc fusion protein approved for the treatment of rheumatoid arthritis and other inflammatory conditions, functions through both blocking the action of the pro-inflammatory cytokine TNF-α and inducing ADCC of TNF-α expressing cells [49]. By contrast, the Fc domain of abatacept (Orencia®), an inhibitor of T-cell co-stimulation, contains mutations that prevent activation of ADCC and CDC [50]. In addition, an engineered Fc domain with reduced effector functions has been utilized for the generation of an IL-21R-Fc fusion protein, which antagonizes the action of the pro-inflammatory cytokine IL-21 and shows therapeutic potential for the prevention of allograft rejection and the treatment of inflammatory and autoimmune conditions [51,52,53].
While Fc fusion technology has been successfully implemented for the blockade of cytokine action via cytokine receptor-Fc fusions, direct fusion of Fc to cytokines is a useful strategy for enhancing cytokine activity. For instance, fusion of Fc to interleukin-10 (IL-10) has been shown to significantly prolong the biological activity of IL-10 in mice [54,55,56,57]. IL-10 is an anti-inflammatory and immunosuppressive homodimeric cytokine mainly produced by monocytes, but also by B-cells, NK cells and a number of T-cell subsets. Treatment with murine IL-10-Fc containing a modified, non-lytic Fc domain (i.e., abrogated ADCC and CDC) resulted in extended protection against endotoxin-induced septic shock [57] and in prevention of spontaneous autoimmune diabetes in non-obese diabetic (NOD) mice [56]. While possessing the same biological functions of unfused IL-10, the prolonged serum half-life of IL-10-Fc resulted in superior therapeutic activity [56]. Based on these animal models, similar approaches could be attempted to enhance human IL-10 activity, since this cytokine displayed insufficient therapeutic efficacy in clinical trials of rheumatoid arthritis [58]. Other developed cytokine-Fc fusions displaying enhanced biological activity in mice include a cytolytic IL-2-Fc, which targets destruction of activated T-cells in autoimmune conditions [59] and a nonlytic IL-7-Fc with potential as a vaccine adjuvant for induction of T-cell mediated immune responses [60]. However, whether analogous fusion proteins have the potential of promoting comparable biological effects in humans remains unclear.
5.2. Fusion to Serum Albumin
Similarly to IgG antibodies, serum albumin (SA) displays an unusually long circulation half-life (~3 weeks in humans). Interestingly, half-life prolongation of these functionally and structurally unrelated proteins results primarily from interaction with FcRn. Although SA binds FcRn at a different site than IgG, both interactions are pH-dependent and result in FcRn-mediated rescue from cellular catabolism [61]. Due to its high potential for half-life prolongation, the use of SA as a carrier protein has been implemented in a number of therapeutics [13]. Some of the strategies utilized for this purpose include genetic fusion to SA, conjugation to SA-binding moieties and fusion to SA-binding antibodies or antibody fragments [13]. Notably, cytokines that have been genetically fused to SA include IL-2 [62], G-CSF [63], IFN-α [64,65,66] and IFN-β [67].
Albinterferon-α2b (albIFN), a genetic fusion of IFN-α2b and SA, is a prominent example of cytokine pharmacokinetic enhancement through combination with albumin (Figure 1 and Table 2). Phase III clinical trials demonstrated that the therapeutic efficacy of albIFN was superior to that of IFN-α2b and not inferior to PEGylated IFN-α2b (PEGintron®) in mediating sustained virologic responses in chronic hepatitis C patients [64,66]. Furthermore, fusion to SA extended the elimination half-life of IFN-α2b to ~200 h, thus allowing administration of albIFN in two- or four-week dose intervals, as opposed to weekly (PEGintron, t1/2 ~40 h) or tri-weekly dosing regimes (IFN-α2b, t1/2 ~4 h) [21,65].
Recently, cytokines have been fused to domain antibodies (dAbs) that bind SA (Figure 1). Often referred to as nanobodies, dAbs are single heavy (VH) or light (VL) chain variable domains, which are only 11–13 kDa in size and capable of binding antigen [68]. Albumin-binding dAbs (AlbudAbsTM) have been used to generate genetic fusions to IFN-α2b and IL-1 receptor antagonist (IL-1Ra), a naturally occurring anti-inflammatory cytokine that blocks IL-1 signalling (Anakinra®, approved for treatment of RA) (Table 1). The resulting fusion proteins displayed a marked increase in half-life, which correlated with superior efficacy [69,70]. IL-1Ra-AlbudAb mediated enhanced reduction of arthritis scores in a mouse model of collagen-induced arthritis, when compared to IL1-Ra [69], while IFN-α2b-AlbudAb showed superior activity than that of an IFN-α2b-SA fusion protein, as measured by in vitro anti-viral activity and mediation of tumour regression in a mouse model of melanoma [70].
5.3. Fusion to Human Transferrin
Genetic fusion to serum transferrin (Tf) represents another strategy for cytokine optimization primarily resulting in enhanced pharmacokinetics. Serum transferrin is a 80 kDa glycoprotein that mediates iron transport from systemic circulation into cells and tissues [71]. When bound to ferric ions, transferrin displays high affinity for the transferrin receptors (TfRs) displayed on the surface of most cell types [71]. Upon interaction, the Tf/TfR complex is internalized via receptor-mediated endocytosis into endosomes, where iron is released and Tf/TfR is then recycled to the cell surface [71,72]. Fusion of protein therapeutics to Tf or TfR-binding antibodies has been utilized for a number of applications, including half-life extension, targeting of malignant cells overexpressing TfRs and targeting of the brain capillary endothelium for transport of therapeutics across the brain blood barrier [47,72,73,74]. Moreover, expression of TfRs in the gastrointestinal epithelium provides potential for oral delivery of Tf-associated drugs [75].
In a study from Bai et al. in 2005 [76], subcutaneous administration of a G-CSF-Tf fusion protein to BDF1 mice resulted in comparable biological activity to unfused G-CSF (filgrastim), as measured by increase in absolute neutrophil counts (ANC). Given that the G-CSF-Tf fusion only displayed one tenth of the activity of filgrastim in vitro, half-life prolongation was suggested to elevate in vivo biological activity to match that of filgrastim. Notably, oral administration of G-CSF-Tf resulted in significant increases in ANC, while ANC after oral administration of unfused G-CSF remained at baseline levels. More recently, introduction of a helical linker between G-CSF and Tf resulted in further improvements in biological activity [77].
5.4. Fusion to Agonistic Receptors
While most soluble cytokine receptors act as natural cytokine antagonists, some of them can potentiate cytokine activity [78]. One such receptor is the IL-15 receptor α sub-unit, which has been shown to augment the biological activity of IL-15 [79]. IL-15 is a 15 kDa monomeric common γc cytokine that displays many overlapping functions with IL-2, as they share γc and IL-2Rβ for signalling. Notably, IL-15 does not share IL-2Rα with IL-2, but instead has its own high-affinity (KD = 100 pM) receptor α chain, IL-15Rα. The dominant mechanism of IL-15 action in vivo appears to occur via IL-15Rα-mediated trans-presentation of IL-15 from antigen presenting cells to neighbouring cells bearing the dimeric IL-2Rβγ [80]. Based on its immunostimulatory properties, the use of IL-15 is being explored in clinical trials of melanoma and renal cancer (Table 1).
Administration of soluble IL-15 in mouse models stimulates the expansion of MP CD8+ T-cells and NK cells, which express high levels of IL-2Rβγ [81]. Interestingly, this effect was significantly enhanced when IL-15 was administered in combination with a soluble IL-15Rα-Fc [79]. In agreement with this observation, a fusion protein composed of IL-15 and a functional domain (Sushi domain) of IL-15Rα showed significant increases in IL-15 activity [82]. The conversion of IL-15 into a superagonist following IL-15α binding appears to result from conformational changes that increase the interaction with IL-2Rβγ [79].
5.6. Fusion to Toxins
A number of cytokines have been fused to bacterial toxins in order to promote targeted cytotoxicity of specific cell subsets (Figure 1) [86,87]. Ontak® (denileukin diftitox) is a fusion protein composed of IL-2 and the enzymatically active portion of diphtheria toxin (DT) approved for the treatment of cutaneous T-cell lymphoma (CTCL) (Table 2) [88]. Malignant cells in CTCL lesions overexpress IL-2Rβγ and IL-2Rαβγ, with greater denileukin diftitox sensitivity observed in cells displaying the IL-2R heterotrimer [89]. The IL-2 portion of denileukin diftitox directs DT to cells bearing IL-2Rs, ultimately resulting in cell death through inhibition of protein synthesis [90].
Partial and complete responses are observed in approximately 20% and 10% of CTCL patients after denileukin diftitox treatment, respectively [91]. Nevertheless, treatment-associated toxicities are common and include the development of VLS (in ~26% of patients [91]) and, in rare cases, visual loss [92]. Although the mechanisms mediating the latter effect are unknown, a direct effect on Tregs—which express high levels of IL-2Rαβγ—has been proposed to promote autoimmune retinopathy and subsequent visual impairment [93,94].
Other cytokines that have been fused to toxins for targeted cell cytotoxicity include the functionally related cytokines, IL-4 and IL-13. Fusion proteins composed of either IL-4 or IL-13 and a truncated form of Pseudomonas exotoxin (PE) were developed to target the elimination of malignant cells within glioblastoma multiforme, a type of solid brain tumour that overexpresses IL-4 and IL-13 receptors (Table 2) [87]. Notably, IL-13-PE (cintredekin besudotox) reached phase III clinical trials, where it mediated increased progression-free survival in patients receiving drug infusions within resected tumour regions, when compared to patients receiving Gliadel® (carmustine) wafers after tumour resection [95]. However, no overall survival advantage was achieved with cintredekin besudotox treatment, which may have reflected sub-optimal drug delivery [95,96].
5.7. Fusion to Antibodies and Antibody Fragments (Immunocytokines)
The generation of cytokine-antibody fusion proteins is an elegant approach for engineering targeted cytokine activity (Figure 1). The rationale for the development of such fusion proteins, also known as immunocytokines, derives from the superior efficacy and reduced toxicity of localized over systemic administration of therapeutic cytokines [97,98,99]. Since physical delivery of cytokines is not feasible in many instances, immunocytokines provide an alternative avenue for achieving localized cytokine action. As such, immunocytokines are designed to target disease antigens through their antibody moieties in order to induce (or potentiate) effector functions through their cytokine components. A number of immunocytokines are currently undergoing clinical trials (Table 2), with many more being evaluated in pre-clinical studies. The vast number of engineered immunocytokines and their therapeutic potential has been recently reviewed in previous issues [98,100] and elsewhere [47,101,102]. A brief summary of the major factors determining the therapeutic activity of specific immunocytokines, namely, commonly targeted antigens, antibody formats and employed cytokines, is presented below.
5.7.1. Target Antigens
Antibodies (or antigen-binding antibody fragments) mediate the targeted delivery of immunocytokines into disease environments and/or to specific cell subsets. Ideal target antigens should be, therefore, overexpressed in diseased tissues, while remaining at low levels elsewhere. Accordingly, a number of immunocytokines incorporate antigen-binding moieties that target antigens overexpressed on the surface of malignant cells (e.g., epithelial cell adhesion molecule [103,104], GD2 disialoganglioside [105,106,107], HER2/neu [108,109], CD20 [110,111] and CD30 [112]), as well as targeting of neoangiogenic antigens found in tumours and chronic inflammation sites (e.g., fibronectin splice variants EDA/EDB and A1 domain of tenascin C [113,114,115]). Furthermore, targeting of DNA/histone complexes exposed in necrotic tissue provides an additional strategy for directing immunocytokines into tumours and metastases [116,117].
5.7.2. Antibody Format
A large number of genetically engineered antibodies and antibody fragments have been utilized for the generation of immunocytokines (see [47,98,101,102]). Different antibody formats display variations in molecular size, affinity and avidity, which mainly affect pharmacokinetic aspects, such as serum half-life and biodistribution [118]. Antibody formats that have been assessed in clinical trials of immunocytokines include full-length IgG, single-chain Fv (scFv) and diabodies (Figure 1) [98,101]. Fusion to full-length IgG (~150 kDa) provides bivalent antigen binding and prolonged serum half-life, due to reduced renal clearance and FcRn-mediated recycling [13,114]. Furthermore, effector functions mediated through the Fc domain of IgG (ADCC and CDC) can be maintained in order to potentiate targeted cell cytotoxicity [119]. However, IgG-based immunocytokines display slow and non-homogeneous tissue penetration, particularly when targeting solid tumours [120]. Tumour penetration can be enhanced by utilization of the smaller scFv (~30 kDa) fragments, which are composed of single VL and VH domains joined by a ~15 residue flexible polypeptide linker [121]. Nevertheless, the monovalent nature of scFv fragments is correlated with poor tumour retention, while their reduced molecular size results in rapid clearance from blood circulation [122]. Generation of scFv fragments with shorter linkers (~5 residues) prevents VH and VL from the same polypeptide chain from interacting and promotes the formation of bivalent scFv homodimers, also known as diabodies (~55 kDa). Similarly to scFv, diabodies display rapid tissue penetration, while displaying enhanced tumour retention and circulation half-lives [123].
Engineering of novel antibody formats is likely to continue expanding the repertoire of immunocytokines with optimized disease-targeting properties. A recent advance illustrating this trend was the generation of an IFN-α-based immunocytokine displaying multivalent and dual antigen binding, with therapeutic potential for the treatment of non-Hodgkin lymphoma [124,125].
5.7.3. Cytokine Partners
A number of cytokines, including IL-2, IL-7, IL-12, IL-15, GM-CSF, IFN-α, IFN-γ and members of the TNF-superfamily, have been utilized for the development of immunocytokines with potential for cancer immunotherapy [47,101]. While targeted delivery of most of these cytokines into cancerous lesions aims for the recruitment and activation of cytotoxic immune cells, such as T-cells and NK cells; some others also promote anti-proliferative activities (IFN-α) [111] or induce cell death upon binding to their cognate receptors (TNF-superfamily members) [126]. By contrast, the anti-inflammatory cytokine IL-10, which suppresses the expression of inflammatory mediators by immune cells, has been utilized for the development of immunocytokines targeting the neo-vasculature of chronic inflammation sites [58,127].
6. Cytokine Mutagenesis
Mutagenesis can be employed to engineer cytokines with enhanced stability, half-life, specificity and activity (Figure 1). In early studies, scanning and deletion mutagenesis allowed the development of cytokines with modified activities [128,129,130,131,132]. These approaches have gradually been replaced by protein engineering techniques, such as rational design, computational modelling and directed evolution, which allow for more efficient cytokine optimization.
6.1. Stability
Most protein therapeutics suffer from sub-optimal stability and cytokines are no exception [133]. Poor stability can result in high levels of protein unfolding, degradation and aggregation, which can lead to many issues affecting formulation, shelf life, immunogenicity and therapeutic efficacy [134,135,136]. A simple strategy for improving cytokine stability is the mutation of free cysteines to serines, thereby preventing undesired disulphide bonds that could lead to protein aggregation. This approach has been applied to a number of cytokines, including the commercially available form of recombinant human IL-2, Proleukin® (Table 2) [134,137,138,139]. Cytokine stability can also be enhanced by the introduction of stabilizing mutations within α-helices. This strategy is particularly relevant to the 4-helix bundle cytokines and requires the mutation of residues with low α-helical propensity (e.g., proline, glycine) to residues with high α-helical propensity (e.g., alanine). Using this approach, the introduction of double and triple glycine-to-alanine substitutions resulted in the generation of a number of G-CSF variants displaying significantly enhanced resistance to chemical denaturation [140]. Furthermore, a computational method developed to identify optimal sequences for a defined three-dimensional structure was used to engineer a G-CSF variant with 10 core amino acid substitutions that displayed decreased aggregation and increased thermal stability [141].
6.2. Half-Life Extension
Degradation via receptor-mediated endocytosis plays a major role in the clearance of therapeutic cytokines [13,142]. Evidence suggests that after internalization, cytokines that remain bound to their receptors in endosomal compartments (pH ~ 5–6) are destined to lysosomal degradation, while those that are released from their receptors are recycled back to the cell surface [143,144,145]. Based on this premise, rationally designed G-CSF variants with reduced affinity for G-CSF receptor at endosomal pH, but unaffected binding to the receptor at physiological pH, showed increased cytokine recycling correlated with prolonged half-life and enhanced potency in vitro [146]. This pH- specific reduction in affinity was achieved by mutating a number of residues involved in receptor binding to histidine, a process known as “histidine switching”. Since histidine (pKa = 6.5) has a net positive charge at endosomal pH, but is neutral at physiological pH, these substitutions exclusively targeted disruption of receptor binding in endosomes, thereby promoting G-CSF recycling.
6.3. Modulation of Specificity
Modulating the specificity and activity of pleiotropic cytokines is necessary for increasing therapeutic efficacy and preventing the development of adverse effects. Accordingly, a number of cytokines have been engineered through rational design and directed evolution in order to mediate selective stimulation of specific cell subsets.
Generated by yeast display, variants of IL-2 with greater than 100-fold increases in affinity to IL-2Rα were shown to mediate enhanced expansion of T-cells in vitro [147,148]. Further modification of these variants to disrupt binding to IL-2Rβ and IL2Rγ resulted in the generation of signalling-deficient analogues that act as competitive antagonists of IL-2 in cells displaying IL-2Rαβγ. The therapeutic application of such IL-2 mutants is mainly directed towards the inhibition of immunosuppressive Tregs in malignant conditions [149].
Recently, an IL-2 variant displaying ~250-fold increased affinity to IL-2Rβ was developed through in vitro evolution using yeast display [150]. Interestingly, most of the five mutations resided in the core of IL-2 and resulted in a conformational change similar to the one induced after IL-2Rα binding. This mutant, also known as super-2, displayed increased ability to stimulate cells displaying IL-2Rβγ, when compared to unmodified IL-2. Accordingly, super-2 mediated more potent expansion of MP CD8+ T-cells and NK cells, which correlated with increased anti-tumour activity in mice. By shifting the balance of IL-2 stimulation towards cells displaying IL-2Rβγ, super-2 is predicted to mediate tumour regression with a reduced incidence of treatment-associated adverse effects [150].
Modulation of the activity of another common γc cytokine, IL-4, was achieved by using a combination of rational design and directed evolution [151]. IL-4 is a pleiotropic cytokine that can signal through either type I or type II (also responsive to IL-13) IL-4Rs. Importantly, regulatory functions of IL-4 are mostly mediated by cells displaying type I IL-4Rs (e.g., B-cells and T-cells) and drive the development of humoral immune responses (TH2 responses). In contrast, effector functions of IL-4 are largely mediated by non-hematopoietic cells expressing the type II IL-4Rs and promote the development of allergic inflammation [152].
Interestingly, both types of IL-4Rs display the high affinity (KD = 100 pM) IL-4Rα subunit, differing only in the signalling subunit they incorporate, namely γc in type I and IL-13Rα in type II IL-4Rs. Increasing the affinity of IL-4 for γc or IL-13Rα by mutagenesis produced a number of IL-4 variants that mediated selective in vitro expansion of cells bearing either type I or type II IL-4Rs, respectively [151]. In particular, IL-4 variants with enhanced selectivity for cell subsets displaying type I receptors show therapeutic potential for immune stimulation with reduced effector functions.
6.4. Development of Cytokine Antagonists
Disruption of cytokine binding to signalling receptor sub-units is a useful approach for the development of antagonists with therapeutic potential. An IL-4 variant (pitrakinra) that was rationally designed to have reduced binding to γc and IL-13Rα, but unimpaired binding to IL-4Rα, is currently undergoing phase II clinical trials for the treatment of allergic asthma and atopic eczema (Table 2) [153,154,155]. Pitrakinra effectively targets cells bearing type I and II IL-4Rs without inducing signalling, thereby acting as a dual competitive inhibitor of IL-4 and IL-13 [156]. Similarly, by mutating residues in IL-6 to disrupt binding to gp130, the signalling sub-unit of IL-6R, but not affecting the binding to the IL-6Rα chain, resulted in the development of an IL-6R antagonist with therapeutic potential for the treatment of multiple myeloma and lung fibrosis [157,158,159,160].
7. Concluding Remarks
The immunomodulatory properties of cytokines provide exceptional potential for the treatment of several conditions. However, sub-optimal pharmacokinetics and toxicity have limited their therapeutic potential, as reflected by the reduced number of cytokines currently approved for clinical use. The therapeutic shortcomings displayed by most cytokines have motivated the optimization of these proteins through a number of molecular engineering strategies, including chemical conjugation (e.g., PEGylation), immunocomplexing, development of cytokine fusion proteins and mutagenesis. These strategies have been mainly directed towards the generation of cytokines with prolonged half-life, enhanced specificity/activity and localized action, but also to take advantage of the targeting properties of some cytokines (e.g., cytokine-toxin fusion proteins) and for the generation of cytokine antagonists.
The large number of engineered cytokines currently undergoing pre-clinical and clinical evaluation are likely to constitute the next generation of cytokine therapeutics displaying increased efficacy and reduced incidence of adverse effects. As research continues in this field, we anticipate that optimally designed cytokines will be developed utilizing a combination of molecular engineering strategies in order to simultaneously enhance multiple pharmacokinetic and pharmacodynamic parameters.
Acknowledgements
This work was supported by the Australian National Health and Medical Research Council. We thank C. Loetsch and P. Schofield for proofreading and critique of the manuscript.
Conflict of Interest
The authors declare no conflict of interest.
References
- Dinarello, C.A. IL-1: Discoveries, controversies and future directions. Eur. J. Immunol. 2010, 40, 599–606. [Google Scholar] [CrossRef]
- Beeson, P.B. Temperature-elevating effect of a substance obtained from polymorphonuclear leucocytes. J. Clin. Invest. 1948, 27, 524–524. [Google Scholar]
- Morgan, D.A.; Ruscetti, F.W.; Gallo, R. Selective in vitro growth of T-lymphocytes from normal human bone marrows. Science 1976, 193, 1007–1008. [Google Scholar]
- Gillis, S.; Smith, K.A. Long-term culture of tumor-specific cytotoxic T-cells. Nature 1977, 268, 154–156. [Google Scholar] [CrossRef]
- Gordon, J.; Maclean, L.D. A lymphocyte-stimulating factor produced in vitro. Nature 1965, 208, 795–796. [Google Scholar] [CrossRef]
- Isaacs, A.; Lindenmann, J. Virus interference. 1. The interferon. Proc. Royal Soc. B 1957, 147, 258–267. [Google Scholar] [CrossRef]
- Pestka, S. The interferons: 50 years after their discovery, there is much more to learn. J. Biol. Chem. 2007, 282, 20047–20051. [Google Scholar] [CrossRef]
- Cantell, K.; Hirvonen, S.; Kauppinen, H.L.; Myllyla, G. Production of interferon in human leukocytes from normal donors with the use of Sendai virus. Methods Enzymol. 1981, 78, 29–38. [Google Scholar] [CrossRef]
- Goeddel, D.V.; Yelverton, E.; Ullrich, A.; Heyneker, H.L.; Miozzari, G.; Holmes, W.; Seeburg, P.H.; Dull, T.; May, L.; Stebbing, N.; et al. Human-leukocyte interferon produced by Escherichia-coli is biologically-active. Nature 1980, 287, 411–416. [Google Scholar] [CrossRef]
- Nagata, S.; Taira, H.; Hall, A.; Johnsrud, L.; Streuli, M.; Ecsodi, J.; Boll, W.; Cantell, K.; Weissmann, C. Synthesis in Escherichia-coli of a polypeptide with human-leukocyte interferon activity. Nature 1980, 284, 316–320. [Google Scholar] [CrossRef]
- Devos, R.; Plaetinck, G.; Cheroutre, H.; Simons, G.; Degrave, W.; Tavernier, J.; Remaut, E.; Fiers, W. Molecular-cloning of human interleukin-2 carrier DNA and its expression in Escherichia-coli. Nucleic Acids Res. 1983, 11, 4307–4323. [Google Scholar] [CrossRef]
- Taniguchi, T.; Matsui, H.; Fujita, T.; Takaoka, C.; Kashima, N.; Yoshimoto, R.; Hamuro, J. Structure and expression of a cloned cDNA for human interleukin-2. Nature 1983, 302, 305–310. [Google Scholar] [CrossRef]
- Kontermann, R.E. Strategies for extended serum half-life of protein therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876. [Google Scholar] [CrossRef]
- Boyman, O.; Surh, C.D.; Sprent, J. Potential use of IL-2/anti-IL-2 antibody immune complexes for the treatment of cancer and autoimmune disease. Expert Opin. Biol. Ther. 2006, 6, 1323–1331. [Google Scholar] [CrossRef]
- Atkins, M.B.; Lotze, M.T.; Dutcher, J.P.; Fisher, R.I.; Weiss, G.; Margolin, K.; Abrams, J.; Sznol, M.; Parkinson, D.; Hawkins, M.; et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 1999, 17, 2105–2116. [Google Scholar]
- Rosenberg, S.A.; Yang, J.C.; White, D.E.; Steinberg, S.M. Durability of complete responses in patients with metastatic cancer treated with high-dose interleukin-2—Identification of the antigens mediating response. Ann. Surg. 1998, 228, 307–317. [Google Scholar] [CrossRef]
- Jevševar, S.; Kunstelj, M.; Porekar, V.G. PEGylation of therapeutic proteins. Biotechnol. J. 2010, 5, 113–128. [Google Scholar]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef]
- Maack, T.; Johnson, V.; Kau, S.T.; Figueiredo, J.; Sigulem, D. Renal filtration, transport, and metabolism of low-molecular-weight proteins - Review. Kidney Int. 1979, 16, 251–270. [Google Scholar] [CrossRef]
- Crawford, J. Once-per-cycle pegfilgrastim (Neulasta) for the management of chemotherapy-induced neutropenia. Semin. Oncol. 2003, 30, 24–30. [Google Scholar] [CrossRef]
- Bukowski, R.M.; Tendler, C.; Cutler, D.; Rose, E.; Laughlin, M.M.; Statkevich, P. Treating cancer with PEG intron—Pharmacokinetic profile and dosing guidelines for an improved interferon-alpha-2b formulation. Cancer 2002, 95, 389–396. [Google Scholar] [CrossRef]
- Perry, C.M.; Jarvis, B. Peginterferon-alpha-2a (40kD)—A review of its use in the management of chronic hepatitis C. Drugs 2001, 61, 2263–2288. [Google Scholar] [CrossRef]
- Glue, P.; Fang, J.W.S.; Rouzier-Panis, R.; Raffanel, C.; Sabo, R.; Gupta, S.K.; Salfi, M.; Jacobs, S.; Hepatitis, C.I.T.G. Pegylated interferon-alpha 2b: Pharmacokinetics, pharmacodynamics, safety, and preliminary efficacy data. Clin. Pharmacol. Ther. 2000, 68, 556–567. [Google Scholar] [CrossRef]
- Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Parkinson, D.R.; Marincola, F.M.; Weber, J.S.; Seipp, C.A.; White, D.E.; Rosenberg, S.A. The use of polyethylene glycol-modified interleukin-2 (PEG-IL-2) in the treatment of patients with metastatic renal-cell carcinoma and melanoma—A phase-I study and a randomised prospective study comparing IL-2 alone versus IL-2 combined with PEG-IL-2. Cancer 1995, 76, 687–694. [Google Scholar]
- Katre, N.V.; Knauf, M.J.; Laird, W.J. Chemical modification of recombinant interleukin-2 by polyethilene-glycol increases its potency in the murine Meth-A sarcoma model. Proc. Natl. Acad. Sci. USA 1987, 84, 1487–1491. [Google Scholar] [CrossRef]
- Klein, B.; Brailly, H. Cytokine-binding proteins—Stimulating antagonists. Immunol. Today 1995, 16, 216–220. [Google Scholar] [CrossRef]
- Sato, J.; Hamaguchi, N.; Doken, K.; Gotoh, K.; Ootsu, K.; Iwasa, S.; Ogawa, Y.; Toguchi, H. Enhancement of antitumor-activity of recombinant interleukin-2 (rIL-2) by immunocomplexing with a monoclonal-antibody against rIL-2. Biotherapy 1993, 6, 225–231. [Google Scholar] [CrossRef]
- Rosenblum, M.G.; Unger, B.W.; Gutterman, J.U.; Hersh, E.M.; David, G.S.; Frincke, J.M. Modification of human-leukocyte interferon pharmacology with a monoclonal-antibody. Cancer Res. 1985, 45, 2421–2424. [Google Scholar]
- Finkelman, F.D.; Madden, K.B.; Morris, S.C.; Holmes, J.M.; Boiani, N.; Katona, I.M.; Maliszewski, C.R. Anti-cytokine antibodies as carrier proteins—Prolongation of in-vivo effects of exogenous cytokines by injection of cytokine anti-cytokine antibody complexes. J. Immunol. 1993, 151, 1235–1244. [Google Scholar]
- Boyman, O.; Kovar, M.; Rubinstein, M.P.; Surh, C.D.; Sprent, J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 2006, 311, 1924–1927. [Google Scholar] [CrossRef]
- Schein, C.H. The shape of the messenger: Using protein structure information to design novel cytokine-based therapeutics. Curr. Pharm. Des. 2002, 8, 2113–2129. [Google Scholar] [CrossRef]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar]
- Krieg, C.; Letourneau, S.; Pantaleo, G.; Boyman, O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11906–11911. [Google Scholar] [CrossRef]
- Tomala, J.; Chmelova, H.; Mrkvan, T.; Rihova, B.; Kovar, M. In Vivo Expansion of Activated Naive CD8(+) T Cells and NK Cells Driven by Complexes of IL-2 and Anti-IL-2 Monoclonal Antibody As Novel Approach of Cancer Immunotherapy. J. Immunol. 2009, 183, 4904–4912. [Google Scholar] [CrossRef]
- Kamimura, D.; Sawa, Y.; Sato, M.; Agung, E.; Hirano, T.; Murakami, M. IL-2 in vivo activities and antitumor efficacy enhanced by an anti-IL-2 mAb. J. Immunol. 2006, 177, 306–314. [Google Scholar]
- Webster, K.E.; Walters, S.; Kohler, R.E.; Mrkvan, T.; Boyman, O.; Surh, C.D.; Grey, S.T.; Sprent, J. In vivo expansion of T reg cells with IL-2-mAb complexes: Induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J. Exp. Med. 2009, 206, 751–760. [Google Scholar] [CrossRef]
- Tang, Q.; Adams, J.Y.; Penaranda, C.; Melli, K.; Piaggio, E.; Sgouroudis, E.; Piccirillo, C.A.; Salomon, B.L.; Bluestone, J.A. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 2008, 28, 687–697. [Google Scholar] [CrossRef]
- Liu, R.; Zhou, Q.; La Cava, A.; Campagnolo, D.I.; Van Kaer, L.; Shi, F.-D. Expansion of regulatory T cells via IL-2/anti-IL-2 mAb complexes suppresses experimental myasthenia. Eur. J. Immunol. 2010, 40, 1577–1589. [Google Scholar] [CrossRef]
- Wilson, M.S.; Pesce, J.T.; Ramalingam, T.R.; Thompson, R.W.; Cheever, A.; Wynn, T.A. Suppression of Murine Allergic Airway Disease by IL-2:Anti-IL-2 Monoclonal Antibody-Induced Regulatory T Cells. J. Immunol. 2008, 181, 6942–6954. [Google Scholar]
- Tam Nguyen, D.; Kyaw, T.S.; Kanellakis, P.; To, K.; Tipping, P.; Toh, B.-H.; Bobik, A.; Agrotis, A. Cytokine Therapy With Interleukin-2/Anti-Interleukin-2 Monoclonal Antibody Complexes Expands CD4+CD25+Foxp3+ Regulatory T Cells and Attenuates Development and Progression of Atherosclerosis. Circulation 2012, 126, 1256–1266. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Letourneau, S.; van Leeuwen, E.M.; Krieg, C.; Martin, C.; Pantaleo, G.; Sprent, J.; Surh, C.D.; Boyman, O. IL-2/anti-IL-2 antibody complexes show strong biological activity by avoiding interaction with IL-2 receptor alpha subunit CD25. Proc. Natl. Acad. Sci. USA 2010, 107, 2171–2176. [Google Scholar] [CrossRef]
- Carter, P.J. Introduction to current and future protein therapeutics: A protein engineering perspective. Exp. Cell Res. 2011, 317, 1261–1269. [Google Scholar] [CrossRef]
- Czajkowsky, D.M.; Hu, J.; Shao, Z.; Pleass, R.J. Fc-fusion proteins: New developments and future perspectives. EMBO Mol. Med. 2012, 4, 1015–1028. [Google Scholar] [CrossRef]
- Bitonti, A.J.; Dumont, J.A.; Low, S.C.; Peters, R.T.; Kropp, K.E.; Palombella, V.J.; Stattel, J.M.; Lu, Y.C.; Tan, C.A.; Song, J.J.; et al. Pulmonary delivery of an erythropoietin Fc fusion protein in non-human primates through an immunoglobulin transport pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 9763–9768. [Google Scholar] [CrossRef]
- Dumont, J.A.; Bitonti, A.J.; Clark, D.; Evans, S.; Pickford, M.; Newman, S.P. Delivery of an erythropoietin-Fc fusion protein by inhalation in humans through an immunoglobulin transport pathway. J. Aerosol Med. 2005, 18, 294–303. [Google Scholar] [CrossRef]
- Chang, C.H.; Gupta, P.; Goldenberg, D.M. Advances and challenges in developing cytokine fusion proteins as improved therapeutics. Expert Opin. Drug Discov. 2009, 4, 181–194. [Google Scholar] [CrossRef]
- Strohl, W.R. Optimization of Fc-mediated effector functions of monoclonal antibodies. Curr. Opin. Biotechnol. 2009, 20, 685–691. [Google Scholar] [CrossRef]
- Shoji-Hosaka, E.; Kobayashi, Y.; Wakitani, M.; Uchida, K.; Niwa, R.; Nakamura, K.; Shitara, K. Enhanced Fc-dependent cellular cytotoxicity of Fc fusion proteins derived from TNF receptor II and LFA-3 by fucose removal from Asn-linked oligosaccharides. J. Biochem. (Tokyo) 2006, 140, 777–783. [Google Scholar] [CrossRef]
- Davis, P.M.; Abraham, R.; Xu, L.; Nadler, S.G.; Suchard, S.J. Abatacept binds to the fc receptor CD64 but does not mediate complement-dependent cytotoxicity or antibody-dependent cellular cytotoxicity. J. Rheumatol. 2007, 34, 2204–2210. [Google Scholar]
- McGuire, H.M.; Walters, S.; Vogelzang, A.; Lee, C.M.Y.; Webster, K.E.; Sprent, J.; Christ, D.; Grey, S.; King, C. Interleukin-21 Is Critically Required in Autoimmune and Allogeneic Responses to Islet Tissue in Murine Models. Diabetes 2011, 60, 867–875. [Google Scholar] [CrossRef]
- Bubier, J.A.; Bennett, S.M.; Sproule, T.J.; Lyons, B.L.; Olland, S.; Young, D.A.; Roopenian, D.C. Treatment of BXSB-Yaa mice with IL-21R-Fc fusion protein minimally attenuates systemic lupus erythematosus. Ann. NY Acad. Sci. 2007, 1110, 590–601. [Google Scholar]
- Young, D.A.; Hegen, M.; Margery Ma, H.L.; Whitters, M.J.; Albert, L.M.; Lowe, L.; Senices, M.; Wu, P.W.; Sibley, B.; Leathurby, Y.; et al. Blockade of the interleukin-21/interleukin-21 receptor pathway ameliorates disease in animal models of rheumatoid arthritis. Arthritis Rheum. 2007, 56, 1152–1163. [Google Scholar] [CrossRef]
- Feng, X.M.; Zheng, X.X.; Yi, S.N.; Lehnert, A.M.; Strom, T.B.; O'Connell, P.J. IL-10/Fc inhibits macrophage function and prolongs pancreatic islet xenograft survival. Transplantation 1999, 68, 1775–1783. [Google Scholar] [CrossRef]
- FloresVillanueva, P.O.; Zheng, X.X.; Strom, T.B.; Stadecker, M.J. Recombinant IL-10 and IL-10/Fc treatment down-regulate egg antigen-specific delayed hypersensitivity reactions and egg granuloma formation in schistosomiasis. J. Immunol. 1996, 156, 3315–3320. [Google Scholar]
- Zheng, X.X.; Steele, A.W.; Hancock, W.W.; Stevens, A.C.; Nickerson, P.W.; RoyChaudhury, P.; Tian, Y.; Strom, T.B. A noncytolytic IL-10/Fc fusion protein prevents diabetes, blocks autoimmunity, and promotes suppressor phenomena in NOD mice. J. Immunol. 1997, 158, 4507–4513. [Google Scholar]
- Zheng, X.X.; Steele, A.W.; Nickerson, P.W.; Steurer, W.; Steiger, J.; Strom, T.B. Administration of noncytolytic IL-10/Fc in murine models of lipopolysaccharide-induced septic shock and allogeneic islet transplantation. J. Immunol. 1995, 154, 5590–5600. [Google Scholar]
- Schwager, K.; Kaspar, M.; Bootz, F.; Marcolongo, R.; Paresce, E.; Neri, D.; Trachsel, E. Preclinical characterization of Dekavil (F8-IL10), a novel clinical-stage immunocytokine which inhibits the progression of collagen-induced arthritis. Arthritis Res. Ther. 2009, 11. Article No. R142. [Google Scholar]
- Zheng, X.X.; Steele, A.W.; Hancock, W.W.; Kawamoto, K.; Li, X.C.; Nickerson, P.W.; Li, Y.S.; Tian, Y.; Strom, T.B. IL-2 receptor-targeted cytolytic IL-2/Fc fusion protein treatment blocks diabetogenic autoimmunity in nonobese diabetic mice. J. Immunol. 1999, 163, 4041–4048. [Google Scholar]
- Nam, H.J.; Song, M.-Y.; Choi, D.-H.; Yang, S.-H.; Jin, H.-T.; Sung, Y.-C. Marked enhancement of antigen-specific T-cell responses by IL-7-fused nonlytic, but not lytic, Fc as a genetic adjuvant. Eur. J. Immunol. 2010, 40, 351–358. [Google Scholar] [CrossRef]
- Anderson, C.L.; Chaudhury, C.; Kim, J.; Bronson, C.L.; Wani, M.A.; Mohanty, S. Perspective - FcRn transports albumin: Relevance to immunology and medicine. Trends Immunol. 2006, 27, 343–348. [Google Scholar] [CrossRef]
- Osborn, B.L.; Gu, M.; Grzegorzewski, K.J.; Logan, T.F.; Crowder, K.; Weiss, G.R.; Syed, S.; Rowensky, E.; Tolcher, A.; Agarwala, S.S. Preliminary pharmacokinetic evaluation of Albuleukin; an interleukin-2 human serum albumin fusion protein, in solid tumor patients. Proc. Amer. Assoc. Cancer Res. 2004, 2004, 1099. [Google Scholar]
- Halpern, W.; Riccobene, T.A.; Agostini, H.; Baker, K.; Stolow, D.; Gu, M.L.; Hirsch, J.; Mahoney, A.; Carrell, J.; Boyd, E.; et al. Albugranin (TM), a recombinant human granulocyte colony stimulating factor (G-CSF) genetically fused to recombinant human albumin induces prolonged myelopoietic effects in mice and monkeys. Pharm. Res. 2002, 19, 1720–1729. [Google Scholar] [CrossRef]
- Nelson, D.R.; Benhamou, Y.; Chuang, W.-L.; Lawitz, E.J.; Rodriguez-Torres, M.; Flisiak, R.; Rasenack, J.W.F.; Kryczka, W.; Lee, C.-M.; Bain, V.G.; et al. Albinterferon Alfa-2b Was Not Inferior to Pegylated Interferon-alpha in a Randomized Trial of Patients With Chronic Hepatitis C Virus Genotype 2 or 3. Gastroenterology 2010, 139, 1267–1276. [Google Scholar] [CrossRef]
- Subramanian, G.M.; Fiscella, M.; Lamouse-Smith, A.; Zeuzem, S.; McHutchison, J.G. Albinterferon alpha-2b: A genetic fusion protein for the treatment of chronic hepatitis C. Nat. Biotechnol. 2007, 25, 1411–1419. [Google Scholar] [CrossRef]
- Zeuzem, S.; Sulkowski, M.S.; Lawitz, E.J.; Rustgi, V.K.; Rodriguez-Torres, M.; Bacon, B.R.; Grigorescu, M.; Tice, A.D.; Lurie, Y.; Cianciara, J.; et al. Albinterferon Alfa-2b Was Not Inferior to Pegylated Interferon-alpha in a Randomized Trial of Patients With Chronic Hepatitis C Virus Genotype 1. Gastroenterology 2010, 139, 1257–1266. [Google Scholar] [CrossRef]
- Sung, C.; Nardelli, B.; LaFleur, D.W.; Blatter, E.; Corcoran, M.; Olsen, H.S.; Birse, C.E.; Pickeral, O.K.; Zhang, J.; Shah, D. An IFN-β-albumin fusion protein that displays improved pharmacokinetic and pharmacodynamic properties in nonhuman primates. J. Int. Cytokine Res. 2003, 23, 25–36. [Google Scholar] [CrossRef]
- Ward, E.S.; Gussow, D.; Griffiths, A.D.; Jones, P.T.; Winter, G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia-coli. Nature 1989, 341, 544–546. [Google Scholar] [CrossRef]
- Holt, L.J.; Basran, A.; Jones, K.; Chorlton, J.; Jespers, L.S.; Brewis, N.D.; Tomlinson, I.M. Anti-serum albumin domain antibodies for extending the half-lives of short lived drugs. Protein Eng. Des. Sel. 2008, 21, 283–288. [Google Scholar] [CrossRef]
- Walker, A.; Dunlevy, G.; Rycroft, D.; Topley, P.; Holt, L.J.; Herbert, T.; Davies, M.; Cook, F.; Holmes, S.; Jespers, L.; et al. Anti-serum albumin domain antibodies in the development of highly potent, efficacious and long-acting interferon. Protein Eng. Des. Sel. 2010, 23, 271–278. [Google Scholar] [CrossRef]
- Qian, Z.M.; Li, H.Y.; Sun, H.Z.; Ho, K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol. Rev. 2002, 54, 561–587. [Google Scholar] [CrossRef]
- Daniels, T.R.; Delgado, T.; Helguera, G.; Penichet, M.L. The transferrin receptor part II: Targeted delivery of therapeutic agents into cancer cells. Clin. Immunol. 2006, 121, 159–176. [Google Scholar] [CrossRef]
- Daniels, T.R.; Delgado, T.; Rodriguez, J.A.; Helguera, G.; Penichet, M.L. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin. Immunol. 2006, 121, 144–158. [Google Scholar] [CrossRef]
- Kim, B.-J.; Zhou, J.; Martin, B.; Carlson, O.D.; Maudsley, S.; Greig, N.H.; Mattson, M.P.; Ladenheim, E.E.; Wustner, J.; Turner, A.; et al. Transferrin Fusion Technology: A Novel Approach to Prolonging Biological Half-Life of Insulinotropic Peptides. J. Pharmacol. Exp. Ther. 2010, 334, 682–692. [Google Scholar] [CrossRef]
- Banerjee, D.; Flanagan, P.R.; Cluett, J.; Valberg, L.S. Transferrin receptors in the human gastrointestinal-tract - relationship to body iron stores. Gastroenterology 1986, 91, 861–869. [Google Scholar]
- Bai, Y.; Ann, D.K.; Shen, W.C. Recombinant granulocyte colony-stimulating factor-transferrin fusion protein as an oral myelopoietic agent. Proc. Natl. Acad. Sci. USA 2005, 102, 7292–7296. [Google Scholar]
- Bai, Y.; Shen, W.-C. Improving the oral efficacy of recombinant granulocyte colony-stimulating factor and transferrin fusion protein by spacer optimization. Pharm. Res. 2006, 23, 2116–2121. [Google Scholar] [CrossRef]
- Jones, S.A.; Rose-John, S. The role of soluble receptors in cytokine biology: The agonistic properties of the sIL-6R/IL-6 complex. BBA-Mol. Cell Res. 2002, 1592, 251–263. [Google Scholar]
- Rubinstein, M.P.; Kovar, M.; Purton, J.F.; Cho, J.H.; Boyman, O.; Surh, C.D.; Sprent, J. Converting IL-15 to a superagonist by binding to soluble IL-15R alpha. Proc. Natl. Acad. Sci. USA 2006, 103, 9166–9171. [Google Scholar]
- Dubois, S.; Mariner, J.; Waldmann, T.A.; Tagaya, Y. IL-15R alpha recycles and presents IL-15 in trans to neighboring cells. Immunity 2002, 17, 537–547. [Google Scholar] [CrossRef]
- Waldmann, T.A. The biology of interleukin-2 and interleukin-15: Implications for cancer therapy and vaccine design. Nat. Rev. Immunol. 2006, 6, 595–601. [Google Scholar] [CrossRef]
- Mortier, E.; Quemener, A.; Vusio, P.; Lorenzen, I.; Boublik, Y.; Grotzinger, J.; Plet, A.; Jacques, Y. Soluble interleukin-15 receptor alpha (IL-15R alpha)-sushi as a selective and potent agonist of IL-15 action through IL-15R beta/gamma. Hyperagonist IL-15 x IL-15R alpha fusion proteins. J. Biol. Chem. 2006, 281, 1612–1619. [Google Scholar]
- Fischer, M.; Goldschmitt, J.; Peschel, C.; Brakenhoff, J.P.; Kallen, K.J.; Wollmer, A.; Grotzinger, J.; Rose-John, S.I. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat. Biotechnol. 1997, 15, 142–145. [Google Scholar] [CrossRef]
- Pflanz, S.; Tacken, I.; Grotzinger, J.; Jacques, Y.; Dahmen, H.; Heinrich, P.C.; Muller-Newen, G. A fusion protein of interleukin-11 and soluble interleukin-11 receptor acts as a superagonist on cells expressing gp130. FEBS Lett. 1999, 450, 117–122. [Google Scholar] [CrossRef]
- Peters, M.; Blinn, G.; Jostock, T.; Schirmacher, P.; Zum Buschenfelde, K.H.M.; Galle, P.R.; Rose-John, S. Combined interleukin 6 and soluble interleukin 6 receptor accelerates murine liver regeneration. Gastroenterology 2000, 119, 1663–1671. [Google Scholar] [CrossRef]
- Manoukian, G.; Hagemeister, F. Denileukin diftitox: A novel immunotoxin. Expert Opin. Biol. Ther. 2009, 9, 1445–1451. [Google Scholar] [CrossRef]
- Shimamura, T.; Husain, S.R.; Puri, R.K. The IL-4 and IL-13 pseudomonas exotoxins: New hope for brain tumor therapy. Neurosurg. Focus 2006, 20, E11–E11. [Google Scholar]
- Foss, F. Clinical experience with denileukin diftitox (ONTAK). Semin. Oncol. 2006, 33, S11–S16. [Google Scholar] [CrossRef]
- Waters, C.A.; Schimke, P.A.; Snider, C.E.; Itoh, K.; Smith, K.A.; Nichols, J.C.; Strom, T.B.; Murphy, J.R. Interleukin-2 receptor-targeted cytotoxicity—Receptor-binding requirements for entry of a diphtheria toxin-related interleukin-2 fusion protein into cells. Eur. J. Immunol. 1990, 20, 785–791. [Google Scholar] [CrossRef]
- Bacha, P.; Williams, D.P.; Waters, C.; Williams, J.M.; Murphy, J.R.; Strom, T.B. Interleukin-2 receptor targeted cyto-toxicity interleukin-2 receptor mediated action of a diphtheria-toxin related interleukin-2 fusion protein. J. Exp. Med. 1988, 167, 612–622. [Google Scholar] [CrossRef]
- Olsen, E.; Duvic, M.; Frankel, A.; Kim, Y.; Martin, A.; Vonderheid, E.; Jegasothy, B.; Wood, G.; Gordon, M.; Heald, P.; et al. Pivotal phase III trial of two dose levels of denileukin diftitox for the treatment of cutaneous T-cell lymphoma. J. Clin. Oncol. 2001, 19, 376–388. [Google Scholar]
- Ruddle, J.B.; Harper, C.A.; Honemann, D.; Seymour, J.F.; Prince, H.M.; Harper, C.A. A denileukin diftitox (Ontak) associated retinopathy? Br. J. Ophthalmol. 2006, 90, 1070–1071. [Google Scholar]
- Ruddle, J.B.; Prince, H.M. Denileukin diftitox and vision loss. Leuk. Lymphoma 2007, 48, 655–656. [Google Scholar] [CrossRef]
- Takeuchi, M.; Keino, H.; Kezuka, T.; Usui, M.; Taguchi, O. Immune responses to retinal self-antigens in CD25(+)CD4(+) regulatory T-Cell-depleted mice. Invest. Ophthalmol. Visual Sci. 2004, 45, 1879–1886. [Google Scholar] [CrossRef]
- Iwamoto, F.M.; Lamborn, K.R.; Robins, H.I.; Mehta, M.P.; Chang, S.M.; Butowski, N.A.; DeAngelis, L.M.; Abrey, L.E.; Zhang, W.-T.; Prados, M.D.; et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro-Oncology 2010, 12, 855–861. [Google Scholar] [CrossRef]
- Mut, M.; Sherman, J.H.; Shaffrey, M.E.; Schiff, D. Cintredekin besudotox in treatment of malignant glioma. Expert Opin. Biol. Ther. 2008, 8, 805–812. [Google Scholar] [CrossRef]
- Jackaman, C.; Bundell, C.S.; Kinnear, B.F.; Smith, A.M.; Filion, P.; van Hagen, D.; Robinson, B.W.S.; Nelson, D.J. IL-2 intratumoral immunotherapy enhances CD8(+) T cells that mediate destruction of tumor cells and tumor-associated vasculature: A novel mechanism for IL-2(1). J. Immunol. 2003, 171, 5051–5063. [Google Scholar]
- Gutbrodt, K.L.; Neri, D. Immunocytokines. Antibodies 2012, 1, 70–87. [Google Scholar] [CrossRef]
- van Horssen, R.; ten Hagen, T.L.M.; Eggermont, A.M.M. TNF-alpha in cancer treatment: Molecular insights, antitumor effects, and clinical utility. Oncologist 2006, 11, 397–408. [Google Scholar] [CrossRef]
- Sondel, P.M.; Gillies, S.D. Current and Potential Uses of Immunocytokines as Cancer Immunotherapy. Antibodies 2012, 1, 149–171. [Google Scholar] [CrossRef]
- Kontermann, R.E. Antibody-cytokine fusion proteins. Arch. Biochem. Biophys. 2012, 526, 194–205. [Google Scholar] [CrossRef]
- Pasche, N.; Neri, D. Immunocytokines: A novel class of potent armed antibodies. Drug Discov. Today 2012, 17, 583–590. [Google Scholar] [CrossRef]
- Connor, J.P.; Cristea, M.C.; Lewis, N.L.; Lewis, L.D.; Komarnitsky, P.B.; Mattiacci, M.R.; Felder, M.; Stewart, S.; Harter, J.; Henslee-Downey, J.; et al. A phase 1b study of humanized KS-interleukin-2 (huKS-IL2) immunocytokine with cyclophosphamide in patients with EpCAM-positive advanced solid tumors. BMC Cancer 2013, 13. Article No. 20. [Google Scholar]
- Patriarca, C.; Macchi, R.M.; Marschner, A.K.; Mellstedt, H. Epithelial cell adhesion molecule expression (CD326) in cancer: A short review. Cancer Treat. Rev. 2012, 38, 68–75. [Google Scholar] [CrossRef]
- Albertini, M.R.; Hank, J.A.; Gadbaw, B.; Kostlevy, J.; Haldeman, J.; Schalch, H.; Gan, J.; Kim, K.; Eickhoff, J.; Gillies, S.D.; et al. Phase II trial of hu14.18-IL2 for patients with metastatic melanoma. Cancer Immunol. Immunother. 2012, 61, 2261–2271. [Google Scholar] [CrossRef]
- Mujoo, K.; Cheresh, D.A.; Yang, H.M.; Reisfeld, R.A. Disialoganglioside-GD2 on human neuroblastoma-cells—Target antigen for monoclonal antibody-mediated cytolysis and suppression of tumor-growth. Cancer Res. 1987, 47, 1098–1104. [Google Scholar]
- Yamane, B.H.; Hank, J.A.; Albertini, M.R.; Sondel, P.M. The development of antibody-IL-2 based immunotherapy with hu14.18-IL2 (EMD-273063) in melanoma and neuroblastoma. Expert Opin. Investig. Drugs 2009, 18, 991–1000. [Google Scholar] [CrossRef]
- Helguera, G.; Rodriguez, J.A.; Penichet, M.L. Cytokines fused to antibodies and their combinations as therapeutic agents against different peritoneal HER2/neu expressing tumors. Mol. Cancer Ther. 2006, 5, 1029–1040. [Google Scholar] [CrossRef]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; Press, M.F. Studies of the HER-2/neu proto-oncogene in human-breast and ovarian-cancer. Science 1989, 244, 707–712. [Google Scholar]
- Gillies, S.D.; Lan, Y.; Williams, S.; Carr, F.; Forman, S.; Raubitschek, A.; Lo, K.M. An anti-CD20-IL-2 immunocytokine is highly efficacious in a SCID mouse model of established human B lymphoma. Blood 2005, 105, 3972–3978. [Google Scholar] [CrossRef]
- Rossi, E.A.; Goldenberg, D.M.; Cardillo, T.M.; Stein, R.; Chang, C.-H. CD20-targeted tetrameric interferon-alpha, a novel and potent immunocytokine for the therapy of B-cell lymphomas. Blood 2009, 114, 3864–3871. [Google Scholar] [CrossRef]
- Hirsch, B.; Brauer, J.; Fischdick, M.; Loddenkemper, C.; Bulfone-Paus, S.; Stein, H.; Duerkop, H. Anti-CD30 Human IL-2 Fusion Proteins Display Strong and Specific Cytotoxicity In Vivo. Curr. Drug Targets 2009, 10, 110–117. [Google Scholar] [CrossRef]
- Frey, K.; Fiechter, M.; Schwager, K.; Belloni, B.; Barysch, M.J.; Neri, D.; Dummer, R. Different patterns of fibronectin and tenascin-C splice variants expression in primary and metastatic melanoma lesions. Exp. Dermatol. 2011, 20, 685–688. [Google Scholar] [CrossRef]
- Kriegsmann, J.; Berndt, A.; Hansen, T.; Borsi, L.; Zardi, L.; Brauer, R.; Petrow, P.K.; Otto, M.; Kirkpatrick, C.J.; Gay, S.; et al. Expression of fibronectin splice variants and oncofetal glycosylated fibronectin in the synovial membranes of patients with rheumatoid arthritis and osteoarthritis. Rheumatol. Int. 2004, 24, 25–33. [Google Scholar] [CrossRef]
- Schliemann, C.; Wiedmer, A.; Pedretti, M.; Szczepanowski, M.; Klapper, W.; Neri, D. Three clinical-stage tumor targeting antibodies reveal differential expression of oncofetal fibronectin and tenascin-C isoforms in human lymphoma. Leuk. Res. 2009, 33, 1718–1722. [Google Scholar] [CrossRef]
- Gillessen, S.; Gnad-Vogt, U.S.; Gallerani, E.; Beck, J.; Sessa, C.; Omlin, A.; Mattiacci, M.R.; Liedert, B.; Kramer, D.; Laurent, J.; et al. A phase I dose-escalation study of the immunocytokine EMD 521873 (Selectikine) in patients with advanced solid tumours. Eur. J. Cancer 2013, 49, 35–44. [Google Scholar] [CrossRef]
- Epstein, A.L.; Chen, F.M.; Taylor, C.R. A novel method for the detection of necrotic lesions in human cancers. Cancer Res. 1988, 48, 5842–5848. [Google Scholar]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Naramura, M.; Gillies, S.D.; Mendelsohn, J.; Reisfeld, R.A.; Mueller, B.M. Mechanisms of cellular cytotoxicity mediated by a recombinant antibody IL2 fusion protein against human-melanoma cells. Immunol. Lett. 1993, 39, 91–99. [Google Scholar] [CrossRef]
- Jain, R.K.; Baxter, L.T. Mechanisms of heterogeneous distribution of monoclonal-antibodies and other macromolecules in tumors—Significance of elevated interstitial pressure. Cancer Res. 1988, 48, 7022–7032. [Google Scholar]
- Yokota, T.; Milenic, D.E.; Whitlow, M.; Schlom, J. Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. Cancer Res. 1992, 52, 3402–3408. [Google Scholar]
- Milenic, D.E.; Yokota, T.; Filpula, D.R.; Finkelman, M.A.J.; Dodd, S.W.; Wood, J.F.; Whitlow, M.; Snoy, P.; Schlom, J. Construction, binding-properties, metabolism, and tumor targeting of a single-chain Fv derived from the pancarcinoma monoclonal-antibody CC49. Cancer Res. 1991, 51, 6363–6371. [Google Scholar]
- Adams, G.P.; Schier, R.; McCall, A.M.; Crawford, R.S.; Wolf, E.J.; Weiner, L.M.; Marks, J.D. Prolonged in vivo tumour retention of a human diabody targeting the extracellular domain of human HER2/neu. Br. J. Cancer 1998, 77, 1405–1412. [Google Scholar] [CrossRef]
- Rossi, E.A.; Rossi, D.L.; Stein, R.; Goldenberg, D.M.; Chang, C.-H. A Bispecific Antibody-IFN alpha 2b Immunocytokine Targeting CD20 and HLA-DR Is Highly Toxic to Human Lymphoma and Multiple Myeloma Cells. Cancer Res. 2010, 70, 7600–7609. [Google Scholar] [CrossRef]
- Kontermann, R.E. Dual targeting strategies with bispecific antibodies. mAbs 2012, 4, 182–197. [Google Scholar] [CrossRef]
- Bremer, E.; de Bruyn, M.; Wajant, H.; Helfrich, W. Targeted Cancer Immunotherapy Using Ligands of the Tumor Necrosis Factor Super-Family. Curr. Drug Targets 2009, 10, 94–103. [Google Scholar] [CrossRef]
- Galeazzi, M.; Baldi, C.; Prisco, E.; Bardelli, M.; Neri, D.; Giovannoni, L.; Selvi, E.; Caporali, R. A Phase Ib Clinical Trial with F8-IL10, an Anti-Inflammatory Immunocytokine for the Treatment of Rheumatoid Arthritis (RA), Used in Combination with Methotrexate (MTX). Arthritis Rheum. 2012, 64, S553–S554. [Google Scholar]
- ReidhaarOlson, J.F.; DeSouzaHart, J.A.; Selick, H.E. Identification of residues critical to the activity of human granulocyte colony-stimulating factor. Biochemistry (Mosc.) 1996, 35, 9034–9041. [Google Scholar] [CrossRef]
- Young, D.C.; Zhan, H.J.; Cheng, Q.L.; Hou, J.Z.; Matthews, D.J. Characterization of the receptor binding determinants of granulocyte colony stimulating factor. Protein Sci. 1997, 6, 1228–1236. [Google Scholar] [CrossRef]
- Collins, L.; Tsien, W.H.; Seals, C.; Hakimi, J.; Weber, D.; Bailon, P.; Hoskings, J.; Greene, W.C.; Toome, V.; Ju, G. Identification of specific residues of human interleukin-2 that affect binding to the 70-kDa subunit (p70) of the interleukin-2 receptor. Proc. Natl. Acad. Sci. USA 1988, 85, 7709–7713. [Google Scholar] [CrossRef]
- Ju, G.; Collins, L.; Kaffka, K.L.; Tsien, W.H.; Chizzonite, R.; Crowl, R.; Bhatt, R.; Kilian, P.L. Structure-function analysis of human interleukin-2—Identification of amino-acid-residues required for biological-activity. J. Biol. Chem. 1987, 262, 5723–5731. [Google Scholar]
- Weir, M.P.; Chaplin, M.A.; Wallace, D.M.; Dykes, C.W.; Hobden, A.N. Structure activity relationships of recombinant human interleukin-2. Biochemistry (Mosc.) 1988, 27, 6883–6892. [Google Scholar]
- Lee, C.M.Y.; McGuire, H.; Basten, A.; King, C.; Christ, D. Expression, purification and characterization of recombinant interleukin-21. J. Immunol. Methods 2010, 362, 185–189. [Google Scholar] [CrossRef]
- Marshall, S.A.; Lazar, G.A.; Chirino, A.J.; Desjarlais, J.R. Rational design and engineering of therapeutic proteins. Drug Discov. Today 2003, 8, 212–221. [Google Scholar] [CrossRef]
- Lowe, D.; Dudgeon, K.; Rouet, R.; Schofield, P.; Jermutus, L.; Christ, D. Aggregation, stability, and formulation of human antibody therapeutics. Adv. Protein Chem. Struct. Biol. 2011, 84, 41–61. [Google Scholar] [CrossRef]
- Dudgeon, K.; Rouet, R.; Kokmeijer, I.; Schofield, P.; Stolp, J.; Langley, D.; Stock, D.; Christ, D. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc. Natl. Acad. Sci. USA 2012, 109, 10879–10884. [Google Scholar]
- Arakawa, T.; Prestrelski, S.J.; Narhi, L.O.; Boone, T.C.; Kenney, W.C. Cysteine-17 of recombinant human granulocyte-colony-stimulating factor is partially solvent-exposed. J. Protein Chem. 1993, 12, 525–531. [Google Scholar] [CrossRef]
- Doyle, M.V.; Lee, M.T.; Fong, S. Comparison of the biological-activities of human recombinant interleukin-2125 and native interleukin-2. J. Biol. Response Mod. 1985, 4, 96–109. [Google Scholar]
- Lin, L. Betaseron. Dev. Biol. Stand. 1998, 96, 97–104. [Google Scholar]
- Bishop, B.; Koay, D.C.; Sartorelli, A.C.; Regan, L. Reengineering granulocyte colony-stimulating factor for enhanced stability. J. Biol. Chem. 2001, 276, 33465–33470. [Google Scholar]
- Luo, P.; Hayes, R.J.; Chan, C.; Stark, D.M.; Hwang, M.Y.; Jacinto, J.M.; Juvvadi, P.; Chung, H.S.; Kundu, A.; Ary, M.L.; et al. Development of a cytokine analog with enhanced stability using computational ultrahigh throughput screening. Protein Sci. 2002, 11, 1218–1226. [Google Scholar] [CrossRef]
- Tang, L.; Persky, A.M.; Hochhaus, G.; Meibohm, B. Pharmacokinetic aspects of biotechnology products. J. Pharm. Sci. 2004, 93, 2184–2204. [Google Scholar] [CrossRef]
- French, A.R.; Lauffenburger, D.A. Controlling receptor/ligand trafficking: Effects of cellular and molecular properties on endosomal sorting. Ann. Biomed. Eng. 1997, 25, 690–707. [Google Scholar] [CrossRef]
- Lauffenburger, D.A.; Fallon, E.M.; Haugh, J.M. Scratching the (cell) surface: Cytokine engineering for improved ligand/receptor trafficking dynamics. Chem. Biol. 1998, 5, R257–R263. [Google Scholar] [CrossRef]
- Fallon, E.M.; Liparoto, S.F.; Lee, K.J.; Ciardelli, T.L.; Lauffenburger, D.A. Increased endosomal sorting of ligand to recycling enhances potency of an interleukin-2 analog. J. Biol. Chem. 2000, 275, 6790–6797. [Google Scholar]
- Sarkar, C.A.; Lowenhaupt, K.; Horan, T.; Boone, T.C.; Tidor, B.; Lauffenburger, D.A. Rational cytokine design for increased lifetime and enhanced potency using pH-activated "histidine switching". Nat. Biotechnol. 2002, 20, 908–913. [Google Scholar] [CrossRef]
- Rao, B.M.; Driver, I.; Lauffenburger, D.A.; Wittrup, K.D. High-affinity CD25-binding IL-2 mutants potently stimulate persistent T cell growth. Biochemistry (Mosc.) 2005, 44, 10696–10701. [Google Scholar] [CrossRef]
- Rao, B.M.; Girvin, A.T.; Ciardelli, T.; Lauffenburger, D.A.; Wittrup, K.D. Interleukin-2 mutants with enhanced alpha-receptor subunit binding affinity. Protein Eng. 2003, 16, 1081–1087. [Google Scholar] [CrossRef]
- Liu, D.V.; Maier, L.M.; Hafler, D.A.; Wittrup, K.D. Engineered Interleukin-2 Antagonists for the Inhibition of Regulatory T Cells. J. Immunother. 2009, 32, 887–894. [Google Scholar] [CrossRef]
- Levin, A.M.; Bates, D.L.; Ring, A.M.; Krieg, C.; Lin, J.T.; Su, L.; Moraga, I.; Raeber, M.E.; Bowman, G.R.; Novick, P.; et al. Exploiting a natural conformational switch to engineer an interleukin-2 'superkine'. Nature 2012, 484, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Junttila, I.S.; Creusot, R.J.; Moraga, I.; Bates, D.L.; Wong, M.T.; Alonso, M.N.; Suhoski, M.M.; Lupardus, P.; Meier-Schellersheim, M.; Engleman, E.G.; et al. Redirecting cell-type specific cytokine responses with engineered interleukin-4 superkines. Nat. Chem. Biol. 2012, 8, 990–998. [Google Scholar] [CrossRef]
- Hershey, G.K.K. IL-13 receptors and signaling pathways: An evolving web. J. Allergy Clin. Immunol. 2003, 111, 677–690. [Google Scholar]
- Antoniu, S.A. Pitrakinra, a dual IL-4/IL-13 antagonist for the potential treatment of asthma and eczema. Curr. Opin. Investig. Drugs 2010, 11, 1286–1294. [Google Scholar]
- Antoniu, S.A.; Cojocaru, I. Pitrakinra for asthma. Expert Opin. Biol. Ther. 2010, 10, 1609–1615. [Google Scholar] [CrossRef]
- Wenzel, S.; Wilbraham, D.; Fuller, R.; Burmeister Getz, E.; Lonphre, M. Effect of an interieukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: Results of two phase 2a studies. Lancet 2007, 370, 1422–1431. [Google Scholar] [CrossRef]
- Tony, H.P.; Shen, B.J.; Reusch, P.; Sebald, W. Design of human interleukin-4 antagonists inhibiting interleukin-4-dependent and interleukin-13-dependent responses in T-cells and B-cells with high-efficiency. Eur. J. Biochem. 1994, 225, 659–665. [Google Scholar] [CrossRef]
- Malara, N.; Foca, D.; Casadonte, F.; Sesto, M.; Paolino, D.; Tassone, P.; Venuta, S.; Savino, R. Treatment with an adenoviral vector expressing the IL-6 receptor superantagonist sant7 compared with the treatment with the recombinant sant7 protein in MM cell lines. J. Immunother. 2006, 29, 674–675. [Google Scholar]
- Tassone, P.; Neri, P.; Burger, R.; Savino, R.; Shammas, M.; Catley, L.; Podar, K.; Chauhan, D.; Masciari, S.; Gozzini, A.; et al. Combination therapy with interleukin-6 receptor superantagonist Sant7 and dexamethasone induces antitumor effects in a novel SCID-hu In vivo model of human multiple myeloma. Clin. Cancer Res. 2005, 11, 4251–4258. [Google Scholar] [CrossRef]
- Gallelli, L.; Falcone, D.; Pelaia, G.; Renda, T.; Terracciano, R.; Malara, N.; Vatrella, A.; Sanduzzi, A.; D'Agostino, B.; Rossi, F.; et al. Interleukin-6 receptor superantagonist Sant7 inhibits TGF-beta-induced proliferation of human lung fibroblasts. Cell Prolif. 2008, 41, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Savino, R.; Ciapponi, L.; Lahm, A.; Demartis, A.; Cabibbo, A.; Toniatti, C.; Delmastro, P.; Altamura, S.; Ciliberto, G. Rational design of a receptor super-antagonist of human interleukin-6. EMBO J. 1994, 13, 5863–5870. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Vazquez-Lombardi, R.; Roome, B.; Christ, D. Molecular Engineering of Therapeutic Cytokines. Antibodies 2013, 2, 426-451. https://doi.org/10.3390/antib2030426
AMA Style
Vazquez-Lombardi R, Roome B, Christ D. Molecular Engineering of Therapeutic Cytokines. Antibodies. 2013; 2(3):426-451. https://doi.org/10.3390/antib2030426
Chicago/Turabian StyleVazquez-Lombardi, Rodrigo, Brendan Roome, and Daniel Christ. 2013. "Molecular Engineering of Therapeutic Cytokines" Antibodies 2, no. 3: 426-451. https://doi.org/10.3390/antib2030426