Ion Chromatographic Analyses of Sea Waters, Brines and Related Samples

Faculty of Chemistry and Chemical Technology, University of Ljubljana, Aškerčeva 5, SI-1000 Ljubljana, Slovenia
Water 2013, 5(2), 659-676; https://doi.org/10.3390/w5020659
Submission received: 10 April 2013
/
Revised: 14 May 2013
/
Accepted: 20 May 2013
/
Published: 5 June 2013
(This article belongs to the Special Issue Analytical Chemistry of Water)
Abstract
: This review focuses on the ion chromatographic methods for the analyses of natural waters with high ionic strength. At the beginning a natural diversity in ionic composition of waters is highlighted and terminology clarified. In continuation a brief overview of other review articles of potential interest is given. A review of ion chromatographic methods is organized in four sections. The first section comprises articles focused on the determination of ionic composition of water samples as completely as possible. The sections—Selected Anions, Selected Cations and Metals—follow. The most essential experimental conditions used in different methods are summarized in tables for a rapid comparison. Techniques encountered in the reviewed articles comprise: direct determinations of ions in untreated samples with ion- or ion-exclusion chromatography, or electrostatic ion chromatography; matrix elimination with column-switching; pre-concentration with a chelation ion chromatography and purge-and-trap pre-concentration. Different detection methods were used: non-suppressed conductometric or suppressed conductometric, direct spectrometric or spectrometric after a post-column derivetization, and inductively coupled plasma in combination with optical emission or mass spectrometry.
1. Natural Diversity of Waters
We restrict the scope of this review and choose to focus on natural waters with high ionic strength in which concentrations of ions extend over several orders of magnitude and therefore these waters present a special challenge for ion chromatographic (IC) analysis, especially if major and minor ions are to be determined simultaneously. Natural waters of different types differ not only in a total concentration of dissolved solids (TDS) but also in concentration proportions between individual ions.
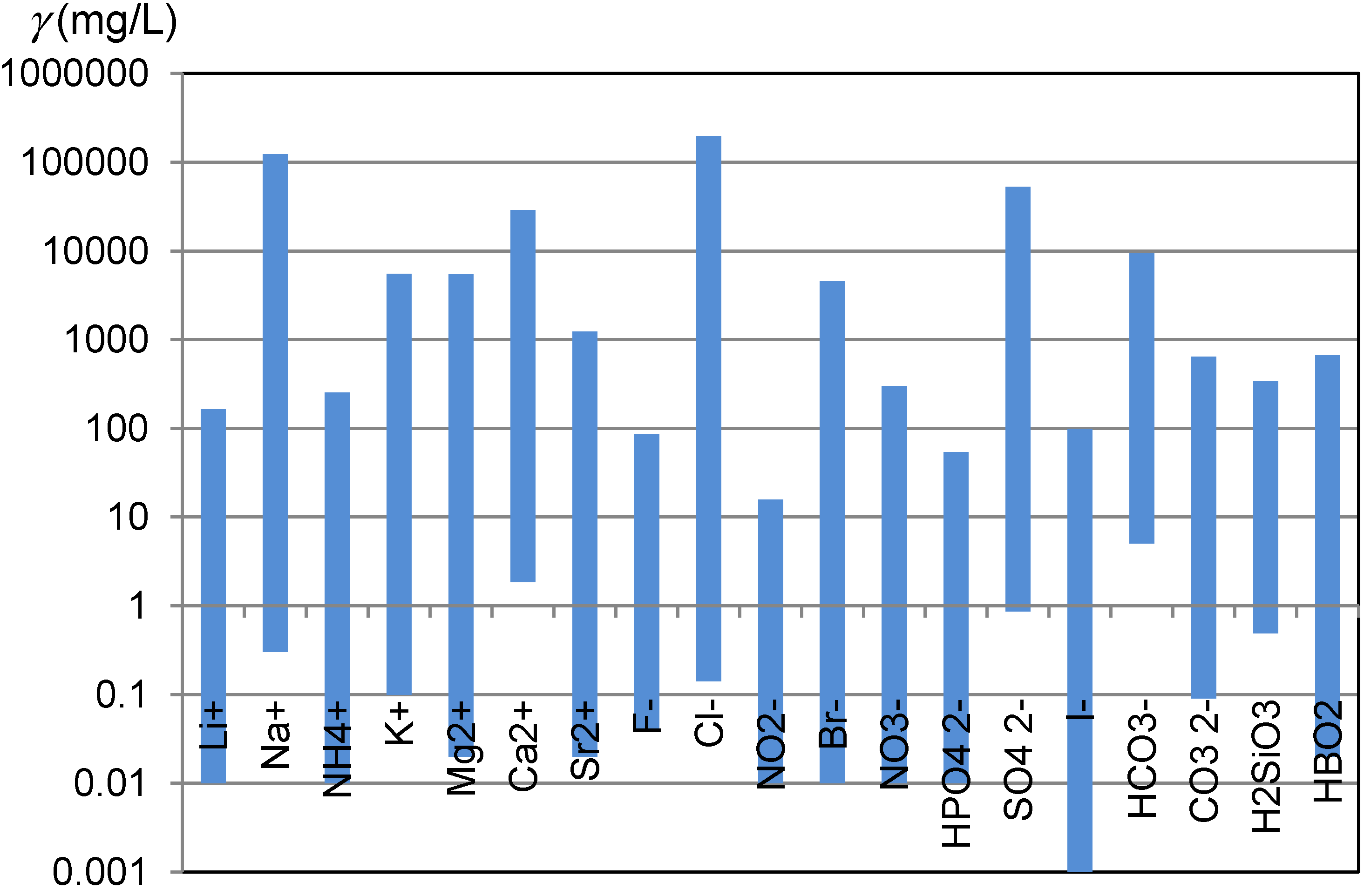
Awareness how diverse are natural waters is not widely shared. Carlé, in his monograph, compiled the results of analyses of more than 700 Central European mineral and thermal waters [1]. An electronic database prepared on the bases of this monograph enables an insight into natural diversity in the ionic composition of waters [2]. One of the aspects of the natural diversity of waters are wide concentration ranges, in which ions which are of interest for ion chromatographic analyses occur in natural waters (Figure 1).
Figure 1.
Concentration ranges of ions and other species which are of interest for ion chromatographic analyses and were observed in the Central European mineral and thermal waters [2].
Figure 1.
Concentration ranges of ions and other species which are of interest for ion chromatographic analyses and were observed in the Central European mineral and thermal waters [2].

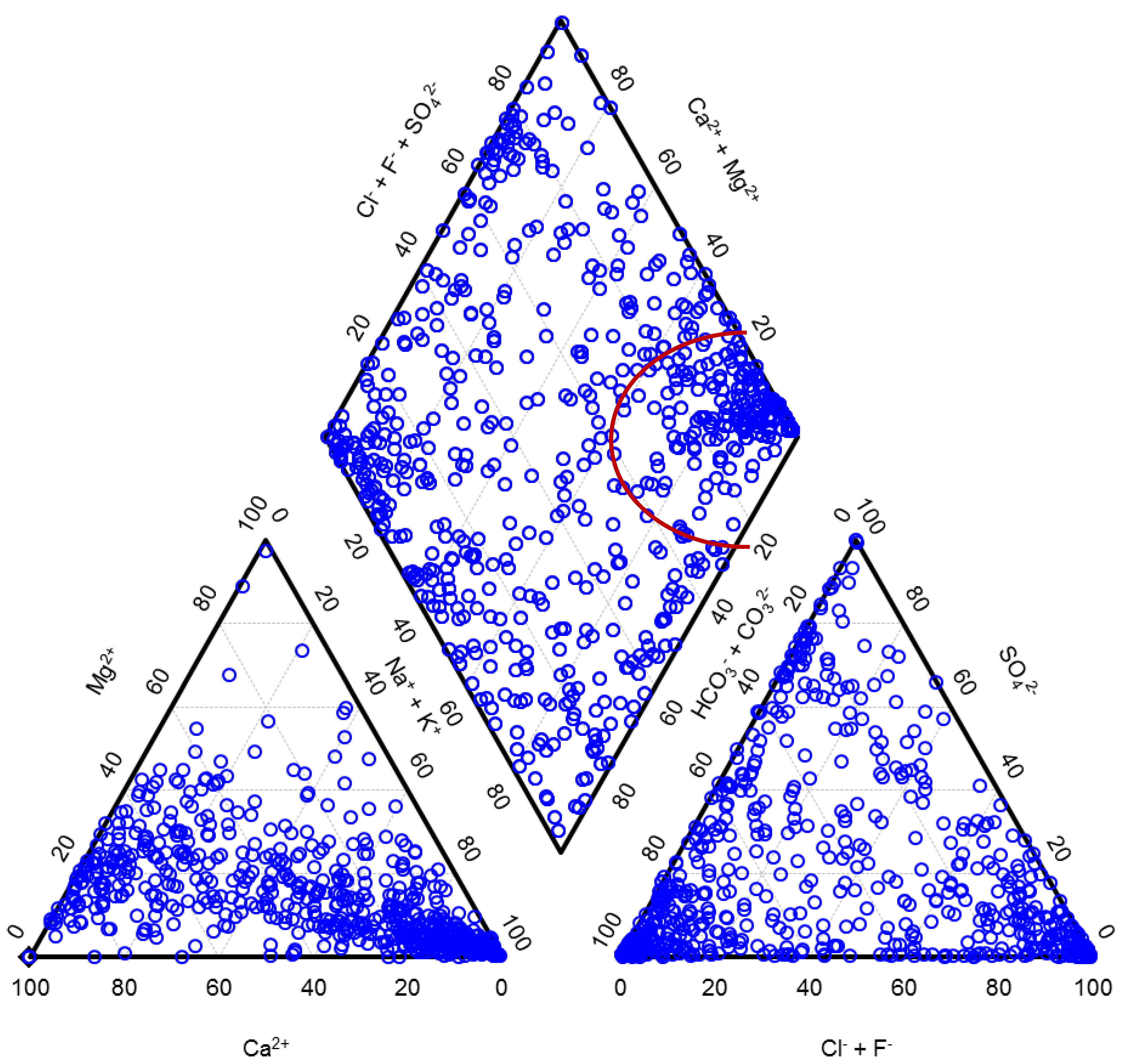
Fractions of the main cations and the main anions expressed in milliequivalent percent (meq%) classify water types. In Figure 2, we present waters from Carlé monograph in the form of a Piper diagram. As can be seen from the diagram, this limited set of water samples already well represents the natural diversity of waters in spite of the fact that waters were from a fairly restricted geographic area. The anionic triangle and a diamond are nearly uniformly covered in dots.
It has to be mentioned that there is some ambiguity in a usage of different terms related to waters. A term mineral water was in the Central European and some Eastern European Countries traditionally understood as a highly mineralized natural water containing TDS in a concentration at least equal to or higher than 1 g/L. Mineral waters comprised not only waters suitable for drinking or bathing but also brines of different types and of a potential industrial relevance, as it is also obvious from a Piper diagram in which the right corner of the diamond is dedicated to brines, also called saline, and well cowered in dots. Sea water with 77.1 meq% of sodium, 18.1 meq% of magnesium, 2.9 meq% of calcium, 1.6 meq% of potassium, 90.9 meq% of chloride and 9.1 meq% of sulfate would have been presented in this same corner of the diagram. Other corners of a diamond of a Piper diagram are dedicated to waters of other distinctive types: the top corner comprises waters with a high permanent hardness, the left corner waters with a high temporary hardness and the bottom corner waters in which alkali carbonates prevail.
Figure 2.
Natural diversity in composition of the Central European mineral and thermal waters. Brines are presented in the right corner of the diamond.
Figure 2.
Natural diversity in composition of the Central European mineral and thermal waters. Brines are presented in the right corner of the diamond.

It has to be pointed out that an entirely different understanding of the term mineral water may be prevailing in some Europe countries and many countries World-wide, where this term is frequently used as a synonymous term for spring waters or ordinary drinking waters, usually bottled but not highly mineralized. In this paper, we use a term with the previously described meaning.
In this review, we provide an overview of ion chromatographic methods developed for analyzing sea waters, brines and other highly mineralized natural waters, e.g., mineral waters and oil-field waters. As it will be presented in the continuation the great majority of applications described in literature deal with ion chromatographic analyses of sea waters or brines, only very few are dedicated to ion chromatographic analyses of mineral waters. One of the possible explanations would have been their natural diversity. Even waters from the same aquifer and from the boreholes, which are not far apart, can be of different types if the boreholes reach into a different section of the aquifer, and waters of different balneological types present distinctive analytical problems. A paper, which tried to give some guidance in this area, demonstrated how ion chromatographic determinations of cations in 116 natural mineral waters can be reduced on only 14 distinctive analytical sub-cases [3].
2. Review Papers
Reviews which cover the topics are not included in this paper, but might be of a potential interest to a reader, are briefly mentioned in this section.
The progress made in the field of ion chromatography has been reviewed from various points of view. The most closely related to the topic of this paper is an early review of the suppressed ion chromatographic analysis of anions in environmental waters containing high salt concentrations, which was published in 1996 [4]. The aspect of determining metal pollutants in environmental matrices using ion chromatography was covered by Shaw and Haddad [5].
Ion chromatographic analyses of ordinary drinking waters, tap or spring waters were presented by Michalski [6], who also provided a brief and comprehensive account of the milestones in the development of ion chromatography. Those more interested in the early development of the area can find more detailed outlines written from two different angles in papers of Fritz [7] and Evans [8] while Haddad et al focused on the development of the suppressor technology [9] and more recently on the recent development and emerging directions in ion chromatography [10].
Paull and Nesterenko provided an overview of applications involving the novel ion chromatographic stationary phases for ion chromatographic analyses of complex matrices [11]. Nakatani et al. presented an overview of one of more recent challenges of ion-exclusion/ion-exchange chromatography in determining simultaneously inorganic anions and cations [12]. Monolitic columns, which open up some new opportunities for ion chromatography, were also reviewed [13,14].
3. Ion Chromatographic Analyses of Sea Waters, Brines and Related Samples
This review is divided into subsections in a similar way, as it is a common practice in reporting the results of an extended water analysis, which among other sections usually at the beginning comprises the following sections: anions, cations and metals.
Even though papers focusing on the determinations of a total ionic composition of waters with ion chromatography as completely as possible are very few we start the overview with them, since they well illustrate the potential of this analytical technique, and an anion-cation balance or a charge balance is an important introductory analytical data quality check in any extended water analysis.
3.1. Total Anionic and/or Cationic Composition of Water
3.1.1. Anions and Cations
In ten natural mineral waters of different composition and different total mineralization F−, Cl−, Br−, SO42−, I−, Na+, K+, Mg2+, Ca2+ were successfully determined within three runs on three different columns, one for iodide, another for the remaining anions and the third for the cations. More details are given in Table 1. Taking into account that NO3−, NO2− and HPO42− were not detected, 12 ions of the 20 usually included in an extended chemical analysis of natural waters were covered by ion chromatography alone representing at least 98.60% and up to 99.96% of total cation composition of water samples and between 98.90% and 99.96% of the remaining anion composition after hydrogen carbonate was determined titrimetricaly. Very good agreement of the anion-cation balance or a charge balance was obtained for all mineral waters confirming an excellent performance of ion chromatography for the basic and extended chemical analysis of highly mineralized water samples [15]. Typical chromatograms of a sample are in the original publication presented in Figure 3.

{kind=link}
{kind=link}
Table 1.
Anionic and/or cationic composition of waters with high ionic strength determined as completely as possible with ion chromatography.
| Analyte/Sample | Column | Eluent | Detection | Reference |
|---|---|---|---|---|
| F−, Cl−, Br−, SO42−, I−, Na+, K+, Mg2+, Ca2+ Mineral waters | AG4A, AS4A 1 (20 µeq, BD 15) AG5, AS5 1 (20 µeq, BD 15, LD 120) AG12, CS12 1 (2.8 meq, BD 8) (Dionex, IonPac) | NaHCO3 (15 mM) a NaHCO3, Na2CO3 (1.7 mM, 1.8 mM) a Methanesulphonic Acid (20 mM) b | UV + suppressed conductivity Suppressed conductivity | [15] |
| Cl−, NO3−, SO42−, Na+, K+, Mg2+, Ca2+, Sr2+, Ba2+ Oil field water | Anionic Column 2 Cationic Column 2 Cationic Column 2 (Laboratory prepared, BD 5) | Na benzoate (0.5 mM), Na citrate (0.1 mM) c HNO3 (0.1 mM) c Oxalic acid (1.5 mM), ethylene diamine (1.5 mM) c | Non-suppressed conductivity | [16] |
| F−, Cl−, Br−, NO3−, SO42−, Na+, K+, Mg2+, Ca2+, Sr2+ Sea, estuary water | AG9-HC, AS9-HC 1 (190 µeq, BD 9, LD 90) AG12A, CS12A 1 (2.8 meq, BD 8) (Dionex, IonPac) | Na2CO3 (9 mM) b Methanesulphonic Acid (20 mM) b | Suppressed conductivity | [17] |
| Na+, K+, Mg2+, Ca2+ Brine | AG12A, CS12A 1 (2.8 meq, BD 8) (Dionex, IonPac) | H2SO4 (8.25 mM) d | Suppressed conductivity | [18] |
| Li+, Na+, NH4+, K+, Mg2+, Ca2+, Sr2+ Eutrophic sea, fish otoliths (+Mn2+) | CS12A 1 (2.8 meq, BD 8) (Dionex, IonPac) | Methanesulphonic acid (18 mM) b | Suppressed conductivity | [19] |
Notes: BD, Bead diameter/µm; LD, Latex diameter/nm; mM, mmol/L; 1 Column size, I. D. × Length = (4 × 250) mm; 2 Column size, I. D. × Length = (4.6 × 100) mm; a Flow rate = 2 mL/min; b Flow rate = 1 mL/min; c Flow rate = 1.5 mL/min; d Flow rate = 1.2 mL/min.
Determination of Cl−, NO3−, SO42−, Na+, K+, Mg2+, Ca2+, Sr2+ and Ba2+ in oilfield water was made possible by three runs under the conditions specified in Table 1. The anionic and the cationic column were prepared in the laboratory by chemically modifying 5 μm silica beads. The anionic exchange functionality was achieved by using γ-chloropropyl triethoxy silane, hexamethyl disilazane and N,N-dimethyl benzylamine, phenyl trichlorosilane, chlorosulfonic acid and hexamethyl disilazane were used to obtain the cationic exchange functionality. A good agreement with the results obtained with other conventional methods was confirmed [16]. Typical chromatograms of a sample are in the original publication presented in Figure 2.
Concentrations of F−, Cl−, Br, NO3, SO42−, Na+, K+, Mg2+, Ca2+ and Sr2+ in seawater and derived saline solutions were determined in two single IC runs, one for anions and another one for cations (Table 1) and the results so obtained were critically assessed in terms of anion-cation balance and further related to conductivity and salinity measurements aiming to evaluate the quality/completeness of ion chromatographic analyses and to search for other meaningful relations for efficient recognition/distinction between saline solutions of different types [17]. Typical chromatograms of the samples are in the original publication presented in Figure 3.
3.1.2. Cations
Even though lithium, sodium, ammonium, potassium, rubidium, caesium, calcium, magnesium and strontium were confirmed to be well separated with a gradient elution program the concentrations of Na+, K+, Mg2+ and Ca2+ were later successfully determined in brine samples under isocratic conditions (Table 1) in 1 µL injection volumes which required only minimal dilution and for which good recoveries were confirmed [18].
By successfully determining Li+, Na+, NH4+, K+, Mg2+, Ca2+, Sr2+ in coastal, off-shore and sediment waters and Mn2+ and other already mentioned cations in fish otoliths (Table 1) in the Adriatic Sea and the Canal of Sicily the authors enabled an insight into relation between fish growth and environmental conditions. By observing higher strontium concentrations in coastal waters than off-shore and recognizing a decrease in the Sr/Ca ratio with increasing weight of the otolith they confirmed the hypothesis that juveniles grow in proximity to eutrophic coastline waters [19]. Typical chromatograms of the samples are in the original publication presented in Figures 2 and 3.
3.2. Selected Anions
Among all ion chromatographic analyses of waters with a high ionic strength, the determinations of iodide concentration received the highest attention. An overview comprising the most essential experimental conditions is summarized in Table 2 and continues with the applications in which iodide was determined simultaneously with other anions, and further proceeds with the determinations of bromide as a single ion and ends with bromide and bromate. Another group of related applications, which are summarized in Table 3, is dedicated to nutrients: nitrate, nitrite and phospahate. Methods for ion chromatographic determinations of thiosulfate and polythionates; and perchlorate are described under separate headings at the end of this section and are not included in tables.
Table 2.
Ion chromatographic determinations of iodide or bromide, alone or simultaneously with some other ions in waters with high ionic strength.
| Analyte, Sample | Column | Eluent | Detection | Ref. |
|---|---|---|---|---|
| I− Saline (untreated) | AS11 1 (45 µeq, BD 13, LD 85) (Dionex, IonPac) | Methanesulphonic acid 0.4 mL/L), NaCl (5.84 g/L), 4,4'-bis(dimethylamino)-diphenylmethane (0.8 g/L), Methanol (200 mL/L), water a | Post-column derivatization: N-chlorosuccinimide, succinimide, succinate buffer VIS λ = 605 nm | [20] |
| I−, Sea water (untreated) | AG4A-SC, AS4A-SC 1(20 µeq, BD 13, LD 160) (Dionex, IonPac) | NaOH (100 mM) b | UV λ = 226 nm | [21] |
| I− Sea water (untreated, 2 mL) | Semi-microcolumn 2 Stationary phase: TSKgel SAX (Tosoh) (3.7 meq/g, BD 5) | NaClO4 (30 mM), NaCl (500 mM), sodium phosphate buffer pH 6.0 (5 mM) c | UV λ = 226 nm | [22] |
| I−, Sea water (untreated) | Zwittergent-3-14 modified ODS column 3 (BD 5) | NaClO4 (0.2 mM) Zwittergent-3-14 (0.3 mM) c | UV λ =210 nm | [23] |
| I− Sea water (untreated) | Microcolumn 4: PEG/C30 binary (BD 5) (Laboratory prepared) | Na2SO4 (500 mM), NaCl (50 mM) d | UV λ = 220 nm | [24] |
| I˗ Sea water (untreated) | Microcolumn 4: Polyoxyethylene oleyl ether modified C30 (BD 5) (Laboratory prepared) | NaCl (300 mM) d | UV λ = 220 nm | [25] |
| I−, IO3− Sea water (diluted) | G3154A/102, G3154A/101 5 (50 µeq/g, BD 10) (Agilent) | NH4NO3 (20 mM), pH 5.6 c | ICP-MS | [26] |
| I−, Br−, SO42− Oilfield waters (diluted) | AG9-SC, AS9-SC 1 ((30 to 35) µeq, BD 13, LD 110) (Dionex, IonPac) | Na2CO3 (3 mM) e | Suppressed conductivity | [27] |
| I−, Br−, NO3− Sea water | Zwittergent-3-14 modified ODS 3 | Diluted artificial sea water c | UV λ = 210 nm | [28] |
| Br− Sea water (untreated) | Zwittergent-3-14 modified ODS 3 | Pure water c | UV λ = 220 nm Non-suppressed conductivity | [29] |
| Br− Sea water (untreated) | Cetyltrimethylammonium ion modified monolithic silica capillary columns 6 (Laboratory prepared) | NaCl (500 mM), cetyltrimethylammonium chloride (0.1 mM) f | UV λ = 210 nm | [30] |
| Br−, BrO3− Sea water (diluted) | G3154A/101 (guard, separation column 5, 50 µeq/g, BD 10 (Agilent) | NH4NO3 (20 mM), pH 5.8 c | ICP-MS | [31] |
Notes: BD, Bead diameter/µm; LD, Latex diameter/nm; mM, mmol/L; 1 Column size, I. D. × Length = (4 × 250) mm; 2 Column size, I. D. × Length = (1 × 35) mm; 3 Column size, I. D. × Length = (4.6 × 250) mm; 4 Column size, I. D. × Length = (0.32 × 100) mm; 5 Column size, I. D. × Length = (4.6 × 150) mm; 6 Column size, I. D. × Length = (0.1 × 200) mm; a Flow rate = 0.9 mL/min; b Flow rate = 1.5 mL/min; c Flow rate = 1 mL/min; d Flow rate = 2.1 µL/min; e Flow rate = 2 mL/min; f Flow rate = 5.6 µL/min.
Table 3.
Ion chromatographic determinations of concentrations of nutrients in samples with high ionic strength.
| Analyte, Sample | Column | Eluent | Detection | Ref. |
|---|---|---|---|---|
| NO3−, KCl soil extracts (diluted) | Hamilton PRP X-100 (Hamilton) | KCl (1000 mM) a | UV λ =210 nm | [32] |
| NO3−, NO2− Sea water (untreated) | Cetyltrimethylammonium chloride coated ODS monolithic columns Chromolith Speed ROD RP-18e 1, Chromolith RP-18e 2 (Merck) | NaCl (500 mM), sodium phosphate buffer (5 mM) pH 4.7 b | UV λ =225 nm | [33] |
| NO3−, NO2−, PO43− Estuary water (untreated) | AG9-HC, AG9-HC3, Valve AS9-HC 3 (190 µeq, BD 9, LD 90) (Dionex, IonPac) | NaHCO3, Na2CO3 (3 mM, 14 mM) c (elute-off) Na2CO3 (9 mM) c (elute) | Suppressed conductivity UV λ =225 nm | [34] |
| NO3−, NO2−, PO43− Saline Aquarium water | Metrosep ASUPP7-250 3 (108 µeq, BD 5) (Metrohm) | Na2CO3 (3.5 mM) d | Suppressed conductivity UV λ =215 nm | [35] |
| NO3−, PO43− Estuary water (untreated) | AG4A-SC, AS4A-SC3 (20 µeq, BD13, LD 160) (Dionex, IonPac) | NaHCO3, Na2CO3 (0.765 mM, 0.81 mM) a | Suppressed conductivity UV λ = 210 nm | [36] |
| NO3−, PO43− Marine sediment pore water | AG4A, AG4A, Valve AS4A-SC 3 (20 µeq, BD13, LD 160) (Dionex, IonPac) | NaHCO3, Na2CO3 (Two compositions, elute-off/elute) 1.4 mM, pH 9.16 and 20 mM, pH 9.5) b | Suppressed conductivity | [37] |
| PO43− Sea water (untreated) | ICE-AS1 4 (27 meq, BD 7.5) (Dionex, IonPac) | HCl (50 mM ) e | ICP-MS | [38] |
Notes: BD, Bead diameter/μm; LD, Latex diameter/nm; mM, mmol/L; 1 Column size, I. D. × Length = (4.6 × 50) mm; 2 Column size, I. D. × Length = (4.6 × 100) mm; 3 Column size, I. D. × Length = (4 × 250) mm; 4 Column size, I. D. × Length = (9 × 250) mm; a Flow rate = 2.0 mL/min; b Flow rate = 3.0 mL/min; c Flow rate = 1.0 mL/min; d Flow rate = 0.7 mL/min; e Flow rate = 1.2 mL/min.
3.2.1. Iodide
The first six papers listed in Table 2 describe methods for determining iodide in untreated sea water samples [20,21,22,23,24,25]. Brandao et al. [20] used a post-column derivatization with N-chlorosuccinimide followed by a spectrometric detection at the wavelength 605 nm; in contrast to other authors who reported a direct spectrometric detection of iodide at the wavelengths between 210 and 226 nm. Typical chromatogram of a sample is in the original publication presented in Figure 2. Several methods share a similar approach for minimizing the interfering effect of chloride on the determination of iodide. This is most commonly resolved so that sodium chloride is one of the eluent components [20,22,24] or a sole eluent component [25]. In contrast to this approach Hu et al. prevented the interfering effect of chloride by reducing its affinity towards the stationary phase, by the technique termed electrostatic ion chromatography (EIC), in which zwitterionic surfactant layer is applied on the stationary phase and the zwitterionic surfactant was also present in the eluent [23]. Typical chromatograms of an unspiked and spiked sample are in the original publication presented in Figure 1. Ito used a large 2 mL sample injection volume and proved that by replacing a macrocolumn with a semi-microcolumn with an I.D. 1 mm the method sensitivity increased 2.8-fold [22]. Typical chromatogram of a sample is in the original publication presented in Figure 5. Rong et al. successfully used two different in house prepared microcolumns with an I. D. 0.32 mm for determining iodide in sea water [24,25]. Typical chromatograms of the samples are in the original publications presented in Figure 6 and 4, respectively. Limits of detection (S/N = 3) reported for iodide in different papers were 0.2 µg/L [22],0.8 ppb [20], 0.011 µmol/L [23], 13 µg/L [24] and 19 µg/L [25], respectively. Chandramouleeswaran et al. achieved the recoveries of iodide higher than 98% [21], Ito reported the recoveries 98.4% or 97.0% for a 2 mg/L or 5 mg/L iodide addition, respectively [22].
3.2.2. Iodide and Iodate
Linear plots were obtained in a concentration range (5.0 to 500) µg/L and the detection limits 1.5 µg/L and 2.0 µg/L were achieved for a simultaneous determination of iodate and iodide in seawater, respectively with a non-suppressed ion chromatographic method with inductively coupled plasma mass spectrometric detection (ICP-MS). An anion-exchange column (G3154A/101, Agilent) and an NH4NO3eluent at the pH 5.6 were used. The recoveries for iodate and iodide were 101.3% ± 1.8% and 97.2% ± 2.3%, respectively [26].
3.2.3. Iodide, Bromide and Sulfate
Bromide, iodide and sulfate are very important for the characterization of oilfield waters. An Dionex IonPac AS9-SC analytical column, sodium carbonate eluent and a suppressed conductometric detection were used and the limits of detection 0.1 mg/L for bromide and sulfate and 0.2 mg/L for iodide were achieved [27] Typical chromatograms of the samples are in the original publication presented in Figures 3 and 4.
3.2.4. Iodide, Bromide and Nitrate
An octadecylsilica column modified with a zwitterionic surfactant [3-(N,N-dimethylmyristylammonio) propanesulfonate] and a diluted artificial sea water as the eluent were used for simultaneous determination of iodide, bromide and nitrate in sea water with spectrometric detection at the wavelength 210 nm. Limits of detection were 0.75 µg/L for bromide, 0.52 µg/L for nitrate, and 0.8 µg/L for iodide [28]. Typical chromatogram of a sample is in the original publication presented in Figure 5.
3.2.5. Bromide
The concentration of bromide was directly determined in sea water with EIC with spectrometric and conductometric detection, the results obtained with this method were 0.059 mmol/L ± 1.0% and 0.057 mmol/L ± 0.94%, and were comparable with the results of IC. The zwitterionic surfactants immobilized on octadecylsilica surfaces acted as the stationary phase and water was used as the mobile phase [29]. Typical chromatograms of the samples are in the original publication presented in Figures 5 and 6.
Monolithic silica capillary columns, which were prepared with a sol–gel method, were dynamically modified with cetyltrimethylammonium ion and enabled a determination of 63 mg/L of bromide in 20 nL of untreated sea water sample. The eluent contained NaCl and cetyltrimethylammonium chloride (CTAC). Spectrometric detection was performed at the wavelength 210 nm [30]. Typical chromatogram of a sample is in the original publication presented in Figure 4.
3.2.6. Bromide and Bromate
An ion chromatographic method employing a G3154A/101 guard and separation column (Agilent), NH4NO3eluent at pH 5.8 and ICP-MS detection was developed, which enabled a direct determination of bromate and bromide in a diluted sea water sample with a limit of detection which ranged from (2.0 to 3.0) μg/L [31]. Typical chromatogram of a sample is in the original publication presented in Figure 2.
3.2.7. Nitrate
Nitrate was successfully determined in KCl soil extracts with ion chromatography employing KCl solution as an eluent and performing a spectrometric detection at the wavelength 210 nm. Bromide interference was avoided by spiking the diluted soil extracts with a constant amount of nitrate, which shifted a bromide nitrate ratio so that bromide did not interfere any more, and consequently nitrate was reliably determined in concentrations down to 25 µg/L [32].
3.2.8. Nitrite and Nitrate
Nitrite and nitrate were well resolved and eluted within three minutes from the CTAC modified monolithic ODS columns and successfully determined in untreated sea water in concentrations (10.8 ± 0.5) µg/L, and (21.3 ± 0.5) µg/L if the NaCl, sodium phosphate buffer eluent was used and spectrometric detection was performed at the wavelength 225 nm. The limits of detection (S/N = 3) for nitrite and nitrate were 0.8 μg/L and 1.6 µg/L, respectively [33]. Typical chromatogram of a sample is in the original publication presented in Figure 3.
3.2.9. Nitrate, Nitrite and Phosphate
For nitrate, nitrite and phosphate the limits of detection (0.1, 0.3 and 1) mg/L were achieved, respectively with the suppressed conductometric and spectrometric detection in series at the wavelength 225 nm. Chloride was eliminated with a column-switching method. Nitrate at the concentration level 5 mg/L was successfully determined in an estuary water, while nitrite and phosphate were undetectable [34]. Typical chromatogram of a sample is in the original publication presented in Figure 5.
Nitrate-N, nitrite-N and phosphate-P were successfully determined in saline aquarium water in concentrations as low as (0.026, 0.021 and 0.04) mg/L, respectively with the suppressed conductometric and spectrometric detection at the wavelength 215 nm. Concentrations of these anions in sea water were below the limits of detection [35].
3.2.10. Nitrate and Phosphate
An ion chromatographic method using spectrometric detection at the wavelength 210 nm for nitrate determination and suppressed conductometric detection for phosphate determination was reported. A limit of detection for phosphate in sea water was proved to be 0.1 mg/L and this ion was successfully determined in the estuary waters in concentrations between 0.3 and 1.6 mg/L during the winter months when salinity was lower due to dilution caused by the rainfall, and the nutrients were more intensively supplied by the in-coming streams. Concentration of nitrate between 0.3 and 11.1 mg/L were determined in the samples and the limit of detection was found out to be 0.04 mg/L [36]. Typical chromatogram of a sample is in the original publication presented in Figure 3.
The 0.5 µmol/L and 1 µmol/L limits of detections were achieved for nitrate and phosphate determination in the marine sediment pore water with an ion chromatographic method with suppressed conductometric detection. Experimental conditions defined by six factors were optimized using a fractional factorial experimental design. A column-switching method was used for reducing the chloride interference effect [37]. Typical chromatogram of a sample is in the original publication presented in Figure 6.
3.2.11. Phosphate
The 0.06 µmol/L limit of detection (3s) was achieved with ion-exclusion chromatography with ICP-MS detection for phosphate determination in untreated sea water. The concentration of dissolved phosphate determined in sea-water with this ion chromatographic method (1.69 ± 0.04) µmol/L is in good agreement with the result of the colorimetric procedure (1.71 ± 0.04) µmol/L [38]. Typical chromatogram of a sample is in the original publication presented in Figure 4.
3.2.12. Thiosulfate and Polythionates
A pre-concentration technique using an ion-exchange pre-column and a reversed phase analytical column in series and a water-acetonitrile eluent containing tetrabutylammonium ions and carbonate buffer was developed for determining thiosulfate and polythionates in natural saline waters. Limits of detection achieved with a pre-concentration of 6 mL of 50-times diluted seawater were: 1 nmol/L for trithionate, and 0.3 nmol/L for tetrathionate and pentathionate [39].
3.2.13. Perchlorate
The limit of detection 0.77 µg/L (S/N = 3) was reported for an ion chromatographic method intended for perchlorate determination in waters of different conductivities ranging up to 14.7 mS/cm. With a pre-concentration of a 2 mL sample volume and elimination of interfering ions by column-switching, higher recoveries of perchlorate ion were achieved than with the recommended EPA Method [40].
3.3. Selected Cations
The more comprehensive ion chromatographic determinations of cations closer matching the total cationic composition of water samples were presented in Section 1.1. Here we present methods that focus on determinations of selected cations or a single cation. The main experimental conditions defining these methods are summarized in Table 4.
Table 4.
Ion chromatographic determinations of concentrations of selected cations in waters with high ionic strength.
| Analyte, Sample | Column | Eluent | Detection | Ref. |
|---|---|---|---|---|
| Mg2+, Ca2+, Sr2+ Brine, Sea water (diluted) | CG-2, CS-2 (Dionex, IonPac) | Ethylenediamine (2.5 mM),HCl (5 mM), zinc acetate (1 mM) | Suppressed conductivity | [41] |
| Sr2+ Sea water (diluted) | high-speed cationic column (Cat.no.269-25, Wescan) | Ethylenediamine (0.5 mM), oxalic acid (0.5 mM), pH 6 a | Non-suppressed conductivity | [42] |
| NH4+ Sea water(diluted) | CG-12A Valve CS-12A1 (2800 µeq, BD 8) (Dionex, IonPac) | H2SO4 (25 mM) b | Suppressed conductivity | [43] |
| NH4+ Sea water (purge-and-trap) | CG-16A CS-16A2 (8400 µeq, BD 5) (Dionex, IonPac) | Methanesulphonic Acid (45 mM) b | Suppressed conductivity | [44] |
Notes: BD, Bead diameter/μm; LD, Latex diameter/nm; mM, mmol/L; 1 Column size, I. D. × Length = (4 × 250) mm; 2 Column size, I. D. × Length = (5 × 250) mm; a Flow rate = 1.2 mL/min; b Flow rate = 1 mL/min.
3.3.1. Magnesium, Calcium and Strontium
Strontium, magnesium and calcium were successfully simultaneously determined in high salinity sub-surface waters with ion chromatography with suppresses conductometric detection using an eluent which comprised ethylenediamine, HCl and zinc acetate, and the results compared well with those obtained with atomic absorption spectrometry [41].
3.3.2. Strontium
A non-suppressed ion chromatographic method with conductometric detection was developed for determination of strontioum in coastal sea water and the results compared well with those obtained by ICP-OES. The eluent comprised ethylenediamine and oxalic acid. The limit of detection for strontium determination was found to be 1 mg/L [42]. Typical chromatogram of a sample is in the original publication presented in Figure 5.
3.3.3. Ammonium
Ion chromatographic method, with suppressed conductometric detection and an on-line sample pre-concentration with a column-switching, was developed for ammonium determination in sea water. The limit of detection for ammonium ion in the presence of 1000 mg/L sodium was 12.8 µg/L. It was proved that 90% of ammonium was introduced and concentrated if concentration was lower than 1 mg/L [43].Typical chromatogram of a sample is in the original publication presented in Figure 3.
The 1.35 µg/L (75 nmol/L) limit of detection was confirmed for ammonium determination in sea water samples with the approach that combined a purge-and-trap pre-concentration step with ion chromatography with suppressed conductometric detection. A linear range for ammonium extended from 50 nmol/L to 6.0 µmol/L [44]. Typical chromatogram of a sample is in the original publication presented in Figure 7.
3.4. Metals
The main experimental conditions for ion chromatographic determinations of metals in samples with high ionic strength are summarized in Table 5. Chelation ion chromatography as a pre-concentration step in combination with spectrometric detection in the visible range after post-column derivatization is most frequently reported. More details are given in continuation.
| Analyte, Sample | Column | Eluent | Detection | Ref. |
|---|---|---|---|---|
| Zn, Pb, Ni, Cu Sea water, CASS-2 coastal sea-water certified reference material | Xylenol Orange-impregnated polystyrene-divinylbenzene neutral PLPRS resin (Polymer Laboratories) (BD 5) | KNO3 (500 mM), lactic acid (50 mM), pH adjustment NH3 or HNO3a | Post-column derivatization: Pyridyl-azo-resorcinol, NH3, NH4NO3 pH 10.2 VIS λ = (490 to 520) nm | [45] |
| Cu, Ni, Zn, Co Reference materials for trace metals estuarine water (SLEW-2) (diluted, pH 3) | MetPac-CC1 (iminodiacetate chelating concentrator column) CG5, CS5 1(BD 13, LD 110, 150 μeq sulfonic, 70 μeq alkanol quaternary ammonium) (Dionex, IonPac) | Pyridine-2, 6-dicarboxylic acid (6 mM), acetate buffer CH3COOH, (50 mM), CH3COONa (50 mM), pH 4.5 a | Post-column derivatization: Pyridyl-azo-resorcinolNH3, CH3COOH, pH 9.7 VIS λ = 520 nm | [46] |
| Selenium species Artificial sea water | AS10, AS10 1 (170 µeq, BD 8.5, LD 65) AG11, AS11 1 (45 µeq, BD 13, LD 85) (Dionex, IonPac) | Na2CO3 (10 mM) a NaOH (10 mM) a | ICP-AES | [47] |
| La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er Sea water | N-methyl-γ-amino-butyrohydroxamate resin guard and concentrator column 2, (1.5 to l.9) mM/g CS5 3(BD 13, LD 110, 150 μeq sulfonic, 70 μeq alkanol quaternary ammonium) (Dionex, IonPac) | Oxalic acid (30 mM, pH 4.5), diglycolic acid (5 mM) | Post-column derivatization: 4(2-pyridylazo)resorcinol ZnEDTA, NH3, NH4Cl, pH 10, VIS λ = 495 nm | [48] |
| Mn, Co, Ni, Cu, Zn, No, Cd, Sb, Pb, U Sea-Water Ref. Material NASS-4 | Nitrilotriacetate (NTA)-type chelating poly (styrene-divinylbenzene) resin 4 | KNO3 (500 mM) a | ICP-MS | [49] |
| Be Artificial sea water | Iminodiacetic acid functionalised silica gel (Bio-ChemMack) (BD 8)1 | KNO3 (400 mM), adjusted to pH 2.5 using HNO3 a | Post-column deriv.: chrome azurol S, Triton X-100, 2-(N-morpholino) ethane-sulfonic acid, pH 6.0, VIS λ = 590 nm | [50] |
| V, Mo, W Artificial sea water | Chelated polystyrene-divinylbenzene resins: BAETM (bis (2-aminoethylthio) methylated) 5 Chelated acrylonitrile and divinylbenzene resin: γ-ABHX (γ-aminobutyrohydroxamate) 6 | HNO3 pH 1.5 a | ICP-MS | [51] |
| Cd, Pb, Cu Mineral waters | Aurin tricarboxylic acid impregnated polystyrene divinylbenzene resin 3 (BD 7) | KNO3 (100 mM) pH gradient 1 | Post-column derivatiz.: borate buffer, 4-(2-pyridylazo)resorcinol VIS λ = 520 nm | [52] |
Notes: BD, Bead diameter/μm; LD, Latex diameter/nm; mM, mmol/L; 1 Column size, I. D. × Length = (4 × 250) mm; 2 Column size, I. D. × Length = (2 × 50) mm; 3 Column size, I. D. × Length = (4.6 × 250) mm; 4 Column size, I. D. × Length = (4.6 × 50) mm; 5 Column size, I. D. × Length = (4 × 50) mm; 6 Column size, I. D. × Length = (6 × 30) mm; a Flow rate = 1 mL/min.
Low µg/L levels of Zn-II, Pb-II, Ni-II and Cu-II were determined in CASS-2 coastal sea-water certified reference material with chelation ion chromatography with a pre-concentration and separation on a Xylenol Orange-impregnated resin, and spectrometric detection in the visible range after post-column derivatization [45]. Typical chromatograms of the samples are in the original publication presented in Figures 1 and 2.
The limits of detection 0.025 µg/L, 0.036 µg/L, 0.064 µg/L and 0.022 µg/L were achieved for copper, nickel, zinc and cobalt, respectively with a pre-concentration of a 25 mL-sample on an iminodiacetate chelating column. Desorption from the concentrator column and separation in an ion exchange column was achieved with an eluent containing pyridine-2,6-dicarboxylic acid in acetate buffer. Detection was spectrometric in the visible range after post-column derivatization, samples had to be diluted and pH set to 3. The reference material for trace metals in estuarine water (SLEW-2) was successfully analysed [46]. Typical chromatograms of the samples are in the original publication presented in Figure 8.
Inorganic and amino acid forms of selenium were determined by ion chromatography with inductively coupled plasma atomic emission spectroscopic (ICP-AES) detection. Three different columns were tested. The columns AS10 and AS11 (Dionex) both proved useful, but limits of detection were lower for the later and ranged between 0.32 mg/L (Se-methionine) and 0.62 mg/L (Se-cystine) and were close to 0.5 mg/L for HSeO3− and HSeO42− [47]. Typical chromatograms of the samples are in the original publication presented in Figures 1 and 2.
The 2.5 μg/L detection limit was achieved for the determination of lanthanides in sea water by a pre-concentration of 25 mL of sea water on a chelation stationary phase followed by ion chromatographic separation and spectrometric detection in the visible range after a post-column derivatization [48]. Typical chromatogram of a sample is in the original publication presented in Figure 5.
The results of the determination of the concentrations of metals (Mn, Co, Ni, Cu, Zn, No, Cd, Sb, Pb and U) in the Open Ocean Sea-Water Reference Material NASS-4 obtained with a chelation ion chromatographic method with ICP-MS detection were in good agreement with the certified values. With the efficient preconcentration and simultaneous matrix elimination, the detection limits ranged from 4 ng/L for Cu to 0.7 µg/L for Mo [49].
Beryllium was successfully determined in artificial sea water using an iminodiacetic acid functionalized silica gel column and spectrometric detection in the visible range after a post-column derivatization. Detection limits for Be(II) in a standard solution and a typical tap water sample were 3 μg/L and 4 µg/L, respectively [50]. Typical chromatograms of the samples are in the original publication presented in Figures 5 and 6. Two kinds of chelating resin, bis (2-aminoethylthio) methylated resin (BAETM) and gamma-aminobutyrohydroxamate resin (gamma-ABHX) were synthesized and tested. The detection limit 0.01 µg/L was achieved for the determination of vanadium, molybdenum and tungsten with a BAETM resin column. The γ-ABHX resin column demonstrated efficient separation between target metals and sea water matrix cations [51]. Typical chromatogram of a sample is in the original publication presented in Figure 7.
Metal ions Cd (II), Pb (II) and Cu (II) were baseline separated from other transition metals and the alkaline earth metals within 30 min and their concentrations determined in highly mineralized water with a chromatographic system comprising aurin tricarboxylic acid impregnated polystyrene divinylbenzene resin, pH gradien elution and post-column derivatization with spectrometric detection in the visible range. The limits of detection were 1 µg/L for Cd (II) and 5 µg/L for both Pb (II) and Cu (II),respectively [52]. Typical chromatogram of a sample is in the original publication presented in Figure 7.
4. Conclusions
Even though ion chromatography is four decades after its introduction in the mid-seventies a well-established analytical technique with very many applications, it can be concluded that applications dedicated to analyses of natural waters with high ionic strength are not so numerous, confirming that they still represent an analytical challenge. Less frequent are applications targeting the determination of the total ionic composition of highly mineralized waters with ion chromatography as completely as possible, and those trying to master ion chromatographic analyses of highly mineralized waters of different types.
References
- Carlé, W. Die Mineral- und Thermalwässer von Mitteleuropa, Geologie, Chemismus, Genese; Wissenschaftliche Verlagsgesellschaft MBH: Stuttgart, Germany, 1975. [Google Scholar]
- Gros, N. The comparison between slovene and central european mineral and thermal waters. Acta Chim.Slov. 2003, 50, 57–66. [Google Scholar]
- Gros, N.; Gorenc, B. Simple recognition of similar samples for the ion-chromatographic determination of the main cations. J. Chromatogr. A 1997, 789, 323–327. [Google Scholar] [CrossRef]
- Singh, R.P.; Abbas, N.M.; Smesko, S.A. Suppressed ion chromatographic analysis of anions in environmental waters containing high salt concentrations. J. Chromatogr. A 1996, 733, 73–91. [Google Scholar] [CrossRef]
- Shaw, M.J.; Haddad, P.R. The determination of trace metal pollutants in enviromental matrices using ion chromatography. Environ. Int. 2004, 30, 403–431. [Google Scholar] [CrossRef]
- Michalski, R. Ion chromatography as a reference method for determination of inorganic ions in water and wastewater. Crit. Rev. Anal. Chem. 2006, 36, 107–127. [Google Scholar] [CrossRef]
- Fritz, J.S. Early milestones in the development of ion-exchange chromatography: A personal account. J. Chromatogr. A 2004, 1039, 3–12. [Google Scholar] [CrossRef]
- Evans, B. The history of ion chromatography: The engineering perspective. J. Chem. Educ. 2004, 81, 1285–1292. [Google Scholar] [CrossRef]
- Haddad, P.R.; Jackson, P.E.; Shaw, M.J. Developments in suppressor technology for inorganic ion analysis by ion chromatography using conductivity detection. J. Chromatogr. A 2003, 1000, 725–742. [Google Scholar] [CrossRef]
- Haddad, P.R.; Nesterenko, P.N.; Buchberger, W. Recent developments and emerging directions in ion chromatography. J. Chromatogr. A 2008, 1184, 456–473. [Google Scholar] [CrossRef]
- Paull, B.; Nesterenko, P.N. Novel ion chromatographic stationary phases for the analysis of complex matrices. Analyst 2005, 130, 134–146. [Google Scholar] [CrossRef]
- Nakatani, N.; Kozaki, D.; Mori, M.; Tanaka, K. Recent progress and applications of ion-exclusion/ion-exchange chromatography for simultaneous determination of inorganic anions and cations. Anal. Sci. 2012, 28, 845–852. [Google Scholar] [CrossRef]
- Chambers, S.D.; Glenn, K.M.; Lucy, C.A. Developments in ion chromatography using monolithic columns. J. Sep. Sci. 2007, 30, 1628–1645. [Google Scholar] [CrossRef]
- Nordborg, A.; Hilder, E.F. Recent advances in polymer monoliths for ion-exchange chromatography. Anal. Bioanal. Chem. 2009, 394, 71–84. [Google Scholar] [CrossRef]
- Gros, N.; Gorenc, B. Performance of ion chromatography in the determination of anions and cations in various natural waters with elevated mineralization. J. Chromatogr. A 1997, 770, 119–124. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, S.X.; Chen, L.R.; Xu, Y.Q.; Ma, P. Determination of inorganic ions in oil field waters by single-column ion chromatography. J. Chromatogr. A 1997, 789, 569–573. [Google Scholar] [CrossRef]
- Gros, N.; Camoes, M.F.; Oliveira, C.; Silva, M.C.R. Ionic composition of seawaters and derived saline solutions determined by ion chromatography and its relation to other water quality parameters. J. Chromatogr. A 2008, 1210, 92–98. [Google Scholar] [CrossRef]
- Hodge, E.M.; Martinez, P.; Sweetin, D. Determination of inorganic cations in brine solutions by ion chromatography. J. Chromatogr. A 2000, 884, 223–227. [Google Scholar] [CrossRef]
- Marini, M.; Campanelli, A.; Abballe, F. Measurement of alkaline and earthy ions in fish otolith and sea water using a high performance ion chromatography. Mar. Chem. 2006, 99, 24–30. [Google Scholar] [CrossRef]
- Brandao, A.C.M.; Buchberger, W.W.; Butler, E.C.V.; Fagan, P.A.; Haddad, P.R. Matrix-elimination ion chromatography with postcolumn reaction detection for the determination of iodide in saline waters. J. Chromatogr. A 1995, 706, 271–275. [Google Scholar] [CrossRef]
- Chandramouleeswaran, S.; Vijayalakshmi, B.; Kartihkeyan, S.; Rao, T.P.; Iyer, C.S.P. Ion-chromatographic determination of iodide in sea water with uv detection. Mikrochim. Acta 1998, 128, 75–77. [Google Scholar] [CrossRef]
- Ito, K. Semi-micro ion chromatography of iodide in seawater. J. Chromatogr. A 1999, 830, 211–217. [Google Scholar] [CrossRef]
- Hu, W.Z.; Yang, P.J.; Hasebe, K.; Haddad, P.R.; Tanaka, K. Rapid and direct determination of iodide in seawater by electrostatic ion chromatography. J. Chromatogr. A 2002, 956, 103–107. [Google Scholar] [CrossRef]
- Rong, L.; Lim, L.W.; Takeuchi, T. Determination of iodide in seawater samples by ion chromatography with chemically-bonded poly(ethylene glycol) stationary phase. J. Chromatogr. A 2006, 1128, 68–72. [Google Scholar] [CrossRef]
- Rong, L.; Lim, L.W.; Takeuchi, T. Determination of iodide in seawater using c30 column modified with polyoxyethyleneoleyl ether in ion chromatography. Talanta 2007, 72, 1625–1629. [Google Scholar] [CrossRef]
- Chen, Z.L.; Megharaj, M.; Naidu, R. Speciation of iodate and iodide in seawater by non-suppressed ion chromatography with inductively coupled plasma mass spectrometry. Talanta 2007, 72, 1842–1846. [Google Scholar]
- Kadnar, R.; Rieder, J. Determination of anions in oil-field waters by ion chromatography. J. Chromatogr. A 1995, 706, 301–305. [Google Scholar] [CrossRef]
- Hu, W.Z.; Haddad, P.R.; Hasebe, K.; Tanaka, K.; Tong, P.; Khoo, C. Direct determination of bromide, nitrate, and iodide in saline matrixes using electrostatic ion chromatography with an electrolyte as eluent. Anal. Chem. 1999, 71, 1617–1620. [Google Scholar] [CrossRef]
- Hu, W.Z.; Cao, S.A.; Tominaga, M.; Miyazaki, A. Direct determination of bromide ions in sea water by ion chromatography using water as the mobile phase. Anal. Chim. Acta 1996, 322, 43–47. [Google Scholar] [CrossRef]
- Suzuki, A.; Lim, L.W.; Hiroi, T.; Takeuchi, T. Rapid determination of bromide in seawater samples by capillary ion chromatography using monolithic silica columns modified with cetyltrimethylammonium ion. Talanta 2006, 70, 190–193. [Google Scholar] [CrossRef]
- Chen, Z.L.; Megharaj, M.; Naidu, R. Determination of bromate and bromide in seawater by ion chromatography, with an ammonium salt solution as mobile phase, and inductively coupled plasma mass spectrometry. Chromatographia 2007, 65, 115–118. [Google Scholar] [CrossRef]
- Raessler, M.; Hilke, I. Ion-chromatographic determination of low concentrations of nitrate in solutions of high salinity. Microchim. Acta 2006, 154, 27–29. [Google Scholar] [CrossRef]
- Ito, K.; Takayama, Y.; Makabe, N.; Mitsui, R.; Hirokawa, T. Ion chromatography for determination of nitrite and nitrate in seawater using monolithic ods columns. J. Chromatogr. A 2005, 1083, 63–67. [Google Scholar] [CrossRef]
- Bruno, P.; Caselli, M.; de Gennaro, G.; de Tommaso, B.; Lastella, G.; Mastrolitti, S. Determination of nutrients in the presence of high chloride concentrations by column-switching ion chromatography. J. Chromatogr. A 2003, 1003, 133–141. [Google Scholar] [CrossRef]
- Wilson, B.; Gandhi, J.; Zhang, C.L. Analysis of inorganic nitrogen and related anions in high salinity water using ion chromatography with tandem UV and conductivity detectors. J. Chromatogr. Sci. 2011, 49, 596–602. [Google Scholar] [CrossRef]
- Carrozzino, S.; Righini, F. Ion-chromatographic determination of nutrients in sea-water. J. Chromatogr. A 1995, 706, 277–280. [Google Scholar] [CrossRef]
- Dahllof, I.; Svensson, O.; Torstensson, C. Optimising the determination of nitrate and phosphate in sea water with ion chromatography using experimental design. J. Chromatogr. A 1997, 771, 163–168. [Google Scholar] [CrossRef]
- Yang, L.; Sturgeon, R.E.; Lam, J.W.H. On-line determination of dissolved phosphate in sea-water by ion-exclusion chromatography inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom. 2001, 16, 1302–1306. [Google Scholar] [CrossRef]
- Weir, S.I.; Butler, E.C.V.; Haddad, P.R. Ion chromatography with UV detection for the determination of thiosulfate and polythionates in saline waters. J. Chromatogr. A 1994, 671, 197–203. [Google Scholar] [CrossRef]
- Tian, K.; Dasgupta, P.K.; Anderson, T.A. Determination of trace perchlorate in high-salinity water samples by ion chromatography with on-line preconcentration an preelution. Anal. Chem. 2003, 75, 701–706. [Google Scholar] [CrossRef]
- Singh, R.P.; Pambid, E.R.; Debayle, P.; Abbas, N.M. Ethylenediamine hydrochloric-acid zinc(II) eluent for the suppressed ion chromatographic-separation of strontium(II) from a large amount of calcium(II)—Application of the method to the simultaneous determination of magnesium(II), calcium(II) and strontium(II) in high salinity subsurface waters. Analyst 1991, 116, 409–414. [Google Scholar] [CrossRef]
- Butt, S.B.; Farhat, W.; Jan, S.; Ahmed, S.; Mohammad, B.; Akram, N. Optimization of a mobile phase for monitoring strontium in seawater using non-suppressed ion chromatography. J. Liq. Chromatogr. Relat. Technol. 2004, 27, 1729–1742. [Google Scholar] [CrossRef]
- Huang, Y.; Mou, S.; Riviello, J.M. Determination of ammonium in seawater by column-switching ion chromatography. J. Chromatogr. A 2000, 868, 209–216. [Google Scholar] [CrossRef]
- Wang, P.Y.; Wu, J.Y.; Chen, H.J.; Lin, T.Y.; Wu, C.H. Purge-and-trap ion chromatography for the determination of trace ammonium ion in high-salinity water samples. J. Chromatogr. A 2008, 1188, 69–74. [Google Scholar] [CrossRef]
- Paull, B.; Foulkes, M.; Jones, P. High-performance chelation ion-chromatographic determination of trace-metals in coastal sea-water using dye-impregnated resins. Analyst 1994, 119, 937–941. [Google Scholar] [CrossRef]
- Motellier, S.; Pitsch, H. Simultaneous analysis of some transition metals at ultra-trace level by ion-exchange chromatography with on-line preconcentration. J. Chromatogr. A 1996, 739, 119–130. [Google Scholar] [CrossRef]
- Harwood, J.J.; Su, W. Analysis of organic and inorganic selenium anions by ion chromatography inductively coupled plasma atomic emission spectroscopy. J. Chromatogr. A 1997, 788, 105–111. [Google Scholar] [CrossRef]
- Liu, C.Y.; Lee, N.M.; Wang, T.H. Chelation ion chromatography as a technique for trace elemental analysis in complex matrix samples. Anal. Chim. Acta 1997, 337, 173–182. [Google Scholar] [CrossRef]
- Kumagai, H.; Yamanaka, M.; Sakai, T.; Yokoyama, T.; Suzuki, T.M.; Suzuki, T. Determination of trace metals in sea-water by inductively coupled plasma mass spectrometry interfaced with an ion chromatographic separation system: Effectiveness of nitrilotriacetate chelating resin as the column stationary phase for preconcentration and elimination of matrix effects. J. Anal. At. Spectrom. 1998, 13, 579–582. [Google Scholar] [CrossRef]
- Bashir, W.; Paull, B. Sensitive and selective ion chromatographic method for the determination of trace beryllium in water samples. J. Chromatogr. A 2001, 910, 301–309. [Google Scholar] [CrossRef]
- Huang, C.Y.; Lee, N.M.; Lin, S.Y.; Liu, C.Y. Determination of vanadium, molybdenum and tungsten in complex matrix samples by chelation ion chromatography and on-line detection with inductively coupled plasma mass spectrometry. Anal. Chim. Acta 2002, 466, 161–174. [Google Scholar] [CrossRef]
- Shaw, M.J.; Cowan, J.; Jones, P. Fabrication of an aurin tricarboxylic acid immobilized chelating polymer for the ion chromatographic determination of trace metal ions in highly mineralized waters. Anal. Lett. 2003, 36, 423–439. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Gros, N. Ion Chromatographic Analyses of Sea Waters, Brines and Related Samples. Water 2013, 5, 659-676. https://doi.org/10.3390/w5020659
AMA Style
Gros N. Ion Chromatographic Analyses of Sea Waters, Brines and Related Samples. Water. 2013; 5(2):659-676. https://doi.org/10.3390/w5020659
Chicago/Turabian StyleGros, Nataša. 2013. "Ion Chromatographic Analyses of Sea Waters, Brines and Related Samples" Water 5, no. 2: 659-676. https://doi.org/10.3390/w5020659