Sonochemical Treatment of Water Polluted by Chlorinated Organocompounds. A Review

Abstract
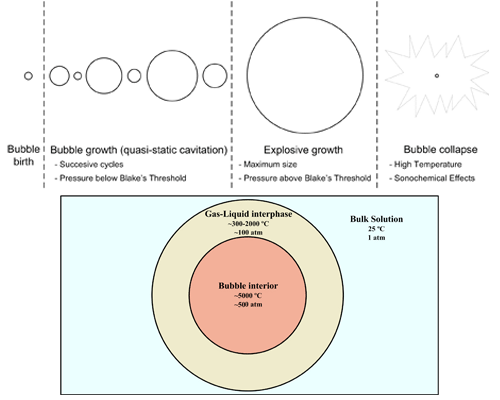
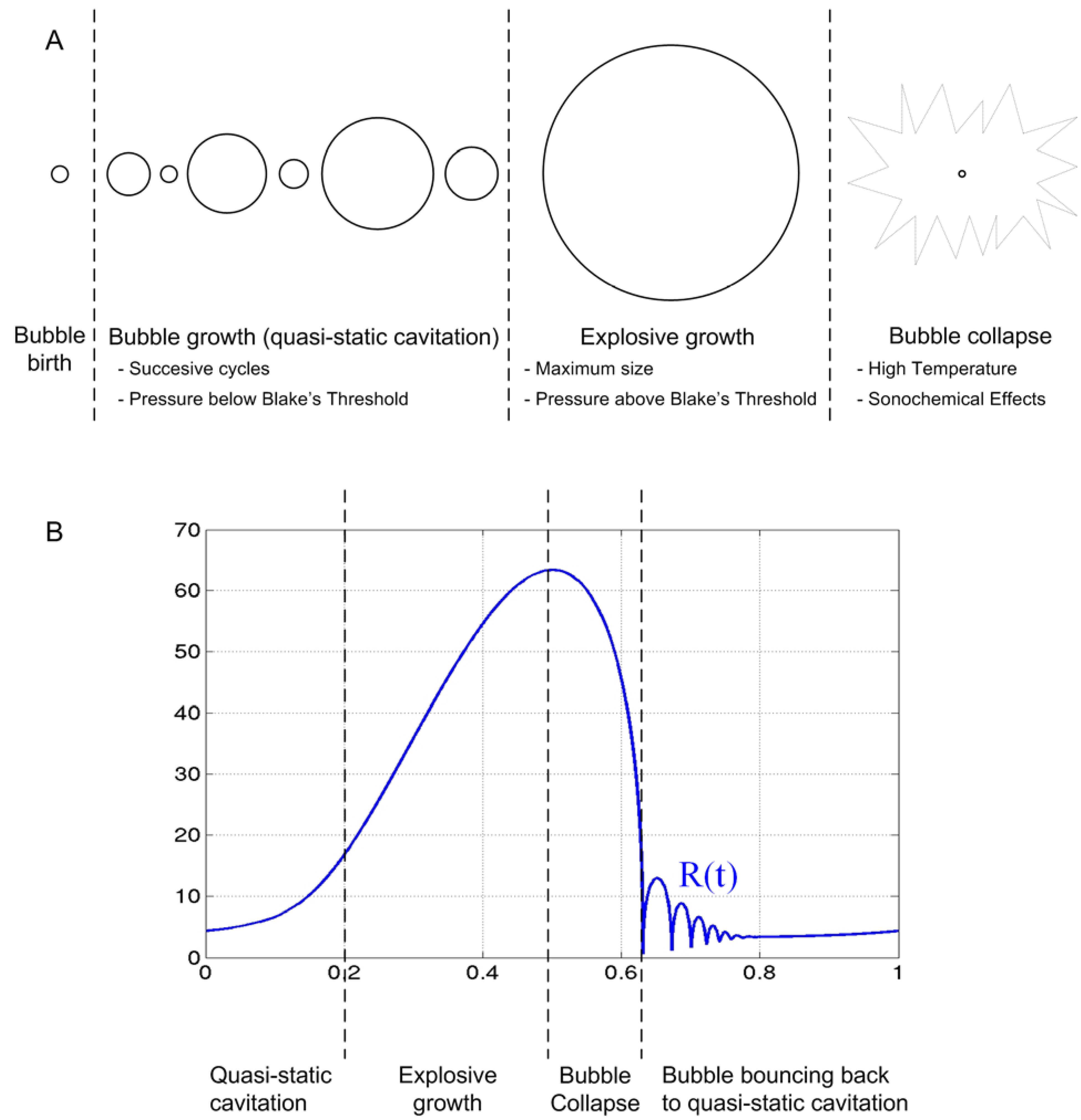
:
1. Introduction
2. Experimental devices and methodology
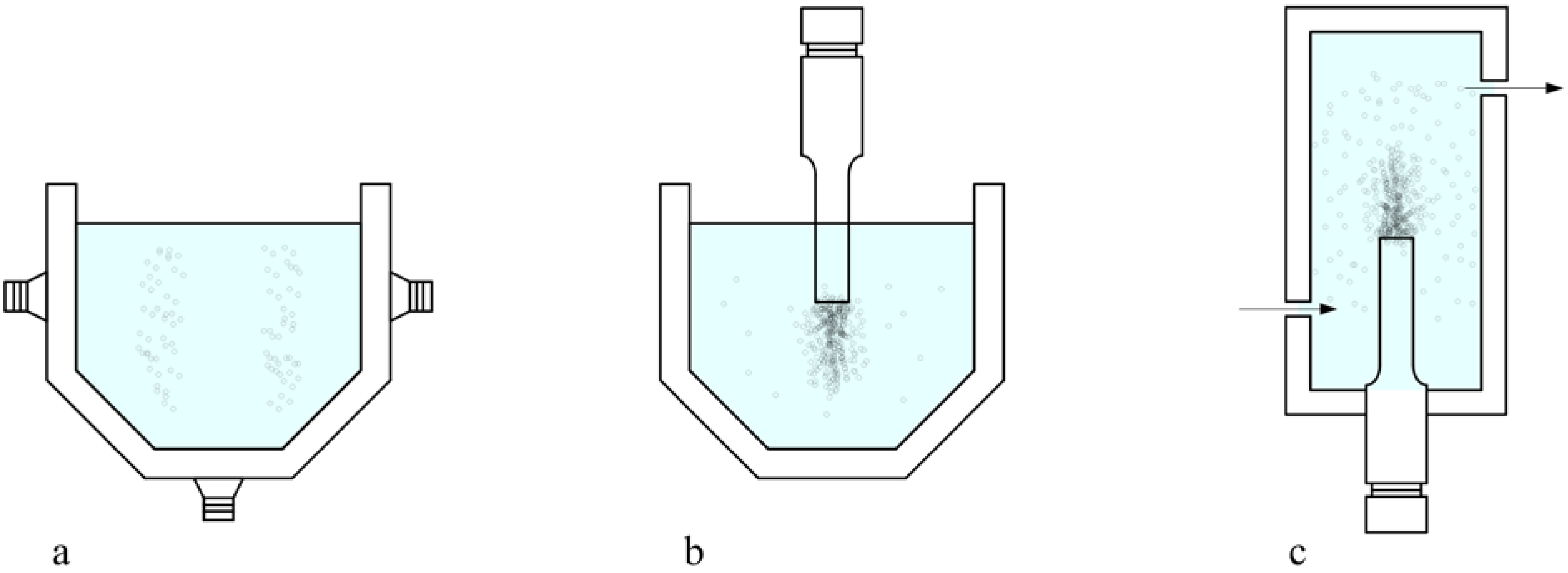
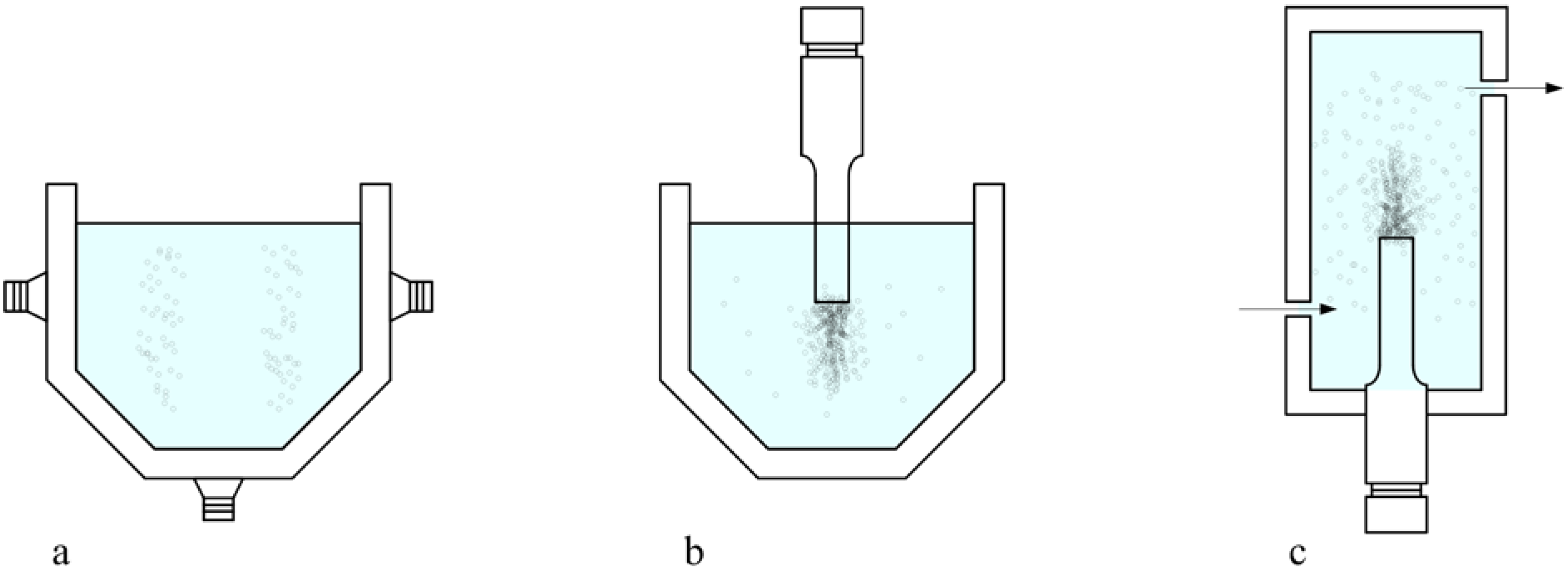
3. Influence of the operational variables
3.1. Influence of the initial concentration
3.2. Influence of the ultrasonic frequency

3.3. Influence of the ultrasonic power
3.4. Influence of the dissolved gas
3.5. Interrelation between the operational variables
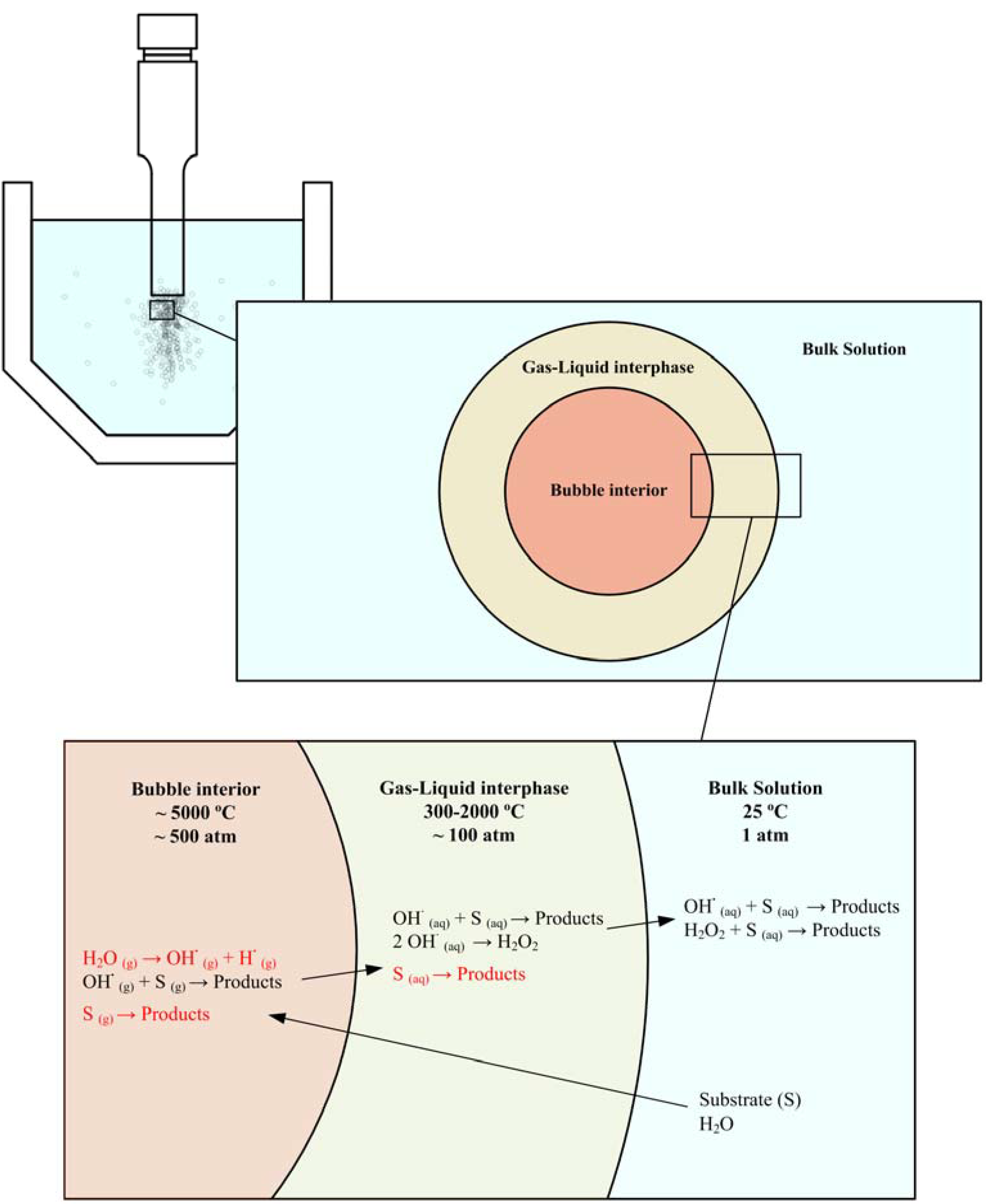
4. Mechanisms and Kinetics
4.1. General statements

4.2. Contributions from the studies with chlorinated compounds
4.3. Specific mechanisms
5. Mass balance and speciation. Technical and Environmental viability
6. Conclusions and perspectives. Economical viability
- (i)
- The temperature monitoring system should be carefully designed to ensure strict control. This allows us to fix values for the solubility, octanol-water partition coefficient, Henry constant, vapor pressure, density and other properties in our chlorinated organocompound + water system. A summary of the theoretical values of these properties is advised to design properly not only the experimental set-up but also the analytical procedure.
- (ii)
- Previous trials should be carried in the preparation of the solutions to check the chemical inertness of the materials used, i.e., glass, teflon and septum taps, although these materials have usually been used in chlorinated compound sonolysis [34,38]. Solutions of the chlorinated compound should be prepared by stirring, and maintained at 25 ºC in volumetric flasks with (a) Teflon or glass covered magnetic bars and/or (b) glass or septum taps. Liquid and gas samples have to be sequentially withdrawn and analyzed for the possible combinations, and no more than 3% of the initial material should be lost during the chosen experiment time. Keeping in mind that headspaces should be as small as possible in order to avoid complications in the experiments [52], the real headspace above the aqueous solution in the closed sonoreactor should also be studied. To do that, the sonoreactor must be airtight in order to prevent any compound loss by air stripping, and separate control experiments should be carried out in the absence of ultrasonic irradiation, in order to measure a % of volatilization, to be compared with the estimation made from data given in the literature [37,139]. Schwarzenbach et al. [140] reported the following equation to estimate the partitioning of organic chemicals due to Henry's law:where Vw represents the volume of water phase, Vg represents the volume of gas phase; and K'H represents the dimensionless Henry's constant. The acceptance of high values of volatilization percentage is not unusual [38]. This is because it is stated [31] that in a closed system volatile solutes re-enter the treated solution and the observed losses are due to chemical reaction and not to volatilization.
- (iii)
- Due to the fact that many buffers are effective radical scavengers [141], we have to analyze very carefully the options for adjusting the pH of the solutions.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| k min-1 | F kHz | C μM | T K | Π W cm-3 | pH | gas | Intermediates and by products | Mechanism model | Ref | |
|---|---|---|---|---|---|---|---|---|---|---|
| CCl4 | 0.025 | 20 | 200 | 286 | 0.65 | nat. to 3.5 | --- | C2Cl6, C2Cl4, Cl-, HOCl HCl, CO2 | pyrolysis | [30] |
| CCl4 | 0.070 | 500 | 200 | 286 | 0.1 | nat.to 3.5 | --- | C2Cl6, C2Cl4, Cl-, HOCl HCl, CO2 | pyrolysis | [30] |
| CCl4 | 0.044 | 205 | 200 | 286 | 0.06 | nat. to 3.5 | --- | C2Cl6, C2Cl4, Cl-, HOCl HCl, CO2 | pyrolysis | [30] |
| CCl4 | 0.049 | 358 | 200 | 286 | 0.06 | nat. to 3.5 | --- | C2Cl6, C2Cl4, Cl-, HOCl HCl, CO2 | pyrolysis | [30] |
| CCl4 | 0.055 | 618 | 200 | 286 | 0.06 | nat. to 3.5 | --- | C2Cl6, C2Cl4, Cl-, HOCl HCl, CO2 | pyrolysis | [30] |
| CCl4 | 0.039 | 1078 | 200 | 286 | 0.06 | nat. to 3.5 | --- | C2Cl6, C2Cl4, Cl-, HOCl HCl, CO2 | pyrolysis | [30] |
| CCl4 | 0.043 ± 0.002 | 20 | 396 | 293-298 | 0.23 | nat. | --- | not detected | pyrolysis | [37] |
| CCl4 | 0.198 | 20 | 195 | 298 | 1.23 | 11.8 | Ar | C2Cl6, C2Cl4, Cl-, HOCl | pyrolysis | [31] |
| CCl4 | 0.234 | 20 | 19.5 | 298 | 1.23 | 11.8 | Ar | C2Cl6, C2Cl4, Cl-, HOCl | pyrolysis | [31] |
| CHCl3 | 0.028 | 205 | 200 | 286 | 0.08 | nat. to 3.5 | --- | C2Cl4, Cl-, HOCl HCl, CO2 | pyrolysis | [30] |
| CHCl3 | 0.043 ± 0.005 | 20 | 1590 | 293-298 | 0.23 | nat. | --- | not detected | pyrolysis | [37] |
| CHCl3 | 0.028 | 205 | 150 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| CHCl3 | 0.0248 ± 0.0008 | 20 | 84 | 298 | 0.1843.75 | 5.4-5.8unbf | air | not studied | pyrolysis | [43] |
| CH2Cl2 | 0.016 | 205 | 200 | 286 | 0.08 | nat. to 3.5 | --- | C2Cl4, Cl-, HOCl HCl, CO2 | pyrolysis | [30] |
| CH2Cl2 | 0.033 ± 0.002 | 20 | 2471 | 293-298 | 0.23 | nat. | --- | not detected | pyrolysis | [37] |
| CH2Cl2 | 0.0393 ± 0.0027 | --- | 1176-11764 | 288-293 | 0.23 | nat. to 3 | --- | not studied, only HCl | not commented | [39] |
| CH2Cl2 | 0.016 | 205 | 150 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,2-DCA | 0.021 ± 0.003 | 20 | 1364 | 293-298 | 0.23 | nat. | --- | not detected | pyrolysis | [37] |
| TCE | 0.021 ± 0.001 | 20 | 1255 | 293-298 | 0.23 | nat. | --- | not detected | pyrolysis | [37] |
| TCE | 0.0455 ± 0.0011 | 520 | 5 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0364 ± 0.0012 | 520 | 50 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0345 ± 0.0013 | 520 | 250 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0232 ± 0.0008 | 520 | 500 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0230 ± 0.0009 | 520 | 1000 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0208 ± 0.0009 | 520 | 2000 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| CH2Cl2 | 0.016 | 205 | 150 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,2-DCA | 0.021 ± 0.003 | 20 | 1364 | 293-298 | 0.23 | nat. | --- | not detected | pyrolysis | [37] |
| TCE | 0.021 ± 0.001 | 20 | 1255 | 293-298 | 0.23 | nat. | --- | not detected | pyrolysis | [37] |
| TCE | 0.0455 ± 0.0011 | 520 | 5 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0364 ± 0.0012 | 520 | 50 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0345 ± 0.0013 | 520 | 250 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0232 ± 0.0008 | 520 | 500 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0230 ± 0.0009 | 520 | 1000 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0208 ± 0.0009 | 520 | 2000 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0127 ± 0.0008 | 520 | 3440 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0617 ± 0.0012 | 520 | 1670 | 302 | 0.095 | 7 | --- | not studied | pyrolysis | [50] |
| TCE | 0.0389 ± 0.0011 | 520 | 3440 | 302 | 0.095 | 7 | --- | not studied | pyrolysis | [50] |
| TCE | 0.0194 ± 0.0014 | 520 | 6680 | 302 | 0.095 | 7 | --- | not studied | pyrolysis | [50] |
| TCE | 0.026 ± 0.004 | 20 | 3340 | 305 | 0.43 | 7-2.9 | air | C2Cl2, C2Cl4 C2HCl, C4Cl2, C4HCl3, C4Cl4, C4HCl5, C4Cl6 | pyrolysis | [38] |
| TCE | 0.033 ± 0.005 | 520 | 3340 | 302 | 0.095 | 7-2.4 | air | C2Cl2, C2Cl4 C2HCl, C4Cl2, C4HCl3, C4Cl4, C4HCl5, C4Cl6 | pyrolysis | [38] |
| TCE | 0.031 ± 0.003 | 520 | 3340 | 302 | 0.095 | 4.7 | air | C2Cl2, C2Cl4 C2HCl, C4Cl2, C4HCl3, C4Cl4, C4HCl5, C4Cl6 | pyrolysis | [38] |
| TCE | 0.039 ± 0.003 | 520 | 3340 | 302 | 0.095 | 7 | air | C2Cl2, C2Cl4 C2HCl, C4Cl2, C4HCl3, C4Cl4, C4HCl5, C4Cl6 | pyrolysis | [38] |
| TCE | 0.043 ± 0.004 | 520 | 3340 | 302 | 0.095 | 10 | air | C2Cl2, C2Cl4 C2HCl, C4Cl2, C4HCl3, C4Cl4, C4HCl5, C4Cl6 | pyrolysis | [38] |
| TCE | 0.062 ± 0.0034 | 520 | 3340 | 302 | 0.095 | --- | Ar | C2Cl2, C2Cl4 C2HCl, C4Cl2, C4HCl3, C4Cl4, C4HCl5, C4Cl6 | pyrolysis | [38] |
| 1,1,1-TCA | 0.046 ± 0.003 | 20 | 824 | 293-298 | 0.23 | nat. | --- | not detected | pyrolysis + radical attack | [37] |
| 1,1,1-TCA | 0.0324 | 205 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,1,1-TCA | 0.0528 | 358 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,1,1-TCA | 0.0582 | 618 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,1,1-TCA | 0.0378 | 1078 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,1,2-TCA | 0.0126 | 205 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,1,2-TCA | 0.192 | 358 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,1,2-TCA | 0.192 | 618 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,1,2-TCA | 0.0114 | 1078 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| C2Cl6 | 0.0246 | 205 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| C2Cl6 | 0.0378 | 358 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| C2Cl6 | 0.0438 | 618 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| C2Cl6 | 0.027 | 1078 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 2Cl-PhOH | 0.0048 ± 0.0004 | 20 | 83 | 306 | 0.5 | nat. to 5.7 | air | chlorohydroquinone, catechol, 3-chlorocatechol | solution bulk at <75 μM gas bubble/liquid interface at >75 μM | [57] |
| 3Cl-PhOH | 0.0044 ± 0.0005 | 20 | 78 | 306 | 0.5 | nat. to 5.4 | air | chlorohydroquinone, 4-chlorocatechol, 3-chlorocatechol | solution bulk at low C gas bubble/liquid interface at high C | [57] |
| 4Cl-PhOH | 0.0033 ± 0.0002 | 20 | 75 | 306 | 0.5 | nat. to 5.1 | air | hydroquinone, 4-chlororesorcinol, 4-chlorocatechol, Cl- | solution bulk at low C gas bubble/liquid interface at high C | [57] |
| 4Cl-PhOH | 0.00156 | 20 | 0.5 | 293 | 0.12 | nat. | air | 4-chlorocatechol, hydroquinone, Cl- formic, oxalic acid, CO2 | radical attack | [52] |
| 4Cl-PhOH | 0.0291 | 500 | 0.5 | 293 | 0.12 | nat. | air | 4-chlorocatechol, hydroquinone, Cl- formic, oxalic acid, CO2 | radical attack | [52] |
| 4Cl-PhOH | 0.0039 | 1.7 MHz | 27 | 298 | 0.18 | --- | --- | C6H5O3Cl | pyrolysis | [59] |
| 4Cl-PhOH | 0.0017 ± 0.00008 | 20 | 100 | 298±5 | 0.24 | --- | O2 | 4-chlorocatechol | pyrolysis + radical attack | [55] |
| 4Cl-PhOH | 0.021 ± 0.0005 | 515 | 100 | 298±5 | 0.24 | --- | O2 | 4-chlorocatechol | pyrolysis + radical attack | [55] |
| 2,4- Cl-PhOH | 0.0189 | 360 | 800-950 | 293±5 | 0.2 | 4 | air | ---- | ---- | [48] |
| 2,3,5- Cl-PhOH | 0.0111 | 360 | 800-950 | 293±5 | 0.2 | 4 | air | ------ | ------ | [61] |
| ClC6H5 | 0.0072 | 20 | 0.5 | 293 | 0.12 | nat. | air | HCl, CO, CO2, C2H2, soot | pyrolysis | [52] |
| ClC6H5 | 0.0486 | 500 | 0.5 | 293 | 0.12 | nat. | air | HCl, CO, CO2, C2H2, soot | pyrolysis | [52] |
| ClC6H5 | 0.0177 ± 0.0008 | 520 | 1720 | 302 | 0.095 | 7 | air | main by-products: methane, acetlyene, butenyne, butadiyne, benzene, phenylacetylene, styrene, C7H7Cl, chlorophenol | pyrolysis | [46] |
| ClC6H5 | 0.0184 ± 0.0007 | 520 | 1720 | 302 | 0.095 | 4.7 | air | main by-products: methane, acetlyene, butenyne, butadiyne, benzene, phenylacetylene, styrene, C7H7Cl, chlorophenol | pyrolysis | [46] |
| ClC6H5 | 0.0181 ± 0.0004 | 520 | 1720 | 302 | 0.095 | 10 | air | main by-products: methane, acetlyene, butenyne, butadiyne and benzene, phenylacetylene, styrene, C7H7Cl, chlorophenol | pyrolysis | [46] |
| ClC6H5 | 0.0190 ± 0.0007 | 520 | 1720 | 302 | 0.095 | unbf | air | main by-products: methane, acetlyene, butenyne, butadiyne and benzene, phenylacetylene, styrene, C7H7Cl, chlorophenol | pyrolysis | [46] |
| ClC6H5 | 0.0285 ± 0.00108 | 520 | 1720 | 302 | 0.095 | 7 | Ar | main by-products: methane, acetlyene, butenyne, butadiyne and benzene, phenylacetylene, styrene, C7H7Cl, chlorophenol | pyrolysis | [46] |
| ClC6H5 | 0.0243 ± 0.0007 | 520 | 860 | 302 | 0.095 | 7 | --- | not studied | pyrolysis | [50] |
| ClC6H5 | 0.0176 ± 0.0006 | 520 | 1720 | 302 | 0.095 | 7 | --- | not studied | pyrolysis | [50] |
| ClC6H5 | 0.0057 ± 0.0005 | 520 | 3440 | 302 | 0.095 | 7 | --- | not studied | pyrolysis | [50] |
| ClC6H5 | 0.0160 ± 0.0004 | 520 | 500 | 302 | 0.06 | --- | --- | benzene, phenylacetylene, styrene, biphenyl, byphenylene, dihalogenated benzene, monoahlogenated phenol, 1-halo-4-ethynyl-benzene and monohalogenated bypehnyl | pyrolysis | [50] |
| ClC6H5 | 0.0127 ± 0.0004 | 520 | 1000 | 302 | 0.06 | --- | --- | benzene, phenylacetylene, styrene, biphenyl, byphenylene, dihalogenated benzene, monoahlogenated phenol, 1-halo-4-ethynyl-benzene and monohalogenated bypehnyl | pyrolysis | [50] |
| ClC6H5 | 0.0065 ± 0.0003 | 520 | 2000 | 302 | 0.06 | --- | --- | benzene, phenylacetylene, styrene, biphenyl, byphenylene, dihalogenated benzene, monoahlogenated phenol, 1-halo-4-ethynyl-benzene and monohalogenated bypehnyl | pyrolysis | [50] |
| ClC6H5 | 0.0365 ± 0.0027 | 520 | 1 | 302 | 0.06 | 7 | air | ------ | pyrolysis + radical attack | [50] |
| ClC6H5 | 0.0335 ± 0.0009 | 520 | 5 | 302 | 0.06 | 7 | air | ------ | pyrolysis + radical attack | [50] |
| ClC6H5 | 0.0329 ± 0.0011 | 520 | 25 | 302 | 0.06 | 7 | air | ------ | pyrolysis + radical attack | [50] |
| ClC6H5 | 0.0302 ± 0.0010 | 520 | 50 | 302 | 0.06 | 7 | air | ------ | pyrolysis + radical attack | [50] |
| ClC6H5 | 0.0190 ± 0.0012 | 520 | 100 | 302 | 0.06 | 7 | air | ------ | pyrolysis + radical attack | [50] |
| ClC6H5 | 0.0154 ± 0.0006 | 520 | 250 | 302 | 0.06 | 7 | air | ----- | pyrolysis + radical attack | [50] |
| ClC6H5 | 0.0025 ± 0.0001 | 520 | 3440 | 302 | 0.06 | 7 | air | ------ | pyrolysis + radical attack | [50] |
| ClC6H5 | 0.020 ± 0.001 | 500 | 500 | 293 | 0.1 | --- | air | CO, C2H2, CH4, CO2, soot | pyrolysis + radical attack | [49] |
| ClC6H5 | 0.035 ± 0.001 | 500 | 40 | 293 | 0.1 | --- | air | CO, C2H2, CH4, CO2, soot | pyrolysis + radical attack | [49] |
| ClC6H5 | 0.026 ± 0.001 | 500 | 200 | 293 | 0.1 | --- | air | CO, C2H2, CH4, CO2, soot | pyrolysis + radical attack | [49] |
| 1,4-Cl2C6H4 | 0.054 ± 0.001 | 500 | 200 | 293 | 0.2 | --- | air | not studied | pyrolysis + radical attack | [49] |
| 1,4-Cl2C6H4 | 0.009 ± 0.001 | 500 | 200 | 293 | 0.04 | --- | air | not studied | pyrolysis + radical attack | [49] |
| 1,4-Cl2C6H4 | 0.022 ± 0.001 | 500 | 500 | 293 | 0.1 | --- | air | not studied | pyrolysis + radical attack | [49] |
| 1,4-Cl2C6H4 | 0.036 ± 0.001 | 500 | 40 | 293 | 0.1 | --- | air | not studied | pyrolysis + radical attack | [49] |
| 1,4-Cl2C6H4 | 0.028 ± 0.001 | 500 | 300 | 293 | 0.1 | --- | air | not studied | pyrolysis + radical attack | [49] |
| pCBA | ----- | 20 | 5, 10, 20 | 298±2 | 0.095 | --- | air | ----- | radical attack | [53] |
| 1Cl-naphthalene | 0.036 ± 0.001 | 500 | 40 | 293 | 0.1 | --- | air | not studied | pyrolysis + radical attack | [49] |
| 1Cl-naphthalene | 0.038 ± 0.001 | 500 | 200 | 293 | 0.2 | --- | air | not studied | pyrolysis + radical attack | [49] |
| 1Cl-naphthalene | 0.008 ± 0.001 | 500 | 200 | 293 | 0.04 | --- | air | not studied | pyrolysis + radical attack | [49] |
| ClC6H5CH3 | 0.02637 ±0.00138 | 520 | 680 | 302 | 0.097 | --- | --- | ------ | pyrolysis | [44] |
| ClC6H5CH3 | 0.02923 ±0.00225 | 520 | 340 | 302 | 0.097 | --- | --- | ------- | pyrolysis | [44] |
| ClC6H5CH3 | 0.04145 ±0.00223 | 520 | 170 | 302 | 0.097 | --- | --- | --------- | pyrolysis | [44] |
| 2-PCB | 0.123 ± 0.002 | 20 | 4.6 | 288 | 0.24340.1/1.3240.1/165 | --- | Ar | biphenyl, toluene, ethylbenzene, diethylbiphenyl, dibutenylbiphenyl phenol, propylphenol, di-tert-butylphenol, cyclohexenyl diphenol | pyrolysis + radical attack | [54] |
| 2-PCB | 3.6 10-3 ± 7.8 10-5 | 205 | 4.6 | 288 | 0.243160/29160/400 | --- | Ar | biphenyl, toluene, ethylbenzene, diethylbiphenyl, dibutenylbiphenyl phenol, propylphenol, di-tert-butylphenol, cyclohexenyl diphenol | pyrolysis + radical attack | [54] |
| 2-PCB | 8.5 10-3 ± 4.2 10-5 | 358 | 4.6 | 288 | 0.243 | --- | Ar | biphenyl, toluene, ethylbenzene, diethylbiphenyl, dibutenylbiphenyl phenol, propylphenol, di-tert-butylphenol, cyclohexenyl diphenol | pyrolysis + radical attack | [54] |
| 2-PCB | 6.9 10-3 ± 1.4 10-5 | 618 | 4.6 | 288 | 0.243 | --- | Ar | biphenyl, toluene, ethylbenzene, diethylbiphenyl, dibutenylbiphenyl phenol, propylphenol, di-tert-butylphenol, cyclohexenyl diphenol | pyrolysis + radical attack | [54] |
| 2-PCB | 4.1 10-3 ± 1.2 10-5 | 1071 | 4.6 | 288 | 0.243 | --- | Ar | biphenyl, toluene, ethylbenzene, diethylbiphenyl, dibutenylbiphenyl phenol, propylphenol, di-tert-butylphenol, cyclohexenyl diphenol | pyrolysis + radical attack | [54] |
| 4-PCB | 0.096 ± 0.002 | 20 | 5.4 | 288 | 0.243 | --- | Ar | biphenyl, toluene, ethylbenzene, diethylbiphenyl, dibutenylbiphenyl phenol, propylphenol, di-tert-butylphenol, cyclohexenyl diphenol | pyrolysis + radical attack | [54] |
| 2,4,5-PCB | 0.156 ± 0.002 | 20 | 0.076 | 288 | 0.243 | --- | Ar | ethyl benzene, diethylbiphenyl, trichlorophenol | pyrolysis + radical attack | [54] |
| Alachlor | 0.0016 | 20 | 370 | 291 | 0.25 | --- | Air | MW=149, MW=225, MW=241, MW=253, MW=285 | pyrolysis + radical attack | [62] |
| Alachlor | 0.0375 | 300 | 370 | 291 | 0.25 | --- | Ar | MW=149, MW=225, MW=241, MW=253, MW=285 | pyrolysis + radical attack | [62] |
| MCPA | 0.0066 | 500 | 500 | 298 | 0.214 | --- | N2 | 4-chloro-2-methylphenol | pyrolysis + radical attack | [63] |
| MCPA | 0.047 | 500 | 500 | 298 | 0.214 | --- | O2 | 4-chloro-2-methylphenol | pyrolysis + radical attack | [63] |
| MCPA | 0.029 | 500 | 500 | 298 | 0.214 | --- | Ar | 4-chloro-2-methylphenol | pyrolysis + radical attack | [63] |
| MCPA | 0.042 | 500 | 500 | 298 | 0.214 | --- | Air | 4-chloro-2-methylphenol | pyrolysis + radical attack | [63] |
Acknowledgements
References and Notes
- Vogel, T.M.; Criddle, C.S.; McCarty, P.L. Transformations of halogenated aliphatic compounds. Environ. Sci. Technol. 1987, 21, 722–737. [Google Scholar] [CrossRef]
- Hitchman, M.L.; Spackman, R.A.; Ross, N.C.; Agra, C. Disposal methods for chlorinated aromatic waste. Chem. Soc. Rev. 1995, 24, 423–430. [Google Scholar]
- Entezari, M.H.; Mostafai, M.; Sarafraz-yazdi, A. A combination of ultrasound and a bio-catalyst: removal of 2-chlorophenol from aqueous solution. Ultrason. Sonochem. 2005, 12, 137–141. [Google Scholar] [CrossRef]
- Adewuyi, Y.G. Sonochemistry in environmental remediation. 1. Combinative and hybrid sonophotochemical oxidation processes for the treatment of pollutants in water. Environ. Sci. Technol. 2005, 39, 3409–3420. [Google Scholar]
- Shannon, M.A.; Bohn, P.W.; Elimelech, M.; Georgiadis, J.G.; Mariñas, B.J.; Mayes, A.M. Science and technology for water purification in the coming decades. Nature 2008, 452, 301–310. [Google Scholar] [CrossRef]
- Suslick, K.S. Sonochemistry. Science 1990, 247, 1439–1445. [Google Scholar] [CrossRef]
- Griffing, V. The Chemical effects of ultrasonics. J. Chem. Phys. 1952, 20, 939–942. [Google Scholar]
- Fitzgerald, M.E.; Griffing, V.; Sullivan, J. Chemical effects of ultrasonics-“Hot Spot” Chemistry. J. Chem. Phys. 1956, 25, 926–933. [Google Scholar] [CrossRef]
- Sehgal, C.M.; Wang, S.Y. Threshold intensities and kinetics of sonoreaction of thymine in aqueous solutions at low ultrasonic intensities. J. Am. Chem. Soc. 1981, 103, 6606–6611. [Google Scholar]
- Levy, S.V.; Low, C.R.R. Ultrasound in Synthesis; Springer-Verlag: London, UK, 1989. [Google Scholar]
- Kimura, T.; Sakamoto, T.; Leveque, J.M.; Sohmiya, H.; Fujita, M.; Ikeda, S.; Ando, T. Standarization of ultrasonic power for sonochemical reaction. Ultrason. Sonochem. 1996, 3, S157–S161. [Google Scholar] [CrossRef]
- Weissler, A.; Cooper, H.W.; Snyder, S. Chemical effect of ultrasonic waves: oxidation of potassium iodide solution by carbon tetrachloride. J. Am. Chem. Soc. 1950, 72, 1769–1775. [Google Scholar] [CrossRef]
- Jennings, B.H.; Townsend, S.N. The sonochemical reactions of carbon tetrachloride and chloroform in aqueous suspension in an inert atmosphere. J. Phys. Chem. 1961, 65, 1574–1579. [Google Scholar] [CrossRef]
- Paniwnyk, L.; Cai, H.; Albu, S.; Mason, T.J.; Cole, R. The enhancement and scale up of the extraction of anti-oxidants from Rosmarinus officinalis using ultrasound. Ultrason. Sonochem. 2009, 16, 287–292. [Google Scholar] [CrossRef]
- Mason, T.J.; Cordemans, E. Practical considerations for process optimization in Synthetic Organic Sonochemistry. In Synthetic Organic Sonochemistry; Luche, J.L., Ed.; Plenum Press: New York, NY, USA, 1998. [Google Scholar]
- Grönroos, A.; Kyllönen, H.; Korpijärvi, K.; Pirkonen, P.; Paavola, T.; Jokela, J.; Rintala, J. Ultrasound assisted method to increase soluble chemical oxygen demand (SCOD) of sewage sludge for digestion. Ultrason. Sonochem. 2005, 12, 115–120. [Google Scholar] [CrossRef]
- Kyllönen, H.; Pirkonen, P.; Hintikka, V.; Parvinen, P.; Grönroos, A.; Sekki, H. Ultrasonically aided mineral processing technique for remediation of soil contaminated by heavy metals. Ultrason. Sonochem. 2004, 11, 211–216. [Google Scholar] [CrossRef]
- Gallego-Juárez, J.A.; Rodríguez Corral, G.; Riera, E.; Vázquez-Martinez, F.; Acosta-Aparicio, V.M.; Campos-Pozuelo, C. Development of industrial models of high power stepped-plate sonic and ultrasonic transducer for use in fluids. IEEE Ultrasonic Sympos. Proc. 2001, 571–578. [Google Scholar]
- Hua, I.; Höchemer, R.; Hoffmann, M.R. Sonochemical degradation of p-Nitrophenol in a Parallel-Plate near-field acoustical processor. Environ. Sci. Technol. 1995, 29, 2790–2795. [Google Scholar] [CrossRef]
- Xie, R.; Xing, Y.; Ghani, Y.A.; Ooi, K.-E.; Ng, S.-W. Full-scale demostration of an ultrasonic disintegration technology in enhancing anaerobic digestion of mixed primary and thickened secondary sewage sludge. J. Environ. Eng. Sci. 2007, 6, 533–541. [Google Scholar]
- Nickel, K.; Neis, U. Ultrasonic disintegration of biosolids for improved biodegradation. Ultrason. Sonochem. 2007, 14, 450–455. [Google Scholar] [CrossRef]
- Khanal, S.K.; Grewell, D.; Sung, S.; van Leeuwen, J. Ultrasound applications in wastewater sludge pretreatment: A review. Crit. Rev. Env. Sci. Technol. 2007, 37, 277–313. [Google Scholar] [CrossRef]
- Appels, L.; Bayens, J.; Degrève, J.; Dewil, R. Principles and potential of the anaerobic digestion of waste-activated sludge. Prog. Energy Combust. Sci. 2008, 34, 755–781. [Google Scholar] [CrossRef]
- Laborde, J.L.; Bouyer, C.; Caltagirone, J.P.; Gérard, A. Acoustic cavitation field prediction at low and high frequency ultrasound. Ultrasonics 1998, 36, 581–587. [Google Scholar] [CrossRef]
- Servant, G.; Caltagirone, J.P.; Gérard, A.; Laborde, J.L.; Hita, A. Numerical simulation of cavitation bubble dynamics induced by ultrasound waves in a high frequency reactor. Ultrason. Sonochem. 2000, 7, 217–227. [Google Scholar] [CrossRef]
- Sáez, V.; Frías-Ferrer, A.; Iniesta, J.; González-García, J.; Aldaz, A.; Riera, E. Characterization of a 20 kHz sonoreactor. Part I: Analysis of mechanical effects by classical and numerical methods. Ultrason. Sonochem. 2005, 12, 59–65. [Google Scholar]
- Mandroyan, A.; Doche, M.L.; Hihn, J.Y.; Viennet, R.; Bailly, Y.; Simonin, L. Modification of the ultrasound induced activity by the presence of an electrode in a sono-reactor working at two low frequencies (20 and 40 kHz). Part II: Mapping flow velocities by particle image velocimetry (PIV). Ultrason. Sonochem. 2009, 16, 97–104. [Google Scholar]
- Sáez, V.; Frías-Ferrer, A.; Iniesta, J.; González-García, J.; Aldaz, A.; Riera, E. Characterization of a 20 kHz sonoreactor. Part II: Analysis of chemical effects by classical and electrochemical methods. Ultrason. Sonochem. 2005, 12, 67–72. [Google Scholar]
- Chendke, P.K.; Fogler, H.S. Sonoluminescence and Sonochemical reactions of aqueous carbon tetrachloride solutions. J. Phys. Chem. 1983, 87, 1362–1369. [Google Scholar] [CrossRef]
- Hung, H.M.; Hoffmann, M.R. Kinetics and Mechanism of the sonolytic degradation of chlorinated hydrocarbons: frequency effects. J. Phys. Chem. A 1999, 103, 2734–2739. [Google Scholar] [CrossRef]
- Hua, I.; Hoffmann, M.R. Kinetics and Mechanism of the sonolytic degradation of CCl4 intermediates and byproducts. Environ. Sci. Technol. 1996, 30, 864–871. [Google Scholar] [CrossRef]
- Rajan, R; Kumar, R.; Gandhi, K.S. Modelling of sonochemical decomposition of CCl4 in aqueous solution. Environ. Sci. Technol. 1998, 32, 1128–1133. [Google Scholar]
- Spurlock, L.A.; Reifsneider, S.B. Chemistry of ultrasound. I. A reconsideration of first principles and the aplications to a Dialkyl sulfide. J. Am. Chem. Soc. 1970, 92, 6112–6117. [Google Scholar]
- Francony, A.; Pétrier, C. Sonochemical degradation of carbon tetrachloride in aqueous solution at two frequencies: 20 kHz and 500 kHz. Ultrason. Sonochem. 1996, 3, S77–S82. [Google Scholar] [CrossRef]
- Petrier, C.; Francony, A. Incidence of wave-frequency on the reaction rates during ultrasonic wastewater treatment. Wat. Sci. Tech. 1997, 35, 175–180. [Google Scholar] [CrossRef]
- Colussi, A.J.; Hung, H.-M.; Hoffmann, M.R. Sonochemical degradation rates of volatiles solutes. J. Phys. Chem. A. 1999, 103, 2696–2699. [Google Scholar]
- Bhatnagar, A.; Cheung, H.M. Sonochemical destruction of chlorinated C1 and C2 volatile organic compounds in dilute aqueous solution. Environ. Sci. Technol. 1994, 28, 1481–1486. [Google Scholar] [CrossRef]
- Drijvers, D.; De Baets, R.; De Visscher, A.; van Langenhove, H. Sonolysis of trichloroethylene in aqueous solution: volatile organic intermediates. Ultrason. Sonochem. 1996, 3, S83–S90. [Google Scholar] [CrossRef]
- Cheung, H.M.; Bhatnagar, A.; Jansen, G. Sonochemical destruction of Chlorinated hydrocarbons in dilute aqueous solution. Environ. Sci. Technol. 1991, 25, 1510–1512. [Google Scholar] [CrossRef]
- Inazu, K.; Nagata, Y.; Maeda, Y. Decomposition of Chlorinated hydrocarbons in aqueous solutions by ultrasonic irradiation. Chem. Lett. 1993, 22, 57–60. [Google Scholar] [CrossRef]
- Gaddam, K.; Cheung, H.-M. Effects of pressure, temperature, and pH on the sonochemical destruction of 1,1,1-trichloroethane in dilute aqueous solution. Ultrason. Sonochem. 2001, 8, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.M.; Kurup, S. Sonochemical destruction of CFC 11 and CFC 13 in dilute aqueous solution. Environ. Sci. Technol. 1994, 28, 1619–1622. [Google Scholar] [CrossRef]
- Shemer, H.; Narkis, N. Sonochemical removal of trihalomethanes from aqueous solutions. Ultrason. Sonochem. 2005, 12, 495–499. [Google Scholar] [CrossRef]
- De Visscher, A.; Van Eenoo, P.; Drijvers, D.; Van Langenhove, H. Kinetics Model for the sonochemical degradation of monocyclic aromatic compounds in aqueous solution. J. Phys. Chem. 1996, 100, 11636–11642. [Google Scholar]
- Kruus, P.; Burk, R.C.; Entezari, M.H.; Otson, R. Sonication of aqueous solutions of chlorobenzene. Ultrason. Sonochem. 1997, 4, 229–233. [Google Scholar] [CrossRef]
- Drijvers, D.; van Langenhove, H.; Vervaet, K. Sonolysis of chlorobenzene in aqueous solution: organic intermediates. Ultrason. Sonochem. 1998, 5, 13–19. [Google Scholar] [CrossRef]
- Drijvers, D.; van Langenhove, H.; Herrygers, V. Sonolysis of fluoro-, chloro-, bromo- and iodobenzene: a comparative study. Ultrason. Sonochem. 2000, 7, 87–95. [Google Scholar]
- Tiehm, A.; Kohnagel, I.; Neis, U. Removal of chlorinated pollutants by a combination of ultrasound and biodegradation. Water. Sci. Technol. 2001, 43, 297–303. [Google Scholar]
- Jiang, Y.; Pétrier, C.; David Waite, T. Kinetics and mechanisms of ultrasonic degradation of volatile chlorinated in aqueous solutions. Ultrason. Sonochem. 2002, 9, 317–323. [Google Scholar] [CrossRef]
- Drijvers, D.; van Langenhove, H.; Nguyen Thi Kim, L.; Bray, L. Sonolysis of an aqueous mixture of trichloroethylene and chlorobenzene. Ultrason. Sonochem. 1999, 6, 115–121. [Google Scholar] [CrossRef]
- Dewulf, J.; van Langenhove, H.; De Visscher, A.; Sabbe, S. Ultrasonic degradation of trichloroethylene and chlorobenzene at micromolar concentrations: kinetics and modelling. Ultrason. Sonochem. 2001, 8, 143–150. [Google Scholar] [CrossRef]
- Petrier, C.; Jiang, Y.; Lamy, M.-F. Ultrasound and Environment: sonochemical destruction of chloroaromatic derivatives. Environ. Sci. Technol. 1998, 32, 1316–1318. [Google Scholar] [CrossRef]
- Nam, S.-N.; Han, S.-K.; Kang, J.-W.; Choi, H. Kinetics and mechanisms of the sonolytic destruction of non-volatile organic compounds: investigation of the sonochemical reaction zone using several OH· monitoring techniques. Ultrason. Sonochem. 2003, 10, 139–147. [Google Scholar]
- Zhang, G.; Hua, I. Cavitation chemistry of polychlorinated biphenyls: decomposition mechanisms and rates. Environ. Sci. Technol. 2000, 34, 1529–1534. [Google Scholar] [CrossRef]
- Weavers, L.K.; Ling, F.H.; Hoffmann, M.R. Aromatic compound degradation in water using a combination of sonolysis and ozonolysis. Environ. Sci. Technol. 1998, 32, 2727–2733. [Google Scholar] [CrossRef]
- Petrier, C.; Micolle, M.; Merlin, G.; Luche, J.-L.; Reverdy, G. Characteristics of pentachlorophenate degradation in aqueous solution by means of ultrasound. Environ. Sci. Technol. 1992, 26, 1639–1642. [Google Scholar]
- Serpone, N.; Terzian, R.; Hidaka, H.; Pelizzetti, E. Ultrasonic induced dehalogenation and oxidation of 2-, 3-,and 4-chlorophenol in air-equilibrated aqueous media. Similarities with irradiated semiconductor particulates. J. Phys. Chem. 1994, 98, 2634–2640. [Google Scholar]
- Petrier, C.; David, B.; Laguian, S. Ultrasonic degradation at 20 kHz and 500 kHz of atrazine and pentachlorophenol in aqueous solution: preliminary results. Chemosphere 1996, 32, 1709–1718. [Google Scholar] [CrossRef]
- Gondrexon, N.; Renaudin, V.; Petrier, C.; Boldo, P.; Bernis, A.; Gonthier, Y. Degradation of pentachlorophenol aqueous solutions using a continuous flow ultrasonic reactor: experimental performance and modelling. Ultrason. Sonochem. 1999, 5, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Chen, Y.; Wu, M.; Wang, H.; Yingwu, Y.; Lü, Z. Sonochemistry of degrading p-chlorophenol in water by high frequency ultrasound. Ultrason. Sonochem. 2004, 11, 43–46. [Google Scholar] [CrossRef]
- Tiehm, A.; Neis, U. Ultrasonic dehalogenation and toxicity reduction of trichlorophenol. Ultrason. Sonochem. 2005, 12, 121–125. [Google Scholar] [CrossRef]
- Wayment, D.G.; Casadonte, D.J., Jr. Frequency effect on the sonochemical remediation of alachlor. Ultrason. Sonochem. 2002, 9, 251–257. [Google Scholar] [CrossRef]
- Kojima, Y.; Fujita, T.; Ona, E.P.; Matsuda, H.; Koda, S.; Tanahashi, N.; Asakura, Y. Effects of disolved gas species on ultrasonic degradation of (4-chloro-2-methylphenoxy) acetic acid (MCPA) in aqueous solution. Ultrason. Sonochem. 2005, 12, 359–365. [Google Scholar] [CrossRef]
- Pétrier, C.; Lamy, M.-F.; Francony, A.; Renaudin, V.; Gondrexon, N.; David, B.; Benahcene, A. Sonochemical degradation of phenol in dilute aqueous solutions: comparison of the reaction rates at 20 and 487 kHz. J. Phys. Chem. 1994, 98, 10514–10520. [Google Scholar] [CrossRef]
- Kotronarou, A.; Mills, G.; Hoffmann, M.R. Oxidation of hydrogen sulfide in aqueous solution by ultrasonic irradiation. Environ. Sci. Technol. 1992, 26, 2420–2428. [Google Scholar] [CrossRef]
- Kotronarou, A.; Mills, G.; Hoffmann, M.R. Ultrasonic irradiation of p-nitrophenol in aqueous solution. J. Phys. Chem. 1991, 95, 3630–3638. [Google Scholar] [CrossRef]
- Hart, E.J.; Fischer, Ch.-H.; Henglein, A. Sonolysis of hydrocarbons in aqueous solution. Radiat. Phys. Chem. 1990, 36, 511–516. [Google Scholar]
- Kotronarou, A.; Mills, G.; Hoffmann, M.R. Decomposition of parathion in aqueous solution by ultrasonic irradiation. Environ. Sci. Technol. 1992, 26, 1460–1462. [Google Scholar] [CrossRef]
- Kondo, T.; Krishna, C.M.; Riesz, P. Sonolysis of concentrated aqueous solutions of nonvolatile solutes: spin-trapping evidence for free radicals formed by pyrolysis. Radiat. Res. 1998, 118, 211–229. [Google Scholar]
- Suslick, K.S.; Didenko, Y.; Fang, M.M.; Hyeon, T.; Kolbeck, K.J.; McNamara III, W.B.; Mdeleleni, M.M.; Wong, M. Acoustic cavitation and its chemcal consequences. Phil. Trans. R. Soc. Lond. A. 1999, 357, 335–353. [Google Scholar] [CrossRef]
- Petrier, C.; Jeunet, A.; Luche, J.-L.; Reverdy, G. Unexpected frequency effects on the rate of oxidative processes induced by ultrasound. J. Am. Chem. Soc. 1992, 114, 3148–3150. [Google Scholar] [CrossRef]
- Hua, I.; Hoffmann, M.R. Optimization of ultrasonic irradiation as an advanced oxidation technology. Environ. Sci. Technol. 1997, 31, 2237–2243. [Google Scholar] [CrossRef]
- Gogate, P.R.; Pandit, A.B. Engineering design method for cavitational reactors: I.Sonochemical reactors. Aiche J. 2000, 46, 372–379. [Google Scholar] [CrossRef]
- Hoffmann, M.R.; Hua, I.; Höchemer, R. Application of ultrasonic irradiation for the degradation of chemical contaminants in water. Ultrason. Sonochem. 1996, 3, S163–S172. [Google Scholar] [CrossRef]
- Zheng, W.; Maurin, M.; Tarr, M.A. Enhancement of sonochemical degradation of phenol using hydrogen atom scavengers. Ultrason. Sonochem. 2005, 12, 313–317. [Google Scholar] [CrossRef]
- Suslick, K.S.; Hammerton, D.A.; Cline, R.E. The sonochemical hot spot. J. Am. Chem. Soc. 1986, 108, 5641–5642. [Google Scholar] [CrossRef]
- Lepoint, T.; Mullie, F. What exactly is cavitation chemistry? Sonochem. 1994, 1, S13–S22. [Google Scholar] [CrossRef]
- Toegel, R.; Gompf, R.; Pecha, R.; Lohse, D. Does water vapor prevent upscaling sonoluminescence? Phys. Rev. Lett. 2000, 85, 3165–3168. [Google Scholar]
- Petrier, C.; Francony, A. Ultrasonic waste-water treatment: incidence of ultrasonic frequency on the rate of phenol and carbon tetrachloride degradation. Ultrason. Sonochem. 1997, 4, 295–300. [Google Scholar] [CrossRef]
- Mettin, R. Bubble structures in acoustic cavitation. In Bubble and Particle Dynamics in Acoustic Fields: Modern Trends and Applications; Doinikov, A.A., Ed.; Research Signpost: Kerala, India, 2005; pp. 1–36. [Google Scholar]
- Mettin, R. From a single bubble to bubble structures in acoustic cavitation. In Oscillations, Waves and Interactions; Kurz, T., Parlitz, U., Kaatze, U., Eds.; Universitätsverlag Göttingen: Göttingen, Germany, 2007; pp. 171–198. [Google Scholar]
- Yasui, K.; Tuziuti, T.; Kozuka, T.; Towata, A.; Iida, Y. Influence of ambient bubble radius on sonoluminescence and sonochemical reactions. In Revista de Acústica (Special Issue) Volume 38, 19th International Congress on Acoustics, Madrid, Spain, 2-7 September 2007; NLA-01-013. pp. 1–6.
- Mettin, R. Bubble motion and instability. In Proceedings of 11th Meeting of the European Society of Sonochemistry, La Grande-Motte, France, 1-5 June 2008; p. 95.
- Calvisi, M.L.; Lindua, O.; Blake, J.R.; Szeri, A.J. Shape stability and violent collapse of microbubbles in acoustic traveling waves. Phys. Fluids 2007, 19, 047101. [Google Scholar] [CrossRef]
- Storey, B.D.; Szeri, A.J. Water vapour, sonoluminescence and sonochemistry. Proc. R. Soc. London, Ser. A 2000, 456, 1685–1709. [Google Scholar]
- Kang, J.-W.; Hung, H.-M.; Lin, A.; Hoffmann, M.R. Sonolytic destruction of methyl tert-butyl ether by ultrasonic irradiation: the role of O3, H2O2, frequency and power density. Environ. Sci. Technol. 1999, 33, 3199–3205. [Google Scholar] [CrossRef]
- Gutierrez, M.; Henglein, A. Chemical action of pulsed ultrasound: observation of an unprecedented intensity effect. J. Phys. Chem. 1990, 94, 3625–3628. [Google Scholar] [CrossRef]
- Gogate, P.R.; Shirgaonkar, I.Z.; Sivakurmar, M.; Senthilkumar, P.; Vichare, N.P.; Pandit, A.B. Cavitation reactors: efficiency assessment using a model reaction. Aiche J. 2001, 47, 2526–2538. [Google Scholar] [CrossRef]
- Ondruska, B.; Lifka, J.; Hoffmann, J. Aquasonolysis of Ether - Effect of frequency and acoustic power of ultrasound. Chem. Eng. Tech. 2000, 23, 588–592. [Google Scholar] [CrossRef]
- Entezari, M.H.; Kruus, P. Effect of frequency on sonochemical reactions II. Temperature and intensity effects. Ultrason. Sonochem. 1996, 3, 19–24. [Google Scholar] [CrossRef]
- Apfel, R.E. Acoustic cavitation prediction. J. Acoust. Soc. Am. 1981, 69, 1624–1633. [Google Scholar]See also the chapter 4 The forced bubble in The Acoustic Bubble by T. G. Leighton; Academic Press: London, UK, 1994.
- Storey, B.D.; Szeri, A.J. A reduced model of cavitation physics for use in sonochemistry. Proc. R. Soc. London, Ser. A 2001, 457, 1685–1700. [Google Scholar]
- Mark, G.; Tauber, A.; Laupert, R.; Schuchmann, H.; Schulz, D.; Mues, A.; von Sonntag, C. OH - radical formation by ultrasound in aqueous solution - Part II: Terephthalate and Fricke dosimetry and the influence of various conditions on the sonolytic yield. Ultrason. Sonochem. 1998, 5, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Vanhille, C.; Campos-Pozuelo, C. Nonlinear ultrasonic waves in bubbly liquids with unhomogeneous bubble distribution: numerical experiments. Ultrason. Sonochem. 2009, 19, 669–685. [Google Scholar] [CrossRef]
- Louisnard, O. Nonlinear attenuation of sound waves by inertial cavitation bubbles. In To appear in Physics Procedia, International Congress on Ultrasonics, Santiago de Chile, Chile, 11-17 January 2009.
- Schmitt, F.O.; Johnson, C.H.; Olson, A.R. Oxidations promoted by ultrasonic radiation. J. Am. Chem. Soc. 1929, 51, 370–375. [Google Scholar] [CrossRef]
- Makino, K.; Mossoba, M.; Riesz, P. Chemical effects of ultrasound on aqueous solution. Formation of hydroxyl radicals and hydrogen atoms. J. Phys. Chem. 1983, 87, 1369–1377. [Google Scholar]
- Riesz, P.; Berdhal, D.; Christman, C.L. Free radical generation by ultrasound in aqueous and nonaqueous solutions. Environ. Health Perspect. 1985, 64, 233–252. [Google Scholar] [CrossRef]
- Fischer, C.H.; Hart, E.J.; Henglein, A. H/D Isotope exchange in the HD-H2O system under the influence of ultrasound. J. Phys. Chem. 1986, 90, 3059–3060. [Google Scholar] [CrossRef]
- Gutierrez, M.; Henglein, A.; Ibañez, F. Radical Scavenging in the sonolysis of aqueous solutions of I-, Br- and N3-. J. Phys. Chem. 1991, 95, 6044–6047. [Google Scholar] [CrossRef]
- Fischer, Ch.-H.; Hart, E.J.; Henglein, A. Ultrasonic irradiation of water in the presence of 18,18O2: Isotope exchange and isotopic distribution of H2O2. J. Phys. Chem. 1986, 90, 1954–1956. [Google Scholar] [CrossRef]
- Hart, E.J.; Henglein, A. Free radical and free atom reactions in the sonolysis of aqueous iodide and formate solutions. J. Phys. Chem. 1985, 89, 4342–4347. [Google Scholar] [CrossRef]
- Hart, E.J.; Fischer, Ch.-H.; Henglein, A. Isotopic exchange in the sonolysis of aqueous solutions containing 14,14N2 and 15,15N2. J. Phys. Chem. 1986, 90, 5989–5991. [Google Scholar] [CrossRef]
- Misik, V.; Riesz, P. Recent application of EPR and spin trapping to sonochemical studies of organic liquids and aqueous solutions. Ultrason. Sonochem. 1996, 3, S173–S186. [Google Scholar]
- Kondo, T.; Krishna, C.M.; Riesz, P. Pyrolysis radicals formed by ultrasound in aqueous solutions of nucleotides: a spin trapping study. Int. J. Radiat. Biol. 1990, 57, 23–33. [Google Scholar] [CrossRef]
- Guo, A.; Zheng, Z.; Zheng, S.; Hu, W.; Feng, R. Effects of various sono-oxidation parameters on the removal of aqueous 2,4-dinitrophenol. Ultrason. Sonochem. 2005, 12, 461–465. [Google Scholar] [CrossRef]
- Spinks, J.W.T.; Woods, R.J. An introduction to radiation chemistry, 3rd ed.; Wiley: New York, NY, USA, 1990. [Google Scholar]
- Hart, E.J.; Fischer, C.-H.; Henglein, A. Pyrolysis of acetylene in sonolytic cavitation bubbles in aqueous solution. J. Phys. Chem. 1990, 94, 284–290. [Google Scholar] [CrossRef]
- Alegria, A.E.; Lion, Y.; Kondo, T.; Riesz, P. Sonolysis of aqueous surfactant solutions. Probing the interfacial region of cavitation bubbles by spin trapping. J. Phys. Chem. 1989, 93, 4908–4913. [Google Scholar]
- Krishna, C.M.; Lion, Y.; Kondo, T.; Riesz, P. Thermal decomposition of methanol in the sonolysis of methanol-water mixtures. Spin-trapping evidence for isotope exchange reactions. J. Phys. Chem. 1987, 91, 5847–5850. [Google Scholar]
- Hart, E.J.; Fischer, Ch.-H.; Henglein, A. Isotopic exchange in the sonolysis of aqueous solutions containing D2 and CH4. J. Phys. Chem. 1987, 91, 4166–4169. [Google Scholar] [CrossRef]
- Gutierrez, M.; Henglein, A.; Fischer, Ch.-H. Hot spot kinetics of the sonolysis of aqueous acetate solutions. Int. J. Radiat. Biol. 1986, 50, 313–321. [Google Scholar] [CrossRef]
- Riesz, P.; Kondo, T.; Krishna, C.M. Free radical formation by ultrasound in aqueous solutions. A spin trapping study. Free Radical Res. Commun. 1990, 10, 27–35. [Google Scholar] [CrossRef]
- Riesz, P.; Kondo, T. Free radical formation induced by ultrasound and its biological implications. Free Radical Biol. Med. 1992, 13, 247–270. [Google Scholar] [CrossRef]
- Riesz, P.; Kondo, T.; Krishna, C.M. Sonochemistry of volatile and non-volatile solutes in aqueous solutions: e.p.r. and spin trapping studies. Ultrasonics 1990, 28, 295–303. [Google Scholar]
- Henglein, A. Sonochemistry: historical developments and modern aspects. Ultrasonics 1987, 25, 6–16. [Google Scholar] [CrossRef]
- Currell, D.L.; Wilheim, G.; Nagy, S. The effect of certain variables on the ultrasonic cleavage of phenol and of pyridine. J. Am. Chem. Soc. 1963, 85, 127–130. [Google Scholar] [CrossRef]
- Weissler, A. Formation of hydrogen peroxide by ultrasonic waves: free radicals. J. Am. Chem. Soc. 1959, 81, 1077–1081. [Google Scholar] [CrossRef]
- Anbar, M.; Pecht, I. On the sonochemical formation of hydrogen peroxide in water. J. Phys. Chem. 1964, 68, 352–355. [Google Scholar] [CrossRef]
- Henglein, A.; Kormann, C. Scavenging of OH radicals produced in the sonolysis of water. Int. J. Radiat. Biol. 1985, 48, 251–258. [Google Scholar] [CrossRef]
- Krishna, C.M.; Kondo, T.; Riesz, P. Sonochemistry of alcohol-water mixtures: spin-trapping evidence for thermal decomposition and isotope-exchange reactions. J. Phys. Chem. 1989, 93, 5166–5172. [Google Scholar] [CrossRef]
- Atkinson, R. Kinetics and Mechanisms of the gas-phase reactions of the hydroxyl radical with organic compounds under atmospheric conditions. Chem. Rev. 1985, 85, 69–201. [Google Scholar]
- Bapat, P.S.; Gogate, P.R.; Pandit, A.B. Theoretical analysis of sonochemical degradation of phenol and its chloro-derivatives. Ultrason. Sonochem. 2008, 15, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.V.; Lim, K.P.; Kumaran, S.S.; Kiefer, J.H. Thermal decomposition of carbon tetrachloride. J. Phys. Chem. 1993, 97, 1914–1919. [Google Scholar] [CrossRef]
- Nohhebel, D.C.; Walton, J.C. Free-Radical Chemistry; Cambridge University Press: Cambridge, UK, 1974; p. 572. [Google Scholar]
- Ellermann, T. Fine structure of the CCl3 UV absorption spectrum and CCl3 kinetics. Chem. Phys. Lett. 1992, 189, 175–181. [Google Scholar]
- Yokoyama, K.; Fujisawa, G.; Yokoyama, A. The mechanism of the unimolecular dissociation of trichloroethylene CHCl=CCl2 in the ground electronic state. J. Chem. Phys. 1995, 102, 7902–7909. [Google Scholar] [CrossRef]
- Ritter, E.R.; Bozelli, J.W.; Dean, A.M. Kinetic study on thermal decomposition of chlorobenzene diluted in H2. J. Phys. Chem. 1990, 94, 2493–2504. [Google Scholar] [CrossRef]
- Kern, R.D.; Xie, K.; Chen, H. A Shock tube study of chlorobenzene pyrolysis. Combust. Sci. Technol. 1992, 85, 77–86. [Google Scholar] [CrossRef]
- Mead, E.L.; Sutherland, R.G.; Verral, R.E. The effect on water in the presence of dissolved gases. Can. J. Chem. 1976, 54, 1114–1120. [Google Scholar] [CrossRef]
- Hart, E.J.; Henglein, A. Sonolytic decomposition of nitrous oxide in aqueous solution. J. Phys. Chem. 1986, 90, 5992–5995. [Google Scholar] [CrossRef]
- Park, J.-Y.; Lee, Y.-N. Solubility and decomposition kinetics of nitrous acid in aqueous solution. J. Phys. Chem. 1988, 92, 6294–6302. [Google Scholar] [CrossRef]
- Janzen, E.G. Electron Spin Resonance. Anal. Chem. 1974, 46, 478R–490R. [Google Scholar] [CrossRef]
- Kruus, P.; Beutel, L.; Aranda, R.; Penchuk, J.; Otson, R. Formation of complex organochlorine species in water due to cavitation. Chemosphere 1998, 36, 1811–1824. [Google Scholar] [CrossRef]
- Gallego-Juárez, J.A.; Rodriguez, G.; Acosta, V.; Riera, E. Power ultrasonic transducers with extensive radiators for industrial processing. Ultrason. Sonochem. [CrossRef]
- Krüger, O.; Schulze, Th.-L.; Peters, D. Sonochemical treatment of natural ground water at different high frequencies: preliminary results. Ultrason. Sonochem. 1999, 6, 123–128. [Google Scholar] [CrossRef]
- Peters, D. Sonolytic degradation of volatile pollutants in natural ground water: conclusions from a model study. Ultrason. Sonochem. 2001, 8, 221–226. [Google Scholar] [CrossRef]
- Guo, Z.; Gu, C.; Zheng, Z.; Feng, R.; Jiang, F.; Gao, G.; Zheng, Y. Sonodegradation of halomethane mixtures in chlorinated drinking water. Ultrason. Sonochem. 2006, 13, 487–492. [Google Scholar] [CrossRef]
- Clark II, C.J.; Annable, M.D.; Rao, P.S. Evaluation of sonochemical destruction of PCE in situ flushing waste fluids. J. Environ. Eng. 2000, 126, 1033–1038. [Google Scholar] [CrossRef]
- Schwarzenbach, R.P.; Gschwend, P.M.; Imboden, D.M. Environmental organic chemistry; Wiley: New York, NY, USA, 1993. [Google Scholar]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (OH·/O·-) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar]
- Louisnard, O.; González-García, J.; Tudela, I.; Klíma, J.; Sáez, V.; Vargas-Hernández, Y. FEM simulation of a sono-reactor accounting for vibrations of the boundaries. Ultrason. Sonochem. 2009, 16, 250–259. [Google Scholar] [CrossRef]
- Ashokkumar, M.; Lee, J.; Iida, Y.; Yasui, K.; Kozuka, T.; Tuziuti, T.; Towata, A. The detection and control of stable and transient acoustic cavitation bubbles. Phys. Chem. Chem. Phys. 2009, 11, 10118–10121. [Google Scholar] [CrossRef]
- Moholkar, V.S. Mechanistic optimization of a dual frequency sonochemical reactor. Chem. Eng. Sci. 2009, 64, 5255–5267. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
González-García, J.; Sáez, V.; Tudela, I.; Díez-Garcia, M.I.; Deseada Esclapez, M.; Louisnard, O. Sonochemical Treatment of Water Polluted by Chlorinated Organocompounds. A Review. Water 2010, 2, 28-74. https://doi.org/10.3390/w2010028
González-García J, Sáez V, Tudela I, Díez-Garcia MI, Deseada Esclapez M, Louisnard O. Sonochemical Treatment of Water Polluted by Chlorinated Organocompounds. A Review. Water. 2010; 2(1):28-74. https://doi.org/10.3390/w2010028
Chicago/Turabian StyleGonzález-García, José, Verónica Sáez, Ignacio Tudela, María Isabel Díez-Garcia, María Deseada Esclapez, and Olivier Louisnard. 2010. "Sonochemical Treatment of Water Polluted by Chlorinated Organocompounds. A Review" Water 2, no. 1: 28-74. https://doi.org/10.3390/w2010028
APA StyleGonzález-García, J., Sáez, V., Tudela, I., Díez-Garcia, M. I., Deseada Esclapez, M., & Louisnard, O. (2010). Sonochemical Treatment of Water Polluted by Chlorinated Organocompounds. A Review. Water, 2(1), 28-74. https://doi.org/10.3390/w2010028