Combining Targeted Metabolites Analysis and Transcriptomics to Reveal Chemical Composition Difference and Underlying Transcriptional Regulation in Maca (Lepidium Meyenii Walp.) Ecotypes

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Sample Preparation
2.3. Gas Chromatography–Mass Spectrometry Analysis
2.4. RNA Extraction, Quantification and Quality Analysis
2.5. Library Construction and RNA-Seq
2.6. Sequence Analysis and De Novo Assembly
2.7. Functional Annotation
2.8. Differential Gene Expression Analysis
2.9. Correlation Analysis between Metabolome and Transcriptome Data
2.10. qPCR Analysis
3. Results
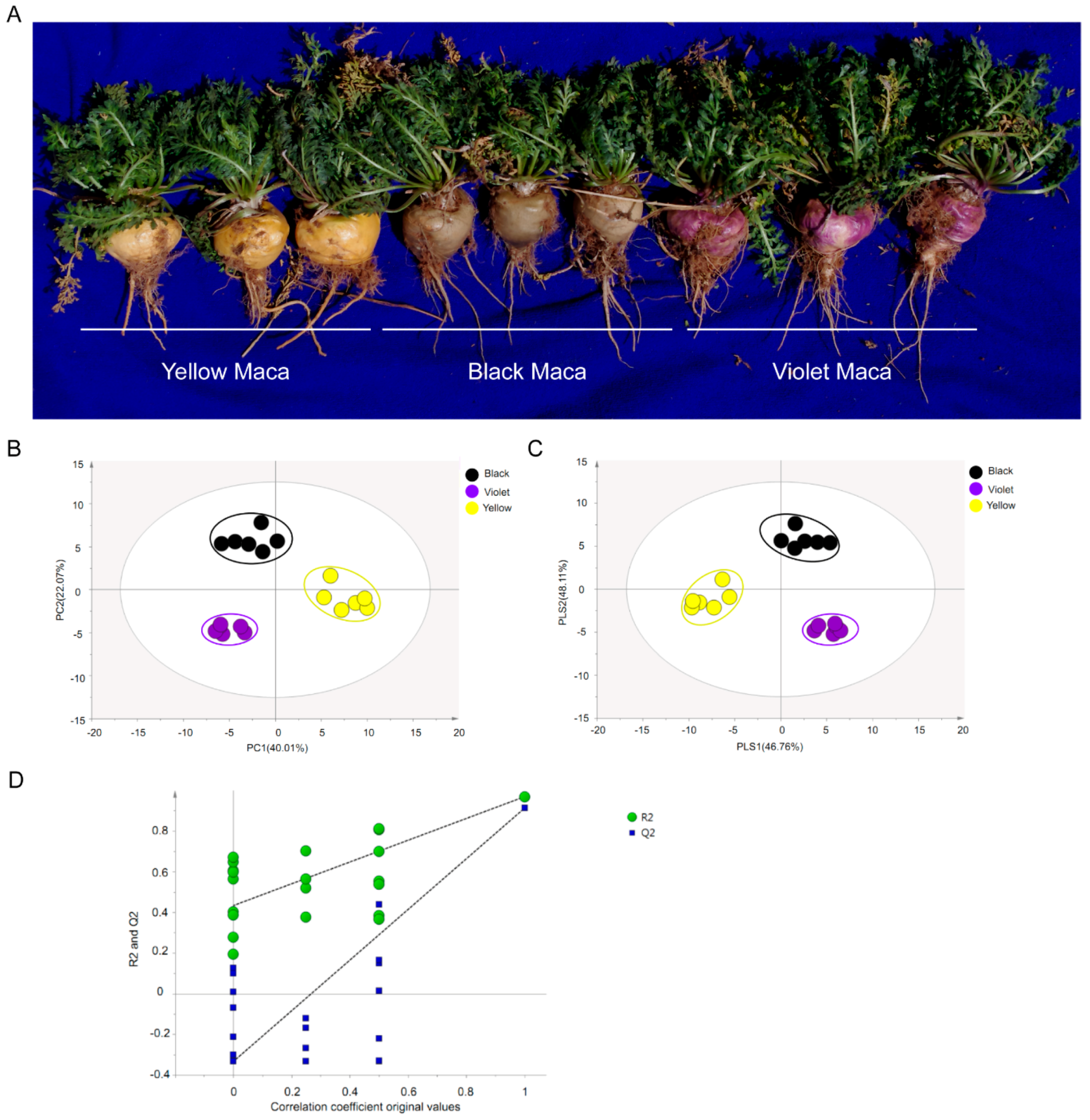
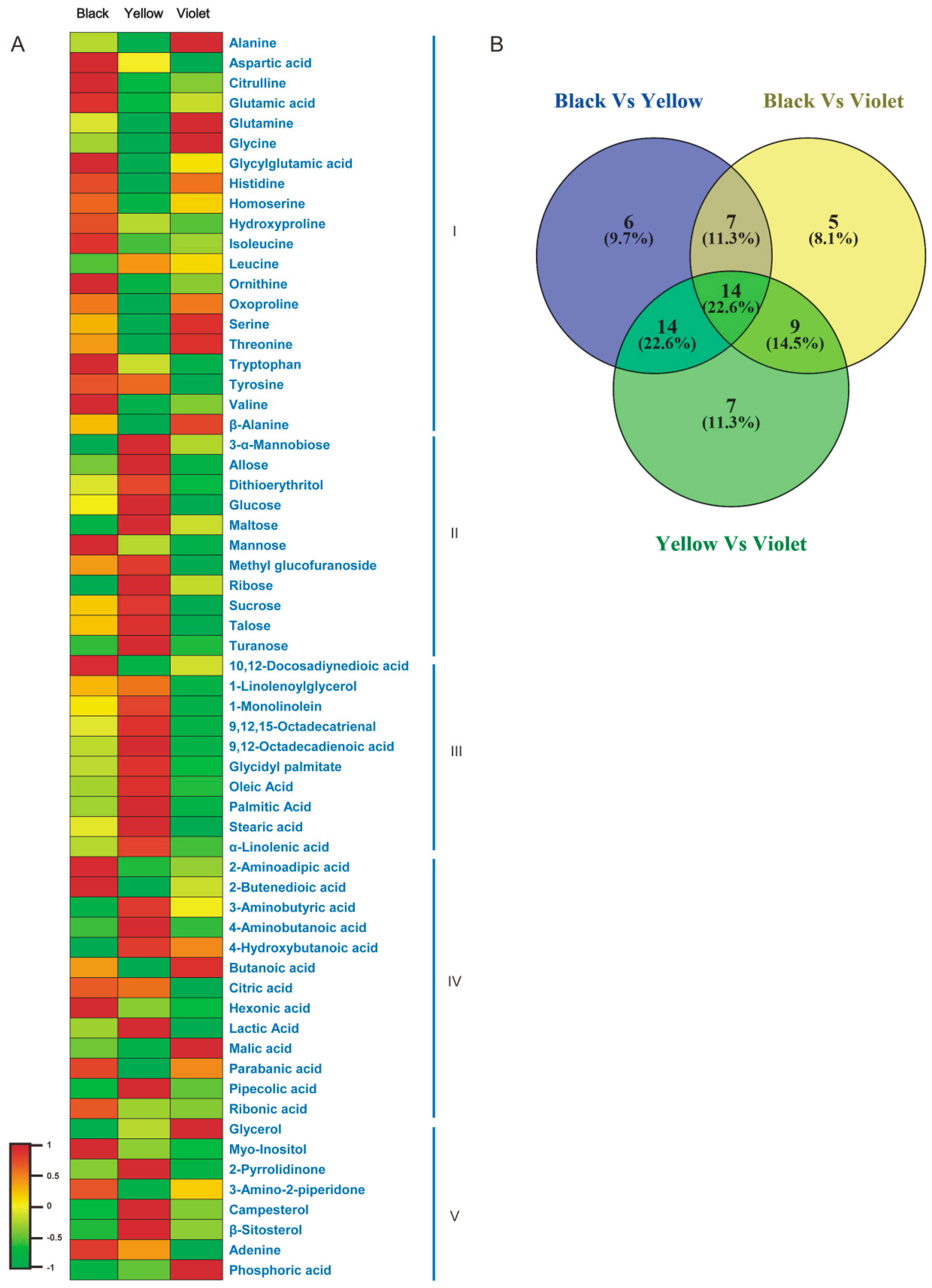
3.1. Metabolic Differences among Three Maca Ecotypes
3.2. Transcriptome Sequencing and De Novo Assembly of Maca
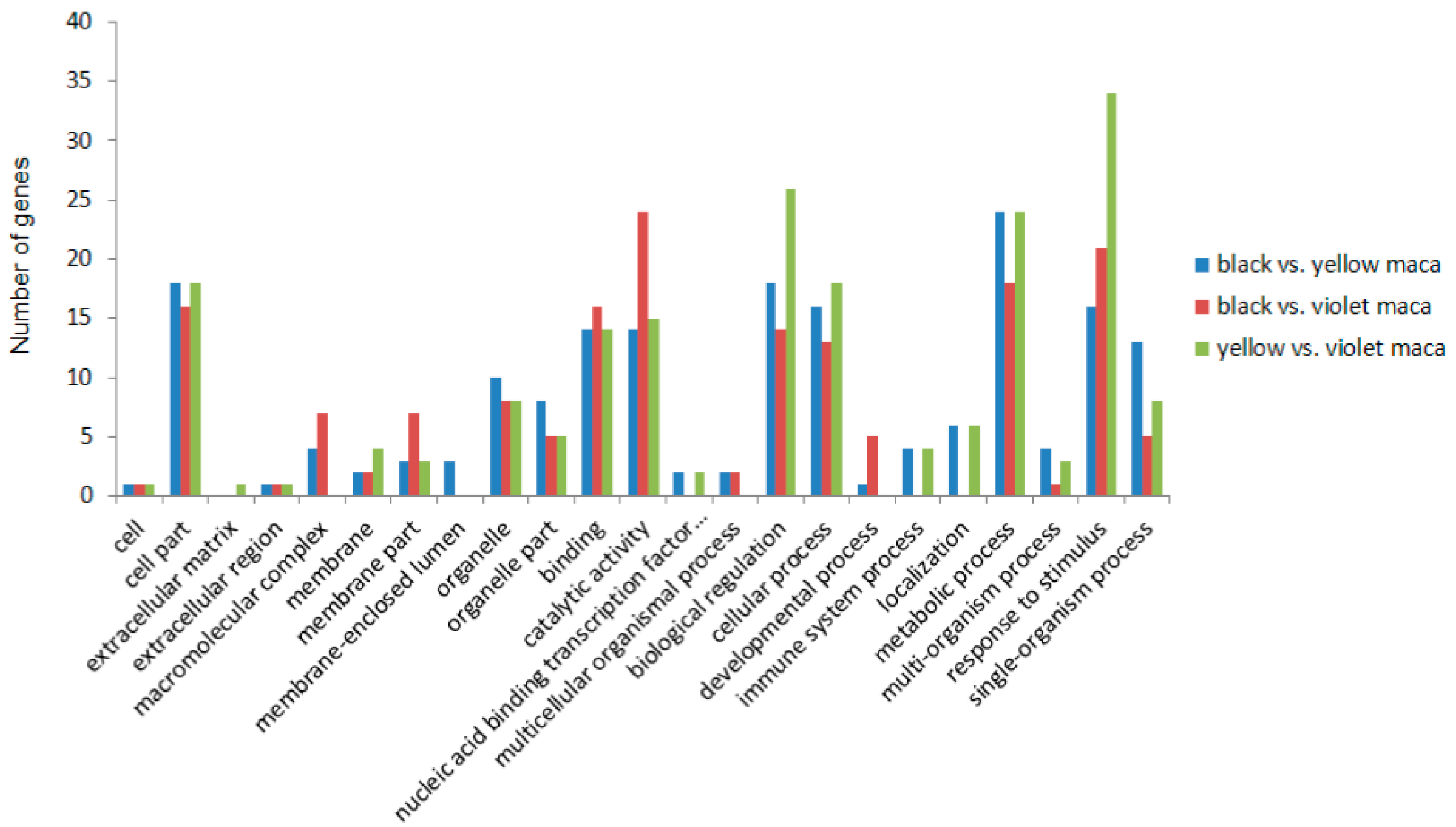
3.3. Functional Annotation
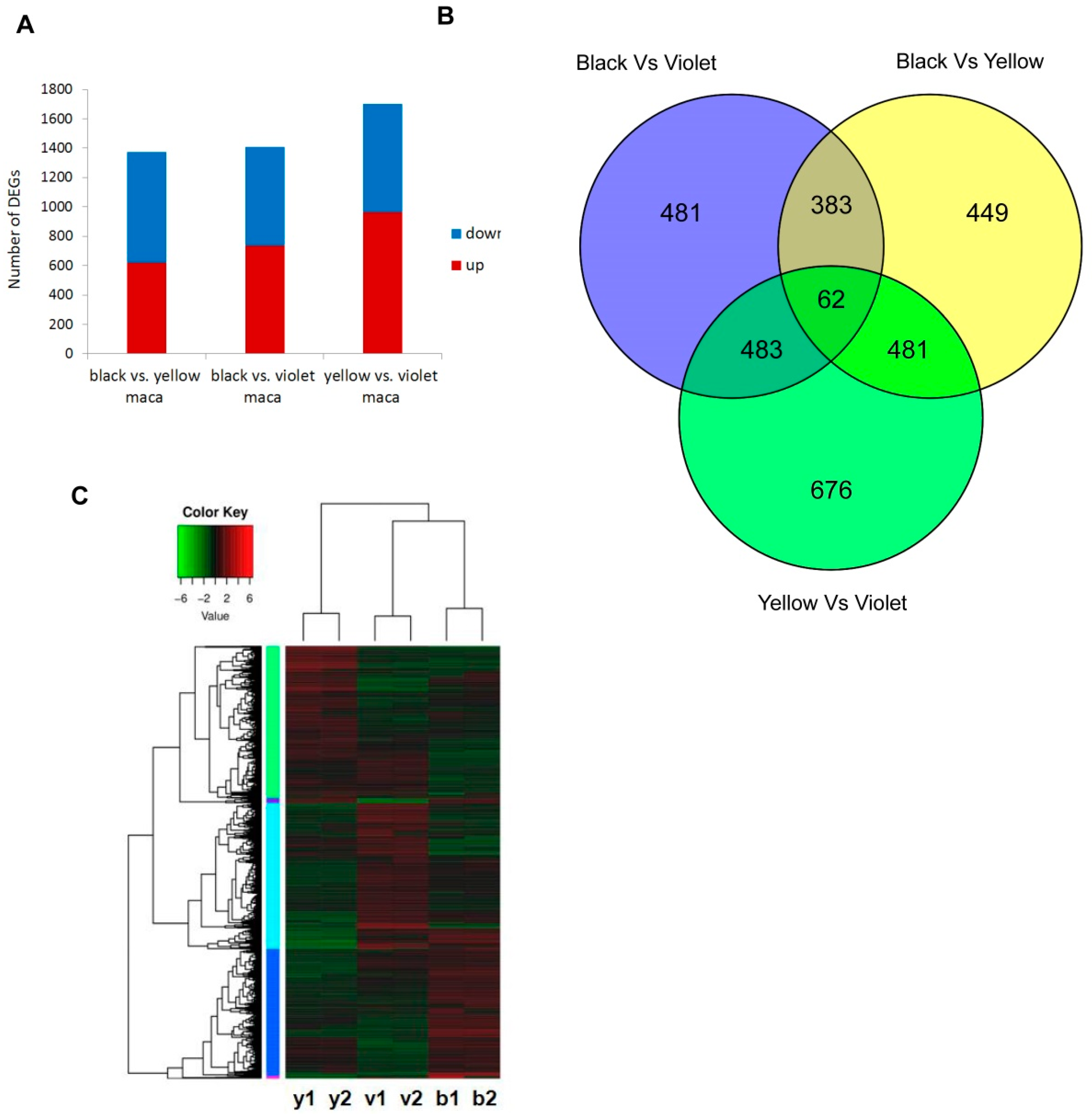
3.4. Comparison of Transcriptome Data among Three Maca Ecotypes
3.5. Experimental Validation of Differentially Expressed Genes
3.6. Correlation Analysis between Differentially Expressed Genes and Metabolites
4. Discussion
4.1. Overview of Metabolome of Maca Ecotypes
4.2. The Trancriptome of Maca
4.3. Different Expression Profiles and Specially Expressed Genes among Maca Ecotypes
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- León, J. The “maca” (Lepidium meyenii), a little known food plant of Peru. Econ. Bot. 1964, 18, 122–127. [Google Scholar] [CrossRef]
- Tello, J.; Hermann, M.; Calderón, A. La maca (Lepidium meyenii walp.): Cultivo alimenticio potencial para las zonas altoandinas. Bot. Lima 1992, 14, 59–66. [Google Scholar]
- Bermejo, J.E.H.; León, J. Neglected Crops: 1492 from a Different Perspective; FAO Plant Production and Protection Series, no. 26; Food and Agriculture Organization of the United Nations: Rome, Italy, 1994. [Google Scholar]
- Quiros, C.F.; Epperson, A.; Hu, J.; Holle, M. Physiological studies and determination of chromosome number in maca, Lepidium meyenii (brassicaceae). Econ. Bot. 1996, 50, 216–223. [Google Scholar] [CrossRef]
- Gonzales, G.F. Ethnobiology and ethnopharmacology of Lepidium meyenii (maca), a plant from the peruvian highlands. J. Evid.-Based Complement. Altern. Med. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dini, A.; Migliuolo, G.; Rastrelli, L.; Saturnino, P.; Schettino, O. Chemical composition of Lepidium meyenii. Food Chem. 1994, 49, 347–349. [Google Scholar] [CrossRef]
- Piacente, S.; Carbone, V.; Plaza, A.; Zampelli, A.; Pizza, C. Investigation of the tuber constituents of maca (Lepidium meyenii walp.). J. Agric. Food Chem. 2002, 50, 5621–5625. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, I.; Zhao, J.; Dunbar, D.C.; Khan, I.A. Constituents of Lepidium meyenii ‘maca’. Phytochemistry 2002, 59, 105–110. [Google Scholar] [CrossRef]
- Cicero, A.F.; Bandieri, E.; Arletti, R. Lepidium meyenii walp. Improves sexual behaviour in male rats independently from its action on spontaneous locomotor activity. J. Ethnopharmacol. 2001, 75, 225–229. [Google Scholar] [CrossRef]
- Gonzales, G.F.; Gasco, M.; Cordova, A.; Chung, A.; Rubio, J.; Villegas, L. Effect of Lepidium meyenii (maca) on spermatogenesis in male rats acutely exposed to high altitude (4340 m). J. Endocrinol. 2004, 180, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Clément, C.; Kneubühler, J.; Urwyler, A.; Witschi, U.; Kreuzer, M. Effect of maca supplementation on bovine sperm quantity and quality followed over two spermatogenic cycles. Theriogenology 2010, 74, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, C.; Leiva-Revilla, J.; Rubio, J.; Gasco, M.; Gonzales, G. Effect of red maca (Lepidium meyenii) on prostate zinc levels in rats with testosterone-induced prostatic hyperplasia. Andrologia 2012, 44, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, G.F.; Miranda, S.; Nieto, J.; Fernández, G.; Yucra, S.; Rubio, J.; Yi, P.; Gasco, M. Red maca (Lepidium meyenii) reduced prostate size in rats. Reprod. Biol. Endocrinol. 2005, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasco, M.; Aguilar, J.; Gonzales, G. Effect of chronic treatment with three varieties of Lepidium meyenii (maca) on reproductive parameters and DNA quantification in adult male rats. Andrologia 2007, 39, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, C.; Rubio, J.; Gasco, M.; Nieto, J.; Yucra, S.; Gonzales, G.F. Effect of short-term and long-term treatments with three ecotypes of Lepidium meyenii (maca) on spermatogenesis in rats. J. Ethnopharmacol. 2006, 103, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Rubio, J.; Caldas, M.; Dávila, S.; Gasco, M.; Gonzales, G.F. Effect of three different cultivars of Lepidium meyenii (maca) on learning and depression in ovariectomized mice. BMC Complement. Altern. Med. 2006, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Gonzales-Castañeda, C.; Rivera, V.; Chirinos, A.L.; Evelson, P.; Gonzales, G.F. Photoprotection against the uvb-induced oxidative stress and epidermal damage in mice using leaves of three different varieties of Lepidium meyenii (maca). Int. J. Dermatol. 2011, 50, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, C.; Cárdenas-Valencia, I.; Leiva-Revilla, J.; Anza-Ramirez, C.; Rubio, J.; Gonzales, G.F. Effects of different varieties of maca (Lepidium meyenii) on bone structure in ovariectomized rats. Complement. Med. Res. 2010, 17, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, L.; Zhao, B. Relationship between glucosinolates and colors in eight accessions of maca. J. Food Eng. Technol. 2005, 4, 45–52. [Google Scholar]
- Zhang, J.; Tian, Y.; Yan, L.; Zhang, G.; Wang, X.; Zeng, Y.; Zhang, J.; Ma, X.; Tan, Y.; Long, N. Genome of plant maca (Lepidium meyenii) illuminates genomic basis for high-altitude adaptation in the central Andes. Mol. Plant 2016, 9, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from rna-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Maslak, P. Blast. ASH Image Bank 2004, 2004, 101039. [Google Scholar] [CrossRef]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the blast2go suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. Kegg for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2011, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by rna-seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 35, D61–D65. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A.; Apweiler, R.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M. The Universal Protein Resource (UniProt). Nucleic Acids Res. 2005, 33, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, A.V.; Sergei, S.; Rao, B.S.; Nikolskaya, A.N.; Mekhedov, S.L.; Raja, M.; Krylov, D.M.; Koonin, E.V.; Boris, K.; Jacobs, A.R. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar]
- Clarke, D.B. Glucosinolates, structures and analysis in food. Anal. Methods 2010, 2, 310–325. [Google Scholar] [CrossRef]
- Li, G.; Ammermann, U.; Quirós, C.F. Glucosinolate contents in maca (Lepidium peruvianum chacon) seeds, sprouts, mature plants and several derived commercial products. Econ. Bot. 2001, 55, 255–262. [Google Scholar] [CrossRef]
- Hill, C.; Harris, R.C.; Kim, H.; Harris, B.; Sale, C.; Boobis, L.; Kim, C.; Wise, J.A. Influence of β-alanine supplementation on skeletal muscle carnosine concentrations and high intensity cycling capacity. Amino Acids 2007, 32, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef] [PubMed]
- Lehnman, I. DNA ligase: Structure, mechanism and function. Science 1974, 186, 790–797. [Google Scholar] [CrossRef]
- Metzlaff, M.; O’dell, M.; Cluster, P.; Flavell, R. Rna-mediated RNA degradation and chalcone synthase a silencing in petunia. Cell 1997, 88, 845–854. [Google Scholar] [CrossRef]
- Chiu, L.-W.; Zhou, X.; Burke, S.; Wu, X.; Prior, R.L.; Li, L. The purple cauliflower arises from activation of a myb transcription factor. Plant Physiol. 2010, 154, 1470–1480. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
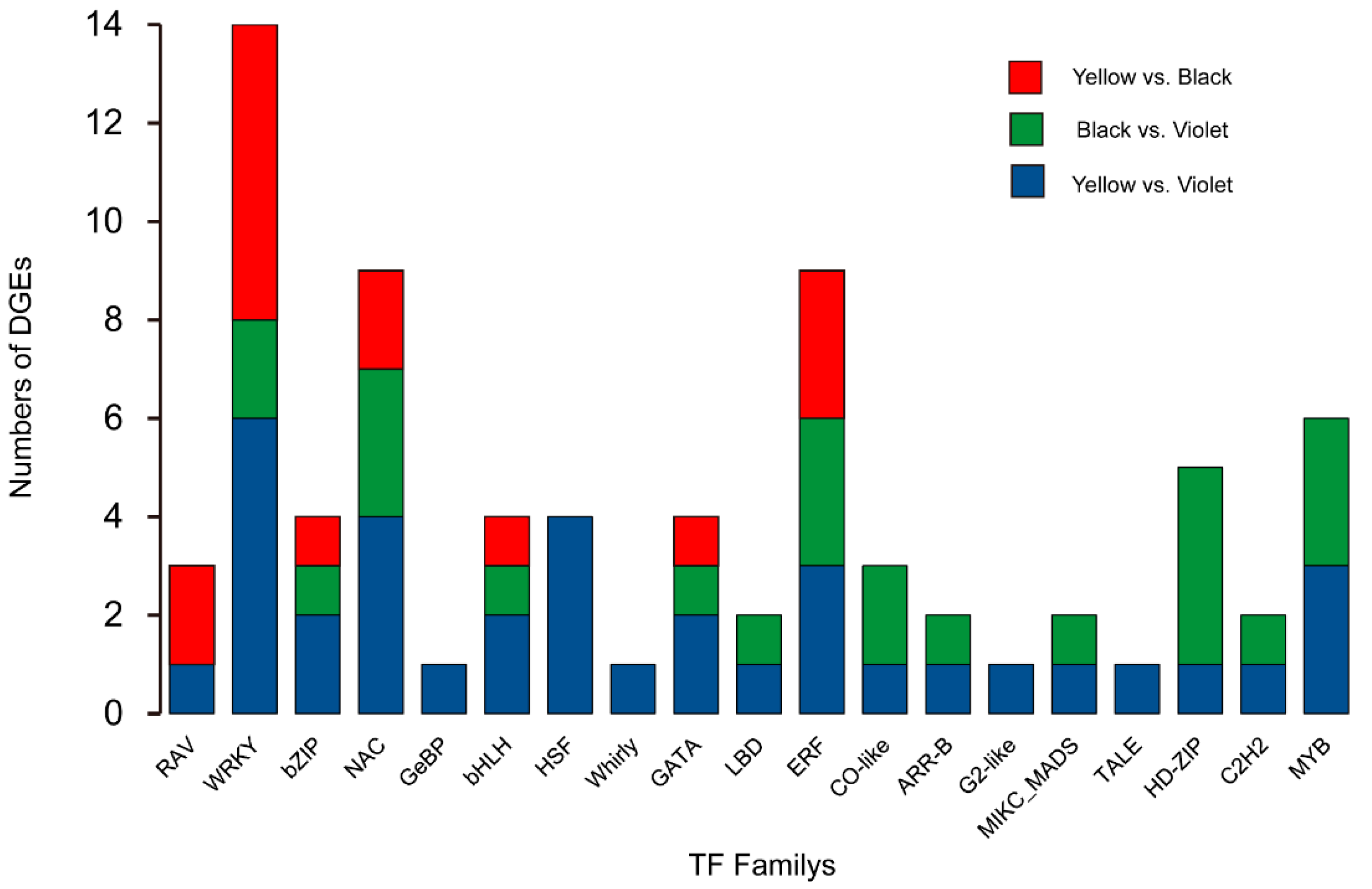{kind=link}
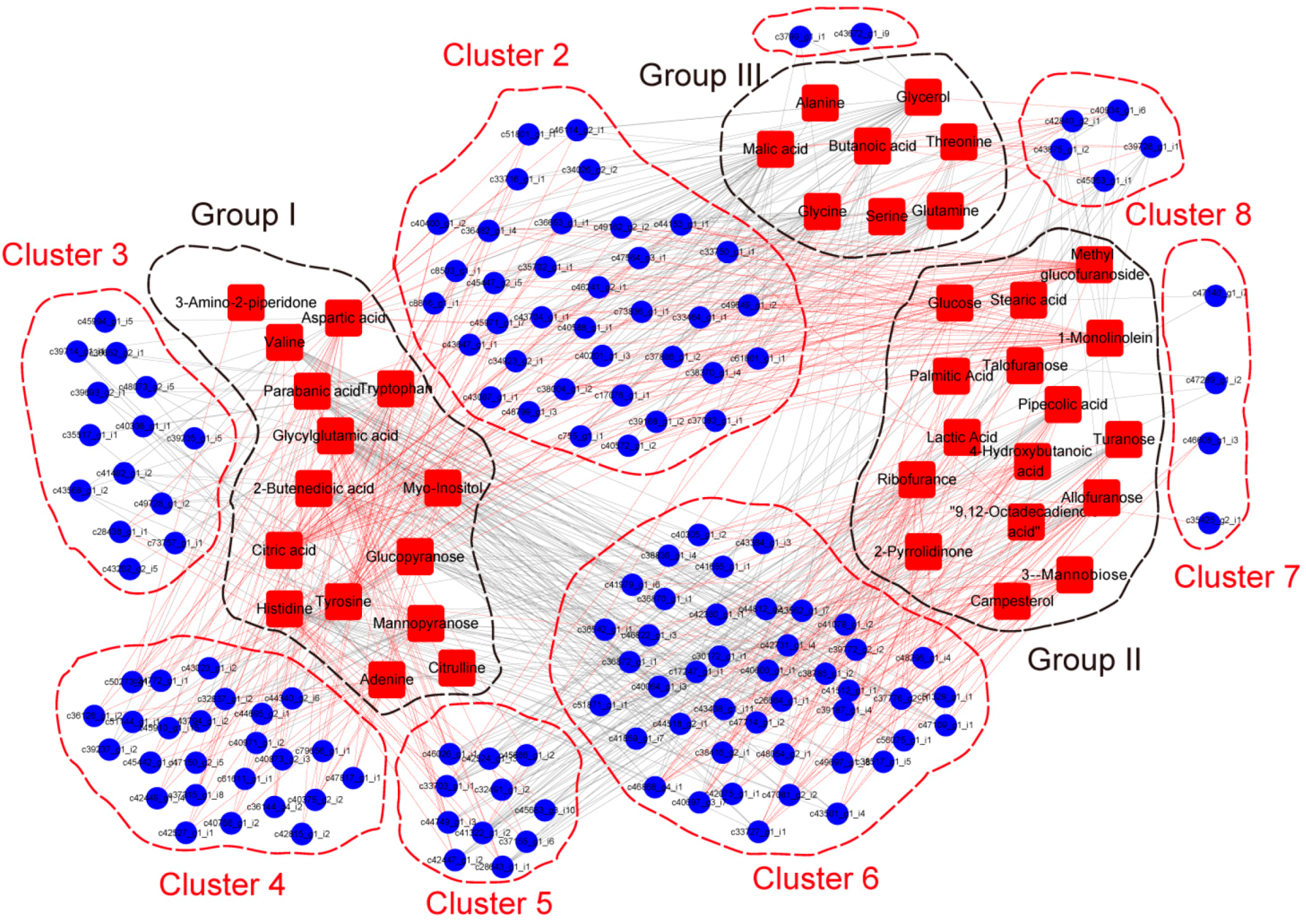{kind=link}
| Annotated Databases | Unigenes | ≥300 nt | ≥1000 nt |
|---|---|---|---|
| Nr | 71,550 | 59,091 | 28,515 |
| COG | 49,846 | 42,993 | 24,192 |
| KEGG | 8287 | 8117 | 6133 |
| GO | 57,433 | 48,655 | 24,362 |
| Swiss-Prot | 51,936 | 43,669 | 23,091 |
| All | 73,113 | 59,614 | 28,548 |
| Gene ID | Specially Expressed In 1 | Swissprot Annotation | GO Annotation |
|---|---|---|---|
| c29661_g1_i1 | B | Unknown | - |
| c34272_g1_i2 | B | Unknown | - |
| c36979_g1_i2 | B | No vein-like protein | - |
| c43742_g2_i1 | B | Plastidic glucose transporter 4 | - |
| c43908_g1_i6 | B | DNA ligase | DNA recombination (GO:0006310) |
| c34364_g1_i4 | Y | Glycine-rich RNA-binding protein | Cell wall (GO:0005618) |
| c36217_g1_i3 | Y | Disease resistance-responsive, dirigent domain-containing protein | Defense response (GO:0006952) |
| c39509_g4_i3 | Y | Zinc ion binding | - |
| c42330_g1_i1 | Y | Probable fructokinase-5 | Ribokinase activity (GO:0004747) |
| c51221_g3_i1 | Y | Transposon Ty3-I Gag-Pol polyprotein | - |
| c34798_g1_i3 | V | Unknown | Microtubule-based movement (GO:0007018) |
| c36120_g1_i1 | V | Retrovirus-related Pol polyprotein from transposon TNT 1-94 | - |
| -c40810_g1_i1 | V | Chalcone synthase | Oxidation-reduction process (GO:0055114) |
| c67803_g1_i1 | V | Unknown | - |
| c8503_g1_i1 | V | Transcription factor MYB75 | DNA binding transcription factor activity (GO:0003700) |
| c41540_g2_i3 | BV | Ubiquitin-conjugating enzyme E2 8 | Proteasome-mediated ubiquitin-dependent protein catabolic process (GO:0043161) |
| c41853_g5_i1 | BV | Unknown | - |
| c42101_g1_i7 | BV | Annexin D8 | Response to water deprivation (GO:0009414) |
| c47327_g1_i3 | BV | Unknown | - |
| c48799_g1_i1 | BV | Aspartic proteinase-like protein 1 | Anchored component of membrane (GO:0031225) |
| c13374_g1_i1 | BY | Unknown | - |
| c20825_g2_i1 | BY | Unknown | - |
| c21560_g1_i1 | BY | Early light-induced protein 1, chloroplastic | Cytoplasm (GO:0005737) |
| c33991_g1_i2 | BY | Unknown | - |
| c46340_g2_i8 | BY | Ultraviolet-B receptor UVR8 | Nucleotide-excision repair (GO:0006289) |
| c36101_g2_i1 | YV | 1-acylglycerol-3-phosphate O-acyltransferase | - |
| c36695_g1_i1 | YV | Retrovirus-related Pol polyprotein from transposon TNT 1-94 | - |
| c40657_g2_i3 | YV | Thioredoxin O1, mitochondrial | Brassinosteroid biosynthetic process (GO:0016132) |
| c43332_g1_i8 | YV | Tetratricopeptide repeat domain-containing protein | Response to sucrose (GO:0009744) |
| c45635_g1_i2 | YV | Haloacid dehalogenase-like hydrolase domain-containing protein 3 | NADP + binding (GO:0070401) |
| c49680_g2_i2 | YV | Retrovirus-related Pol polyprotein from transposon TNT 1-94 | Golgi apparatus (GO:0005794) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Q.; Li, M.; Wang, C.; Li, Z.; Xu, J.; Zheng, Q.; Liu, P.; Zhou, H. Combining Targeted Metabolites Analysis and Transcriptomics to Reveal Chemical Composition Difference and Underlying Transcriptional Regulation in Maca (Lepidium Meyenii Walp.) Ecotypes. Genes 2018, 9, 335. https://doi.org/10.3390/genes9070335
Chen Q, Li M, Wang C, Li Z, Xu J, Zheng Q, Liu P, Zhou H. Combining Targeted Metabolites Analysis and Transcriptomics to Reveal Chemical Composition Difference and Underlying Transcriptional Regulation in Maca (Lepidium Meyenii Walp.) Ecotypes. Genes. 2018; 9(7):335. https://doi.org/10.3390/genes9070335
Chicago/Turabian StyleChen, Qiansi, Meng Li, Chen Wang, Zefeng Li, Jiayang Xu, Qingxia Zheng, Pingping Liu, and Huina Zhou. 2018. "Combining Targeted Metabolites Analysis and Transcriptomics to Reveal Chemical Composition Difference and Underlying Transcriptional Regulation in Maca (Lepidium Meyenii Walp.) Ecotypes" Genes 9, no. 7: 335. https://doi.org/10.3390/genes9070335