Transcriptome Analysis of Adipose Tissue Indicates That the cAMP Signaling Pathway Affects the Feed Efficiency of Pigs

Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. Library Preparation and Sequencing
2.3. RNA Sequencing Analysis
2.4. Differential Expression Analysis and Real-Time Quantitative PCR Validation
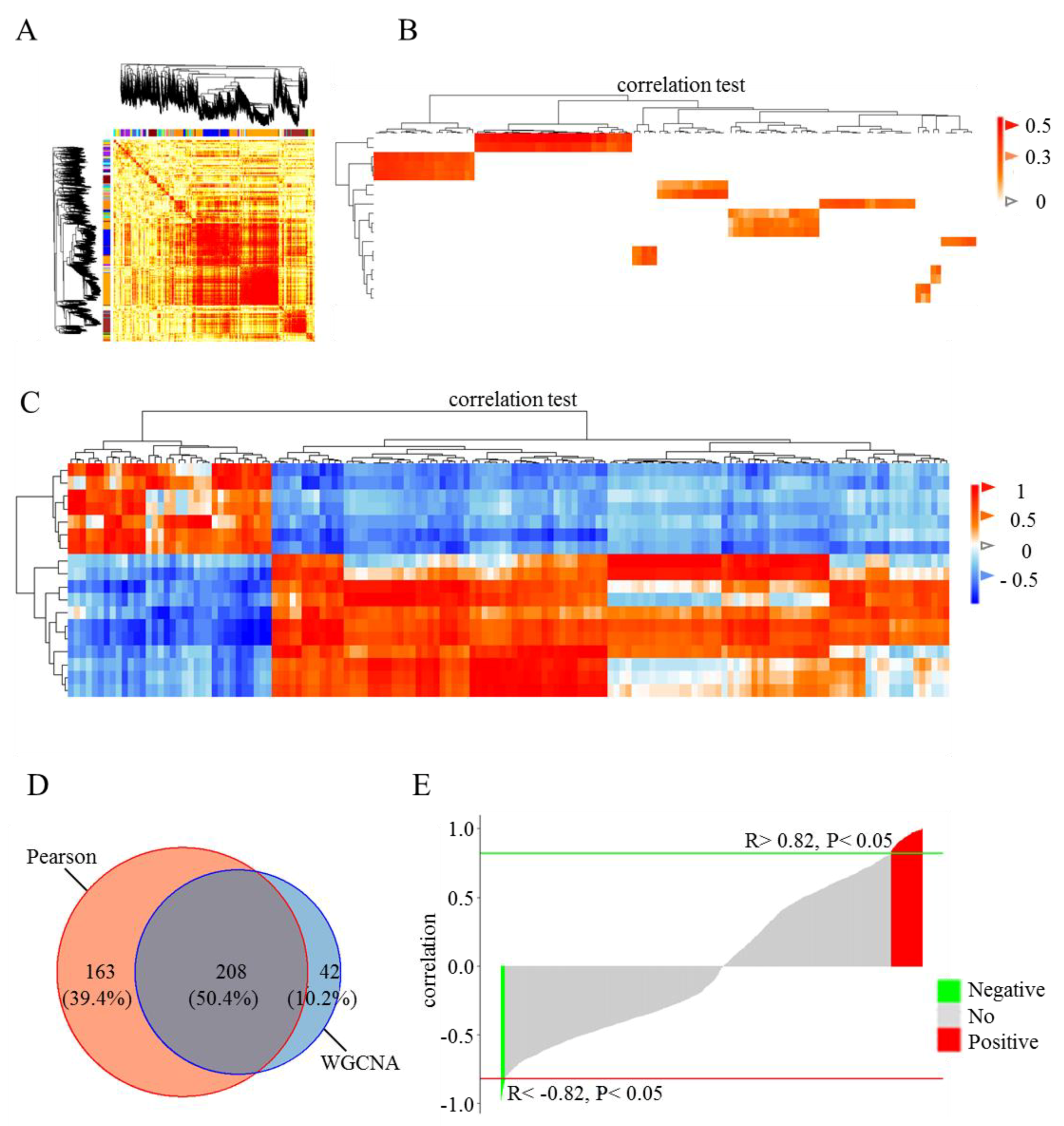
2.5. Correlation Analysis
2.6. Gene Ontology Enrichment and Pathway Analysis
3. Results
3.1. Mapping and Annotation of RNA Sequencing Data
3.2. DE Genes and lincRNAs between High- and Low-FE Pigs
3.3. Correlation Analysis of DE Genes and lincRNAs
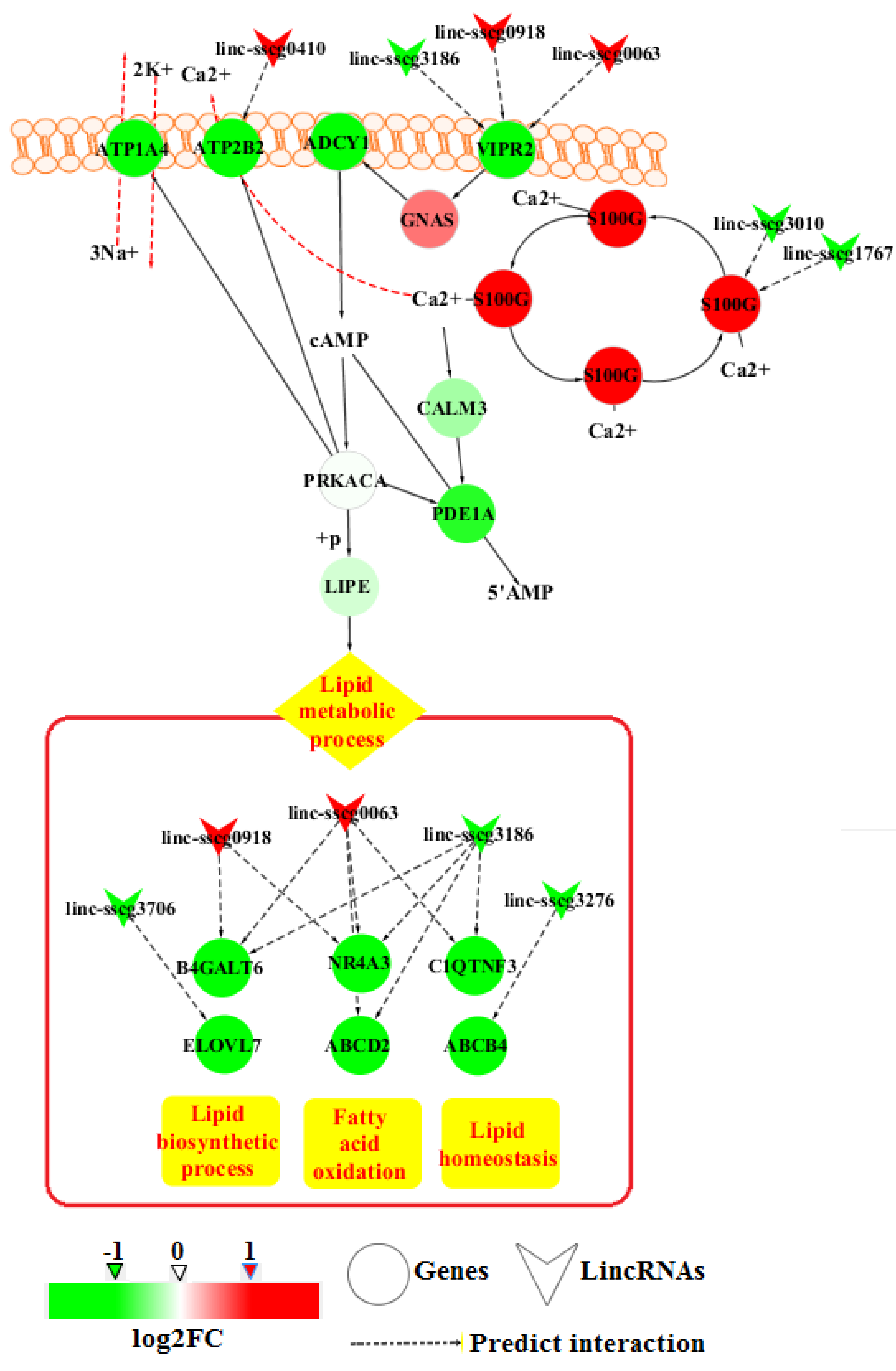
3.4. GO Enrichment and Pathway Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Patience, J.F.; Rossoni-Serao, M.C.; Gutierrez, N.A. A review of feed efficiency in swine: Biology and application. J. Anim. Sci. Biotechnol. 2015, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.A.; Suzuki, K.; Kadowaki, H.; Shibata, T.; Oikawa, T. Genetic parameters for feed efficiency traits and their relationships with growth and carcass traits in Duroc pigs. J. Anim. Breed. Genet. 2007, 124, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Lkhagvadorj, S.; Qu, L.; Cai, W.; Couture, O.P.; Barb, C.R.; Hausman, G.J.; Nettleton, D.; Anderson, L.L.; Dekkers, J.C.; Tuggle, C.K. Gene expression profiling of the short-term adaptive response to acute caloric restriction in liver and adipose tissues of pigs differing in feed efficiency. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R494–R507. [Google Scholar] [CrossRef] [PubMed]
- Louveau, I.; Vincent, A.; Tacher, S.; Gilbert, H.; Gondret, F. Increased expressions of genes and proteins involved in mitochondrial oxidation and antioxidant pathway in adipose tissue of pigs selected for a low residual feed intake. J. Anim. Sci. 2016, 94, 5042–5054. [Google Scholar] [CrossRef] [PubMed]
- Gondret, F.; Vincent, A.; Houee-Bigot, M.; Siegel, A.; Lagarrigue, S.; Causeur, D.; Gilbert, H.; Louveau, I. A transcriptome multi-tissue analysis identifies biological pathways and genes associated with variations in feed efficiency of growing pigs. BMC Genom. 2017, 18, 244. [Google Scholar] [CrossRef] [PubMed]
- Katona, D.; Rajki, A.; Di Benedetto, G.; Pozzan, T.; Spat, A. Calcium-dependent mitochondrial cAMP production enhances aldosterone secretion. Mol. Cell. Endocrinol. 2015, 412, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Kissebah, A.H.; Vydelingum, N.; Tulloch, B.R.; Hope-Gill, H.; Fraser, T.R. The role of calcium in insulin action. I. Purification and properties of enzymes regulating lipolysis in human adipose tissue: Effects of cyclic-AMP and calcium ions. Horm. Metab. Res. 1974, 6, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Goudarzi, F.; Mohammadalipour, A.; Khodadadi, I.; Karimi, S.; Mostoli, R.; Bahabadi, M.; Goodarzi, M.T. The role of calcium in differentiation of human adipose-derived stem cells to adipocytes. Mol. Biotechnol. 2018, 60, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Greenberg, A.G.; Kraemer, F.B.; Zemel, M.B. Mechanism of intracellular calcium ([Ca2+]i) inhibition of lipolysis in human adipocytes. FASEB J. 2001, 15, 2527–2529. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; Souza, S.C.; Zhang, H.H.; Strissel, K.J.; Christoffolete, M.A.; Kovsan, J.; Rudich, A.; Kraemer, F.B.; Bianco, A.C.; Obin, M.S.; et al. Perilipin promotes hormone-sensitive lipase-mediated adipocyte lipolysis via phosphorylation-dependent and -independent mechanisms. J. Biol. Chem. 2006, 281, 15837–15844. [Google Scholar] [CrossRef] [PubMed]
- Holm, C. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochem. Soc. Trans. 2003, 31, 1120–1124. [Google Scholar] [CrossRef] [PubMed]
- Kuo, A.; Lee, M.Y.; Yang, K.; Gross, R.W.; Sessa, W.C. Caveolin-1 regulates lipid droplet metabolism in endothelial cells via autocrine prostacyclin-stimulated, cAMP-mediated lipolysis. J. Biol. Chem. 2018, 293, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Benmansour, N.; Demarne, Y.; Lecourtier, M.J.; Lhuillery, C. Effects of castration, dietary fat and adipose tissue sites on adipocyte plasma-membranes cyclic AMP phosphodiesterase activity in the pig. Int. J. Biochem. 1991, 23, 1205–1211. [Google Scholar] [CrossRef]
- Chen, X.D.; Lei, T.; Xia, T.; Gan, L.; Yang, Z.Q. Increased expression of resistin and tumour necrosis factor-α in pig adipose tissue as well as effect of feeding treatment on resistin and cAMP pathway. Diabetes Obes. Metab. 2004, 6, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.Y.; Li, A.; Wang, L.G.; Irwin, D.M.; Liu, Y.H.; Xu, D.; Han, X.M.; Wang, L.; Wu, S.F.; Wang, L.X.; et al. DNA methylation signatures of long intergenic noncoding RNAs in porcine adipose and muscle tissues. Sci. Rep. 2015, 5, 15435. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Goff, L.A.; Trapnell, C.; Alexander, R.; Lo, K.A.; Hacisuleyman, E.; Sauvageau, M.; Tazon-Vega, B.; Kelley, D.R.; Hendrickson, D.G.; et al. Long noncoding RNAs regulate adipogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 3387–3392. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhang, X.; Li, A.; Xie, L.; Miao, X. Differential regulation of mRNAs and lncRNAs related to lipid metabolism in two pig breeds. Oncotarget 2017, 8, 87539–87553. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Chen, J.; Chen, N. Long noncoding RNA MALAT1 promotes hepatic steatosis and insulin resistance by increasing nuclear SREBP-1c protein stability. Sci. Rep. 2016, 6, 22640. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yang, Z.; Wu, J.; Zhang, L.; Lee, S.; Shin, D.J.; Tran, M.; Wang, L. Long noncoding RNA H19 interacts with polypyrimidine tract-binding protein 1 to reprogram hepatic lipid homeostasis. Hepatology 2018, 67, 1768–1783. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Cheng, M.; Niu, Y.; Chi, X.; Liu, X.; Fan, J.; Fan, H.; Chang, Y.; Yang, W. Identification of a novel human long non-coding RNA that regulates hepatic lipid metabolism by inhibiting SREBP-1c. Int. J. Biol. Sci. 2017, 13, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hou, Y.; Liu, F.; Liu, A.; Jing, L.; Zhao, C.; Luan, Y.; Miao, Y.; Zhao, S.; Li, X. Transcriptome analysis reveals that vitamin A metabolism in the liver affects feed efficiency in pigs. G3 (Bethesda) 2016, 6, 3615–3624. [Google Scholar] [CrossRef] [PubMed]
- Jing, L.; Hou, Y.; Wu, H.; Miao, Y.; Li, X.; Cao, J.; Brameld, J.M.; Parr, T.; Zhao, S. Transcriptome analysis of mRNA and miRNA in skeletal muscle indicates an important network for differential Residual Feed Intake in pigs. Sci. Rep. 2015, 5, 11953. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.Y.; Li, A.M.; Adeola, A.C.; Liu, Y.H.; Irwin, D.M.; Xie, H.B.; Zhang, Y.P. Genome-wide identification of long intergenic noncoding RNA genes and their potential association with domestication in pigs. Genome Biol. Evol. 2014, 6, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Erkens, T.; Van Poucke, M.; Vandesompele, J.; Goossens, K.; Van Zeveren, A.; Peelman, L.J. Development of a new set of reference genes for normalization of real-time RT-PCR data of porcine backfat and longissimus dorsi muscle, and evaluation with PPARGC1A. BMC Biotechnol. 2006, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformat. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Horodyska, J.; Oster, M.; Reyer, H.; Mullen, A.M.; Lawlor, P.G.; Wimmers, K.; Hamill, R.M. Analysis of meat quality traits and gene expression profiling of pigs divergent in residual feed intake. Meat Sci. 2018, 137, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Ramayo-Caldas, Y.; Ballester, M.; Sanchez, J.P.; Gonzalez-Rodriguez, O.; Revilla, M.; Reyer, H.; Wimmers, K.; Torrallardona, D.; Quintanilla, R. Integrative approach using liver and duodenum RNA-Seq data identifies candidate genes and pathways associated with feed efficiency in pigs. Sci. Rep. 2018, 8, 558. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Quan, J.; Yang, M.; Wang, X.; Zheng, E.; Yang, H.; Fu, D.; Yang, Y.; Yang, L.; Li, Z.; et al. Genome-wide association analysis reveals genetic loci and candidate genes for feeding behavior and eating efficiency in Duroc boars. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, H.; Billon, Y.; Brossard, L.; Faure, J.; Gatellier, P.; Gondret, F.; Labussiere, E.; Lebret, B.; Lefaucheur, L.; Le Floch, N.; et al. Review: Divergent selection for residual feed intake in the growing pig. Animal 2017, 11, 1427–1439. [Google Scholar] [CrossRef] [PubMed]
- Schroder, B.; Schlumbohm, C.; Kaune, R.; Breves, G. Role of calbindin-D9k in buffering cytosolic free Ca2+ ions in pig duodenal enterocytes. J. Physiol. 1996, 492, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Yang, H.; Kang, H.S.; Ahn, C.; Hong, E.J.; An, B.S.; Jeung, E.B. Alteration of tight junction gene expression by calcium- and vitamin D-deficient diet in the duodenum of calbindin-null mice. Int. J. Mol. Sci. 2013, 14, 22997–23010. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Lu, Z.; He, M.; Gao, R.; Bai, H.; Shi, B.; Shan, A. Effects of dietary supplementation of fulvic acid on lipid metabolism of finishing pigs. J. Anim. Sci. 2014, 92, 4921–4926. [Google Scholar] [CrossRef] [PubMed]
- Feoktistov, I.; Biaggioni, I. Adenosine A2B receptors. Pharmacol. Rev. 1997, 49, 381–402. [Google Scholar] [PubMed]
- Feoktistov, I.; Murray, J.J.; Biaggioni, I. Positive modulation of intracellular Ca2+ levels by adenosine A2b receptors, prostacyclin, and prostaglandin E1 via a cholera toxin-sensitive mechanism in human erythroleukemia cells. Mol. Pharmacol. 1994, 45, 1160–1167. [Google Scholar] [PubMed]
- Lutz, E.M.; Shen, S.; Mackay, M.; West, K.; Harmar, A.J. Structure of the human VIPR2 gene for vasoactive intestinal peptide receptor type 2. FEBS Lett. 1999, 458, 197–203. [Google Scholar] [CrossRef]
- Carayol, J.; Sacco, R.; Tores, F.; Rousseau, F.; Lewin, P.; Hager, J.; Persico, A.M. Converging evidence for an association of ATP2B2 allelic variants with autism in male subjects. Biol. Psychiatry 2011, 70, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Clausen, M.V.; Nissen, P.; Poulsen, H. The α4 isoform of the Na+, K+-ATPase is tuned for changing extracellular environments. FEBS J. 2016, 283, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Van Roermund, C.W.; Visser, W.F.; Ijlst, L.; Waterham, H.R.; Wanders, R.J. Differential substrate specificities of human ABCD1 and ABCD2 in peroxisomal fatty acid β-oxidation. Biochim. Biophys. Acta 2011, 1811, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Pearen, M.A.; Goode, J.M.; Fitzsimmons, R.L.; Eriksson, N.A.; Thomas, G.P.; Cowin, G.J.; Wang, S.C.; Tuong, Z.K.; Muscat, G.E. Transgenic muscle-specific Nor-1 expression regulates multiple pathways that effect adiposity, metabolism, and endurance. Mol. Endocrinol. 2013, 27, 1897–1917. [Google Scholar] [CrossRef] [PubMed]
- Naganuma, T.; Sato, Y.; Sassa, T.; Ohno, Y.; Kihara, A. Biochemical characterization of the very long-chain fatty acid elongase ELOVL7. FEBS Lett. 2011, 585, 3337–3341. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, N.; Numata, S.; Li, X.; Nomura, T.; Takizawa, M.; Kondo, Y.; Yamashita, Y.; Hashimoto, N.; Kiyono, T.; Urano, T.; et al. β4GalT6 is involved in the synthesis of lactosylceramide with less intensity than β4GalT5. Glycobiology 2013, 23, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Kopp, A.; Bala, M.; Buechler, C.; Falk, W.; Gross, P.; Neumeier, M.; Scholmerich, J.; Schaffler, A. C1q/TNF-related protein-3 represents a novel and endogenous lipopolysaccharide antagonist of the adipose tissue. Endocrinology 2010, 151, 5267–5278. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.Y.; Kobayashi, A.; Takanezawa, Y.; Kioka, N.; Handa, T.; Arai, H.; Matsuo, M.; Ueda, K. Bile salt-dependent efflux of cellular phospholipids mediated by ATP binding cassette protein B4. Hepatology 2007, 46, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, B.; Chen, L.; Gou, L.T.; Li, H.; Fu, X.D. GRID-seq reveals the global RNA-chromatin interactome. Nat. Biotechnol. 2017, 35, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Van Solingen, C.; Scacalossi, K.R.; Moore, K.J. Long noncoding RNAs in lipid metabolism. Curr. Opin. Lipidol. 2018, 29, 224–232. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Sample | Input Reads | Mapped Reads | Aligned Pairs | Uniquely Aligned Pairs |
|---|---|---|---|---|---|
| High | H1 | 35408600 | 31227502 (88.19%) | 14169507 (80.03%) | 13222135 (74.68%) |
| H2 | 33884370 | 30244478 (89.26%) | 13934050 (82.24%) | 13073710 (77.17%) | |
| H3 | 36202560 | 31905042 (88.13%) | 14659340 (80.99%) | 13634859 (75.33%) | |
| Low | L1 | 39969940 | 34795576 (87.05%) | 16005710 (80.09%) | 14889388 (74.50%) |
| L2 | 36467590 | 32541034 (89.23%) | 14934422 (81.91%) | 14000346 (76.78%) | |
| L3 | 38732934 | 34441474 (88.92%) | 15774510 (81.45%) | 14733075 (76.08%) |
| Group | Sample | Input Reads | Mapped Reads | Aligned Pairs | Uniquely Aligned Pairs |
|---|---|---|---|---|---|
| High | H1 | 35408600 | 33379181 (94.27%) | 16218722 (91.61%) | 15853890 (89.55%) |
| H2 | 33884370 | 32306327 (95.34%) | 15789892 (93.20%) | 15485144 (91.40%) | |
| H3 | 36202560 | 34545431 (95.42%) | 16884547 (93.28%) | 16485731 (91.07%) | |
| Low | L1 | 39969940 | 37850914 (94.70%) | 18481876 (92.48%) | 17752593 (88.83%) |
| L2 | 36467590 | 34697774 (95.15%) | 16935896 (92.88%) | 16562230 (90.83%) | |
| L3 | 38732934 | 36654157 (94.63%) | 17854715 (92.19%) | 16734175 (86.41%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Qi, X.; Hu, M.; Lin, R.; Hou, Y.; Wang, Z.; Zhou, H.; Zhao, Y.; Luan, Y.; Zhao, S.; et al. Transcriptome Analysis of Adipose Tissue Indicates That the cAMP Signaling Pathway Affects the Feed Efficiency of Pigs. Genes 2018, 9, 336. https://doi.org/10.3390/genes9070336
Xu Y, Qi X, Hu M, Lin R, Hou Y, Wang Z, Zhou H, Zhao Y, Luan Y, Zhao S, et al. Transcriptome Analysis of Adipose Tissue Indicates That the cAMP Signaling Pathway Affects the Feed Efficiency of Pigs. Genes. 2018; 9(7):336. https://doi.org/10.3390/genes9070336
Chicago/Turabian StyleXu, Yueyuan, Xiaolong Qi, Mingyang Hu, Ruiyi Lin, Ye Hou, Zhangxu Wang, Huanhuan Zhou, Yunxia Zhao, Yu Luan, Shuhong Zhao, and et al. 2018. "Transcriptome Analysis of Adipose Tissue Indicates That the cAMP Signaling Pathway Affects the Feed Efficiency of Pigs" Genes 9, no. 7: 336. https://doi.org/10.3390/genes9070336
APA StyleXu, Y., Qi, X., Hu, M., Lin, R., Hou, Y., Wang, Z., Zhou, H., Zhao, Y., Luan, Y., Zhao, S., & Li, X. (2018). Transcriptome Analysis of Adipose Tissue Indicates That the cAMP Signaling Pathway Affects the Feed Efficiency of Pigs. Genes, 9(7), 336. https://doi.org/10.3390/genes9070336