
Targeting DNA Replication Stress for Cancer Therapy


Abstract
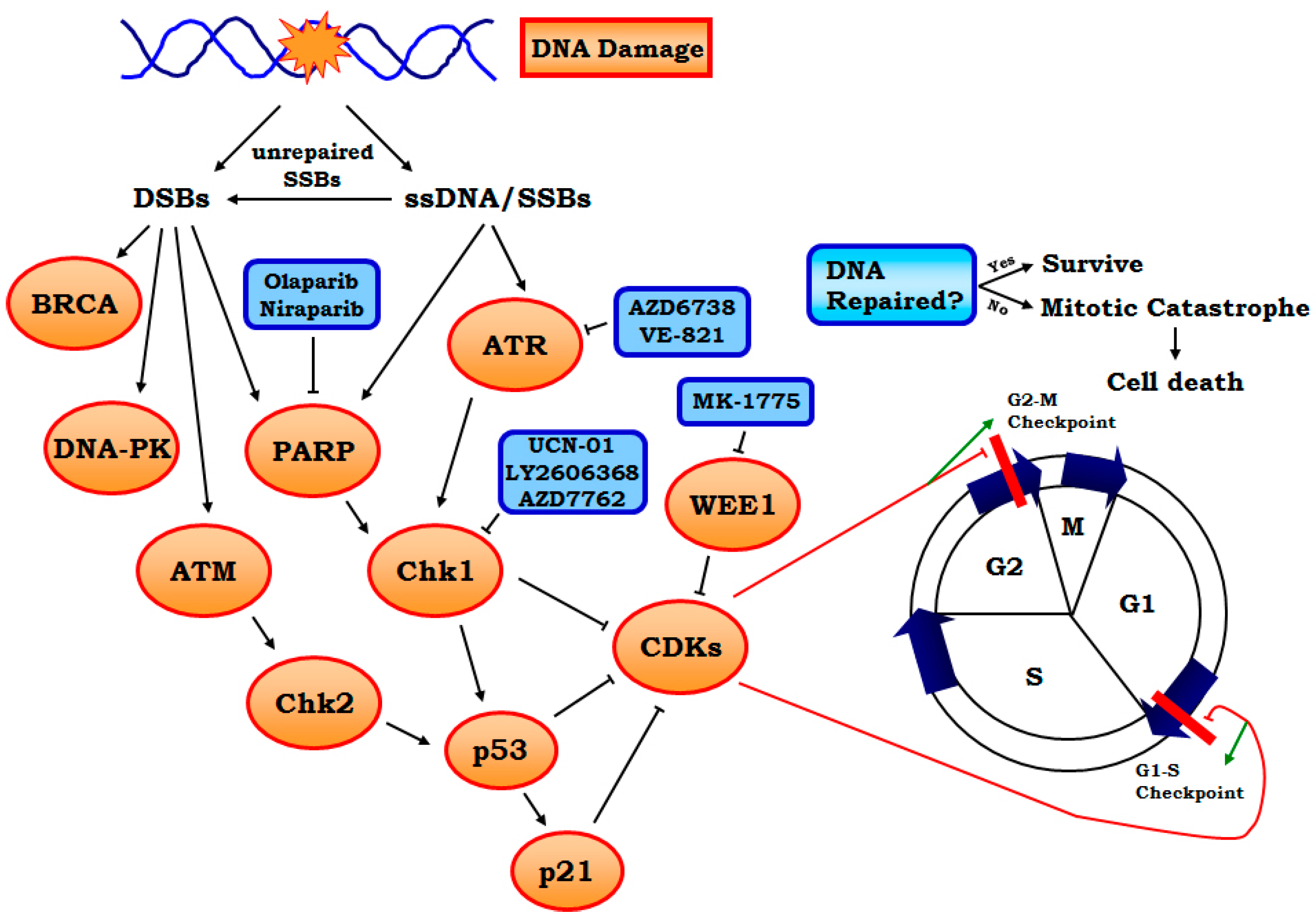
:1. Introduction
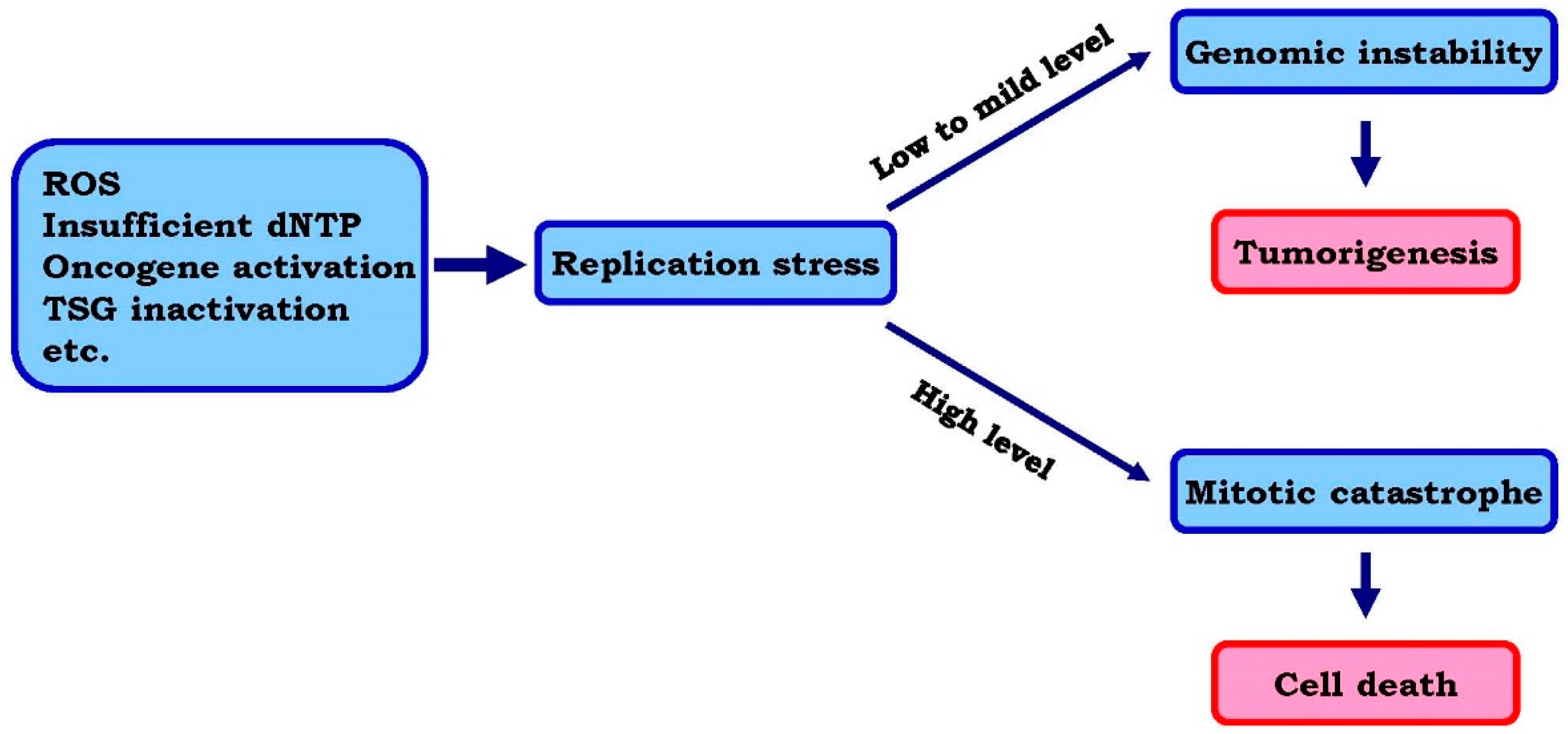
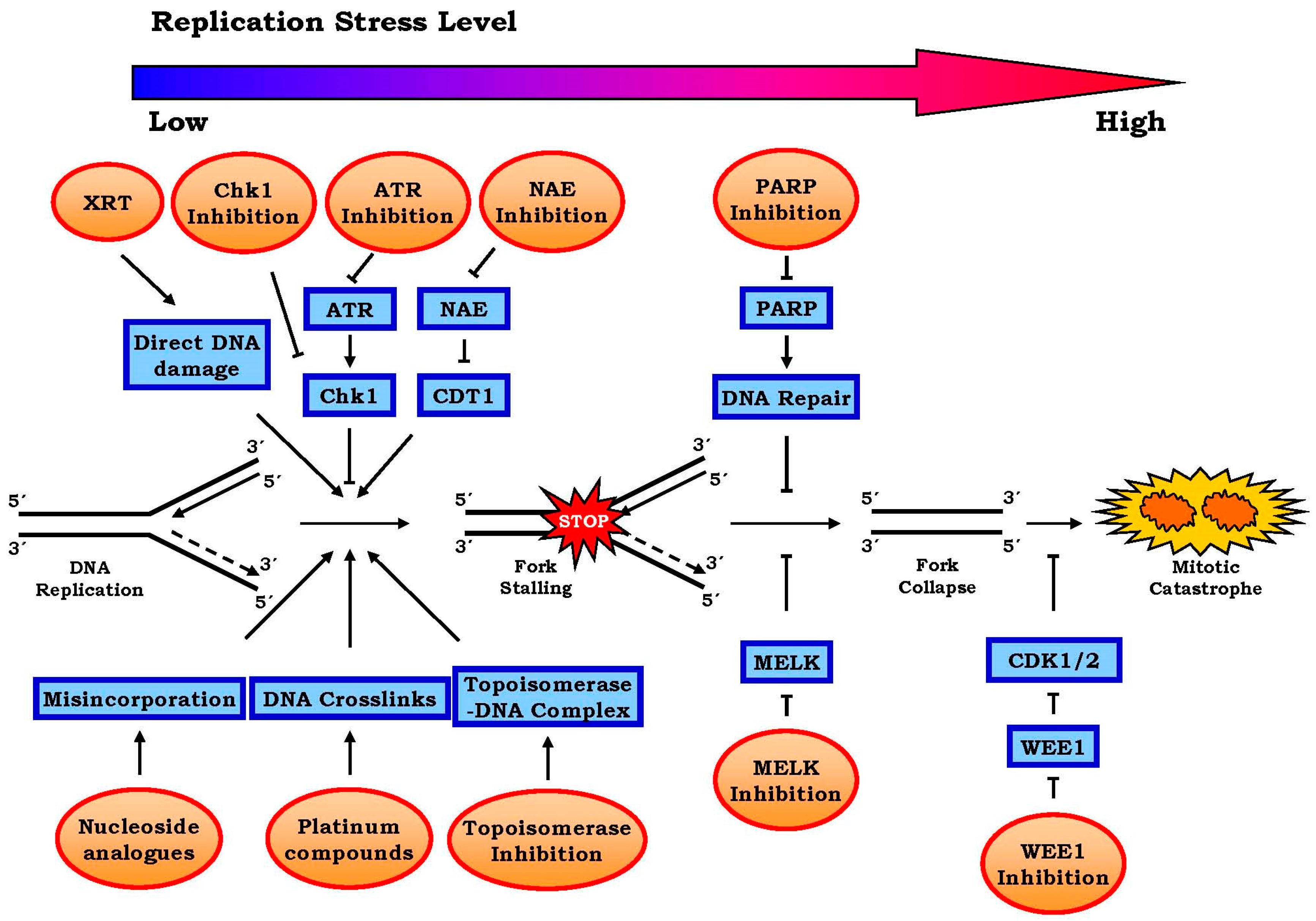
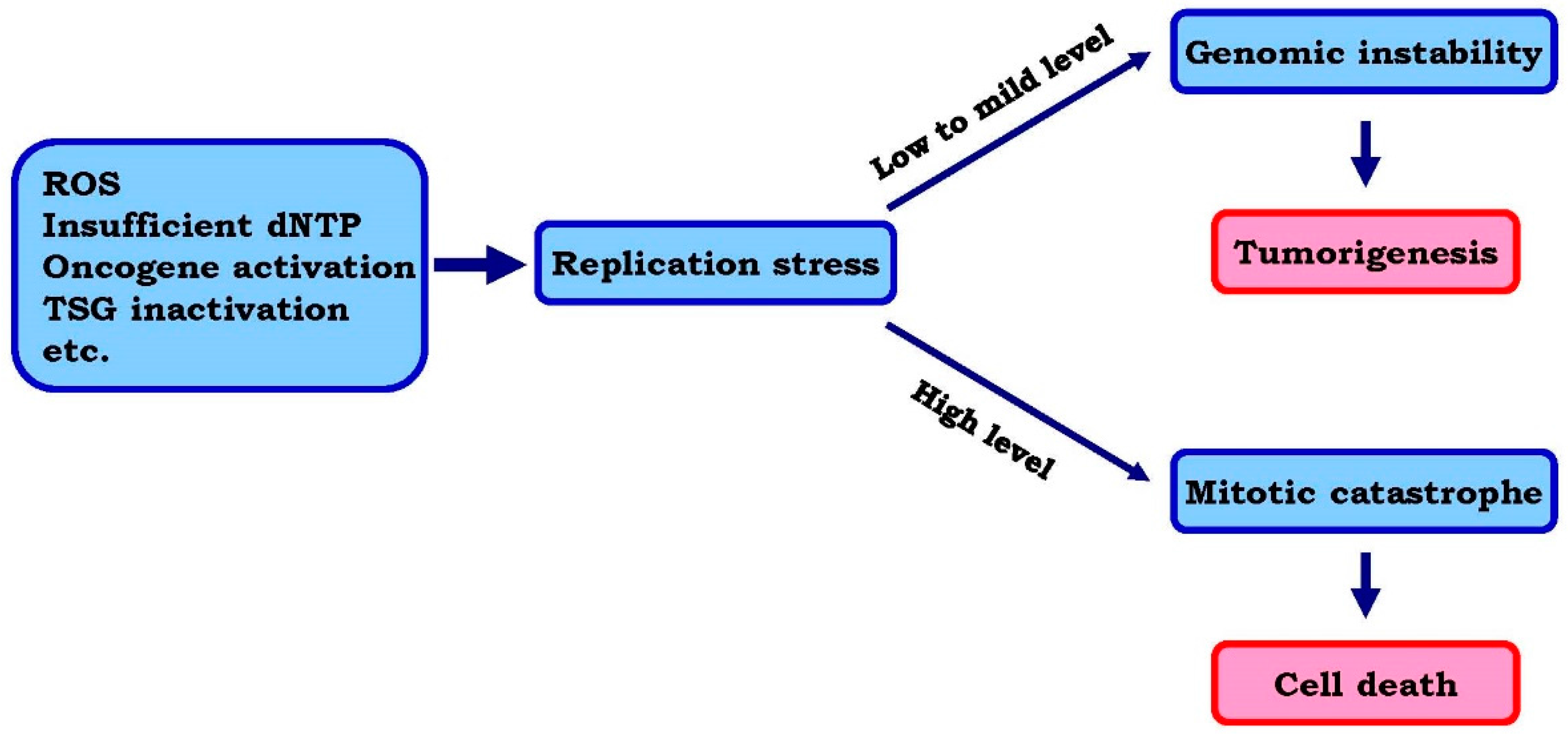
1.1. Replication Stress and Its Underlying Mechanisms
1.2. Rationale for Enhancing Replication Stress to Cause Cancer Cell Death
2. Approaches to Harness Replication Stress for the Treatment of Cancer
2.1. Conventional Approaches
Radiation and chemotherapy
2.2. Emerging Approaches
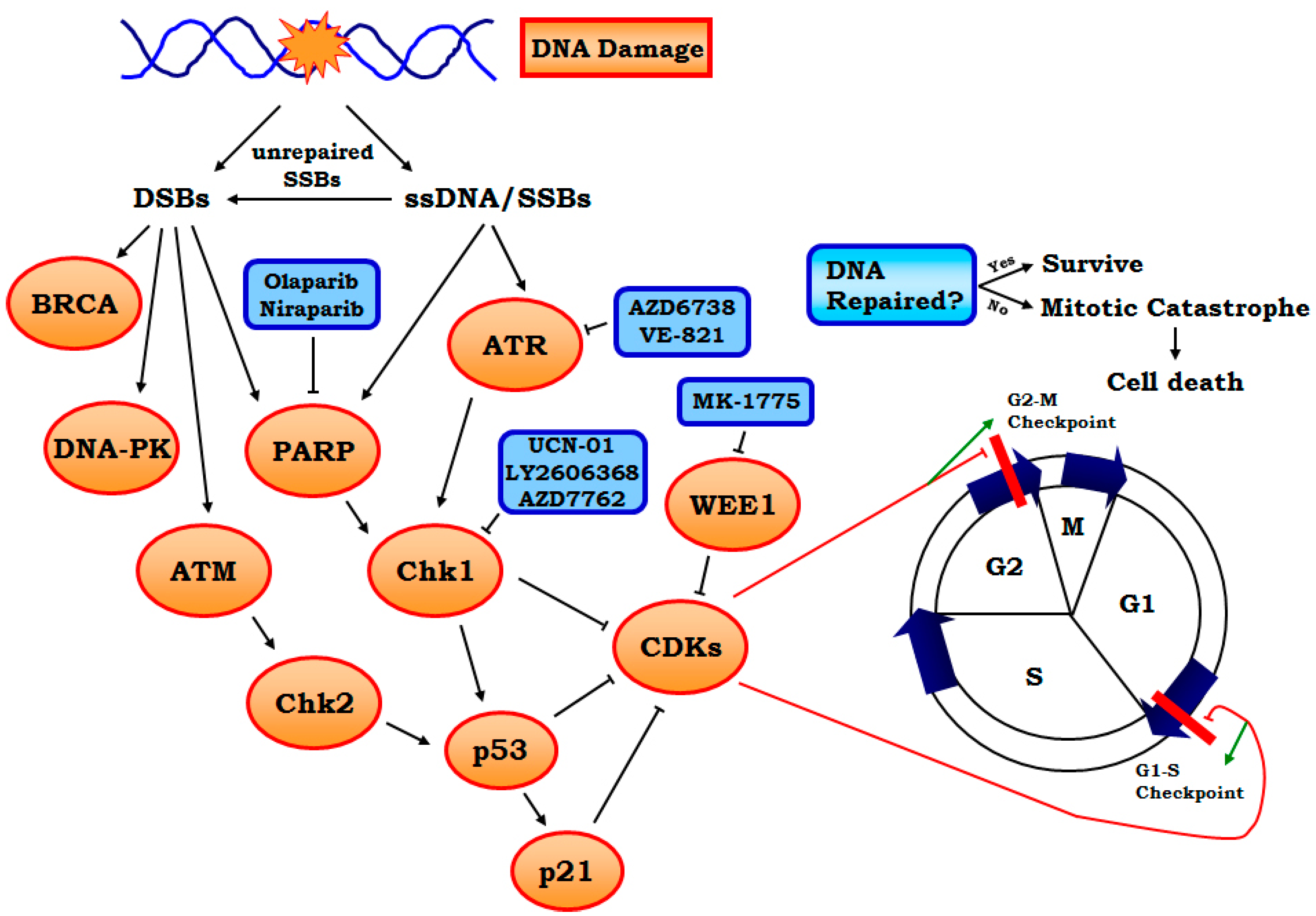
2.2.1. Targeting ATR-Chk1 Signaling
2.2.2. PARP1 Inhibitors
2.2.3. Other Targets Including WEE1, MELK and Neddylation, etc.
3. Future Directions
3.1. Potential of Combination Therapy
3.2. Identification of Future Targets
3.3. Identification of Biomarkers to Detect and Track Replication Stress and Predict Treatment Response
3.4. Identification of Resistance Mechanisms
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R. Oxidative DNA damage, antioxidants, and cancer. Bioessays 1999, 21, 238–246. [Google Scholar] [CrossRef]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Tort, F.; Bartkova, J.; Sehested, M.; Orntoft, T.; Lukas, J.; Bartek, J. Retinoblastoma pathway defects show differential ability to activate the constitutive DNA damage response in human tumorigenesis. Cancer Res. 2006, 66, 10258–10263. [Google Scholar] [CrossRef] [PubMed]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. C-myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ros in cancer: Initiators, amplifiers or an achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Pathania, S.; Nguyen, J.; Hill, S.J.; Scully, R.; Adelmant, G.O.; Marto, J.A.; Feunteun, J.; Livingston, D.M. BRCA1 is required for postreplication repair after uv-induced DNA damage. Mol. Cell. 2011, 44, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, V.; Gautier, J. Single-strand DNA gaps trigger an ATR- and Cdc7-dependent checkpoint. Cell Cycle 2003. [Google Scholar] [CrossRef] [PubMed]
- Eykelenboom, J.K.; Harte, E.C.; Canavan, L.; Pastor-Peidro, A.; Calvo-Asensio, I.; Llorens-Agost, M.; Lowndes, N.F. ATR activates the S-M checkpoint during unperturbed growth to ensure sufficient replication prior to mitotic onset. Cell Rep. 2013, 5, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Canman, C.E. Replication checkpoint: Preventing mitotic catastrophe. Curr. Biol. 2001, 11, R121–R124. [Google Scholar] [CrossRef]
- Lecona, E.; Fernandez-Capetillo, O. Replication stress and cancer: It takes two to tango. Exp. Cell Res. 2014, 329, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.E.; Folkes, L.K.; O’Neill, P. Biological consequences of radiation-induced DNA damage: Relevance to radiotherapy. Clin. Oncol. 2013, 25, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol.Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Kopper, F.; Bierwirth, C.; Schon, M.; Kunze, M.; Elvers, I.; Kranz, D.; Saini, P.; Menon, M.B.; Walter, D.; Sorensen, C.S.; et al. Damage-induced DNA replication stalling relies on mapk-activated protein kinase 2 activity. Proc. Natl. Acad. Sci. USA 2013, 110, 16856–16861. [Google Scholar] [CrossRef] [PubMed]
- Merrick, C.J.; Jackson, D.; Diffley, J.F. Visualization of altered replication dynamics after DNA damage in human cells. J. Biol. Chem. 2004, 279, 20067–20075. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Regairaz, M.; Zhang, Y.W.; Fu, H.; Agama, K.K.; Tata, N.; Agrawal, S.; Aladjem, M.I.; Pommier, Y. Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase i-DNA complexes. J. Cell Biol. 2011, 195, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Seiler, J.A.; Conti, C.; Syed, A.; Aladjem, M.I.; Pommier, Y. The intra-s-phase checkpoint affects both DNA replication initiation and elongation: Single-cell and -DNA fiber analyses. Mol. Cell Biol. 2007, 27, 5806–5818. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Hashimoto, Y.; Herrador, R.; Neelsen, K.J.; Fachinetti, D.; Bermejo, R.; Cocito, A.; Costanzo, V.; Lopes, M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012, 19, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Loegering, D.; Arlander, S.J.; Hackbarth, J.; Vroman, B.T.; Roos-Mattjus, P.; Hopkins, K.M.; Lieberman, H.B.; Karnitz, L.M.; Kaufmann, S.H. Rad9 protects cells from topoisomerase poison-induced cell death. J. Biol. Chem. 2004, 279, 18641–18647. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Watkins, J.L.; Piwnica-Worms, H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc. Natl. Acad. Sci. USA 2002, 99, 14795–14800. [Google Scholar] [CrossRef] [PubMed]
- Syljuasen, R.G.; Sorensen, C.S.; Hansen, L.T.; Fugger, K.; Lundin, C.; Johansson, F.; Helleday, T.; Sehested, M.; Lukas, J.; Bartek, J. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol. Cell. Biol. 2005, 25, 3553–3562. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Woodcock, M.; Helleday, T. Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 16090–16095. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Ellis, M.J.; Petroni, G.R.; Guo, Z.; Cai, S.R.; Ryan, C.E.; Craig Lockhart, A.; Naughton, M.J.; Pluard, T.J.; Brenin, C.M.; et al. A phase II study of UCN-01 in combination with irinotecan in patients with metastatic triple negative breast cancer. Breast Cancer Res.Treat. 2013, 137, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Christensen, S.D.; Frankel, P.H.; Margolin, K.A.; Agarwala, S.S.; Luu, T.; Mack, P.C.; Lara, P.N., Jr.; Gandara, D.R. A phase II study of cell cycle inhibitor UCN-01 in patients with metastatic melanoma: A california cancer consortium trial. Investig. New Drugs 2012, 30, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Infante, J.; Janku, F.; Jones, S.; Nguyen, L.M.; Burris, H.; Naing, A.; Bauer, T.M.; Piha-Paul, S.; Johnson, F.M.; et al. Phase I study of LY2606368, a checkpoint kinase 1 inhibitor, in patients with advanced cancer. J. Clin. Oncol. 2016, 34, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Checkley, S.; MacCallum, L.; Yates, J.; Jasper, P.; Luo, H.; Tolsma, J.; Bendtsen, C. Bridging the gap between in vitro and in vivo: Dose and schedule predictions for the ATR inhibitor AZD6738. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.D.; Maccormick, S.; Charlton, P.A.; Golec, J.M.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012. [Google Scholar] [CrossRef]
- Edelman, M.J.; Bauer, K.S., Jr.; Wu, S.; Smith, R.; Bisacia, S.; Dancey, J. Phase I and pharmacokinetic study of 7-hydroxystaurosporine and carboplatin in advanced solid tumors. Clin. Cancer Res. 2007, 13, 2667–2674. [Google Scholar] [CrossRef] [PubMed]
- Seto, T.; Esaki, T.; Hirai, F.; Arita, S.; Nosaki, K.; Makiyama, A.; Kometani, T.; Fujimoto, C.; Hamatake, M.; Takeoka, H.; et al. Phase I, dose-escalation study of AZD7762 alone and in combination with gemcitabine in japanese patients with advanced solid tumours. Cancer Chemother. Pharmacol. 2013, 72, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Sausville, E.; Lorusso, P.; Carducci, M.; Carter, J.; Quinn, M.F.; Malburg, L.; Azad, N.; Cosgrove, D.; Knight, R.; Barker, P.; et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in us patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, S.K.; Schelman, W.R.; Wilding, G.; Moreno, V.; Baird, R.D.; Miranda, S.; Hylands, L.; Riisnaes, R.; Forster, M.; Omlin, A.; et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2013, 14, 882–892. [Google Scholar] [CrossRef]
- Do, K.; Doroshow, J.H.; Kummar, S. WEE1 kinase as a target for cancer therapy. Cell Cycle 2013, 12, 3159–3164. [Google Scholar] [CrossRef] [PubMed]
- Rajeshkumar, N.V.; de Oliveira, E.; Ottenhof, N.; Watters, J.; Brooks, D.; Demuth, T.; Shumway, S.D.; Mizuarai, S.; Hirai, H.; Maitra, A.; et al. Mk-1775, a potent WEE1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin. Cancer Res. 2011, 17, 2799–2806. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Kang, Y.; Kim, K.; Cha, Y.J.; Cho, H.S. The crystal structure of MPK38 in complex with otssp167, an orally administrative MELK selective inhibitor. Biochem. Biophys. Res. Commun. 2014, 447, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Suzuki, H.; Miyamoto, T.; Takamatsu, N.; Tatsuguchi, A.; Ueda, K.; Kijima, K.; Nakamura, Y.; Matsuo, Y. Development of an orally-administrative MELK-targeting inhibitor that suppresses the growth of various types of human cancer. Oncotarget 2012, 3, 1629–1640. [Google Scholar] [CrossRef] [PubMed]
- Swords, R.T.; Erba, H.P.; DeAngelo, D.J.; Bixby, D.L.; Altman, J.K.; Maris, M.; Hua, Z.; Blakemore, S.J.; Faessel, H.; Sedarati, F.; et al. Pevonedistat (MLN4924), a first-in-class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: A phase 1 study. Br. J. Haematol. 2015, 169, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.S.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Ahel, I.; Ahel, D.; Matsusaka, T.; Clark, A.J.; Pines, J.; Boulton, S.J.; West, S.C. Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature 2008, 451, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate MRE11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [PubMed]
- Min, W.; Bruhn, C.; Grigaravicius, P.; Zhou, Z.W.; Li, F.; Kruger, A.; Siddeek, B.; Greulich, K.O.; Popp, O.; Meisezahl, C.; et al. Poly(ADP-ribose) binding to Chk1 at stalled replication forks is required for S-phase checkpoint activation. Nat. Commun. 2013. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Srihari, S.; Cao, K.A.; Chenevix-Trench, G.; Simpson, P.T.; Ragan, M.A.; Khanna, K.K. A fine-scale dissection of the DNA double-strand break repair machinery and its implications for breast cancer therapy. Nucleic Acids Res. 2014, 42, 6106–6127. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O'Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Ame, J.C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Hoger, T.; Menissier-de Murcia, J.; de Murcia, G. PARP-2, a novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J. Biol. Chem. 1999, 274, 17860–17868. [Google Scholar] [CrossRef] [PubMed]
- Menissier de Murcia, J.; Ricoul, M.; Tartier, L.; Niedergang, C.; Huber, A.; Dantzer, F.; Schreiber, V.; Ame, J.C.; Dierich, A.; LeMeur, M.; et al. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003, 22, 2255–2263. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Hamdy, F.C.; Helleday, T. MRE11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Kalimutho, M.; Parsons, K.; Mittal, D.; Lopez, J.A.; Srihari, S.; Khanna, K.K. Targeted therapies for triple-negative breast cancer: Combating a stubborn disease. Trends Pharmacol. Sci. 2015, 36, 822–846. [Google Scholar] [CrossRef] [PubMed]
- Podo, F.; Buydens, L.M.; Degani, H.; Hilhorst, R.; Klipp, E.; Gribbestad, I.S.; van Huffel, S.; van Laarhoven, H.W.; Luts, J.; Monleon, D.; et al. Triple-negative breast cancer: Present challenges and new perspectives. Mol. Oncol. 2010, 4, 209–229. [Google Scholar] [CrossRef] [PubMed]
- Phuah, S.Y.; Looi, L.M.; Hassan, N.; Rhodes, A.; Dean, S.; Taib, N.A.; Yip, C.H.; Teo, S.H. Triple-negative breast cancer and PTEN (phosphatase and tensin homologue) loss are predictors of BRCA1 germline mutations in women with early-onset and familial breast cancer, but not in women with isolated late-onset breast cancer. Breast Cancer Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Lips, E.H.; Mulder, L.; Oonk, A.; van der Kolk, L.E.; Hogervorst, F.B.; Imholz, A.L.; Wesseling, J.; Rodenhuis, S.; Nederlof, P.M. Triple-negative breast cancer: Brcaness and concordance of clinical features with BRCA1-mutation carriers. Br. J. Cancer 2013, 108, 2172–2177. [Google Scholar] [CrossRef] [PubMed]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Al-Ejeh, F.; Shi, W.; Miranda, M.; Simpson, P.T.; Vargas, A.C.; Song, S.; Wiegmans, A.P.; Swarbrick, A.; Welm, A.L.; Brown, M.P.; et al. Treatment of triple-negative breast cancer using anti-EGFR-directed radioimmunotherapy combined with radiosensitizing chemotherapy and PARP inhibitor. J. Nucl. Med. 2013, 54, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Nowsheen, S.; Cooper, T.; Stanley, J.A.; Yang, E.S. Synthetic lethal interactions between EGFR and PARP inhibition in human triple negative breast cancer cells. PLoS ONE 2012, 7, e46614. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; Sharpe, R.; Garcia-Murillas, I.; Gevensleben, H.; Hurd, M.S.; Shumway, S.D.; Toniatti, C.; Ashworth, A.; Turner, N.C. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012, 2, 524–539. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.; Nahse-Kumpf, V.; Larsen, M.S.; O’Hanlon, K.A.; Patzke, S.; Holmberg, C.; Mejlvang, J.; Groth, A.; Nielsen, O.; Syljuasen, R.G.; et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol. Cell. Biol. 2012, 32, 4226–4236. [Google Scholar] [CrossRef] [PubMed]
- Davezac, N.; Baldin, V.; Blot, J.; Ducommun, B.; Tassan, J.P. Human PEG3 kinase associates with and phosphorylates Cdc25b phosphatase: A potential role for PEG3 in cell cycle regulation. Oncogene 2002, 21, 7630–7641. [Google Scholar] [CrossRef] [PubMed]
- Kig, C.; Beullens, M.; Beke, L.; van Eynde, A.; Linders, J.T.; Brehmer, D.; Bollen, M. Maternal embryonic leucine zipper kinase (MELK) reduces replication stress in glioblastoma cells. J. Biol. Chem. 2013, 288, 24200–24212. [Google Scholar] [CrossRef] [PubMed]
- Paiva, C.; Godbersen, J.C.; Berger, A.; Brown, J.R.; Danilov, A.V. Targeting neddylation induces DNA damage and checkpoint activation and sensitizes chronic lymphocytic leukemia b cells to alkylating agents. Cell Death Dis. 2015. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.Q.; Kentsis, A.; Dias, D.C.; Yamoah, K.; Wu, K. NEDD8 on cullin: Building an expressway to protein destruction. Oncogene 2004, 23, 1985–1997. [Google Scholar] [CrossRef] [PubMed]
- Milhollen, M.A.; Narayanan, U.; Soucy, T.A.; Veiby, P.O.; Smith, P.G.; Amidon, B. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res. 2011, 71, 3042–3051. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Milhollen, M.A.; Smith, P.G.; Narayanan, U.; Dutta, A. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing CDT1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. 2010, 70, 10310–10320. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.J.; Jakubowiak, A.J.; O’Connor, O.A.; Orlowski, R.Z.; Harvey, R.D.; Smith, M.R.; Lebovic, D.; Diefenbach, C.; Kelly, K.; Hua, Z.; et al. Phase I study of the novel investigational NEDD8-activating enzyme inhibitor pevonedistat (MLN4924) in patients with relapsed/refractory multiple myeloma or lymphoma. Clin. Cancer Res. 2016, 22, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, W.; Yan, Z.; Liang, Y.; Li, L.; Yu, X.; Feng, Y.; Fu, S.; Zhang, Y.; Zhao, H.; et al. Radiosensitization by the investigational NEDD8-activating enzyme inhibitor MLN4924 (pevonedistat) in hormone-resistant prostate cancer cells. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.; Tang, Z.; Jin, H.; Sun, Y. Neddylation inhibitor MLN4924 suppresses growth and migration of human gastric cancer cells. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Turrisi, A.T., 3rd; Kim, K.; Blum, R.; Sause, W.T.; Livingston, R.B.; Komaki, R.; Wagner, H.; Aisner, S.; Johnson, D.H. Twice-daily compared with once-daily thoracic radiotherapy in limited small-cell lung cancer treated concurrently with cisplatin and etoposide. N. Engl. J. Med. 1999, 340, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Adelstein, D.J.; Li, Y.; Adams, G.L.; Wagner, H., Jr.; Kish, J.A.; Ensley, J.F.; Schuller, D.E.; Forastiere, A.A. An intergroup phase III comparison of standard radiation therapy and two schedules of concurrent chemoradiotherapy in patients with unresectable squamous cell head and neck cancer. J. Clin. Oncol. 2003, 21, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Loehrer, P.J., Sr.; Einhorn, L.H.; Greco, F.A. Cisplatin plus etoposide in small cell lung cancer. Semin. Oncol. 1988, 15, 2–8. [Google Scholar] [PubMed]
- Okamoto, H.; Watanabe, K.; Nishiwaki, Y.; Mori, K.; Kurita, Y.; Hayashi, I.; Masutani, M.; Nakata, K.; Tsuchiya, S.; Isobe, H.; et al. Phase II study of area under the plasma-concentration-versus-time curve-based carboplatin plus standard-dose intravenous etoposide in elderly patients with small-cell lung cancer. J. Clin. Oncol. 1999, 17, 3540–3545. [Google Scholar] [PubMed]
- Prevo, R.; Fokas, E.; Reaper, P.M.; Charlton, P.A.; Pollard, J.R.; McKenna, W.G.; Muschel, R.J.; Brunner, T.B. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol. Ther. 2012, 13, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Garcia, K.; Blank, J.L.; Bouck, D.C.; Liu, X.J.; Sappal, D.S.; Hather, G.; Cosmopoulos, K.; Thomas, M.P.; Kuranda, M.; Pickard, M.D.; et al. NEDD8-activating enzyme inhibitor MLN4924 provides synergy with mitomycin C through interactions with ATR, BRCA1/BRCA2, and chromatin dynamics pathways. Mol. Cancer Ther. 2014, 13, 1625–1635. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.P.; Lewis, L.D.; Beelen, A.P.; Olszanski, A.J.; Johnston, N.; Rhodes, C.H.; Beaulieu, B.; Ernstoff, M.S.; Eastman, A. Modulation of cell cycle progression in human tumors: A pharmacokinetic and tumor molecular pharmacodynamic study of cisplatin plus the Chk1 inhibitor UCN-01 (NSC 638850). Clin. Cancer Res. 2006, 12, 7079–7085. [Google Scholar] [CrossRef] [PubMed]
- Yazlovitskaya, E.M.; Persons, D.L. Inhibition of cisplatin-induced ATR activity and enhanced sensitivity to cisplatin. Anticancer Res. 2003, 23, 2275–2279. [Google Scholar] [PubMed]
- Russell, M.R.; Levin, K.; Rader, J.; Belcastro, L.; Li, Y.; Martinez, D.; Pawel, B.; Shumway, S.D.; Maris, J.M.; Cole, K.A. Combination therapy targeting the Chk1 and WEE1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. 2013, 73, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Carrassa, L.; Chila, R.; Lupi, M.; Ricci, F.; Celenza, C.; Mazzoletti, M.; Broggini, M.; Damia, G. Combined inhibition of Chk1 and WEE1: In vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle 2012, 11, 2507–2517. [Google Scholar] [CrossRef]
- Chaudhuri, L.; Vincelette, N.D.; Koh, B.D.; Naylor, R.M.; Flatten, K.S.; Peterson, K.L.; McNally, A.; Gojo, I.; Karp, J.E.; Mesa, R.A.; et al. Chk1 and WEE1 inhibition combine synergistically to enhance therapeutic efficacy in acute myeloid leukemia ex vivo. Haematologica 2014, 99, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Burhans, W.C.; Weinberger, M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007, 35, 7545–7556. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Dupre, A.; Boyer-Chatenet, L.; Sattler, R.M.; Modi, A.P.; Lee, J.H.; Nicolette, M.L.; Kopelovich, L.; Jasin, M.; Baer, R.; Paull, T.T.; et al. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol. 2008, 4, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Ashley, A.K.; Hromas, R.; Nickoloff, J.A. More forks on the road to replication stress recovery. J. Mol. Cell Biol. 2011, 3, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Motlekar, N.A.; Burgwin, C.M.; Napper, A.D.; Diamond, S.L.; Mazin, A.V. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem. Biol. 2011, 6, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Budke, B.; Logan, H.L.; Kalin, J.H.; Zelivianskaia, A.S.; McGuire, W.C.; Miller, L.L.; Stark, J.M.; Kozikowski, A.P.; Bishop, D.K.; Connell, P.P. Ri-1: A chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res. 2012, 40, 7347–7357. [Google Scholar] [CrossRef] [PubMed]
- Conti, C.; Leo, E.; Eichler, G.S.; Sordet, O.; Martin, M.M.; Fan, A.; Aladjem, M.I.; Pommier, Y. Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer Res. 2010, 70, 4470–4480. [Google Scholar] [CrossRef] [PubMed]
- Gilmartin, A.G.; Faitg, T.H.; Richter, M.; Groy, A.; Seefeld, M.A.; Darcy, M.G.; Peng, X.; Federowicz, K.; Yang, J.; Zhang, S.Y.; et al. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat. Chem. Biol. 2014, 10, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Conforti, F.; Sayan, A.E.; Sreekumar, R.; Sayan, B.S. Regulation of p73 activity by post-translational modifications. Cell Death Dis. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Dotiwala, F.; Eapen, V.V.; Harrison, J.C.; Arbel-Eden, A.; Ranade, V.; Yoshida, S.; Haber, J.E. DNA damage checkpoint triggers autophagy to regulate the initiation of anaphase. Proc. Natl. Acad. Sci. USA. 2013, 110, E41–E49. [Google Scholar] [CrossRef] [PubMed]
- Gudkov, A.V.; Komarova, E.A. Prospective therapeutic applications of p53 inhibitors. Biochem. Biophys. Res. Commun. 2005, 331, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, M.C.; Iwasaki, O.; Noguchi, C.; Noma, K.; Noguchi, E. Chromatin immunoprecipitation to detect DNA replication and repair factors. Methods Mol. Biol. 2015, 1300, 169–186. [Google Scholar] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat. Med. 2013, 19, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Callen, E.; Kozak, M.L.; Kim, J.M.; Wong, N.; Lopez-Contreras, A.J.; Ludwig, T.; Baer, R.; Faryabi, R.B.; Malhowski, A.; et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 2012, 46, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53bp1 causes PARP inhibitor resistance in BRCA1-mutated mouse mammary tumors. Cancer Dis. 2013, 3, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Zander, S.A.; Derksen, P.W.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA. 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Rehman, F.L.; Bajrami, I.; Brough, R.; Wallberg, F.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Chen, L.; Campbell, J.; et al. A genetic screen using the piggybac transposon in haploid cells identifies PARP1 as a mediator of olaparib toxicity. PLoS ONE 2013, 8, e61520. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Han, E.K.; Anderson, M.; Shi, Y.; Semizarov, D.; Wang, G.; McGonigal, T.; Roberts, L.; Lasko, L.; Palma, J.; et al. Acquired resistance to combination treatment with temozolomide and ABT-888 is mediated by both base excision repair and homologous recombination DNA repair pathways. Mol. Cancer Res. 2009, 7, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Oplustilova, L.; Wolanin, K.; Mistrik, M.; Korinkova, G.; Simkova, D.; Bouchal, J.; Lenobel, R.; Bartkova, J.; Lau, A.; O'Connor, M.J.; et al. Evaluation of candidate biomarkers to predict cancer cell sensitivity or resistance to PARP-1 inhibitor treatment. Cell Cycle 2012, 11, 3837–3850. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Mayor-Ruiz, C.; Lafarga, V.; Murga, M.; Vega-Sendino, M.; Ortega, S.; Fernandez-Capetillo, O. A genome-wide CRISPR Screen identifies Cdc25a as a determinant of sensitivity to ATR inhibitors. Mol. Cell 2016, 62, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Restelli, V.; Chila, R.; Lupi, M.; Rinaldi, A.; Kwee, I.; Bertoni, F.; Damia, G.; Carrassa, L. Characterization of a mantle cell lymphoma cell line resistant to the Chk1 inhibitor PF-00477736. Oncotarget 2015, 6, 37229–37240. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Target/mechanism | Compounds | Stage of development | Reference |
|---|---|---|---|
| DNA misincorporation/modification | Alkylating agents e.g., cyclophosphamide, temozolamide, etc. | Approved | [20] |
| Platinum compounds e.g., cisplatin, carboplatin, etc. | Approved | [21] | |
| Ribonucleotide reductase | Gemcitabine | Approved | [24] |
| Thymidylate synthetase | 5-fluorouracil | Approved | [25] |
| Topoisomerase I | Irinotecan, topotecan | Approved | [29] |
| Topoisomerase II | Etoposide, doxrubicin | Approved | [30] |
| Chk1 | UCN-01 | Phase I/II. Multiple studies completed | [34,40] |
| LY2606368 | Phase I/II, one study completed | [36] | |
| AZD7762 | Phase I. One completed. | [41,42] | |
| ATR | AZD6738 | Phase I, one study completed | [37] |
| PARP1 | Olaparib and niraparib | Approved | [43,44,45] |
| WEE1 | MK-1775 | Phase I | [46,47] |
| MELK | OTS167 | Phase I, 2 studies | [48,49] |
| NAE | MLN4924 | Phase I, multiple studies | [50] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Dai, Q.; Park, D.; Deng, X. Targeting DNA Replication Stress for Cancer Therapy. Genes 2016, 7, 51. https://doi.org/10.3390/genes7080051
Zhang J, Dai Q, Park D, Deng X. Targeting DNA Replication Stress for Cancer Therapy. Genes. 2016; 7(8):51. https://doi.org/10.3390/genes7080051
Chicago/Turabian StyleZhang, Jun, Qun Dai, Dongkyoo Park, and Xingming Deng. 2016. "Targeting DNA Replication Stress for Cancer Therapy" Genes 7, no. 8: 51. https://doi.org/10.3390/genes7080051