Abnormal Base Excision Repair at Trinucleotide Repeats Associated with Diseases: A Tissue-Selective Mechanism

{kind=link}
Abstract
:1. Trinucleotide Repeat Instability in Diseases
2. Trinucleotide Repeat Instability as the Result of Erroneous DNA Repair
3. In Vivo Mechanism of BER in Trinucleotide Repeat Instability
3.1. BER in Various Models of Trinucleotide Repeat Instability
3.2. Level and Accessibility of DNA Lesions at Trinucleotide Repeats
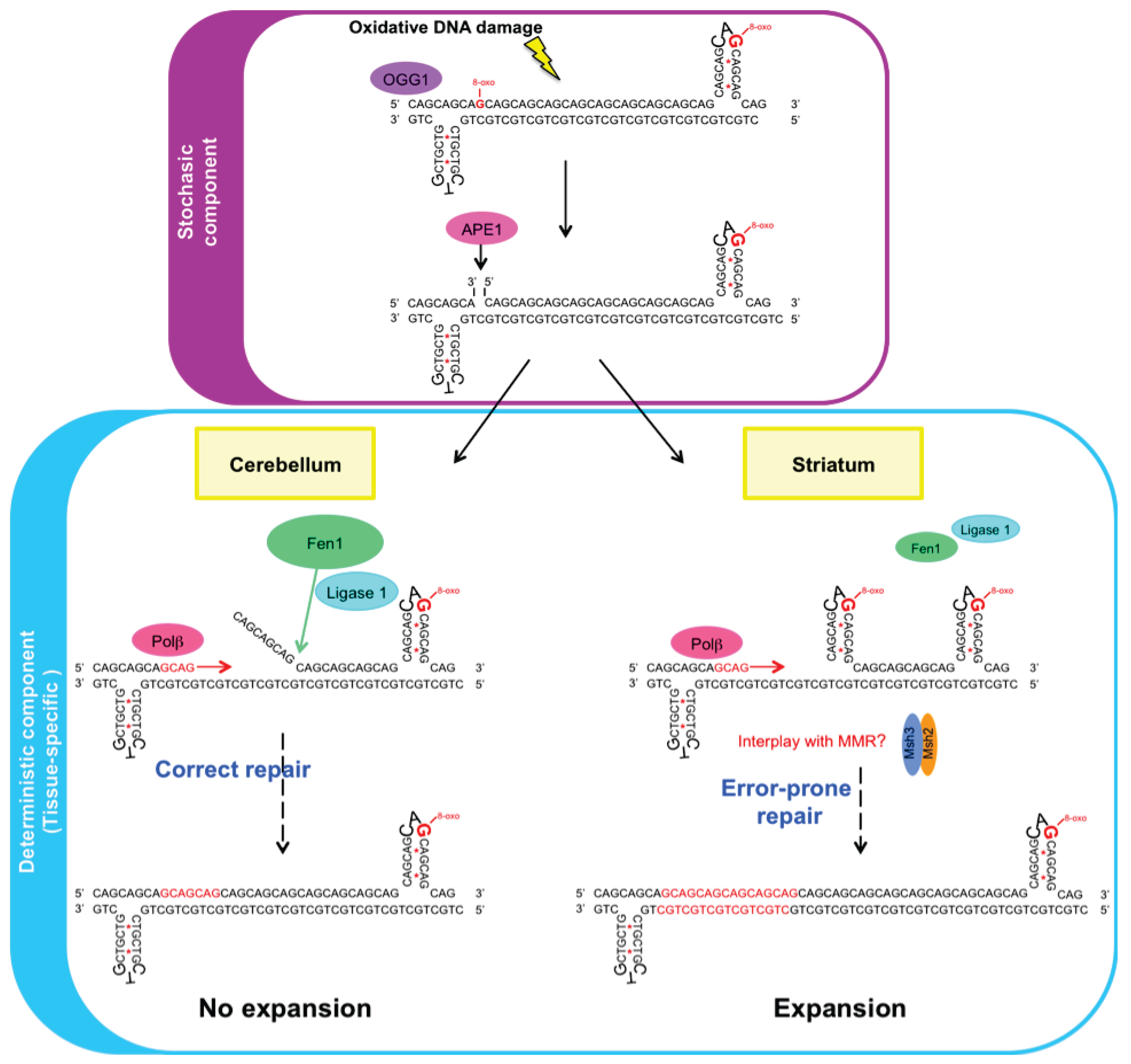
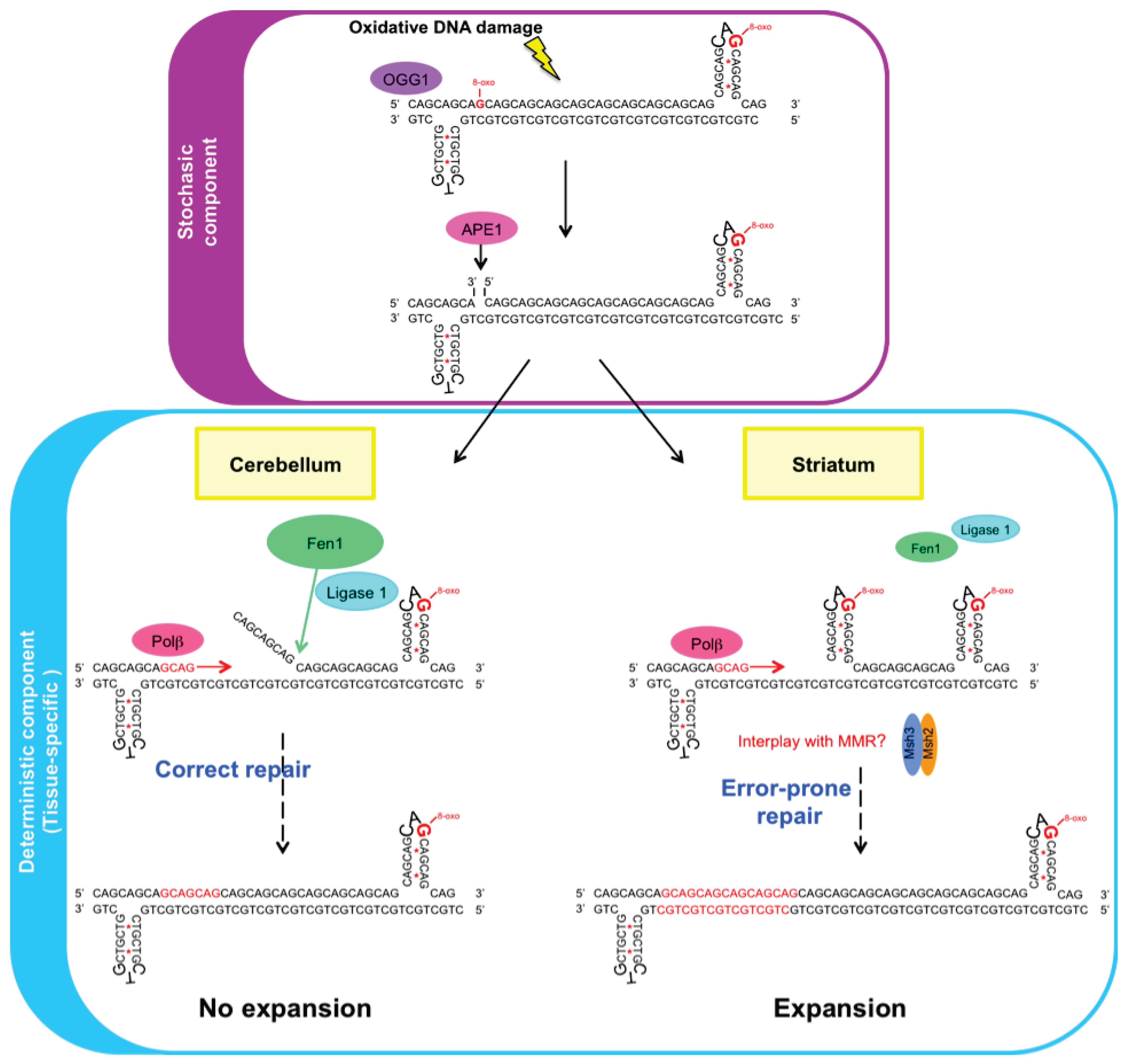
3.3. Mechanism of BER in Tissue-Selective CAG/CTG Repeat Instability
4. Conclusions
Acknowledgements
Conflict of Interest
References
- Mirkin, S.M. Expandable DNA repeats and human disease. Nature 2007, 447, 932–940. [Google Scholar] [CrossRef]
- Pearson, C.E.; Nichol Edamura, K.; Cleary, J.D. Repeat instability: Mechanisms of dynamic mutations. Nat. Rev. Genet. 2005, 6, 729–742. [Google Scholar] [CrossRef]
- Lopez Castel, A.; Cleary, J.D.; Pearson, C.E. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat. Rev. Mol. Cell Biol. 2010, 11, 165–170. [Google Scholar] [CrossRef]
- Pearson, C.E. Slipping while sleeping? Trinucleotide repeat expansions in germ cells. Trends Mol. Med. 2003, 9, 490–495. [Google Scholar] [CrossRef]
- Gonitel, R.; Moffitt, H.; Sathasivam, K.; Woodman, B.; Detloff, P.J.; Faull, R.L.; Bates, G.P. DNA instability in postmitotic neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 3467–3472. [Google Scholar]
- Shelbourne, P.F.; Keller-McGandy, C.; Bi, W.L.; Yoon, S.R.; Dubeau, L.; Veitch, N.J.; Vonsattel, J.P.; Wexler, N.S.; Arnheim, N.; Augood, S.J. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in huntington disease brain. Hum. Mol. Genet. 2007, 16, 1133–1142. [Google Scholar] [CrossRef]
- Swami, M.; Hendricks, A.E.; Gillis, T.; Massood, T.; Mysore, J.; Myers, R.H.; Wheeler, V.C. Somatic expansion of the huntington’s disease cag repeat in the brain is associated with an earlier age of disease onset. Hum. Mol. Genet. 2009, 18, 3039–3047. [Google Scholar] [CrossRef]
- Morales, F.; Couto, J.M.; Higham, C.F.; Hogg, G.; Cuenca, P.; Braida, C.; Wilson, R.H.; Adam, B.; del Valle, G.; Brian, R.; et al. Somatic instability of the expanded ctg triplet repeat in myotonic dystrophy type 1 is a heritable quantitative trait and modifier of disease severity. Hum. Mol. Genet. 2012, 21, 3558–3567. [Google Scholar] [CrossRef]
- Chong, S.S.; McCall, A.E.; Cota, J.; Subramony, S.H.; Orr, H.T.; Hughes, M.R.; Zoghbi, H.Y. Gametic and somatic tissue-specific heterogeneity of the expanded sca1 cag repeat in spinocerebellar ataxia type 1. Nat. Genet. 1995, 10, 344–350. [Google Scholar] [CrossRef]
- Hashida, H.; Goto, J.; Suzuki, T.; Jeong, S.; Masuda, N.; Ooie, T.; Tachiiri, Y.; Tsuchiya, H.; Kanazawa, I. Single cell analysis of cag repeat in brains of dentatorubral-pallidoluysian atrophy (drpla). J. Neurol. Sci. 2001, 190, 87–93. [Google Scholar] [CrossRef]
- Kennedy, L.; Shelbourne, P.F. Dramatic mutation instability in hd mouse striatum: Does polyglutamine load contribute to cell-specific vulnerability in huntington’s disease? Hum. Mol. Genet. 2000, 9, 2539–2544. [Google Scholar] [CrossRef]
- Lopes-Cendes, I.; Maciel, P.; Kish, S.; Gaspar, C.; Robitaille, Y.; Clark, H.B.; Koeppen, A.H.; Nance, M.; Schut, L.; Silveira, I.; et al. Somatic mosaicism in the central nervous system in spinocerebellar ataxia type 1 and machado-joseph disease. Ann. Neurol. 1996, 40, 199–206. [Google Scholar] [CrossRef]
- Telenius, H.; Kremer, B.; Goldberg, Y.P.; Theilmann, J.; Andrew, S.E.; Zeisler, J.; Adam, S.; Greenberg, C.; Ives, E.J.; Clarke, L.A.; et al. Somatic and gonadal mosaicism of the huntington disease gene cag repeat in brain and sperm. Nat. Genet. 1994, 6, 409–414. [Google Scholar] [CrossRef]
- McMurray, C.T. Mechanisms of trinucleotide repeat instability during human development. Nat. Rev. Genet. 2010, 11, 786–799. [Google Scholar] [CrossRef]
- Gacy, A.M.; Goellner, G.; Juranic, N.; Macura, S.; Mcmurray, C.T. Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell 1995, 81, 533–540. [Google Scholar] [CrossRef]
- Pearson, C.E.; Sinden, R.R. Alternative structures in duplex DNA formed within the trinucleotide repeats of the myotonic dystrophy and fragile x loci. Biochemistry 1996, 35, 5041–5053. [Google Scholar] [CrossRef]
- Pearson, C.E.; Tam, M.; Wang, Y.H.; Montgomery, S.E.; Dar, A.C.; Cleary, J.D.; Nichol, K. Slipped-strand dnas formed by long (cag)*(ctg) repeats: Slipped-out repeats and slip-out junctions. Nucleic Acids Res. 2002, 30, 4534–4547. [Google Scholar] [CrossRef]
- Hou, C.; Chan, N.L.; Gu, L.; Li, G.M. Incision-dependent and error-free repair of (cag)(n)/(ctg)(n) hairpins in human cell extracts. Nat. Struct. Mol. Biol. 2009, 16, 869–875. [Google Scholar] [CrossRef]
- Mitas, M.; Yu, A.; Dill, J.; Kamp, T.J.; Chambers, E.J.; Haworth, I.S. Hairpin properties of single-stranded DNA containing a gc-rich triplet repeat: (ctg)15. Nucleic Acids Res. 1995, 23, 1050–1059. [Google Scholar] [CrossRef]
- Panigrahi, G.B.; Lau, R.; Montgomery, S.E.; Leonard, M.R.; Pearson, C.E. Slipped (ctg)*(cag) repeats can be correctly repaired, escape repair or undergo error-prone repair. Nat. Struct. Mol. Biol. 2005, 12, 654–662. [Google Scholar] [CrossRef]
- Pearson, C.E.; Wang, Y.H.; Griffith, J.D.; Sinden, R.R. Structural analysis of slipped-strand DNA (s-DNA) formed in (ctg)n. (cag)n repeats from the myotonic dystrophy locus. Nucleic Acids Res. 1998, 26, 816–823. [Google Scholar] [CrossRef]
- Panigrahi, G.B.; Slean, M.M.; Simard, J.P.; Gileadi, O.; Pearson, C.E. Isolated short ctg/cag DNA slip-outs are repaired efficiently by hmutsbeta, but clustered slip-outs are poorly repaired. Proc. Natl. Acad. Sci. USA 2010, 107, 12593–12598. [Google Scholar]
- Slean, M.M.; Reddy, K.; Wu, B.; Nichol Edamura, K.; Kekis, M.; Nelissen, F.H.; Aspers, R.L.; Tessari, M.; Scharer, O.D.; Wijmenga, S.S.; et al. Interconverting conformations of slipped-DNA junctions formed by trinucleotide repeats affect repair outcome. Biochemistry 2013, 52, 773–785. [Google Scholar] [CrossRef]
- Reddy, K.; Tam, M.; Bowater, R.P.; Barber, M.; Tomlinson, M.; Nichol Edamura, K.; Wang, Y.H.; Pearson, C.E. Determinants of r-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Res. 2010, 39, 1749–1762. [Google Scholar]
- Lin, Y.; Dent, S.Y.; Wilson, J.H.; Wells, R.D.; Napierala, M. R loops stimulate genetic instability of ctg.Cag repeats. Proc. Natl. Acad. Sci. USA 2010, 107, 692–697. [Google Scholar] [CrossRef]
- Libby, R.T.; Hagerman, K.A.; Pineda, V.V.; Lau, R.; Cho, D.H.; Baccam, S.L.; Axford, M.M.; Cleary, J.D.; Moore, J.M.; Sopher, B.L.; et al. Ctcf cis-regulates trinucleotide repeat instability in an epigenetic manner: A novel basis for mutational hot spot determination. PLoS Genet. 2008, 4, e1000257. [Google Scholar] [CrossRef]
- Dion, V.; Lin, Y.; Hubert, L., Jr.; Waterland, R.A.; Wilson, J.H. Dnmt1 deficiency promotes cag repeat expansion in the mouse germline. Hum. Mol. Genet. 2008, 17, 1306–1317. [Google Scholar] [CrossRef]
- Cleary, J.D.; Tome, S.; Lopez Castel, A.; Panigrahi, G.B.; Foiry, L.; Hagerman, K.A.; Sroka, H.; Chitayat, D.; Gourdon, G.; Pearson, C.E. Tissue- and age-specific DNA replication patterns at the ctg/cag-expanded human myotonic dystrophy type 1 locus. Nat. Struct. Mol. Biol. 2010, 17, 1079–1087. [Google Scholar]
- Dion, V.; Wilson, J.H. Instability and chromatin structure of expanded trinucleotide repeats. Trends Genet. 2009, 25, 288–297. [Google Scholar] [CrossRef]
- Dragileva, E.; Hendricks, A.; Teed, A.; Gillis, T.; Lopez, E.T.; Friedberg, E.C.; Kucherlapati, R.; Edelmann, W.; Lunetta, K.L.; MacDonald, M.E.; et al. Intergenerational and striatal cag repeat instability in huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol. Dis. 2009, 33, 37–47. [Google Scholar] [CrossRef]
- Wheeler, V.C.; Lebel, L.A.; Vrbanac, V.; Teed, A.; te Riele, H.; MacDonald, M.E. Mismatch repair gene msh2 modifies the timing of early disease in hdh(q111) striatum. Hum. Mol. Genet. 2003, 12, 273–281. [Google Scholar]
- Manley, K.; Shirley, T.L.; Flaherty, L.; Messer, A. Msh2 deficiency prevents in vivo somatic instability of the cag repeat in huntington disease transgenic mice. Nat. Genet. 1999, 23, 471–473. [Google Scholar]
- Savouret, C.; Garcia-Cordier, C.; Megret, J.; te Riele, H.; Junien, C.; Gourdon, G. Msh2-dependent germinal ctg repeat expansions are produced continuously in spermatogonia from dm1 transgenic mice. Mol. Cell. Biol. 2004, 24, 629–637. [Google Scholar] [CrossRef]
- Van den Broek, W.J.; Nelen, M.R.; Wansink, D.G.; Coerwinkel, M.M.; te Riele, H.; Groenen, P.J.; Wieringa, B. Somatic expansion behaviour of the (ctg)n repeat in myotonic dystrophy knock-in mice is differentially affected by msh3 and msh6 mismatch-repair proteins. Hum. Mol. Genet. 2002, 11, 191–198. [Google Scholar] [CrossRef]
- Kovtun, I.V.; Liu, Y.; Bjoras, M.; Klungland, A.; Wilson, S.H.; McMurray, C.T. Ogg1 initiates age-dependent cag trinucleotide expansion in somatic cells. Nature 2007, 447, 447–452. [Google Scholar] [CrossRef]
- Mollersen, L.; Rowe, A.D.; Illuzzi, J.L.; Hildrestrand, G.A.; Gerhold, K.J.; Tveteras, L.; Bjolgerud, A.; Wilson, D.M., 3rd; Bjoras, M.; Klungland, A. Neil1 is a genetic modifier of somatic and germline cag trinucleotide repeat instability in r6/1 mice. Hum. Mol. Genet. 2012, 21, 4939–4947. [Google Scholar] [CrossRef]
- Hubert, L., Jr.; Lin, Y.; Dion, V.; Wilson, J.H. Xpa deficiency reduces cag trinucleotide repeat instability in neuronal tissues in a mouse model of sca1. Hum. Mol. Genet. 2011, 20, 4822–4830. [Google Scholar] [CrossRef]
- Kovtun, I.V.; Johnson, K.O.; McMurray, C.T. Cockayne syndrome b protein antagonizes ogg1 in modulating cag repeat length in vivo. Aging 2011, 3, 509–514. [Google Scholar]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Lindahl, T. Suppression of spontaneous mutagenesis in human cells by DNA base excision-repair. Mutat. Res. 2000, 462, 129–135. [Google Scholar] [CrossRef]
- Robertson, A.B.; Klungland, A.; Rognes, T.; Leiros, I. DNA repair in mammalian cells: Base excision repair: The long and short of it. Cell Mol. Life Sci. 2009, 66, 981–993. [Google Scholar] [CrossRef]
- Fortini, P.; Dogliotti, E. Base damage and single-strand break repair: Mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair 2007, 6, 398–409. [Google Scholar] [CrossRef]
- Freudenreich, C.H.; Kantrow, S.M.; Zakian, V.A. Expansion and length-dependent fragility of ctg repeats in yeast. Science 1998, 279, 853–856. [Google Scholar] [CrossRef]
- Yang, J.; Freudenreich, C.H. Haploinsufficiency of yeast fen1 causes instability of expanded cag/ctg tracts in a length-dependent manner. Gene 2007, 393, 110–115. [Google Scholar] [CrossRef]
- Subramanian, J.; Vijayakumar, S.; Tomkinson, A.E.; Arnheim, N. Genetic instability induced by overexpression of DNA ligase i in budding yeast. Genetics 2005, 171, 427–441. [Google Scholar] [CrossRef]
- Refsland, E.W.; Livingston, D.M. Interactions among DNA ligase i, the flap endonuclease and proliferating cell nuclear antigen in the expansion and contraction of cag repeat tracts in yeast. Genetics 2005, 171, 923–934. [Google Scholar] [CrossRef]
- Schweitzer, J.K.; Livingston, D.M. The effect of DNA replication mutations on cag tract stability in yeast. Genetics 1999, 152, 953–963. [Google Scholar]
- Lopez Castel, A.; Tomkinson, A.E.; Pearson, C.E. Ctg/cag repeat instability is modulated by the levels of human DNA ligase i and its interaction with proliferating cell nuclear antigen: A distinction between replication and slipped-DNA repair. J. Biol. Chem. 2009, 284, 26631–26645. [Google Scholar] [CrossRef]
- Harrison, C.; Ketchen, A.M.; Redhead, N.J.; O’Sullivan, M.J.; Melton, D.W. Replication failure, genome instability, and increased cancer susceptibility in mice with a point mutation in the DNA ligase i gene. Cancer Res. 2002, 62, 4065–4074. [Google Scholar]
- Tome, S.; Panigrahi, G.B.; Lopez Castel, A.; Foiry, L.; Melton, D.W.; Gourdon, G.; Pearson, C.E. Maternal germline-specific effect of DNA ligase i on ctg/cag instability. Hum. Mol. Genet. 2011, 20, 2131–2143. [Google Scholar] [CrossRef]
- Spiro, C.; McMurray, C.T. Nuclease-deficient fen-1 blocks rad51/brca1-mediated repair and causes trinucleotide repeat instability. Mol. Cell. Biol. 2003, 23, 6063–6074. [Google Scholar] [CrossRef]
- Van den Broek, W.J.; Nelen, M.R.; van der Heijden, G.W.; Wansink, D.G.; Wieringa, B. Fen1 does not control somatic hypermutability of the (ctg)(n)*(cag)(n) repeat in a knock-in mouse model for dm1. FEBS Lett. 2006, 580, 5208–5214. [Google Scholar] [CrossRef]
- Bentley, D.; Selfridge, J.; Millar, J.K.; Samuel, K.; Hole, N.; Ansell, J.D.; Melton, D.W. DNA ligase i is required for fetal liver erythropoiesis but is not essential for mammalian cell viability. Nat. Genet. 1996, 13, 489–491. [Google Scholar] [CrossRef]
- Kucherlapati, M.; Yang, K.; Kuraguchi, M.; Zhao, J.; Lia, M.; Heyer, J.; Kane, M.F.; Fan, K.; Russell, R.; Brown, A.M.; et al. Haploinsufficiency of flap endonuclease (fen1) leads to rapid tumor progression. Proc. Natl. Acad. Sci. USA 2002, 99, 9924–9929. [Google Scholar] [CrossRef]
- Cabelof, D.C.; Guo, Z.; Raffoul, J.J.; Sobol, R.W.; Wilson, S.H.; Richardson, A.; Heydari, A.R. Base excision repair deficiency caused by polymerase beta haploinsufficiency: Accelerated DNA damage and increased mutational response to carcinogens. Cancer Res. 2003, 63, 5799–5807. [Google Scholar]
- Sobol, R.W.; Horton, J.K.; Kuhn, R.; Gu, H.; Singhal, R.K.; Prasad, R.; Rajewsky, K.; Wilson, S.H. Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature 1996, 379, 183–186. [Google Scholar] [CrossRef]
- Xanthoudakis, S.; Smeyne, R.J.; Wallace, J.D.; Curran, T. The redox/DNA repair protein, ref-1, is essential for early embryonic development in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 8919–8923. [Google Scholar] [CrossRef]
- Tebbs, R.S.; Flannery, M.L.; Meneses, J.J.; Hartmann, A.; Tucker, J.D.; Thompson, L.H.; Cleaver, J.E.; Pedersen, R.A. Requirement for the xrcc1 DNA base excision repair gene during early mouse development. Dev. Biol. 1999, 208, 513–529. [Google Scholar] [CrossRef]
- Lin, Y.; Wilson, J.H. Transcription-induced cag repeat contraction in human cells is mediated in part by transcription-coupled nucleotide excision repair. Mol. Cell. Biol. 2007, 27, 6209–6217. [Google Scholar] [CrossRef]
- Dou, H.; Mitra, S.; Hazra, T.K. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases neil1 and neil2. J. Biol. Chem. 2003, 278, 49679–49684. [Google Scholar]
- Entezam, A.; Lokanga, A.R.; Le, W.; Hoffman, G.; Usdin, K. Potassium bromate, a potent DNA oxidizing agent, exacerbates germline repeat expansion in a fragile x premutation mouse model. Hum. Mutat. 2010, 31, 611–616. [Google Scholar]
- Goula, A.V.; Berquist, B.R.; Wilson, D.M., 3rd; Wheeler, V.C.; Trottier, Y.; Merienne, K. Stoichiometry of base excision repair proteins correlates with increased somatic cag instability in striatum over cerebellum in huntington’s disease transgenic mice. PLoS Genet. 2009, 5, e1000749. [Google Scholar] [CrossRef]
- Jarem, D.A.; Wilson, N.R.; Delaney, S. Structure-dependent DNA damage and repair in a trinucleotide repeat sequence. Biochemistry 2009, 48, 6655–6663. [Google Scholar] [CrossRef]
- Jarem, D.A.; Wilson, N.R.; Schermerhorn, K.M.; Delaney, S. Incidence and persistence of 8-oxo-7,8-dihydroguanine within a hairpin intermediate exacerbates a toxic oxidation cycle associated with trinucleotide repeat expansion. DNA Repair 2011, 10, 887–896. [Google Scholar] [CrossRef]
- Hartenstine, M.J.; Goodman, M.F.; Petruska, J. Weak strand displacement activity enables human DNA polymerase beta to expand cag/ctg triplet repeats at strand breaks. J. Biol. Chem. 2002, 277, 41379–41389. [Google Scholar] [CrossRef]
- Liu, Y.; Wilson, S.H. DNA base excision repair: A mechanism of trinucleotide repeat expansion. Trends Biochem. Sci. 2012, 37, 162–172. [Google Scholar] [CrossRef]
- Spiro, C.; Pelletier, R.; Rolfsmeier, M.L.; Dixon, M.J.; Lahue, R.S.; Gupta, G.; Park, M.S.; Chen, X.; Mariappan, S.V.; McMurray, C.T. Inhibition of fen-1 processing by DNA secondary structure at trinucleotide repeats. Mol. Cell 1999, 4, 1079–1085. [Google Scholar] [CrossRef]
- Vallur, A.C.; Maizels, N. Complementary roles for exonuclease 1 and flap endonuclease 1 in maintenance of triplet repeats. J. Biol. Chem. 2010, 285, 28514–28519. [Google Scholar] [CrossRef]
- Liu, Y.; Prasad, R.; Beard, W.A.; Hou, E.W.; Horton, J.K.; McMurray, C.T.; Wilson, S.H. Coordination between polymerase beta and fen1 can modulate cag repeat expansion. J. Biol. Chem. 2009, 284, 28352–28366. [Google Scholar] [CrossRef]
- Liu, Y.; Prasad, R.; Wilson, S.H. Hmgb1: Roles in base excision repair and related function. Biochim. Biophys. Acta 2010, 1799, 119–130. [Google Scholar] [CrossRef]
- Goula, A.V.; Pearson, C.E.; Della Maria, J.; Trottier, Y.; Tomkinson, A.E.; Wilson, D.M., 3rd; Merienne, K. The nucleotide sequence, DNA damage location, and protein stoichiometry influence the base excision repair outcome at cag/ctg repeats. Biochemistry 2012, 51, 3919–3932. [Google Scholar] [CrossRef]
- Lai, Y.; Xu, M.; Zhang, Z.; Liu, Y. Instability of ctg repeats is governed by the position of a DNA base lesion through base excision repair. PLoS One 2013, 8, e56960. [Google Scholar]
- Hou, C.; Zhang, T.; Tian, L.; Huang, J.; Gu, L.; Li, G.M. The role of xpg in processing (cag)n/(ctg)n DNA hairpins. Cell. Biosci. 2011, 1, 11. [Google Scholar] [CrossRef]
- Higham, C.F.; Morales, F.; Cobbold, C.A.; Haydon, D.T.; Monckton, D.G. High levels of somatic DNA diversity at the myotonic dystrophy type 1 locus are driven by ultra-frequent expansion and contraction mutations. Hum. Mol. Genet. 2012, 21, 2450–2463. [Google Scholar] [CrossRef]
- Tome, S.; Simard, J.P.; Slean, M.M.; Holt, I.; Morris, G.E.; Wojciechowicz, K.; te Riele, H.; Pearson, C.E. Tissue-specific mismatch repair protein expression: Msh3 is higher than msh6 in multiple mouse tissues. DNA Repair 2013, 12, 46–52. [Google Scholar] [CrossRef]
- Seriola, A.; Spits, C.; Simard, J.P.; Hilven, P.; Haentjens, P.; Pearson, C.E.; Sermon, K. Huntington’s and myotonic dystrophy hescs: Down-regulated trinucleotide repeat instability and mismatch repair machinery expression upon differentiation. Hum. Mol. Genet. 2011, 20, 176–185. [Google Scholar]
- Hick, A.; Wattenhofer-Donze, M.; Chintawar, S.; Tropel, P.; Simard, J.P.; Vaucamps, N.; Gall, D.; Lambot, L.; Andre, C.; Reutenauer, L.; et al. Induced pluripotent stem cell derived neurons and cardiomyocytes as a model for mitochondrial defects in friedreich’s ataxia. Dis. Model. Mech. 2012, 7. [Google Scholar] [CrossRef]
- Du, J.; Campau, E.; Soragni, E.; Ku, S.; Puckett, J.W.; Dervan, P.B.; Gottesfeld, J.M. Role of mismatch repair enzymes in gaa.Ttc triplet-repeat expansion in friedreich ataxia induced pluripotent stem cells. J. Biol. Chem. 2012, 287, 29861–29872. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Goula, A.-V.; Merienne, K. Abnormal Base Excision Repair at Trinucleotide Repeats Associated with Diseases: A Tissue-Selective Mechanism. Genes 2013, 4, 375-387. https://doi.org/10.3390/genes4030375
Goula A-V, Merienne K. Abnormal Base Excision Repair at Trinucleotide Repeats Associated with Diseases: A Tissue-Selective Mechanism. Genes. 2013; 4(3):375-387. https://doi.org/10.3390/genes4030375
Chicago/Turabian StyleGoula, Agathi-Vasiliki, and Karine Merienne. 2013. "Abnormal Base Excision Repair at Trinucleotide Repeats Associated with Diseases: A Tissue-Selective Mechanism" Genes 4, no. 3: 375-387. https://doi.org/10.3390/genes4030375