High SINE RNA Expression Correlates with Post-Transcriptional Downregulation of BRCA1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Experimental Section
2.1. Cell Culture
2.2. RNA Extraction, Reverse Transcription, and qRT-PCR
2.3. Nuclear Run-on
2.4. Small RNA Enrichment and End Label Small RNA Filter Blots
2.5. Knockdowns
2.6. Plasmid Generation and Nucleofections
2.7. Determination of Hybridization Stringency Conditions
3. Results and Discussion
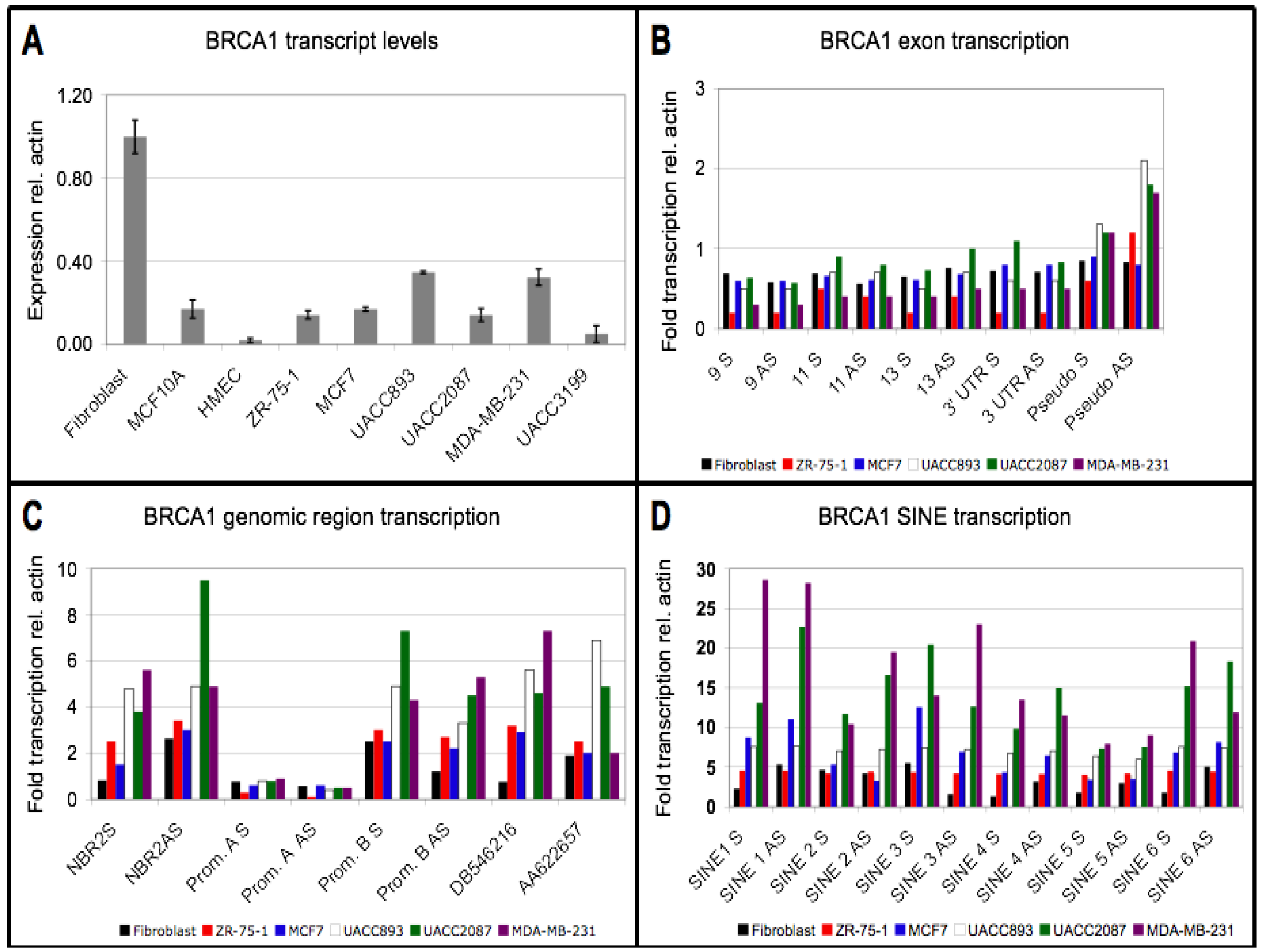
3.1. Reduction in Steady State BRCA1 Transcript Levels is not Associated with Promoter Hypermethylation
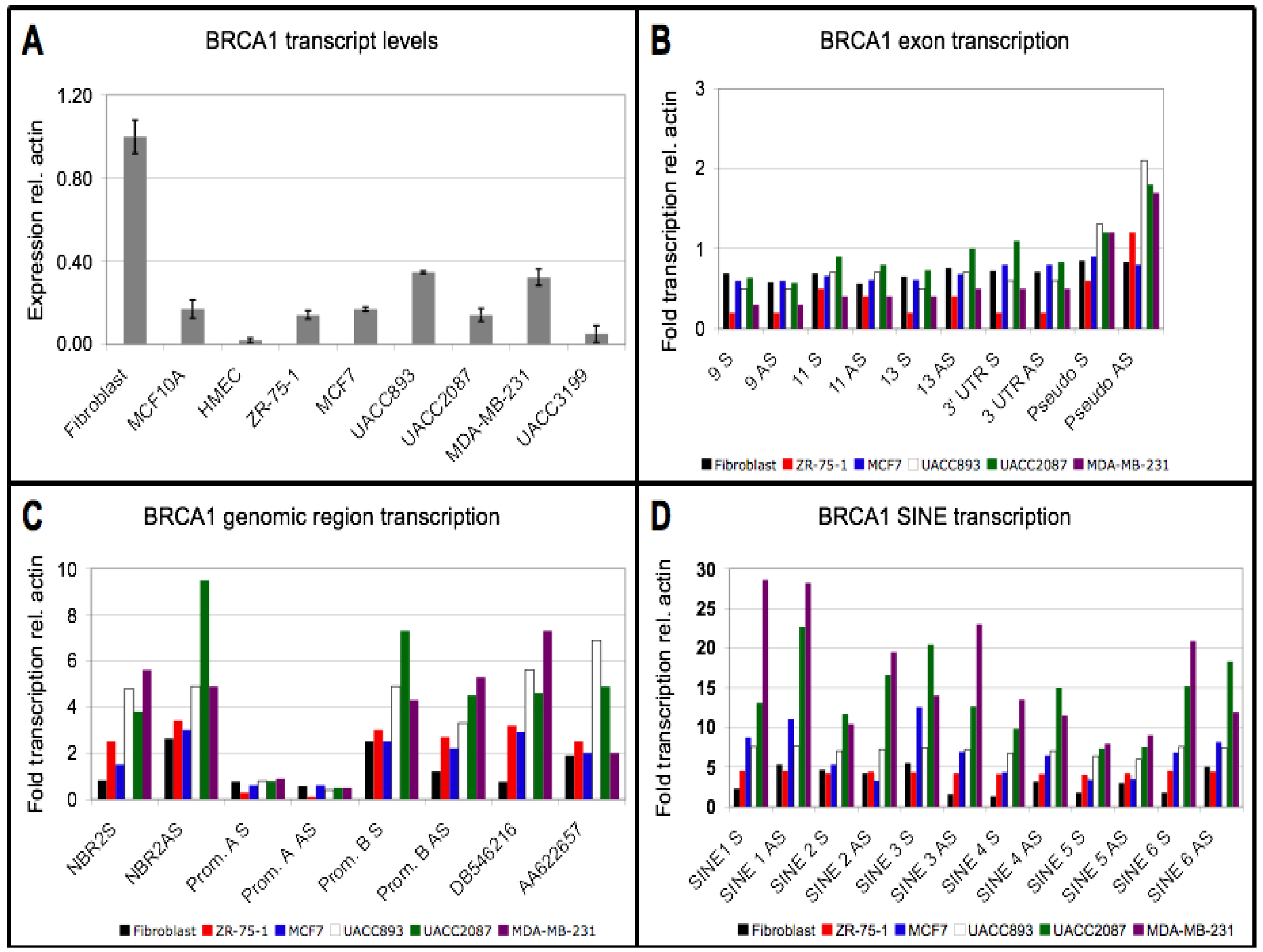
3.2. Downregulation of BRCA1 Transcript Levels is not Associated with a Reduced Transcription Rate
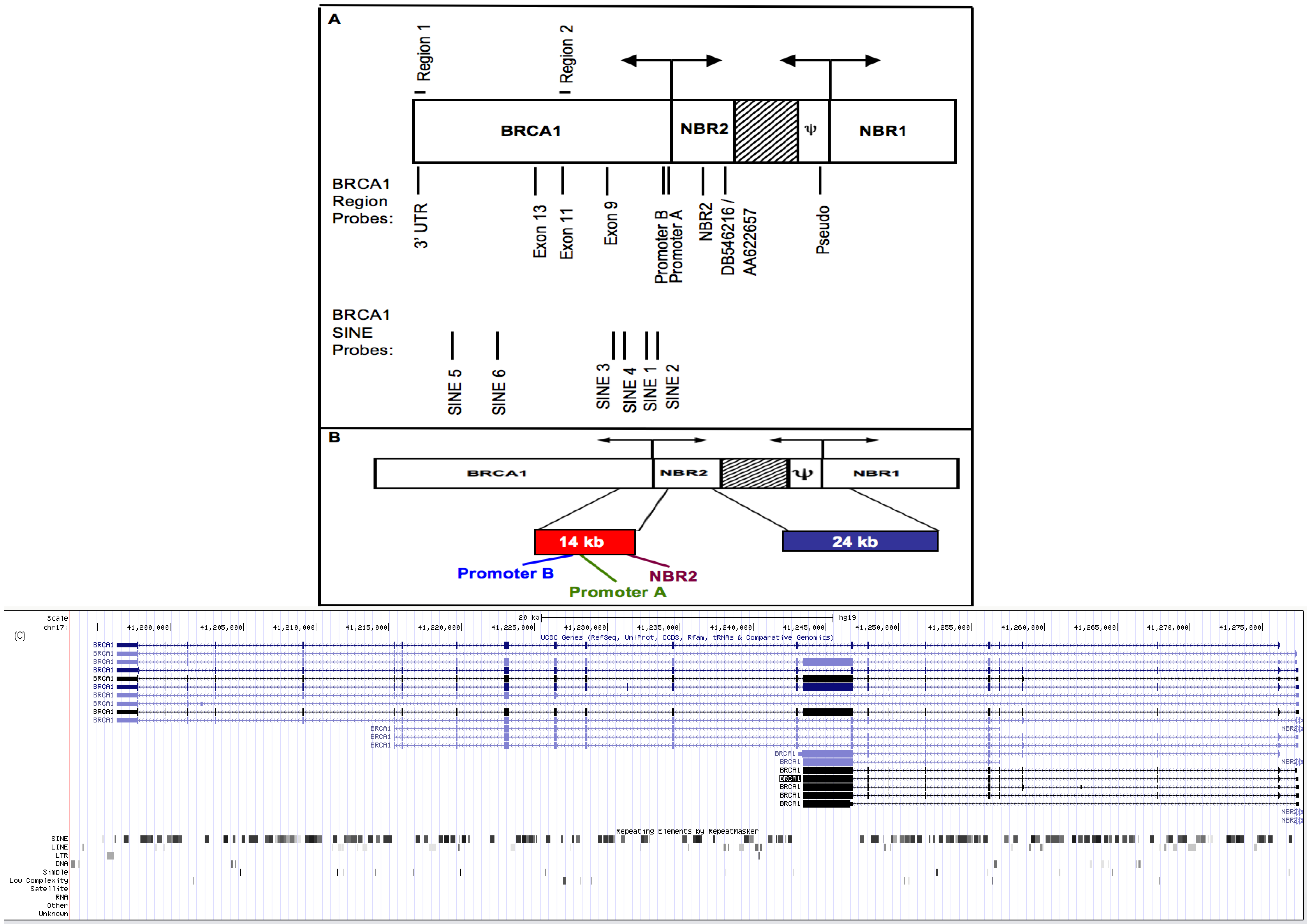
3.3. Repeat Sequences Are Highly Transcribed in Sporadic Breast Cancer Cell Lines
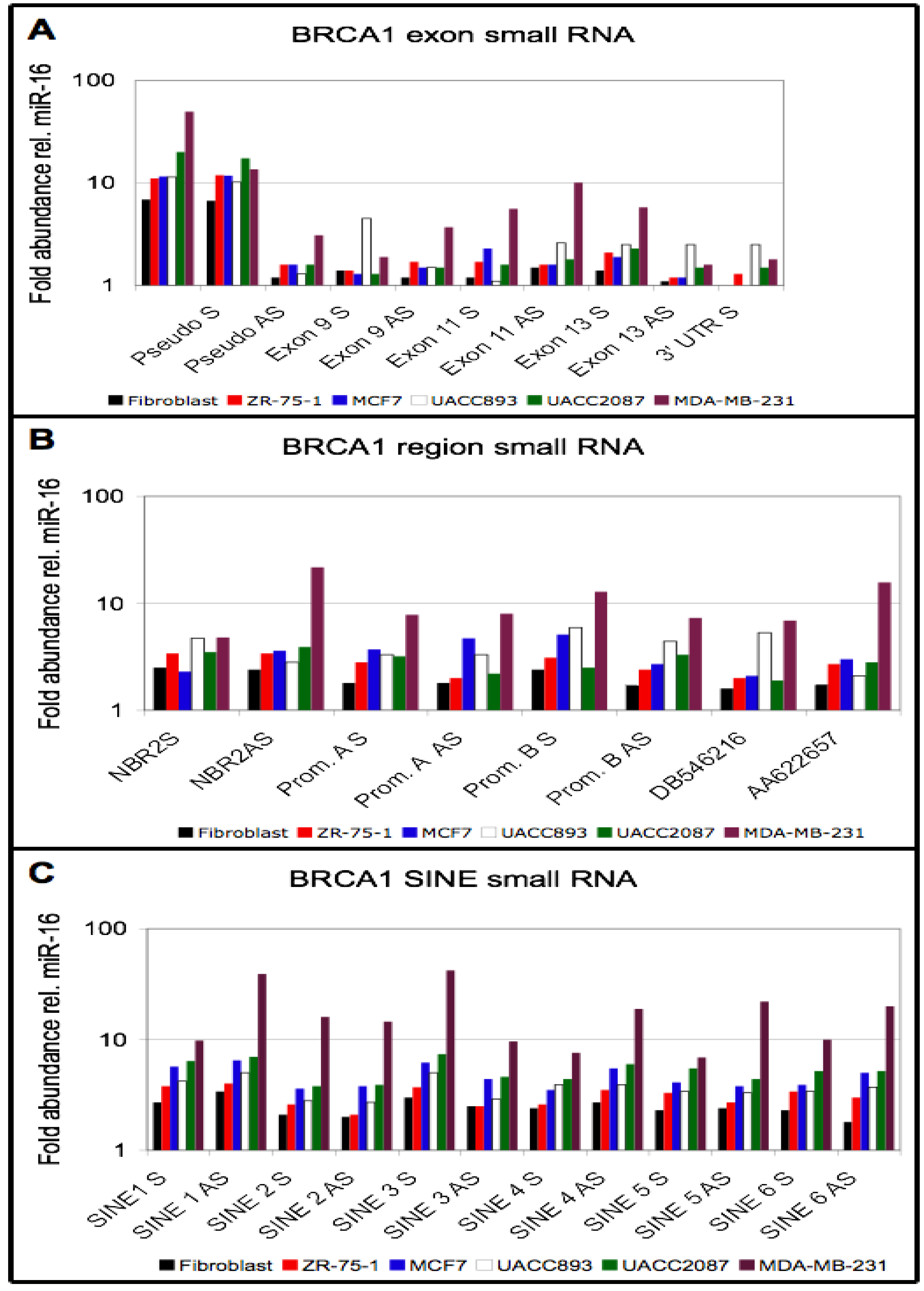
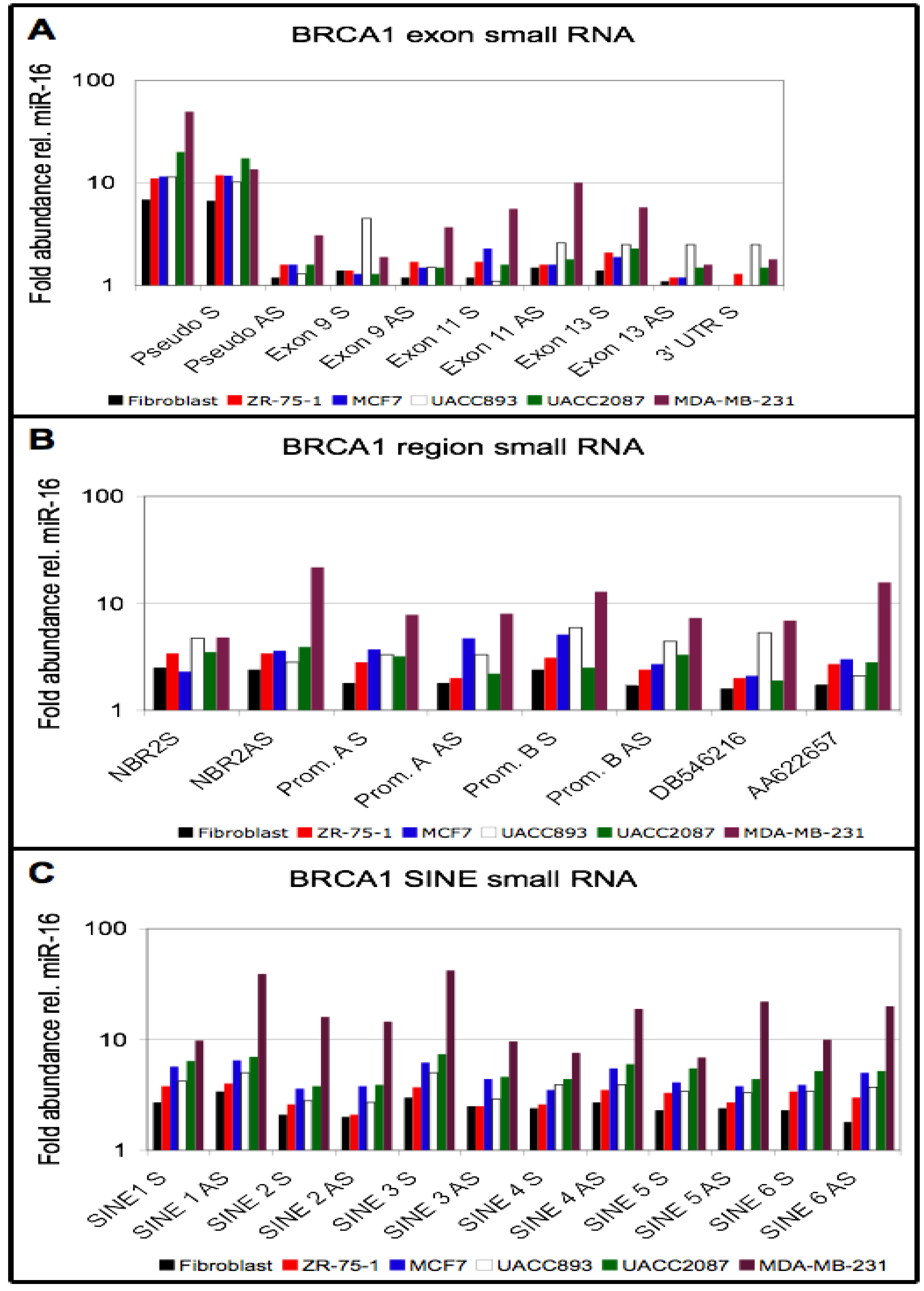
3.4. Small RNA Species from BRCA1 and Associated Retrotransposons Are Elevated in Breast Cancer Cell Lines
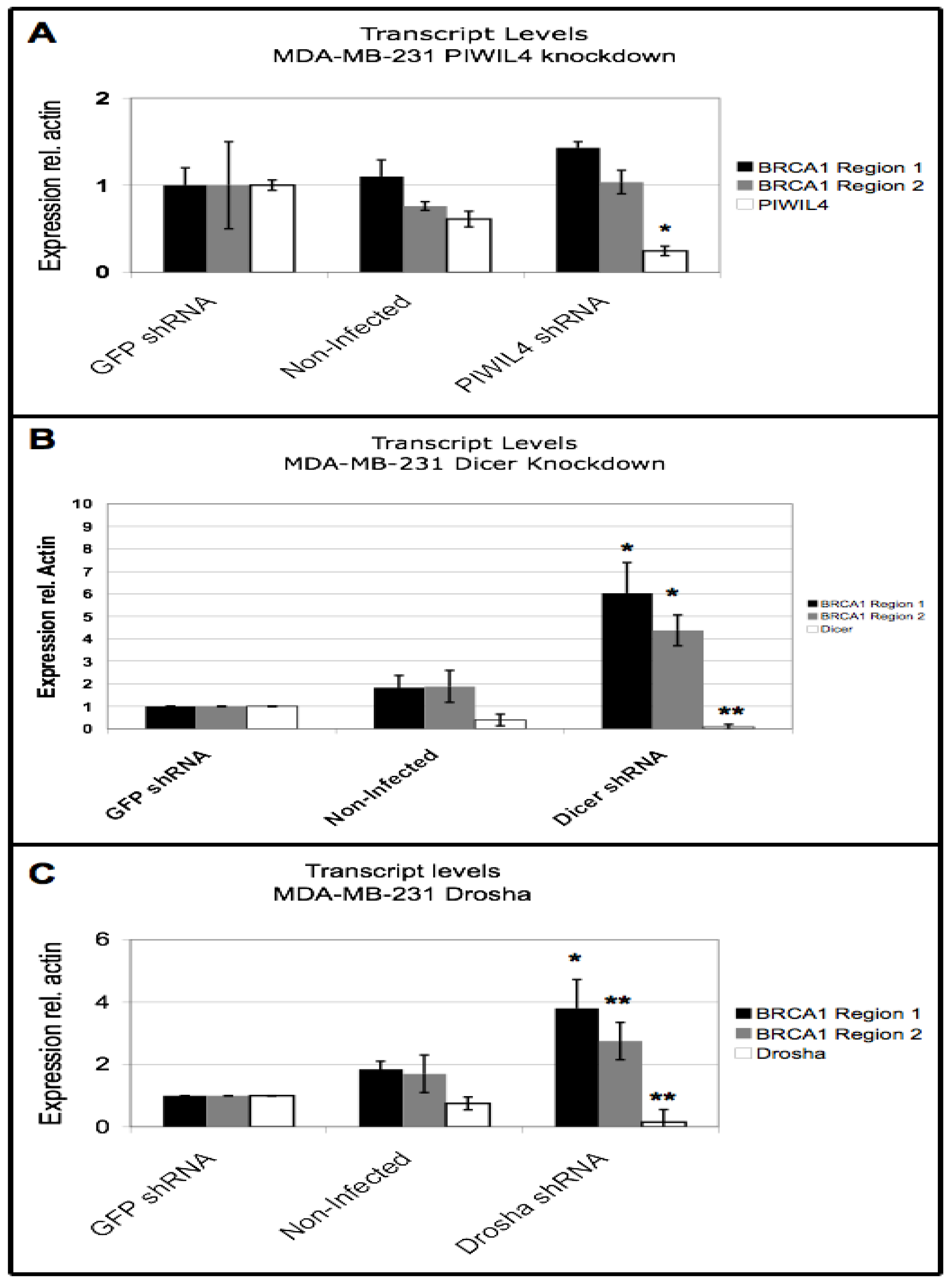
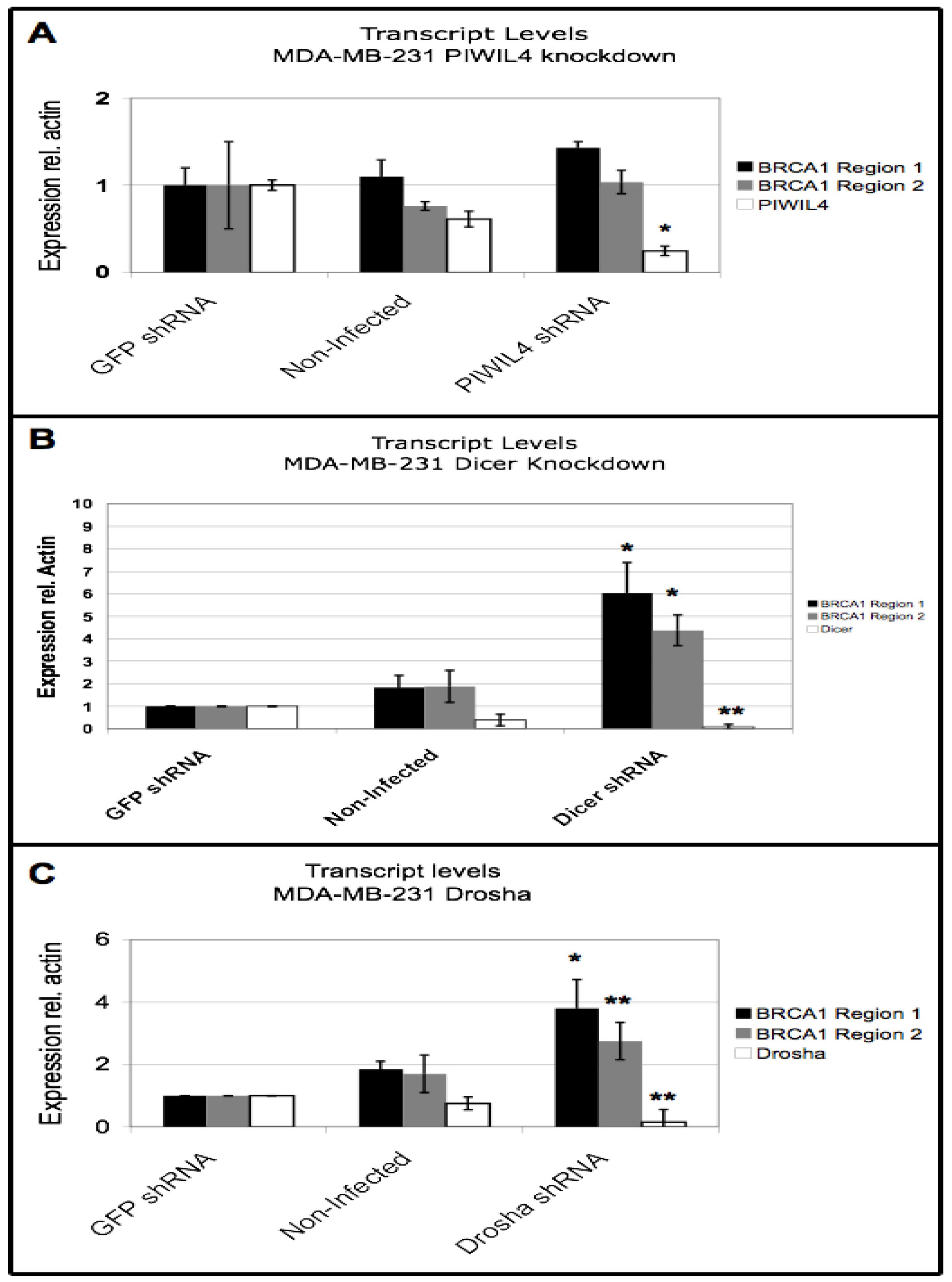
3.5. BRCA1 Downregulation Occurs Through Dicer and Drosha
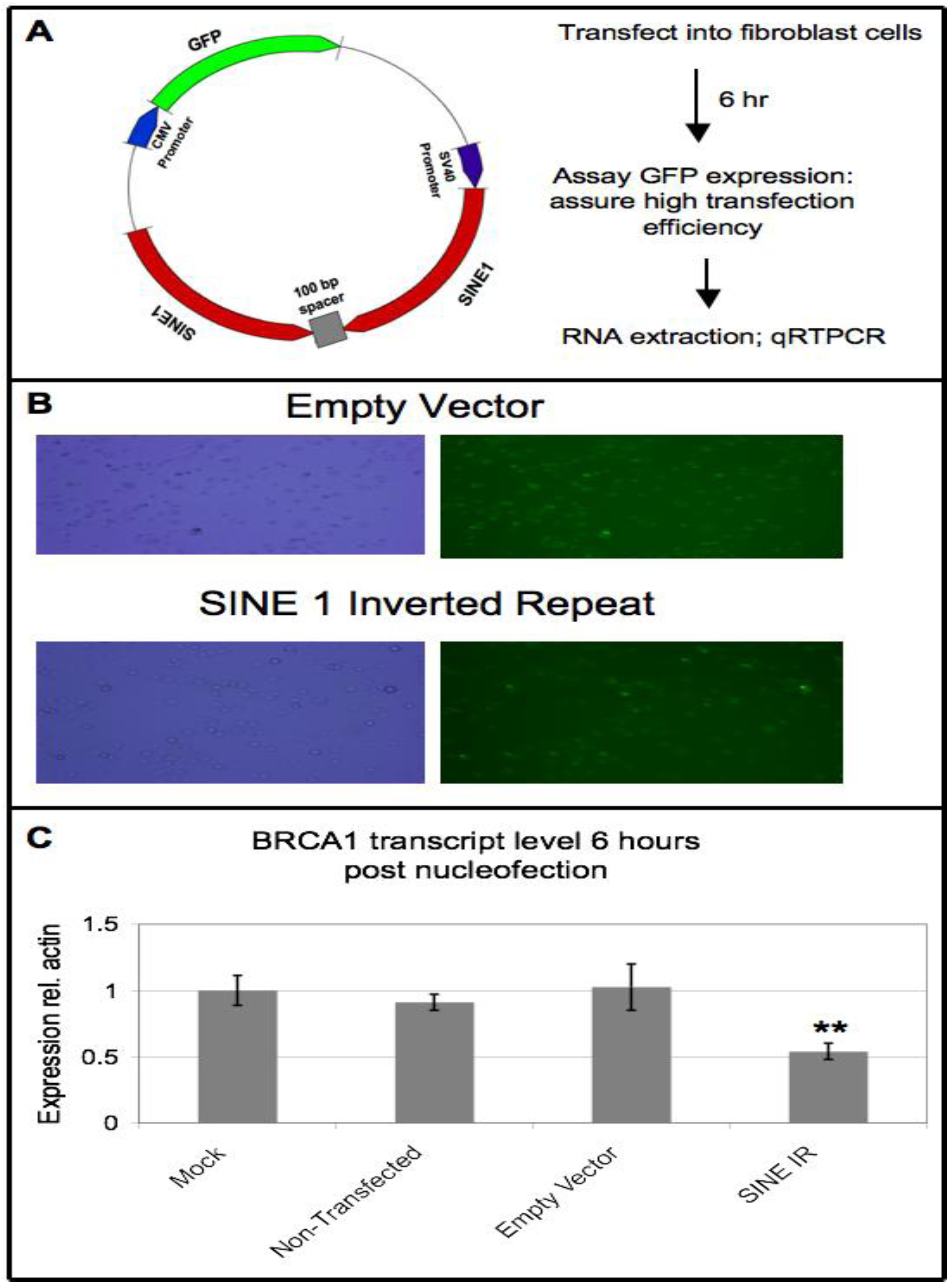
3.6. Ectopic Expression of SINE Sequences Induces Downregulation of BRCA1 in Non-Cancer Cells
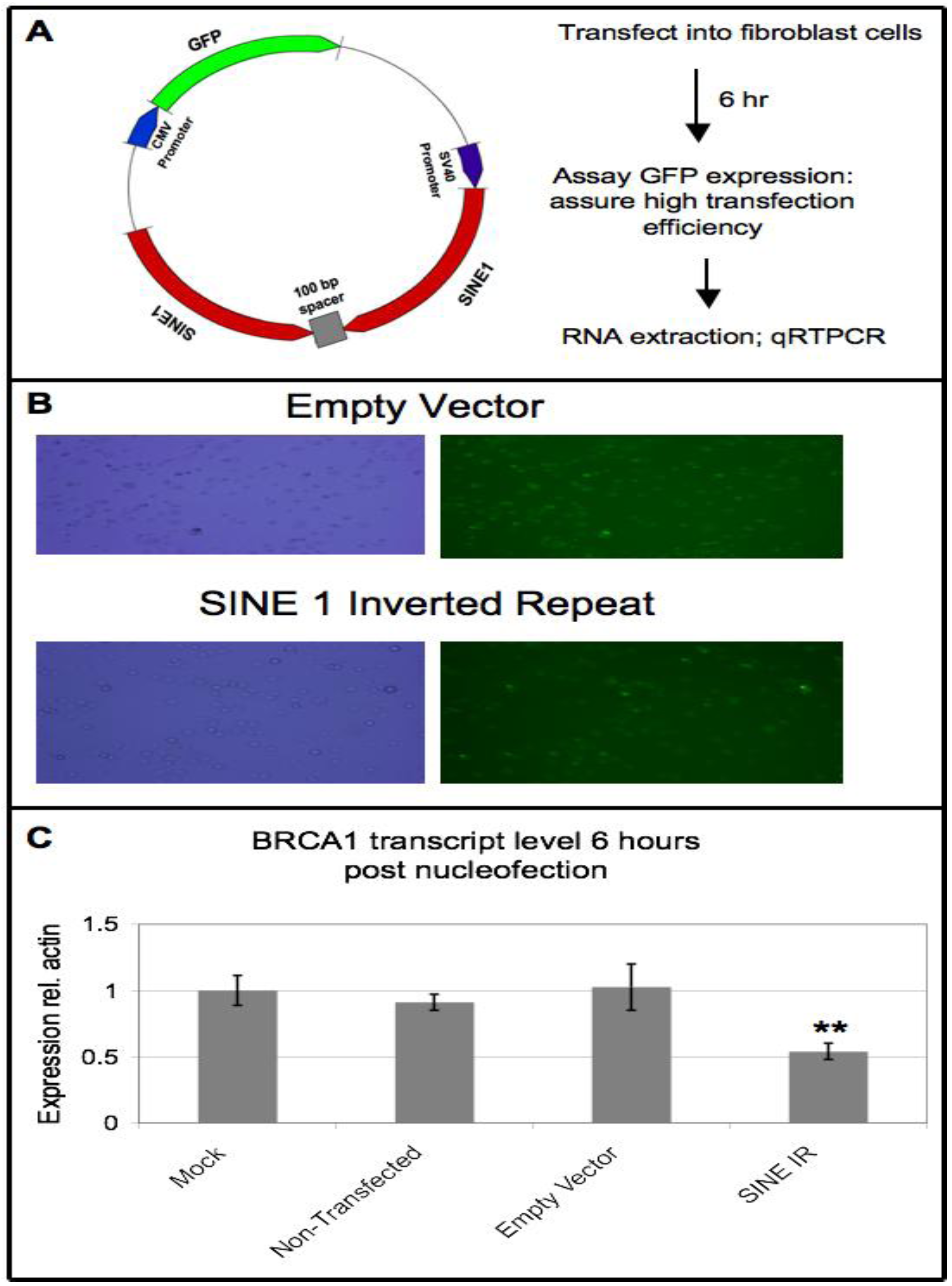
4. Conclusions
Acknowledgments
References and Notes
- Martienssen, R.A.; Colot, V. DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science 2001, 293, 1070–1074. [Google Scholar] [CrossRef]
- Schumann, G.G.; Gogvadze, E.V.; Osanai-Futahashi, M.; Kuroki, A.; Münk, C.; Fujiwara, H.; Ivics, Z.; Buzdin, A.A. Unique functions of repetitive transcriptomes. Int. Rev. Cell. Mol. Biol. 2010, 285, 115–188. [Google Scholar]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Kaikkonen, M.U.; Lam, M.T.; Glass, C.K. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc. Res. 2011, 90, 430–440. [Google Scholar] [CrossRef]
- Levin, H.L.; Moran, J.V. Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 2011, 12, 615–627. [Google Scholar]
- Aravin, A.A.; Sachidanandam, R.; Girard, A.; Fejes-Toth, K.; Hannon, G.J. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science 2007, 316, 744–747. [Google Scholar] [CrossRef]
- Carmell, M.A.; Girard, A.; van de Kant, H.J.; Bourc'his, D.; Bestor, T.H.; de Rooij, D.G.; Hannon, G.J. MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev. Cell. 2007, 12, 503–514. [Google Scholar] [CrossRef]
- Coufal, N.G.; Garcia-Perez, J.L.; Peng, G.E.; Yeo, G.W.; Mu, Y.; Lovci, M.T.; Morell, M.; O'Shea, K.S.; Moran, J.V.; Gage, F.H. LINE-1 retrotransposition in human embryonic stem cells. Hum. Mol. Genet. 2007, 16, 1569–1577. [Google Scholar] [CrossRef]
- Muotri, A.R.; Chu, V.T.; Marchetto, M.C.; Deng, W.; Moran, J.V.; Gage, F.H. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 2005, 435, 903–910. [Google Scholar]
- Coufal, N.G.; Garcia-Perez, J.L.; Peng, G.E.; Yeo, G.W.; Mu, Y.; Lovci, M.T.; Morell, M.; O'Shea, K.S.; Moran, J.V.; Gage, F.H. L1 retrotransposition in human neural progenitor cells. Nature 2009, 460, 1127–1131. [Google Scholar]
- Chenais, B. Transposable elements and human cancer: A causal relationship? Biochim. Biophys. Acta. 2012, 1835, 28–35. [Google Scholar]
- Waterland, R.A.; Jirtle, R.L. Transposable elements: Targets for early nutritional effects on epigenetic gene regulation. Mol. Cell. Biol. 2003, 23, 5293–5300. [Google Scholar]
- Wolff, E.M.; Byun, H.M.; Han, H.F.; Sharma, S.; Nichols, P.W.; Siegmund, K.D.; Yang, A.S.; Jones, P.A.; Liang, G. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 2010, 6, e1000917. [Google Scholar] [CrossRef]
- Hasler, J.; Strub, K. Alu elements as regulators of gene expression. Nucleic Acids Res. 2006, 34, 5491–5497. [Google Scholar] [CrossRef]
- Cordaux, R.; Batzer, M.A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009, 10, 691–703. [Google Scholar] [CrossRef]
- Walters, R.D.; Kugel, J.F.; Goodrich, J.A. InvAluable junk: The cellular impact and function of Alu and B2 RNAs. IUBMB Life 2009, 61, 831–837. [Google Scholar] [CrossRef]
- Thompson, M.E.; Jensen, R.A.; Obermiller, P.S.; Page, D.L.; Holt, J.T. Decreased expression of BRCA1 accelerates growth and is often present during sporadic breast cancer progression. Nat. Genet. 1995, 9, 444–450. [Google Scholar]
- Catteau, A.; Xu, C.F.; Brown, M.A.; Hodgson, S.; Greenman, J.; Mathew, C.G.; Dunning, A.M.; Solomon, E. Identification of a C/G polymorphism in the promoter region of the BRCA1 gene and its use as a marker for rapid detection of promoter deletions. Br. J. Cancer 1999, 79, 759–763. [Google Scholar] [CrossRef]
- Catteau, A.; Harris, W.H.; Xu, C.F.; Solomon, E. Methylation of the BRCA1 promoter region in sporadic breast and ovarian cancer: correlation with disease characteristics. Oncogene. 1999, 18, 1957–1965. [Google Scholar] [CrossRef]
- Girardi, A.J.; Weinstein, D.; Moorhead, P.S. SV40 transformation of human diploid cells. A parallel study of viral and karyologic parameters. Ann. Med. Exp. Biol. Fenn. 1966, 44, 242–254. [Google Scholar]
- Stampfer, M.; Hallowes, R.C.; Hackett, A.J. Growth of normal human mammary cells in culture. In Vitro 1980, 16, 415–425. [Google Scholar] [CrossRef]
- Soule, H.D.; Maloney, T.M.; Wolman, S.R.; Peterson, W.D., Jr.; Brenz, R.; McGrath, C.M.; Russo, J.; Pauley, R.J.; Jones, R.F.; Brooks, S.C. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990, 50, 6075–6086. [Google Scholar]
- Cailleau, R.; Young, R.; Olivé, M.; Reeves, W.J., Jr. Breast tumor cell lines from pleural effusions. J. Natl. Cancer Inst. 1974, 53, 661–674. [Google Scholar]
- Soule, H.D.; Vazguez, J.; Long, A.; Albert, S.; Brennan, M. A human cell line from a pleural effusion derived from a breast carcinoma. J. Natl. Cancer Inst. 1973, 51, 1409–1416. [Google Scholar]
- Trent, J.; Yang, J.M.; Emerson, J.; Dalton, W.; McGee, D.; Massey, K.; Thompson, F.; Villar, H. Clonal chromosome abnormalities in human breast carcinomas. I. Twenty-eight cases with primary disease. Genes Chromosomes Cancer 1993, 7, 185–193. [Google Scholar] [CrossRef]
- Meltzer, P.; Leibovitz, A.; Dalton, W.; Villar, H.; Kute, T.; Davis, J.; Nagle, R.; Trent, J. Establishment of two new cell lines derived from human breast carcinomas with HER-2/neu amplification. Br. J. Cancer 1991, 63, 727–735. [Google Scholar] [CrossRef]
- Engel, L.W.; Young, N.A.; Tralka, T.S.; Lippman, M.E.; O'Brien, S.J.; Joyce, M.J. Establishment and characterization of three new continuous cell lines derived from human breast carcinomas. Cancer Res. 1978, 38, 3352–3364. [Google Scholar]
- Alleman, M.; Sidorenko, L.; McGinnis, K.; Seshadri, V.; Dorweiler, J.E.; White, J.; Sikkink, K.; Chandler, V.L. An RNA-dependent RNA polymerase is required for paramutation in maize. Nature 2006, 442, 295–298. [Google Scholar] [CrossRef]
- Dorweiler, J.E.; Carey, C.C.; Kubo, K.M.; Hollick, J.B.; Kermicle, J.L.; Chandler, V.L. Mediator of paramutation1 is required for establishment and maintenance of paramutation at multiple maize loci. Plant. Cell. 2000, 12, 2101–2118. [Google Scholar]
- Davoren, P.A.; McNeill, R.E.; Lowery, A.J.; Kerin, M.J.; Miller, N. Identification of suitable endogenous control genes for microRNA gene expression analysis in human breast cancer. BMC Mol. Biol. 2008, 9, 76. [Google Scholar] [CrossRef]
- Xu, C.F.; Chambers, J.A.; Solomon, E. Complex regulation of the BRCA1 gene. J. Biol. Chem. 1997, 272, 20994–20997. [Google Scholar] [CrossRef]
- Rice, J.C.; Massey-Brown, K.S.; Futscher, B.W. Aberrant methylation of the BRCA1 CpG island promoter is associated with decreased BRCA1 mRNA in sporadic breast cancer cells. Oncogene. 1998, 17, 1807–1812. [Google Scholar]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigó, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef]
- Ah-Fong, A.M.; Bormann-Chung, C.A.; Judelson, H.S. Optimization of transgene-mediated silencing in Phytophthora infestans and its association with small-interfering RNAs. Fungal Genet. Biol. 2008, 45, 1197–1205. [Google Scholar]
- Kadotani, N.; Nakayashiki, H.; Tosa, Y.; Mayama, S. RNA silencing in the phytopathogenic fungus Magnaporthe oryzae. Mol. Plant. Microbe Interact. 2003, 16, 769–776. [Google Scholar] [CrossRef]
- Robine, N.; Lau, N.C.; Balla, S.; Jin, Z.; Okamura, K.; Kuramochi-Miyagawa, S.; Blower, M.D.; Lai, E.C. A broadly conserved pathway generates 3'UTR-directed primary piRNAs. Curr. Biol. 2009, 19, 2066–2076. [Google Scholar] [CrossRef]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3' UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Peterson, M.; Chandler, V.L.; Bosco, G. High SINE RNA Expression Correlates with Post-Transcriptional Downregulation of BRCA1. Genes 2013, 4, 226-243. https://doi.org/10.3390/genes4020226
Peterson M, Chandler VL, Bosco G. High SINE RNA Expression Correlates with Post-Transcriptional Downregulation of BRCA1. Genes. 2013; 4(2):226-243. https://doi.org/10.3390/genes4020226
Chicago/Turabian StylePeterson, Maureen, Vicki L. Chandler, and Giovanni Bosco. 2013. "High SINE RNA Expression Correlates with Post-Transcriptional Downregulation of BRCA1" Genes 4, no. 2: 226-243. https://doi.org/10.3390/genes4020226