Differential Effects of MicroRNAs on Glioblastoma Growth and Migration

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
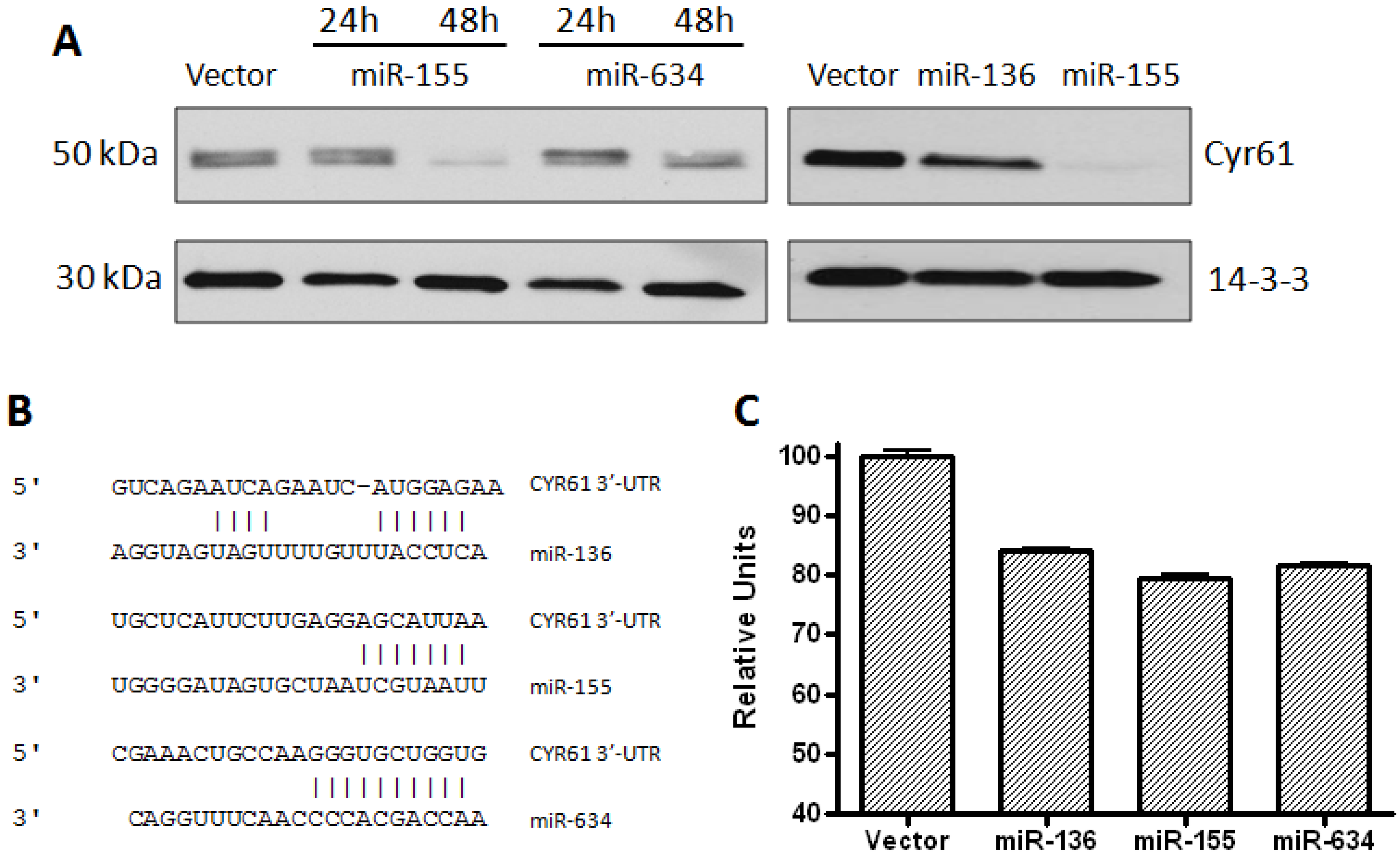
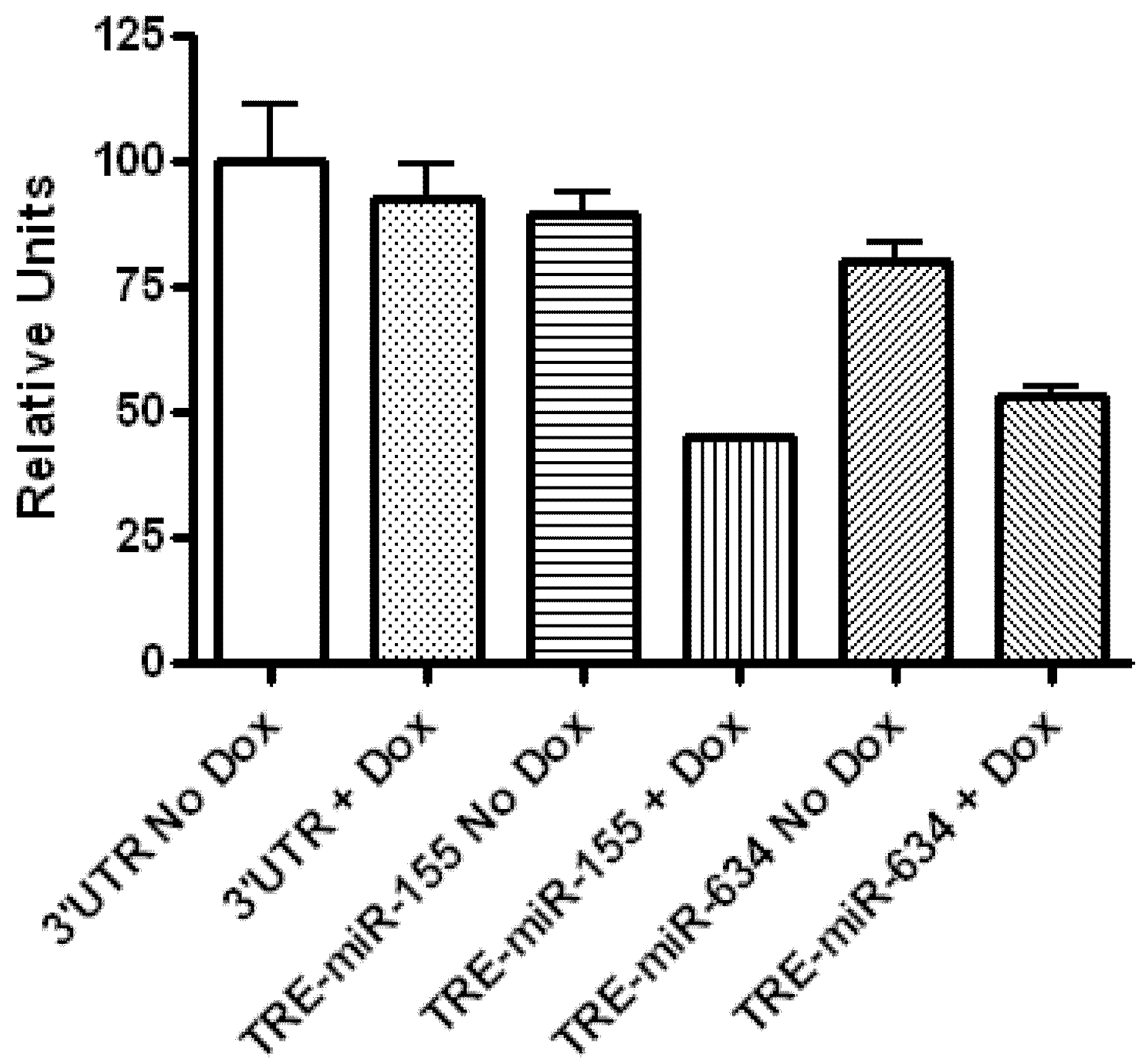
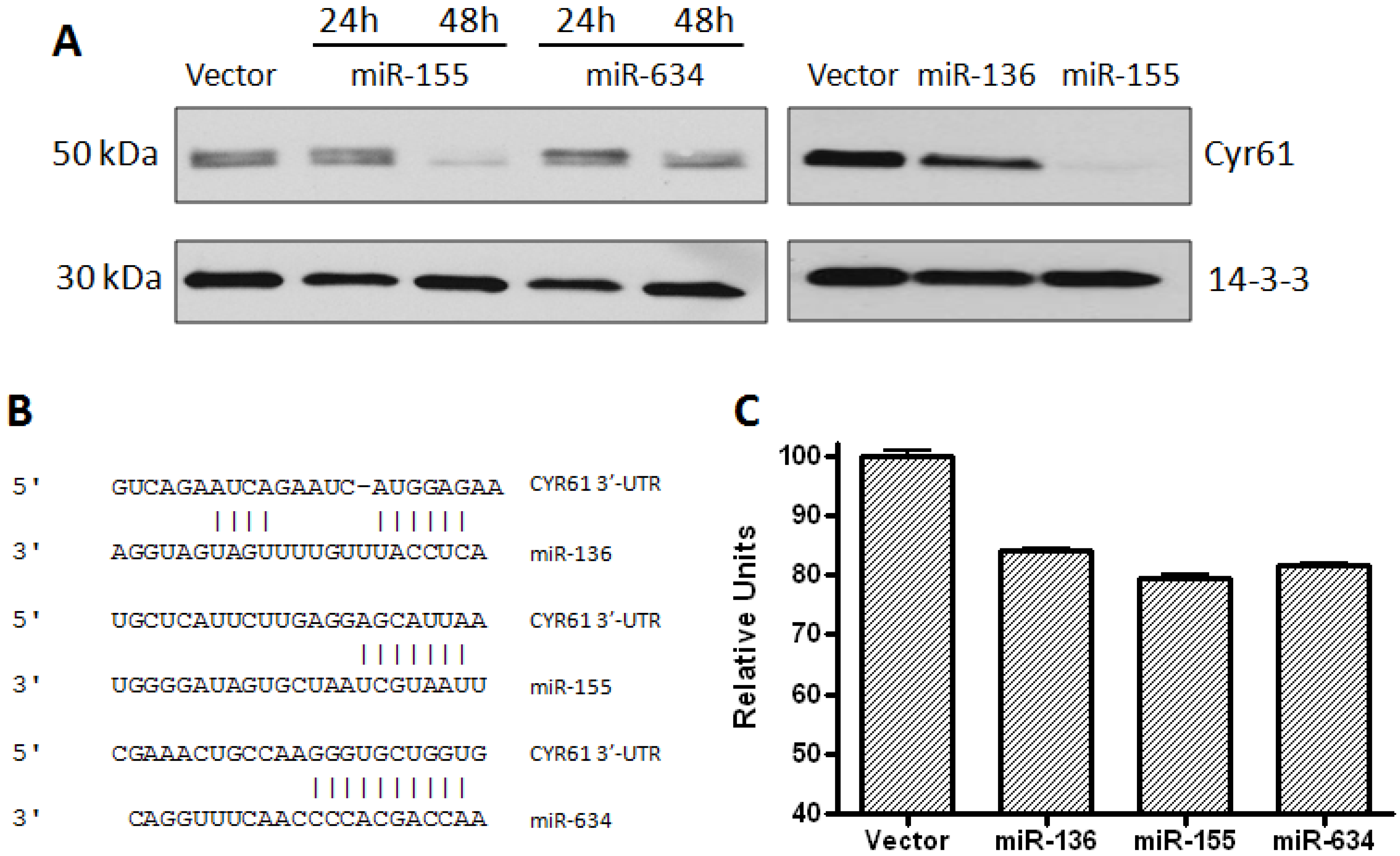
2.1. CYR61 Is a Target for miR-136, miR-155, and miR-634
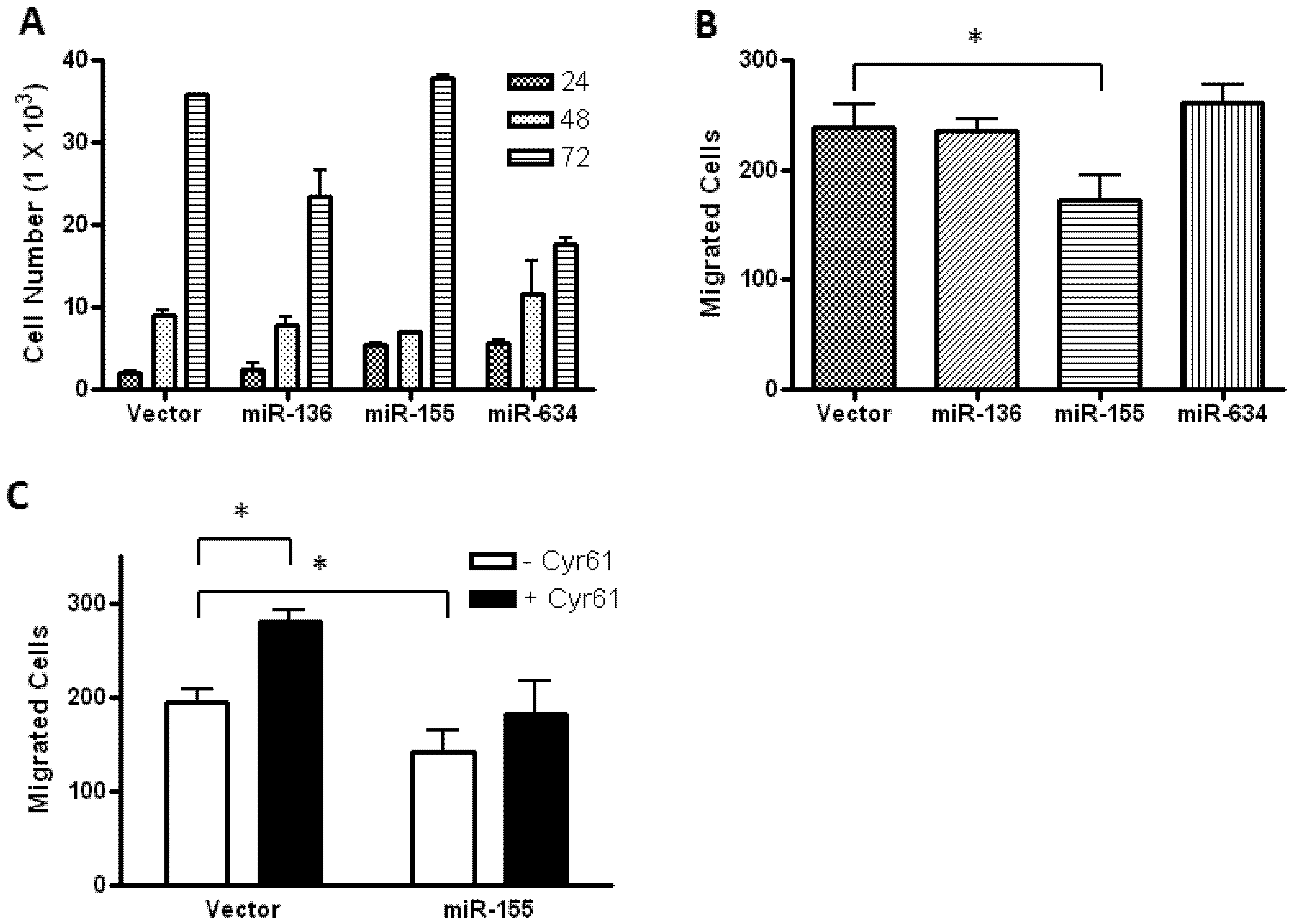
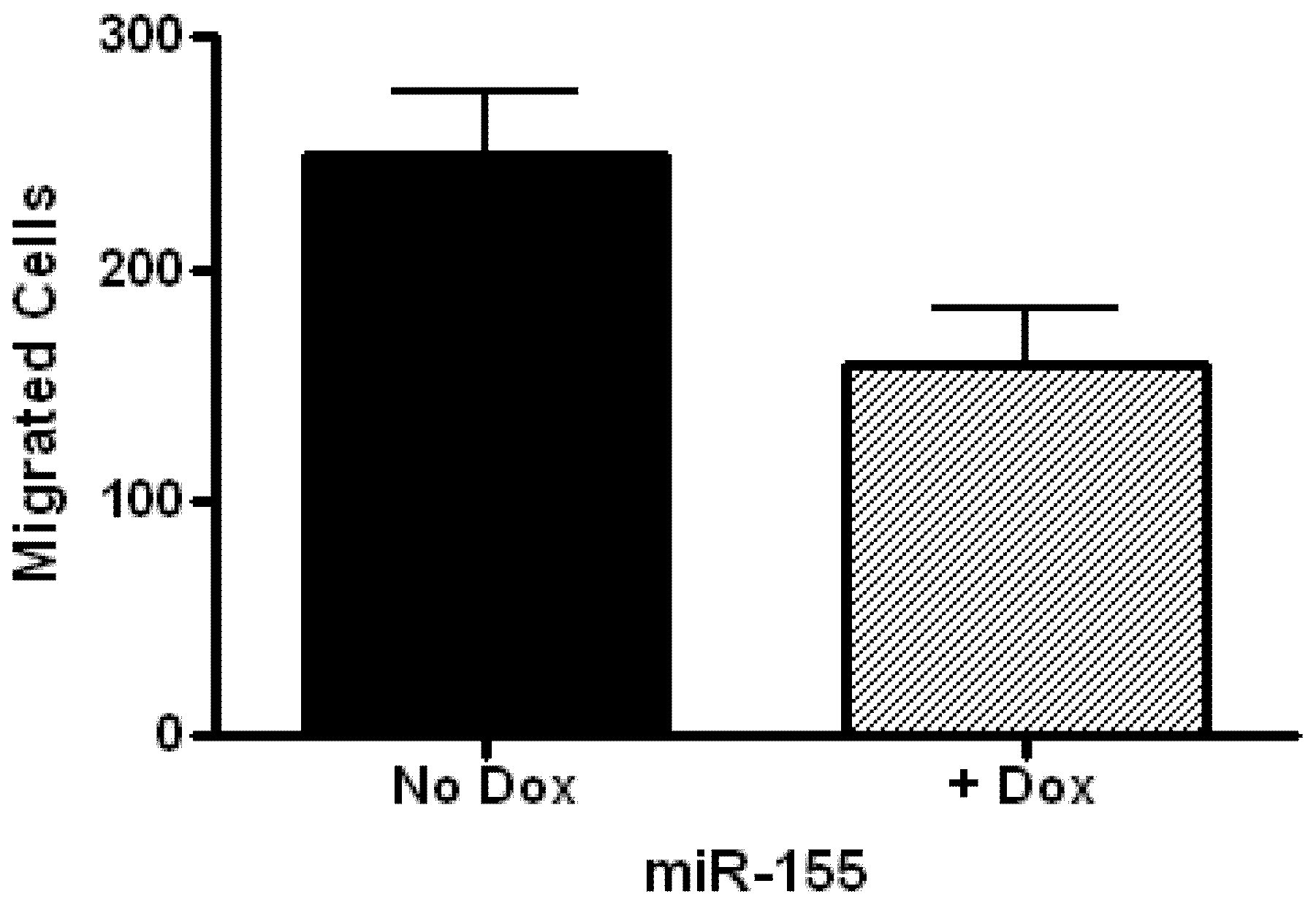
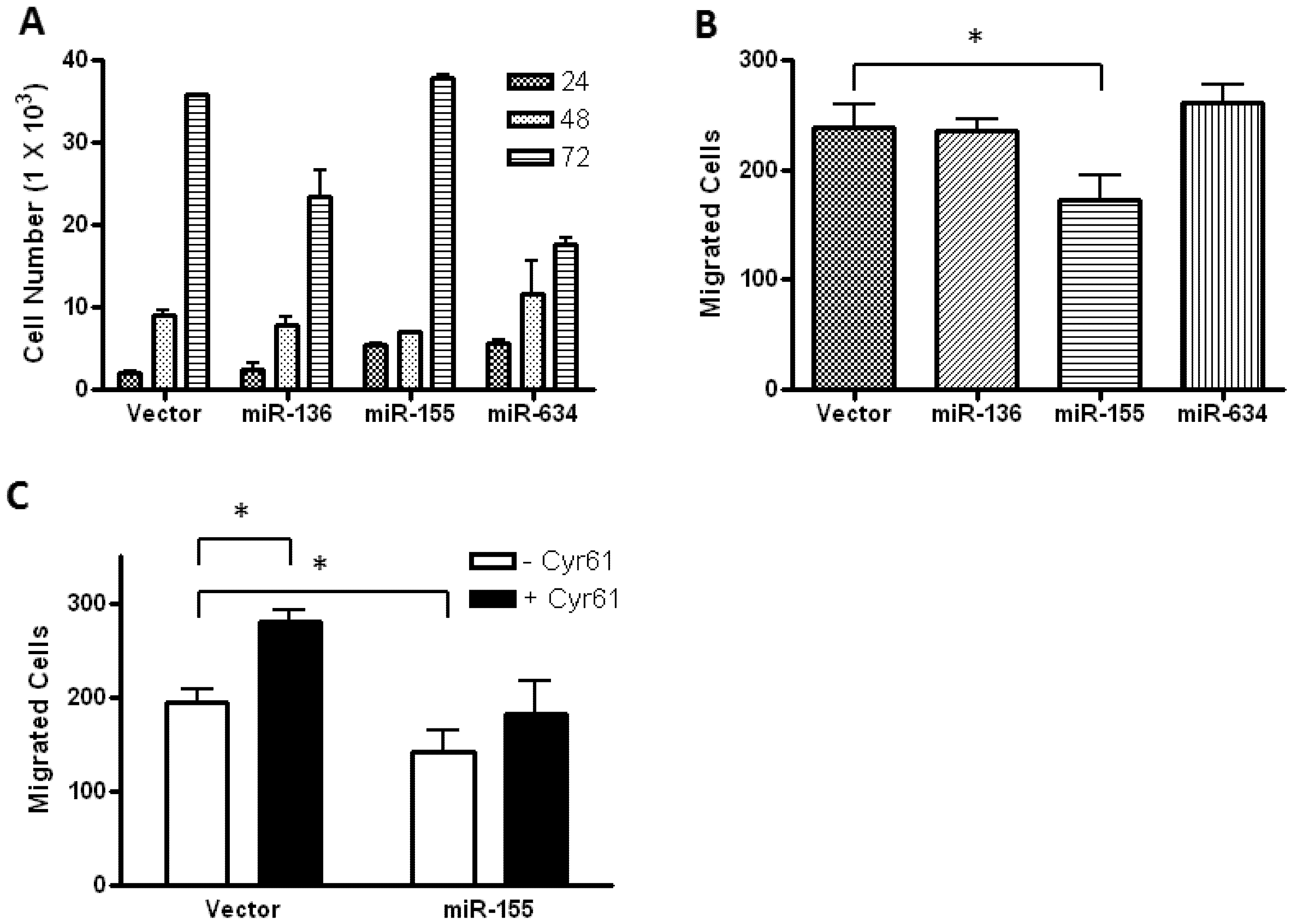
2.2. Glioblastoma Growth and Migration are Differentially Affected by miR-136, miR-155, and miR-634
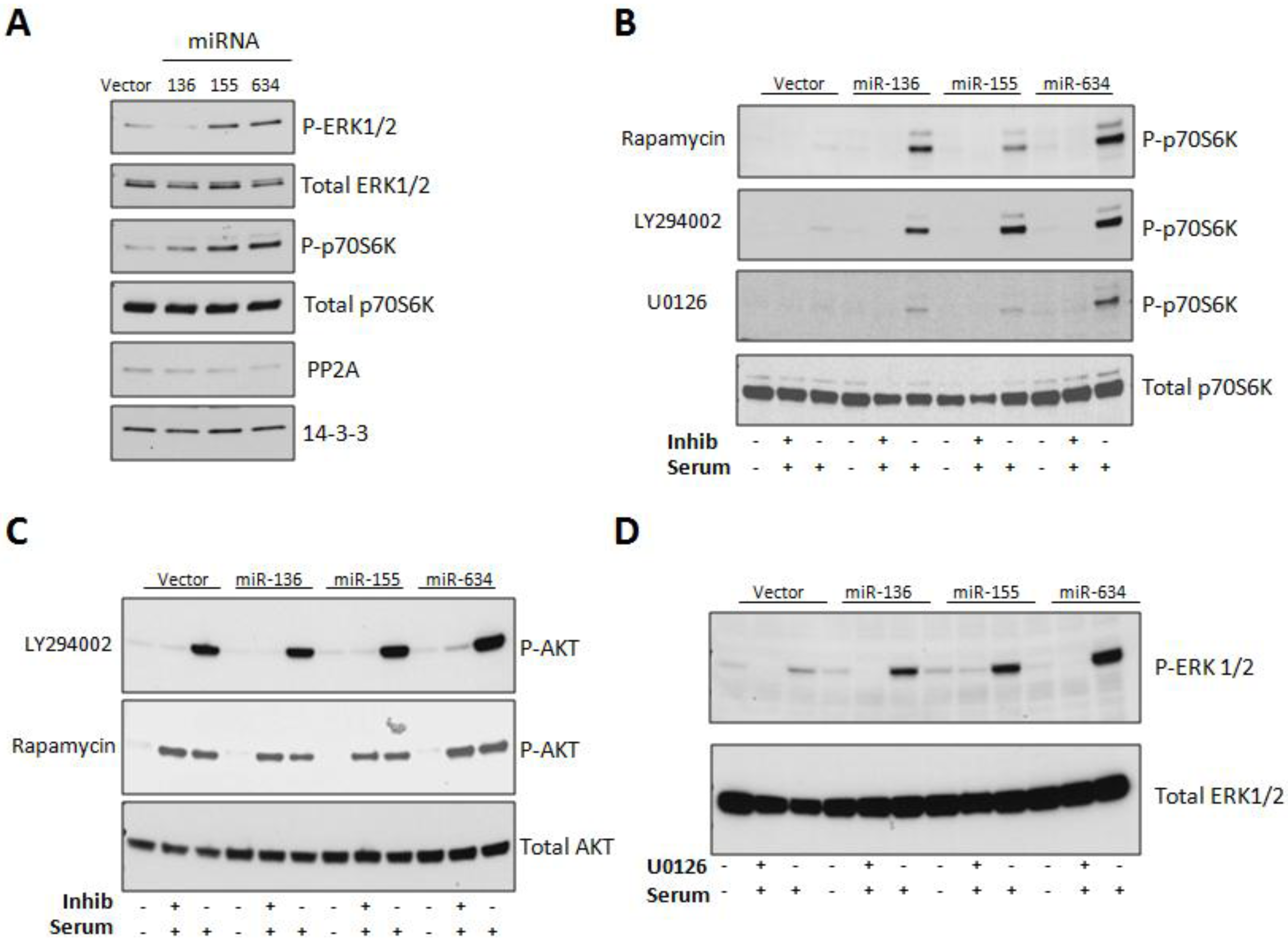
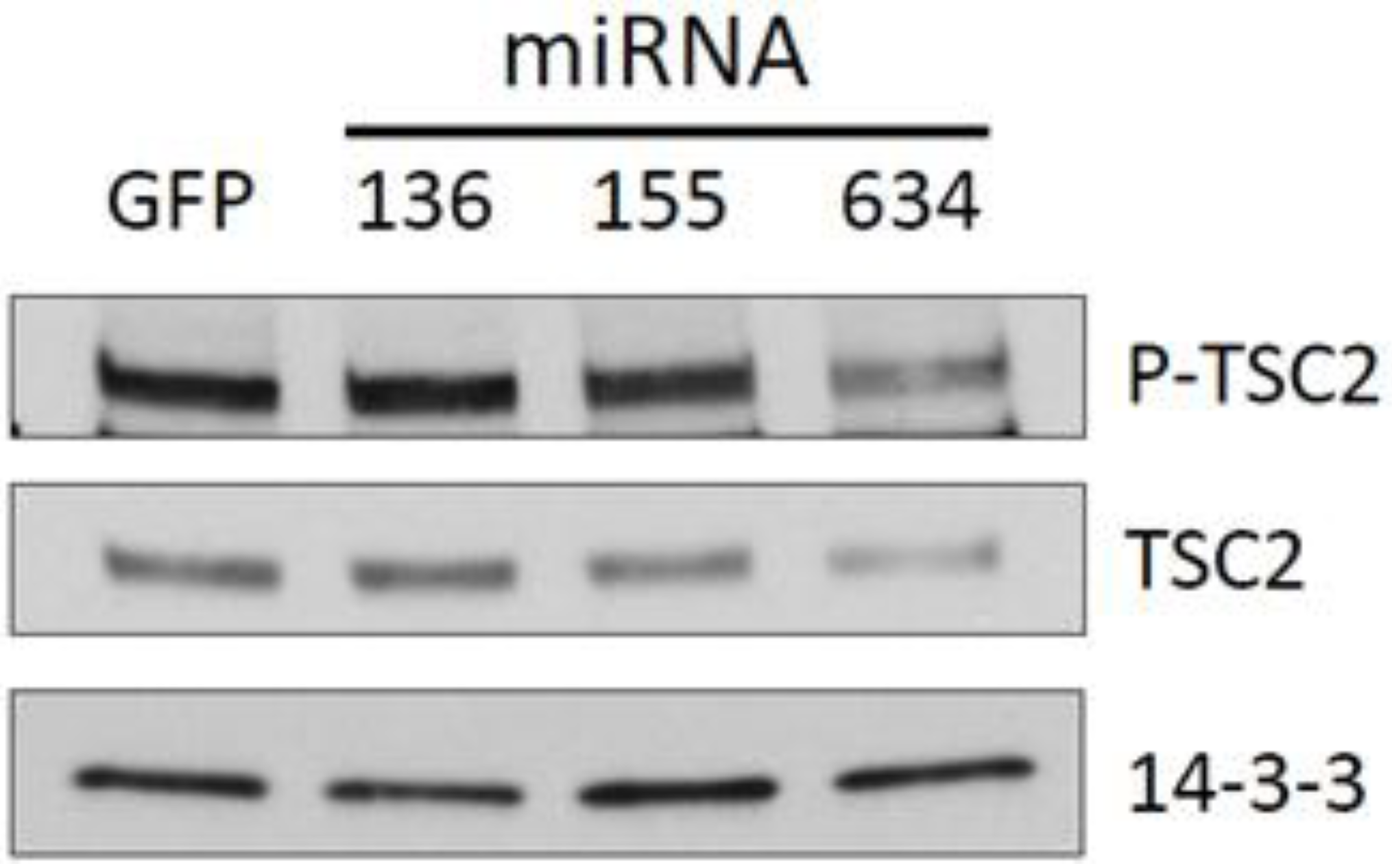
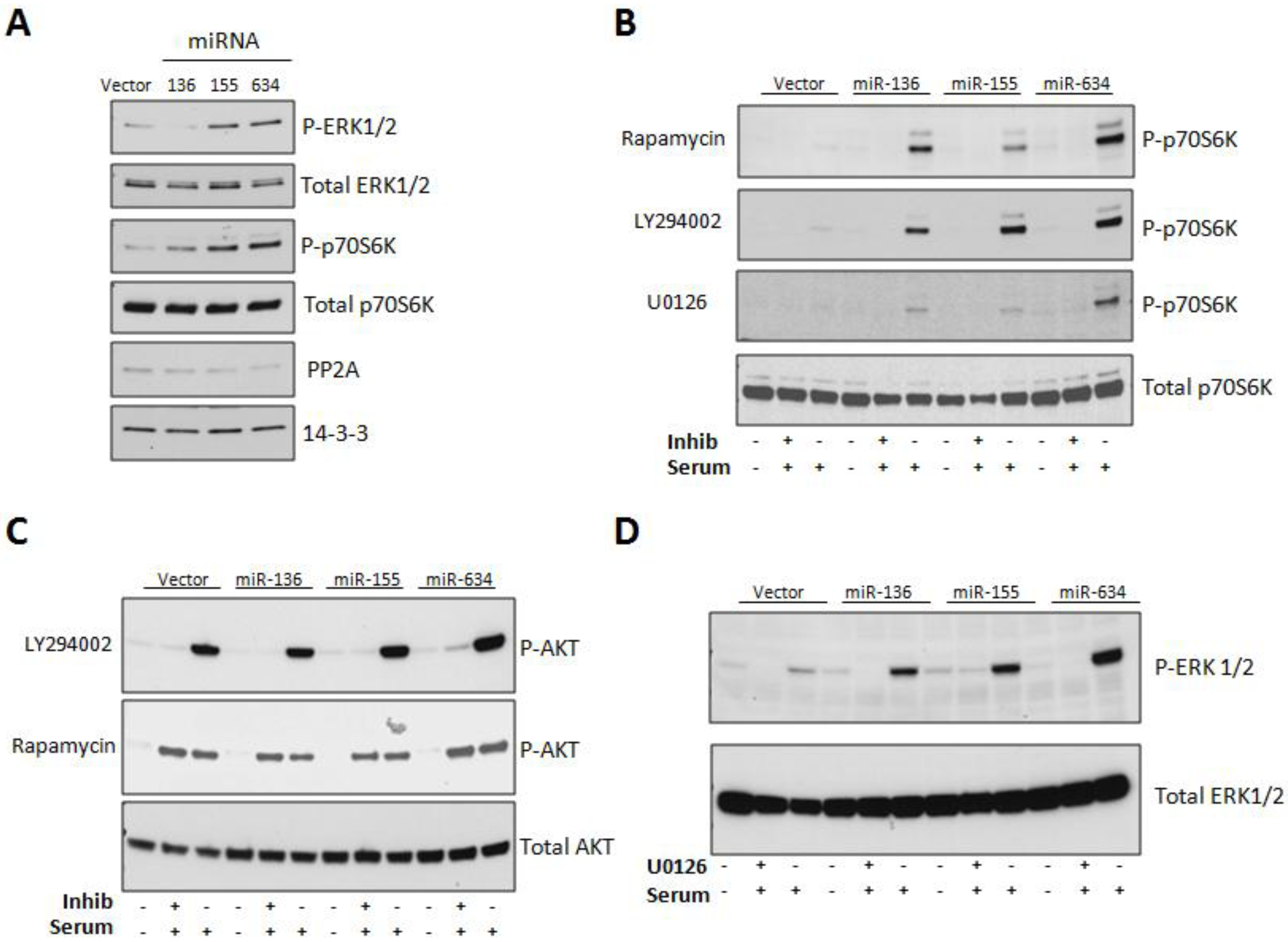
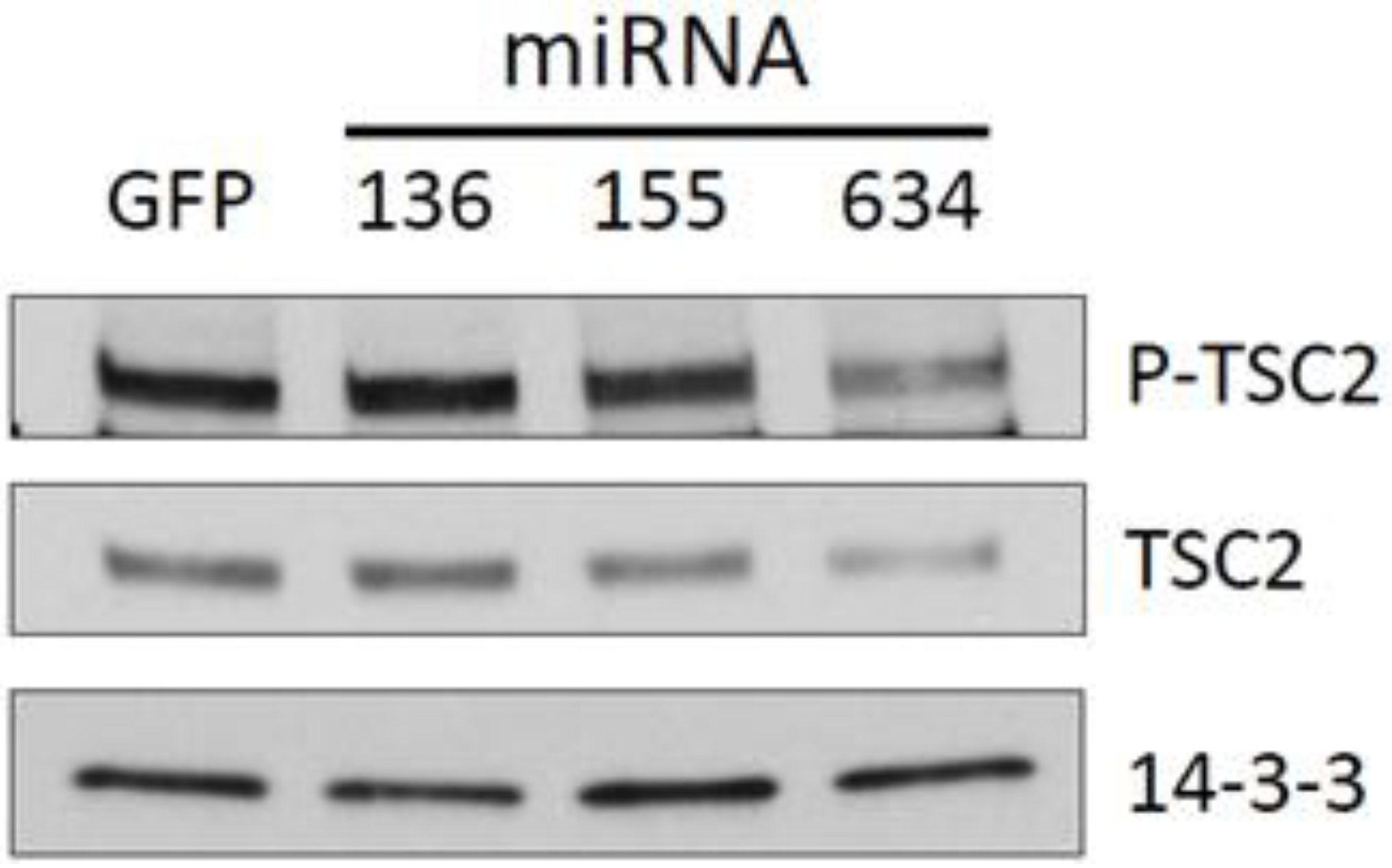
2.3. Expression of miRs-136, -155, and -634 Enhance ERK, AKT, and p70S6K Signaling Pathways
2.4. Fenofibrate Down-Regulates the Expression of CYR61 in LN229 Glioblastoma Cells

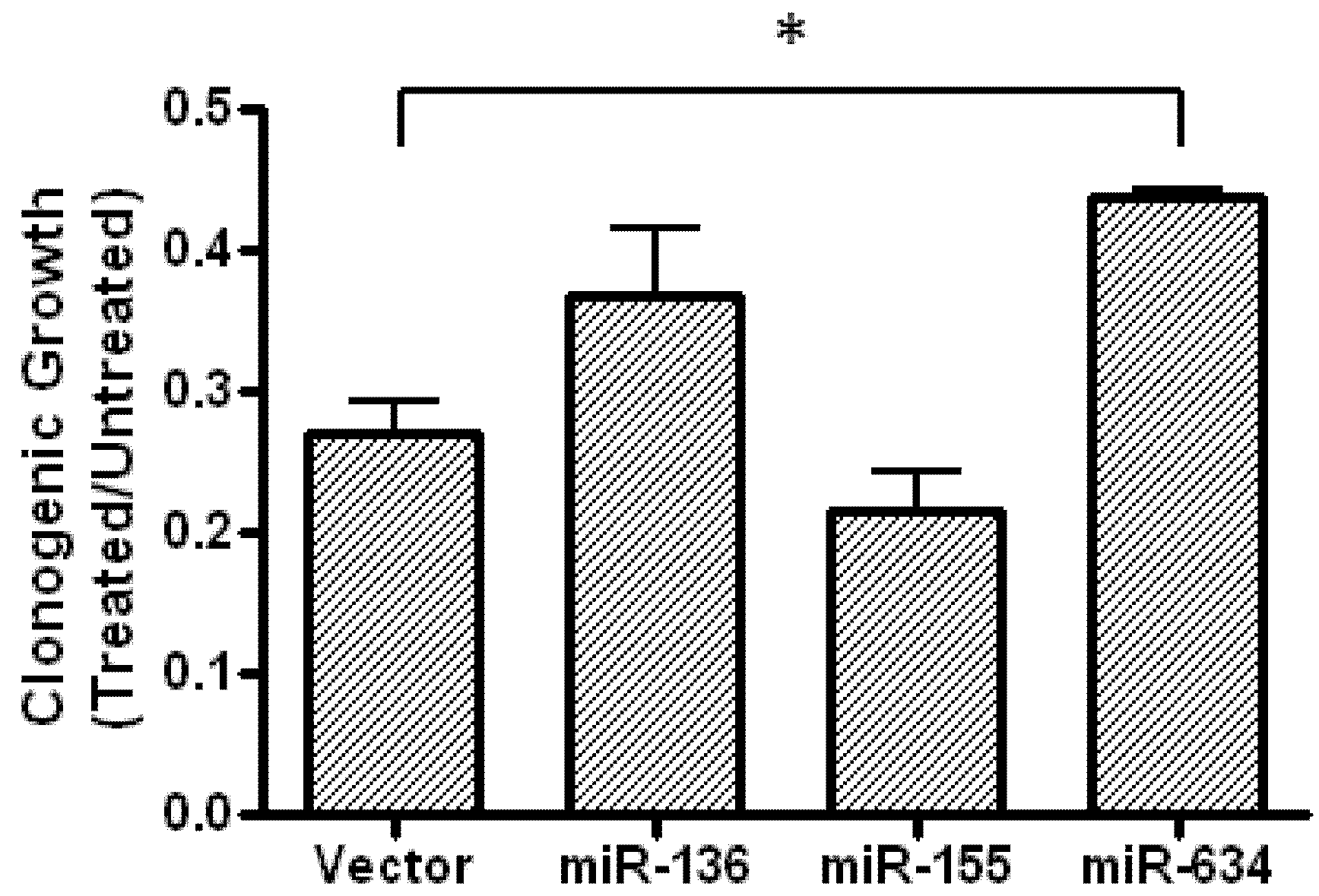
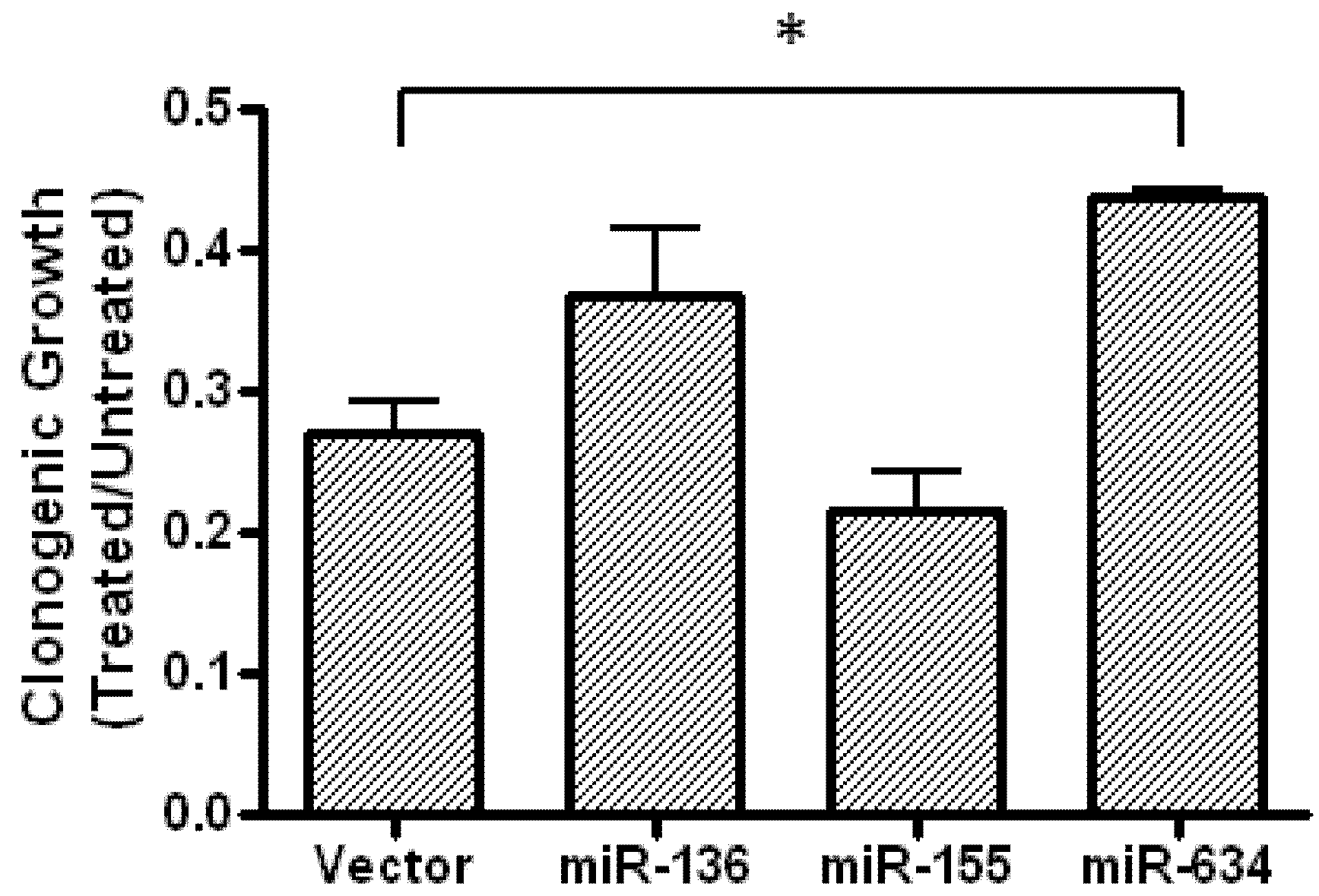
2.5. Expression of miR-634 Partially Protects Cells From Fenofibrate-Treatment in a Clonogenic Assay
3. Experimental Section
3.1. Cell Culture, Transfection, and Reagents
3.2. Western Blot Analysis
3.3. mRNA Microarray Analysis
3.4. Quantitative RT-PCR
3.5. Cloning for miRNA Functional Analysis
3.6. Dual Luciferase Assay
3.7. Cell Growth Assay
3.8. Cell Migration Assay
3.9. Clonogenic Growth Assay
3.10. Statistical analysis
4. Conclusions
Acknowledgments
References
- Nagasawa, D.T.; Chow, F.; Yew, A.; Kim, W.; Cremer, N.; Yang, I. Temozolomide and other potential agents for the treatment of glioblastoma multiforme. Neurosurg. Clin. N Am. 2012, 23, 307–322, ix. [Google Scholar]
- Cavenee, W.K. Accumulation of genetic defects during astrocytoma progression. Cancer 1992, 70, 1788–1793. [Google Scholar] [CrossRef]
- Drukala, J.; Urbanska, K.; Wilk, A.; Grabacka, M.; Wybieralska, E.; Del Valle, L.; Madeja, Z.; Reiss, K. ROS accumulation and IGF-IR inhibition contribute to fenofibrate/PPARalpha—mediated inhibition of glioma cell motility in vitro. Mol. Cancer 2010, 9, 159. [Google Scholar] [CrossRef]
- Furnari, F.B.; Huang, H.J.; Cavenee, W.K. Genetics and malignant progression of human brain tumours. Cancer Surv. 1995, 25, 233–275. [Google Scholar]
- Haas-Kogan, D.; Shalev, N.; Wong, M.; Mills, G.; Yount, G.; Stokoe, D. Protein kinase B (PKB/Akt) activity is elevated in glioblastoma cells due to mutation of the tumor suppressor PTEN/MMAC. Curr. Biol. 1998, 8, 1195–1198. [Google Scholar] [CrossRef]
- Holland, E.C.; Celestino, J.; Dai, C.; Schaefer, L.; Sawaya, R.E.; Fuller, G.N. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat. Genet. 2000, 25, 55–57. [Google Scholar]
- Xie, D.; Yin, D.; Tong, X.; O'Kelly, J.; Mori, A.; Miller, C.; Black, K.; Gui, D.; Said, J.W.; Koeffler, H.P. Cyr61 is overexpressed in gliomas and involved in integrin-linked kinase-mediated Akt and beta-catenin-TCF/Lef signaling pathways. Cancer Res. 2004, 64, 1987–1996. [Google Scholar]
- Lau, L.F. CCN1/CYR61: the very model of a modern matricellular protein. Cell Mol. Life Sci. 2011, 68, 3149–3163. [Google Scholar] [CrossRef]
- Chen, Y.; Du, X.Y. Functional properties and intracellular signaling of CCN1/Cyr61. J. Cell Biochem. 2007, 100, 1337–1345. [Google Scholar] [CrossRef]
- Babic, A.M.; Kireeva, M.L.; Kolesnikova, T.V.; Lau, L.F. CYR61, a product of a growth factor—inducible immediate early gene, promotes angiogenesis and tumor growth. Proc. Natl. Acad. Sci. USA 1998, 95, 6355–6360. [Google Scholar] [CrossRef]
- Chen, N.; Leu, S.J.; Todorovic, V.; Lam, S.C.; Lau, L.F. Identification of a novel integrin alphavbeta3 binding site in CCN1 (CYR61) critical for pro-angiogenic activities in vascular endothelial cells. J. Biol. Chem. 2004, 279, 44166–44176. [Google Scholar]
- Franzen, C.A.; Chen, C.C.; Todorovic, V.; Juric, V.; Monzon, R.I.; Lau, L.F. Matrix protein CCN1 is critical for prostate carcinoma cell proliferation and TRAIL-induced apoptosis. Mol. Cancer Res. 2009, 7, 1045–1055. [Google Scholar] [CrossRef]
- Leask, A. A sticky situation: CCN1 promotes both proliferation and apoptosis of cancer cells. J. Cell. Commun. Signal. 2010, 4, 71–72. [Google Scholar] [CrossRef]
- Haque, I.; Mehta, S.; Majumder, M.; Dhar, K.; De, A.; McGregor, D.; Van Veldhuizen, P.J.; Banerjee, S.K.; Banerjee, S. Cyr61/CCN1 signaling is critical for epithelial-mesenchymal transition and stemness and promotes pancreatic carcinogenesis. Mol. Cancer 2011, 10, 8. [Google Scholar] [CrossRef]
- Holloway, S.E.; Beck, A.W.; Girard, L.; Jaber, M.R.; Barnett, C.C., Jr.; Brekken, R.A.; Fleming, J.B. Increased expression of Cyr61 (CCN1) identified in peritoneal metastases from human pancreatic cancer. J. Am. Coll. Surg. 2005, 200, 371–377. [Google Scholar]
- Jiang, W.G.; Watkins, G.; Fodstad, O.; Douglas-Jones, A.; Mokbel, K.; Mansel, R.E. Differential expression of the CCN family members Cyr61, CTGF and Nov in human breast cancer. Endocr. Relat. Cancer 2004, 11, 781–791. [Google Scholar] [CrossRef]
- Sun, Z.J.; Wang, Y.; Cai, Z.; Chen, P.P.; Tong, X.J.; Xie, D. Involvement of Cyr61 in growth, migration, and metastasis of prostate cancer cells. Br. J. Cancer 2008, 99, 1656–1667. [Google Scholar] [CrossRef]
- Xie, D.; Miller, C.W.; O'Kelly, J.; Nakachi, K.; Sakashita, A.; Said, J.W.; Gornbein, J.; Koeffler, H.P. Breast cancer. Cyr61 is overexpressed, estrogen-inducible, and associated with more advanced disease. J. Biol. Chem. 2001, 276, 14187–14194. [Google Scholar]
- Young, N.; Pearl, D.K.; Van Brocklyn, J.R. Sphingosine-1-phosphate regulates glioblastoma cell invasiveness through the urokinase plasminogen activator system and CCN1/Cyr61. Mol. Cancer Res. 2009, 7, 23–32. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell. Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Duran, R.V.; Hall, M.N. Regulation of TOR by small GTPases. EMBO Rep. 2012, 13, 121–128. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Hummel, R.; Maurer, J.; Haier, J. MicroRNAs in brain tumors : a new diagnostic and therapeutic perspective? Mol. Neurobiol. 2011, 44, 223–234. [Google Scholar] [CrossRef]
- Zhang, Y.; Diao, Z.; Su, L.; Sun, H.; Li, R.; Cui, H.; Hu, Y. MicroRNA-155 contributes to preeclampsia by down-regulating CYR61. Am. J. Obstet. Gynecol. 2012, 202, 466.e1–466.e7. [Google Scholar]
- Yang, Y.; Wu, J.; Guan, H.; Cai, J.; Fang, L.; Li, J.; Li, M. MiR-136 promotes apoptosis of glioma cells by targeting AEG-1 and Bcl-2. FEBS Lett. 2012, 586, 3608–3612. [Google Scholar] [CrossRef]
- Ostling, P.; Leivonen, S.K.; Aakula, A.; Kohonen, P.; Makela, R.; Hagman, Z.; Edsjo, A.; Kangaspeska, S.; Edgren, H.; Nicorici, D.; Bjartell, A.; Ceder, Y.; Perala, M.; Kallioniemi, O. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res. 2011, 71, 1956–1967. [Google Scholar]
- Jin, Y.; Kim, H.P.; Ifedigbo, E.; Lau, L.F.; Choi, A.M. Cyr61 protects against hyperoxia-induced cell death via Akt pathway in pulmonary epithelial cells. Am. J. Respir. Cell Mol. Biol. 2005, 33, 297–302. [Google Scholar] [CrossRef]
- Lin, M.T.; Kuo, I.H.; Chang, C.C.; Chu, C.Y.; Chen, H.Y.; Lin, B.R.; Sureshbabu, M.; Shih, H.J.; Kuo, M.L. Involvement of hypoxia-inducing factor-1alpha-dependent plasminogen activator inhibitor-1 up-regulation in Cyr61/CCN1-induced gastric cancer cell invasion. J. Biol. Chem. 2008, 283, 15807–15815. [Google Scholar]
- Menendez, J.A.; Vellon, L.; Mehmi, I.; Teng, P.K.; Griggs, D.W.; Lupu, R. A novel CYR61-triggered 'CYR61-alphavbeta3 integrin loop' regulates breast cancer cell survival and chemosensitivity through activation of ERK1/ERK2 MAPK signaling pathway. Oncogene 2005, 24, 761–779. [Google Scholar] [CrossRef]
- Faraonio, R.; Salerno, P.; Passaro, F.; Sedia, C.; Iaccio, A.; Bellelli, R.; Nappi, T.C.; Comegna, M.; Romano, S.; Salvatore, G.; Santoro, M.; Cimino, F. A set of miRNAs participates in the cellular senescence program in human diploid fibroblasts. Cell. Death Differ. 2012, 19, 713–721. [Google Scholar] [CrossRef]
- Leontieva, O.V.; Blagosklonny, M.V. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY) 2012, 2, 924–935. [Google Scholar]
- Jozwiak, J.; Jozwiak, S.; Grzela, T.; Lazarczyk, M. Positive and negative regulation of TSC2 activity and its effects on downstream effectors of the mTOR pathway. Neuromolecular. Med. 2005, 7, 287–296. [Google Scholar] [CrossRef]
- Dan, H.C.; Sun, M.; Yang, L.; Feldman, R.I.; Sui, X.M.; Ou, C.C.; Nellist, M.; Yeung, R.S.; Halley, D.J.; Nicosia, S.V.; Pledger, W.J.; Cheng, J.Q. Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J. Biol. Chem. 2002, 277, 35364–35370. [Google Scholar]
- Ma, L.; Teruya-Feldstein, J.; Bonner, P.; Bernardi, R.; Franz, D.N.; Witte, D.; Cordon-Cardo, C.; Pandolfi, P.P. Identification of S664 TSC2 phosphorylation as a marker for extracellular signal-regulated kinase mediated mTOR activation in tuberous sclerosis and human cancer. Cancer Res. 2007, 67, 7106–7112. [Google Scholar]
- Winter, J.N.; Jefferson, L.S.; Kimball, S.R. ERK and Akt signaling pathways function through parallel mechanisms to promote mTORC1 signaling. Am. J. Physiol. Cell. Physiol. 2011, 300, C1172–C1180. [Google Scholar] [CrossRef]
- Grabacka, M.; Placha, W.; Plonka, P.M.; Pajak, S.; Urbanska, K.; Laidler, P.; Slominski, A. Inhibition of melanoma metastases by fenofibrate. Arch. Dermatol. Res. 2004, 296, 54–58. [Google Scholar] [CrossRef]
- Saidi, S.A.; Holland, C.M.; Charnock-Jones, D.S.; Smith, S.K. In vitro and in vivo effects of the PPAR-alpha agonists fenofibrate and retinoic acid in endometrial cancer. Mol. Cancer 2006, 5, 13. [Google Scholar] [CrossRef]
- Urbanska, K.; Pannizzo, P.; Grabacka, M.; Croul, S.; Del Valle, L.; Khalili, K.; Reiss, K. Activation of PPARalpha inhibits IGF-I-mediated growth and survival responses in medulloblastoma cell lines. Int. J. Cancer 2008, 123, 1015–1024. [Google Scholar] [CrossRef]
- Wilk, A.; Urbanska, K.; Grabacka, M.; Mullinax, J.; Marcinkiewicz, C.; Impastato, D.; Estrada, J.J.; Reiss, K. Fenofibrate-induced nuclear translocation of FoxO3A triggers Bim-mediated apoptosis in glioblastoma cells in vitro. Cell Cycle 2012, 11, 2660–2671. [Google Scholar] [CrossRef]
- Eletto, D.; Russo, G.; Passiatore, G.; Del Valle, L.; Giordano, A.; Khalili, K.; Gualco, E.; Peruzzi, F. Inhibition of SNAP25 expression by HIV-1 Tat involves the activity of mir-128a. J. Cell Physiol. 2008, 216, 764–770. [Google Scholar] [CrossRef]
- Pacifici, M.; Delbue, S.; Ferrante, P.; Jeansonne, D.; Kadri, F.; Nelson, S.; Velasco-Gonzalez, C.; Zabaleta, J.; Peruzzi, F. Cerebrospinal fluid miRNA profile in HIV-encephalitis. J. Cell. Physiol. 2012, 228, 1070–1075. [Google Scholar]
- Rom, S.; Rom, I.; Passiatore, G.; Pacifici, M.; Radhakrishnan, S.; Del Valle, L.; Pina-Oviedo, S.; Khalili, K.; Eletto, D.; Peruzzi, F. CCL8/MCP-2 is a target for mir-146a in HIV-1-infected human microglial cells. Faseb. J. 2010, 24, 2292–2300. [Google Scholar]
- Li, C.L.; Nie, H.; Wang, M.; Su, L.P.; Li, J.F.; Yu, Y.Y.; Yan, M.; Qu, Q.L.; Zhu, Z.G.; Liu, B.Y. microRNA-155 is downregulated in gastric cancer cells and involved in cell metastasis. Oncol. Rep. 2012, 27, 1960–1966. [Google Scholar]
- Liu, J.; van Mil, A.; Aguor, E.N.; Siddiqi, S.; Vrijsen, K.; Jaksani, S.; Metz, C.; Zhao, J.; Strijkers, G.J.; Doevendans, P.A.; Sluijter, J.P. MiR-155 inhibits cell migration of human cardiomyocyte progenitor cells (hCMPCs) via targeting of MMP-16. J. Cell. Mol. Med. 2012, 16, 2379–2386. [Google Scholar] [CrossRef]
- Dai, Y.; Qiu, Z.; Diao, Z.; Shen, L.; Xue, P.; Sun, H.; Hu, Y. MicroRNA-155 inhibits proliferation and migration of human extravillous trophoblast derived HTR-8/SVneo cells via down-regulating cyclin D1. Placenta 2012, 33, 824–829. [Google Scholar] [CrossRef]
- Lee, D.Y.; Jeyapalan, Z.; Fang, L.; Yang, J.; Zhang, Y.; Yee, A.Y.; Li, M.; Du, W.W.; Shatseva, T.; Yang, B.B. Expression of versican 3'-untranslated region modulates endogenous microRNA functions. PLoS One 2010, 5, e13599. [Google Scholar]
- Lee, H.Y.; Chung, J.W.; Youn, S.W.; Kim, J.Y.; Park, K.W.; Koo, B.K.; Oh, B.H.; Park, Y.B.; Chaqour, B.; Walsh, K.; Kim, H.S. Forkhead transcription factor FOXO3a is a negative regulator of angiogenic immediate early gene CYR61, leading to inhibition of vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ. Res. 2007, 100, 372–380. [Google Scholar] [CrossRef]
- Bostner, J.; Karlsson, E.; Pandiyan, M.J.; Westman, H.; Skoog, L.; Fornander, T.; Nordenskjold, B.; Stal, O. Activation of Akt, mTOR, and the estrogen receptor as a signature to predict tamoxifen treatment benefit. Breast Cancer Res. Treat. 2013, 137, 397–406. [Google Scholar]
- Grunwald, V.; DeGraffenried, L.; Russel, D.; Friedrichs, W.E.; Ray, R.B.; Hidalgo, M. Inhibitors of mTOR reverse doxorubicin resistance conferred by PTEN status in prostate cancer cells. Cancer Res. 2002, 62, 6141–6145. [Google Scholar]
- Wendel, H.G.; De Stanchina, E.; Fridman, J.S.; Malina, A.; Ray, S.; Kogan, S.; Cordon-Cardo, C.; Pelletier, J.; Lowe, S.W. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 2004, 428, 332–337. [Google Scholar]
- Yoon, H.; Min, J.K.; Lee, J.W.; Kim, D.G.; Hong, H.J. Acquisition of chemoresistance in intrahepatic cholangiocarcinoma cells by activation of AKT and extracellular signal-regulated kinase (ERK)1/2. Biochem. Biophys. Res. Commun. 2011, 405, 333–337. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, F.; Zheng, C.; Sun, S.; Jiang, Y. Knockdown of clusterin sensitizes pancreatic cancer cells to gemcitabine chemotherapy by ERK1/2 inactivation. J. Exp. Clin. Cancer Res. 2012, 31, 73. [Google Scholar] [CrossRef]
Supplemental Materials


© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jeansonne, D.; Pacifici, M.; Lassak, A.; Reiss, K.; Russo, G.; Zabaleta, J.; Peruzzi, F. Differential Effects of MicroRNAs on Glioblastoma Growth and Migration. Genes 2013, 4, 46-64. https://doi.org/10.3390/genes4010046
Jeansonne D, Pacifici M, Lassak A, Reiss K, Russo G, Zabaleta J, Peruzzi F. Differential Effects of MicroRNAs on Glioblastoma Growth and Migration. Genes. 2013; 4(1):46-64. https://doi.org/10.3390/genes4010046
Chicago/Turabian StyleJeansonne, Duane, Marco Pacifici, Adam Lassak, Krzysztof Reiss, Giuseppe Russo, Jovanny Zabaleta, and Francesca Peruzzi. 2013. "Differential Effects of MicroRNAs on Glioblastoma Growth and Migration" Genes 4, no. 1: 46-64. https://doi.org/10.3390/genes4010046