The Replication Fork: Understanding the Eukaryotic Replication Machinery and the Challenges to Genome Duplication


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
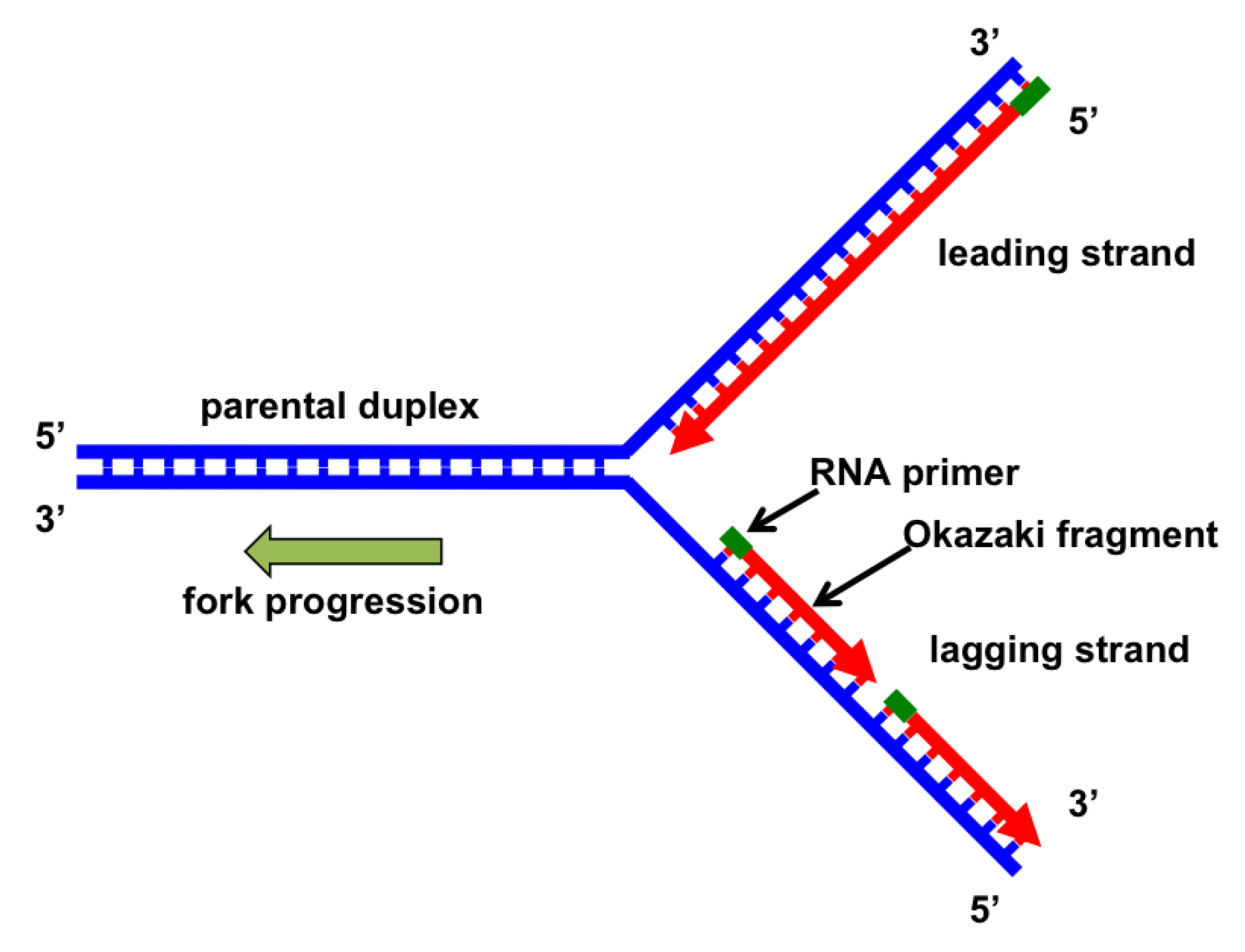
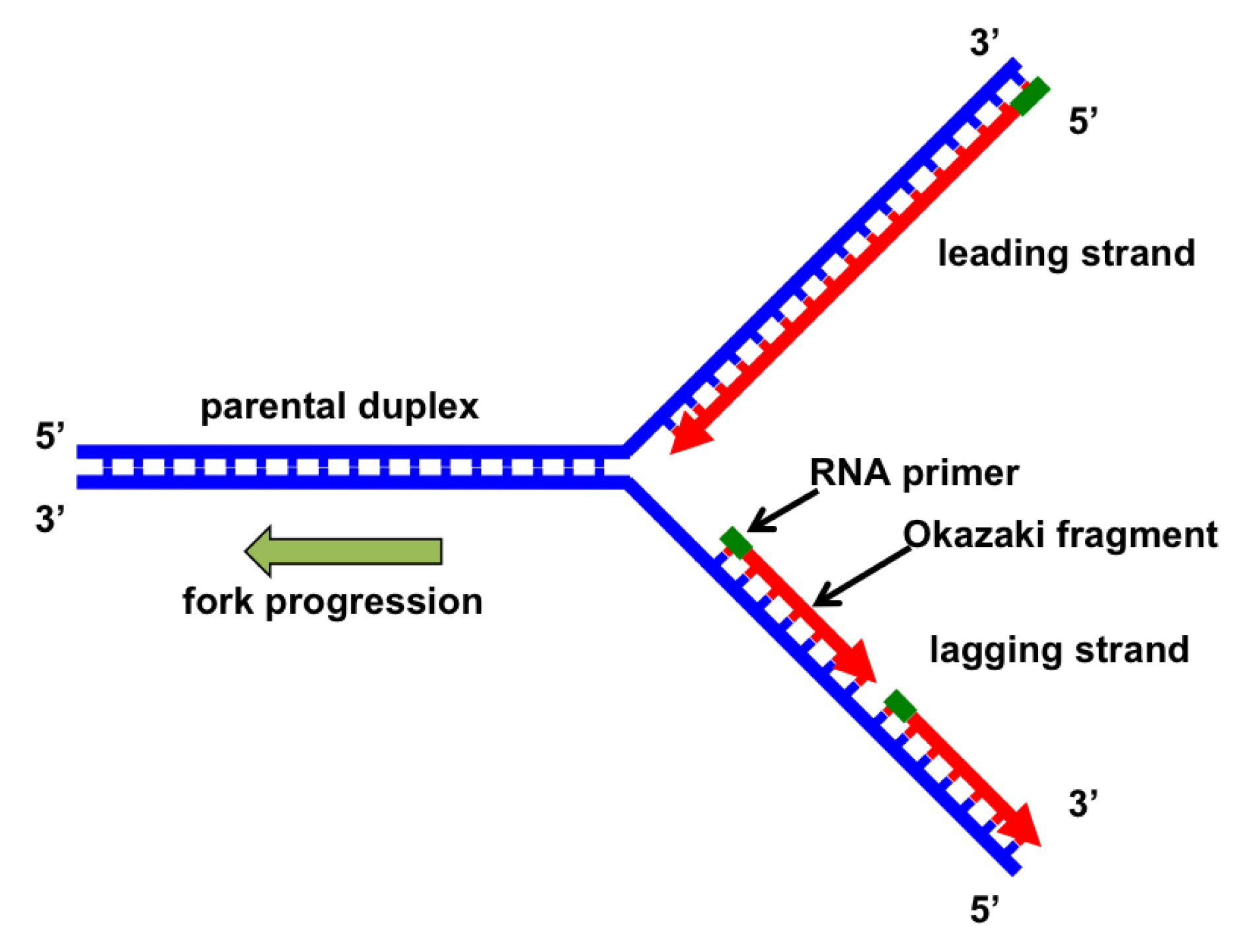
:1. Introduction: Basics of DNA Replication
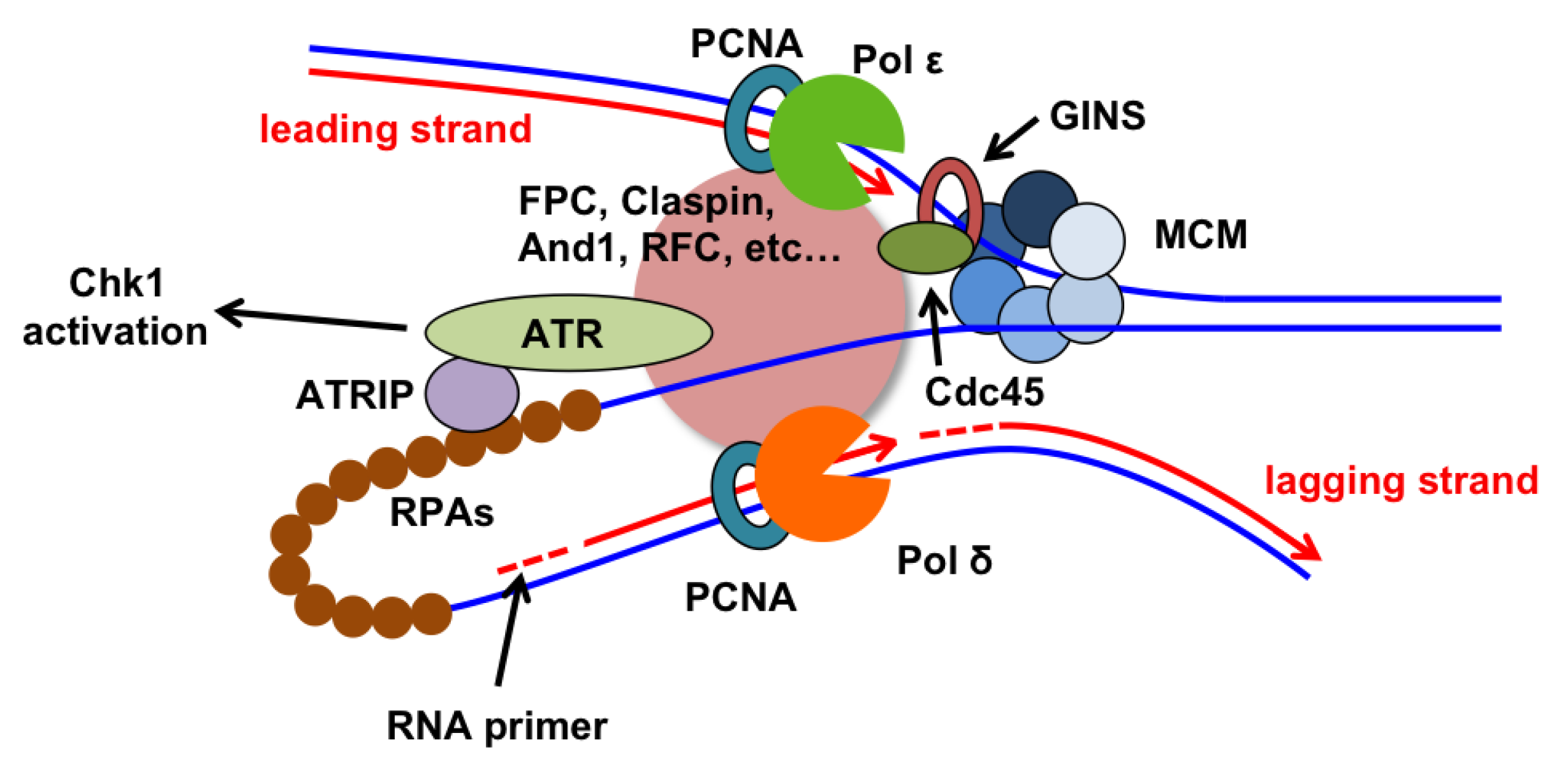
2. DNA Polymerases and Helicases
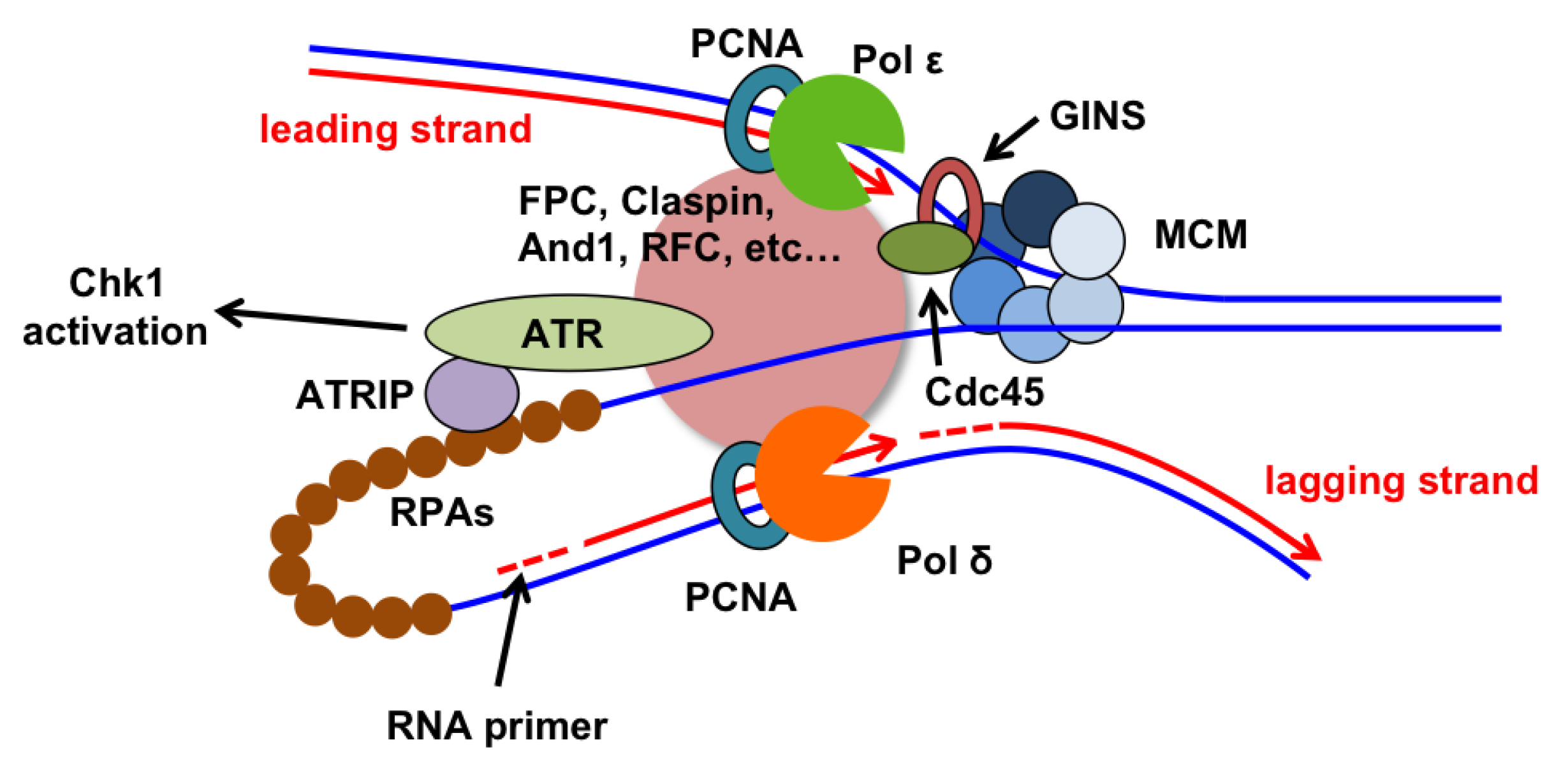
2.1. Replicative DNA Polymerases: Further Subdivision of Labor
2.2. Helicases Unwind DNA for Replication
3. Control of DNA Replication at the Replication Fork
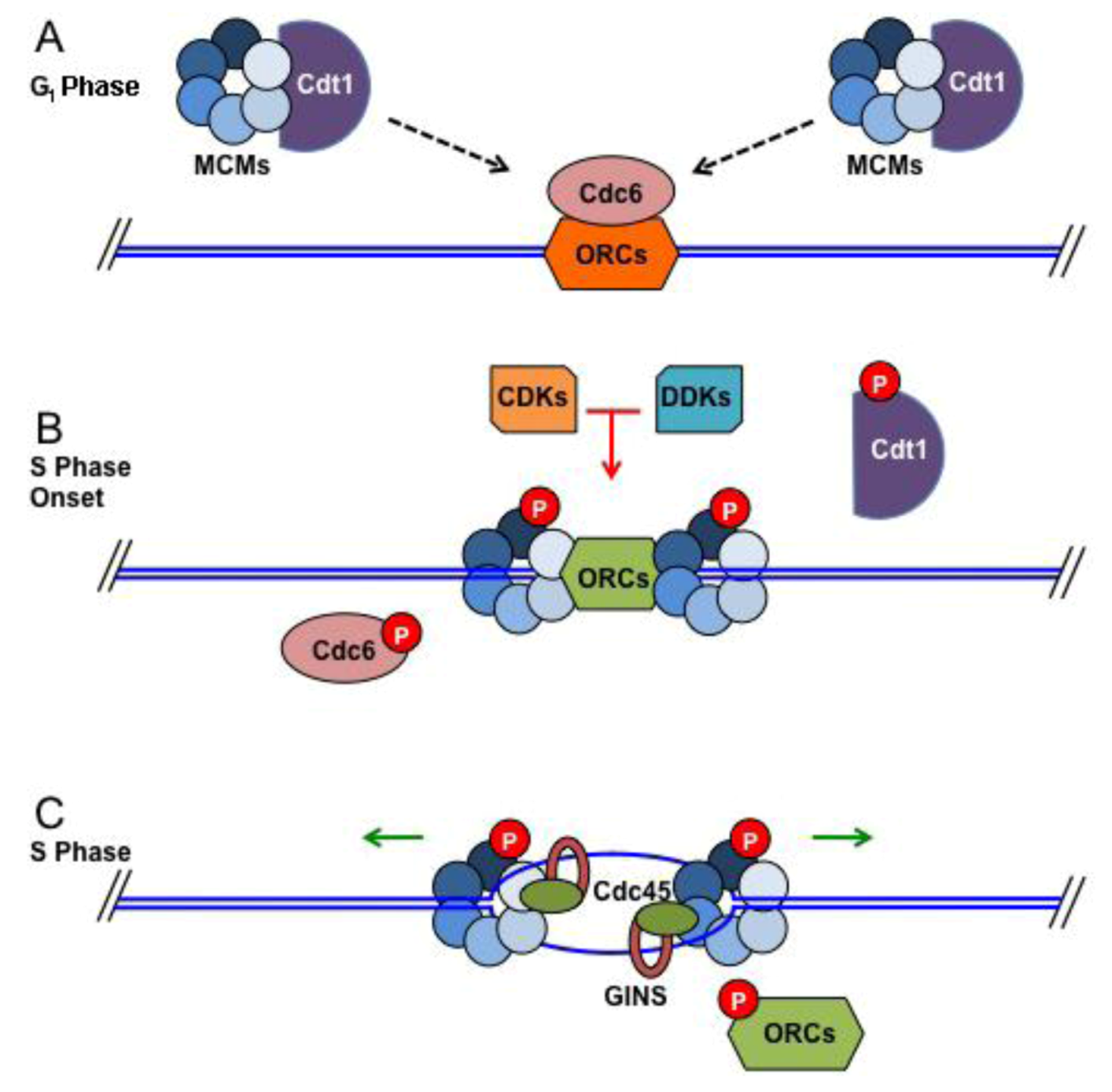
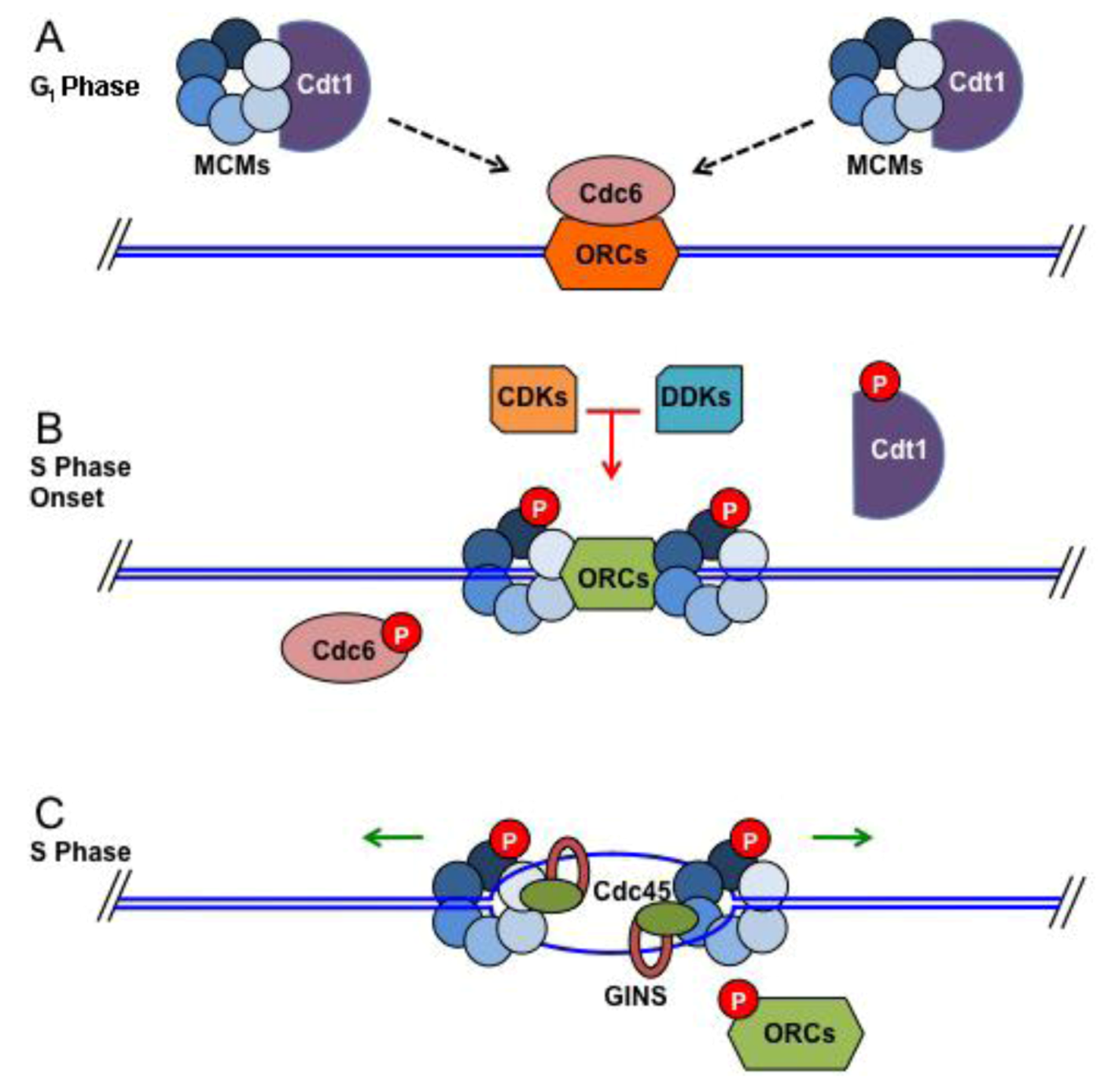
3.1. Replication Initiation at Origins
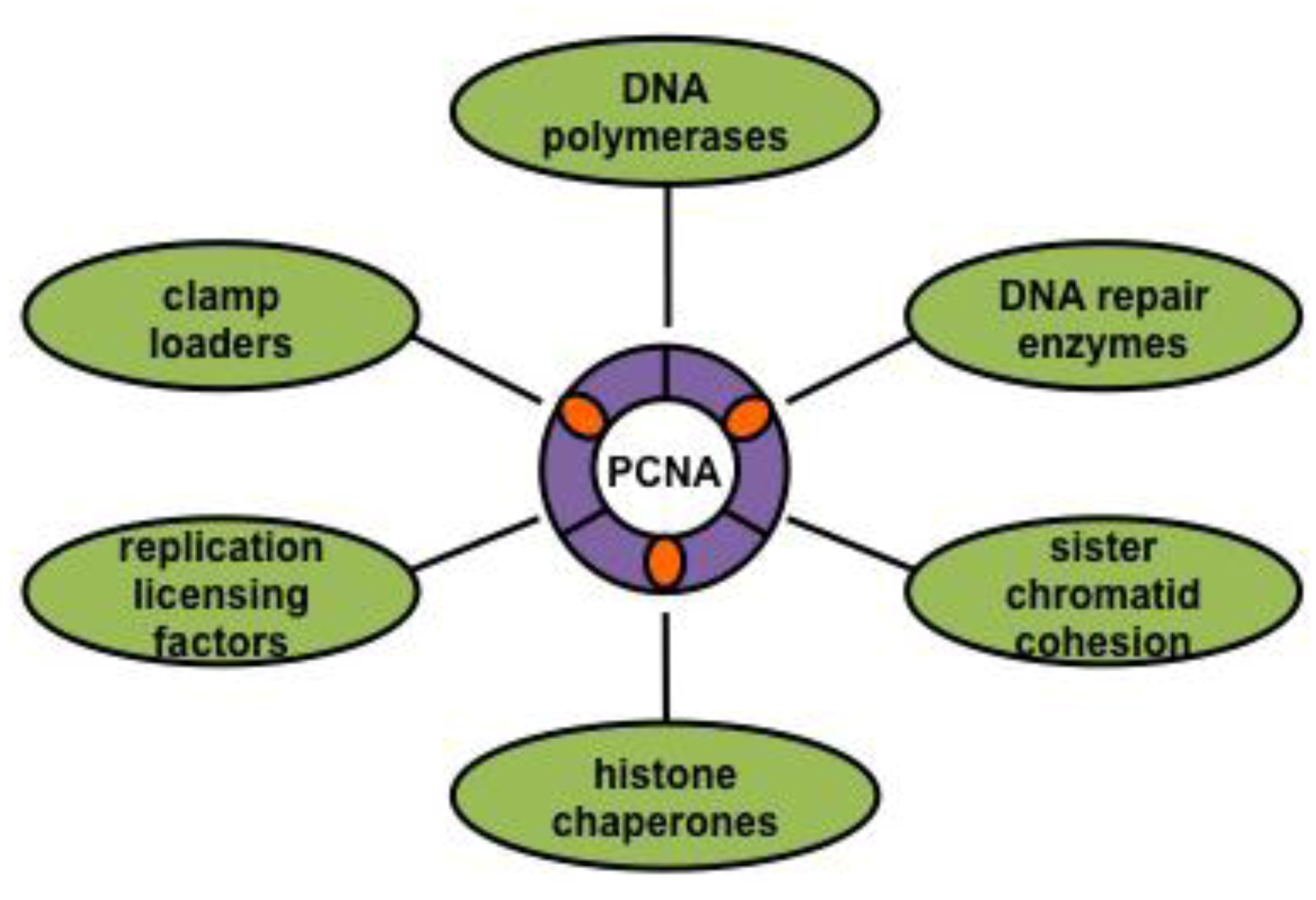
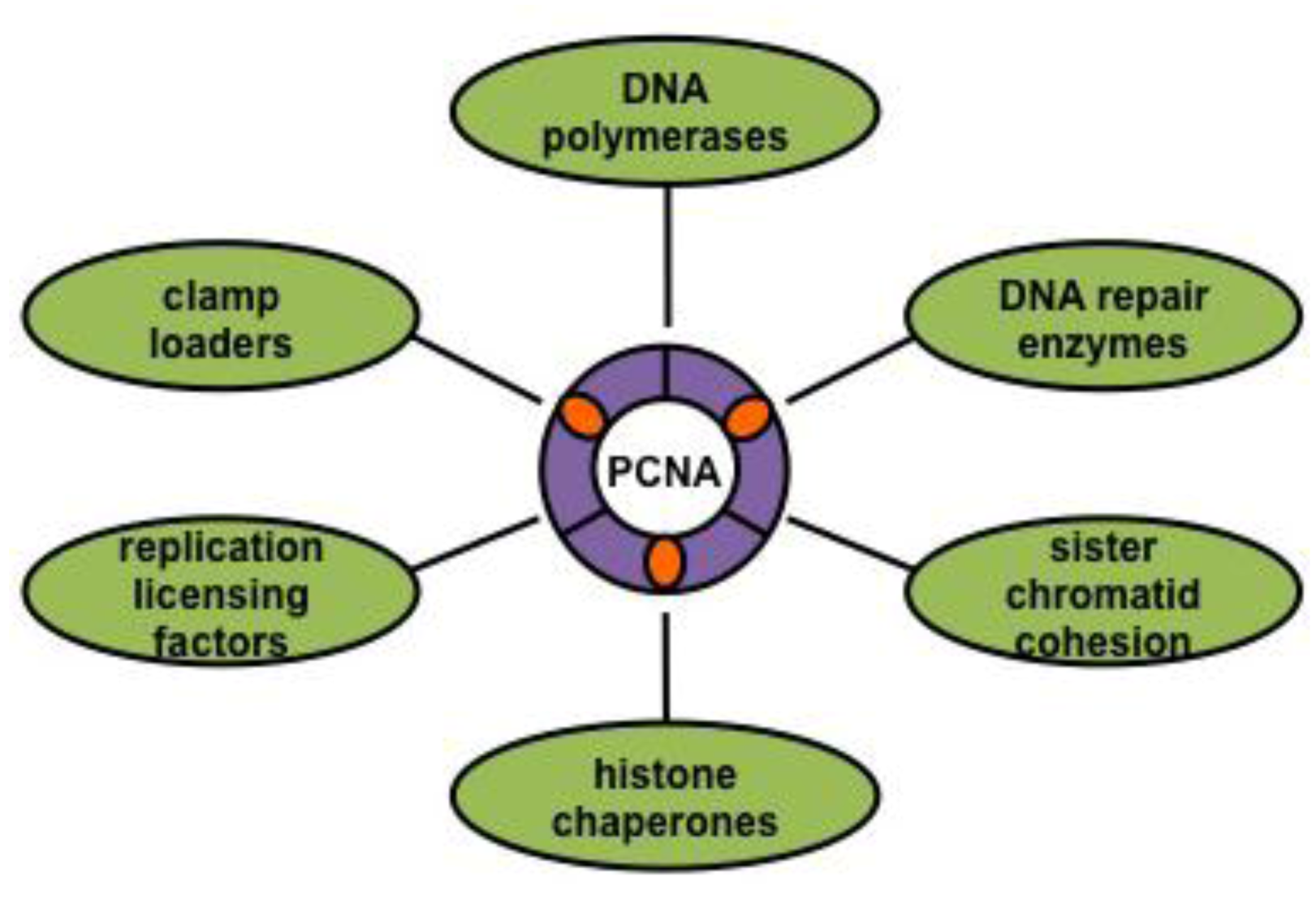
3.2. The DNA Sliding Clamp: PCNA
3.3. Clamp Loaders
3.4. Okazaki Fragment Maturation
3.5. Linking Helicase and Polymerase Functions
3.6. Replication Checkpoint Proteins
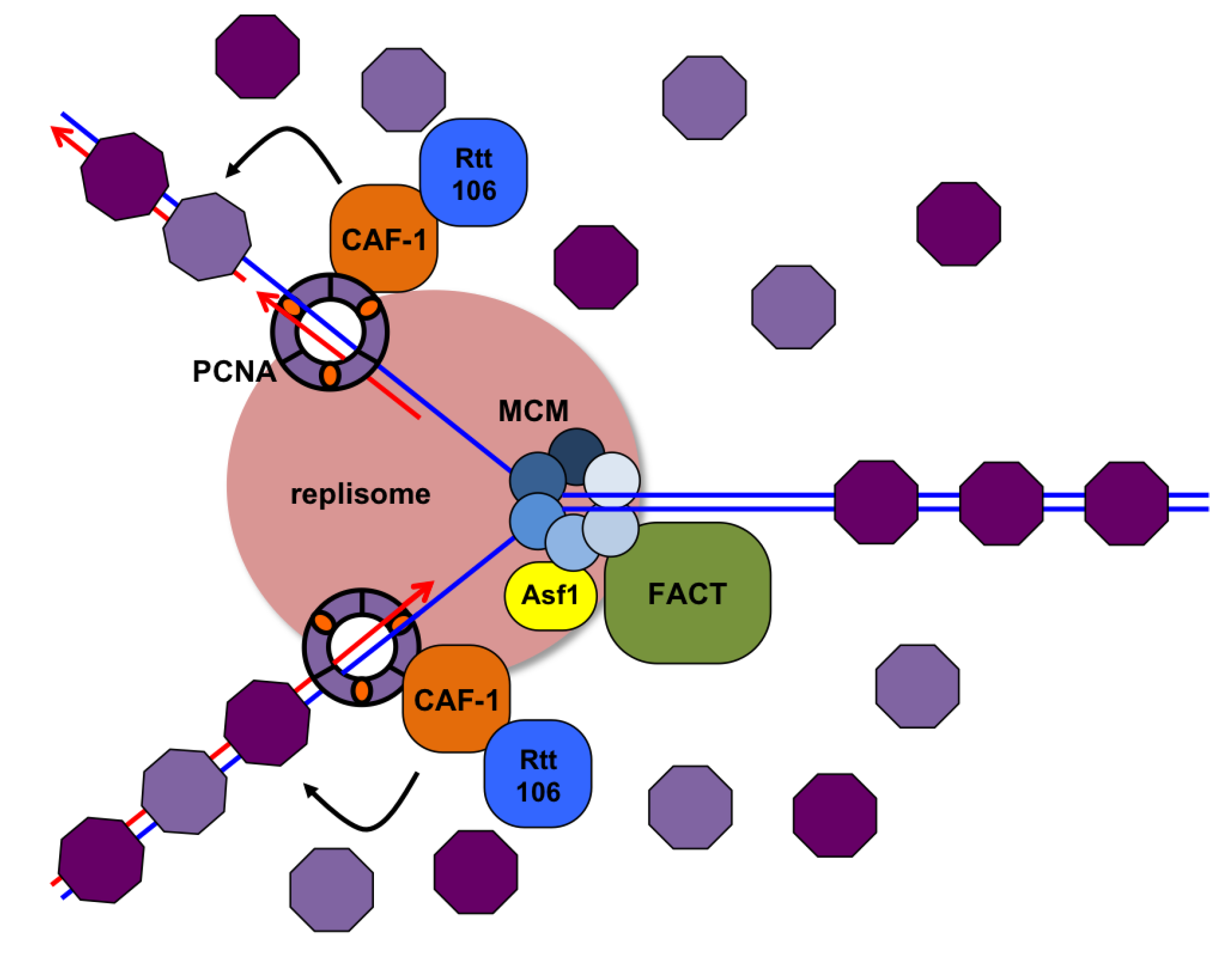
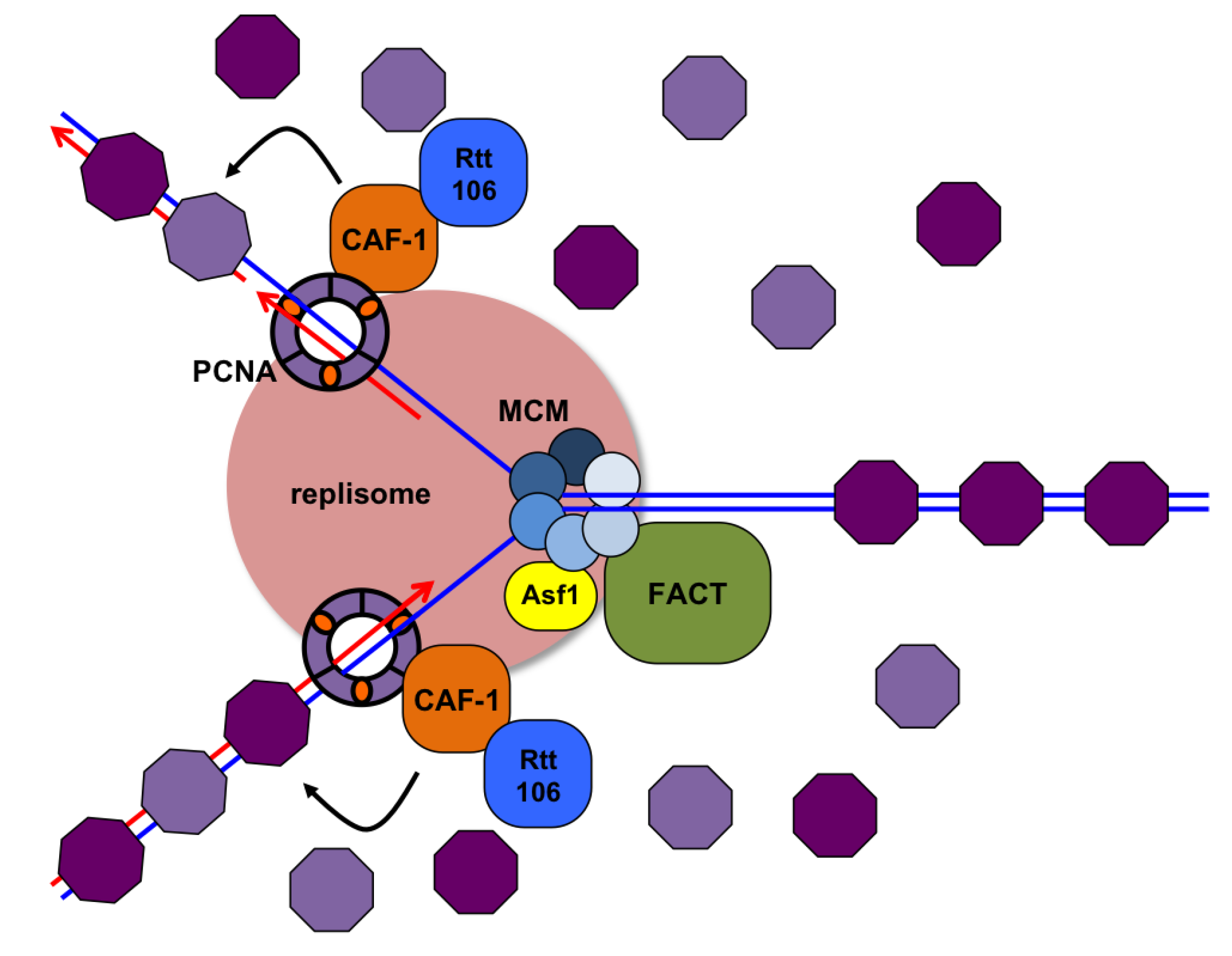
3.7. Replication through Nucleosomes
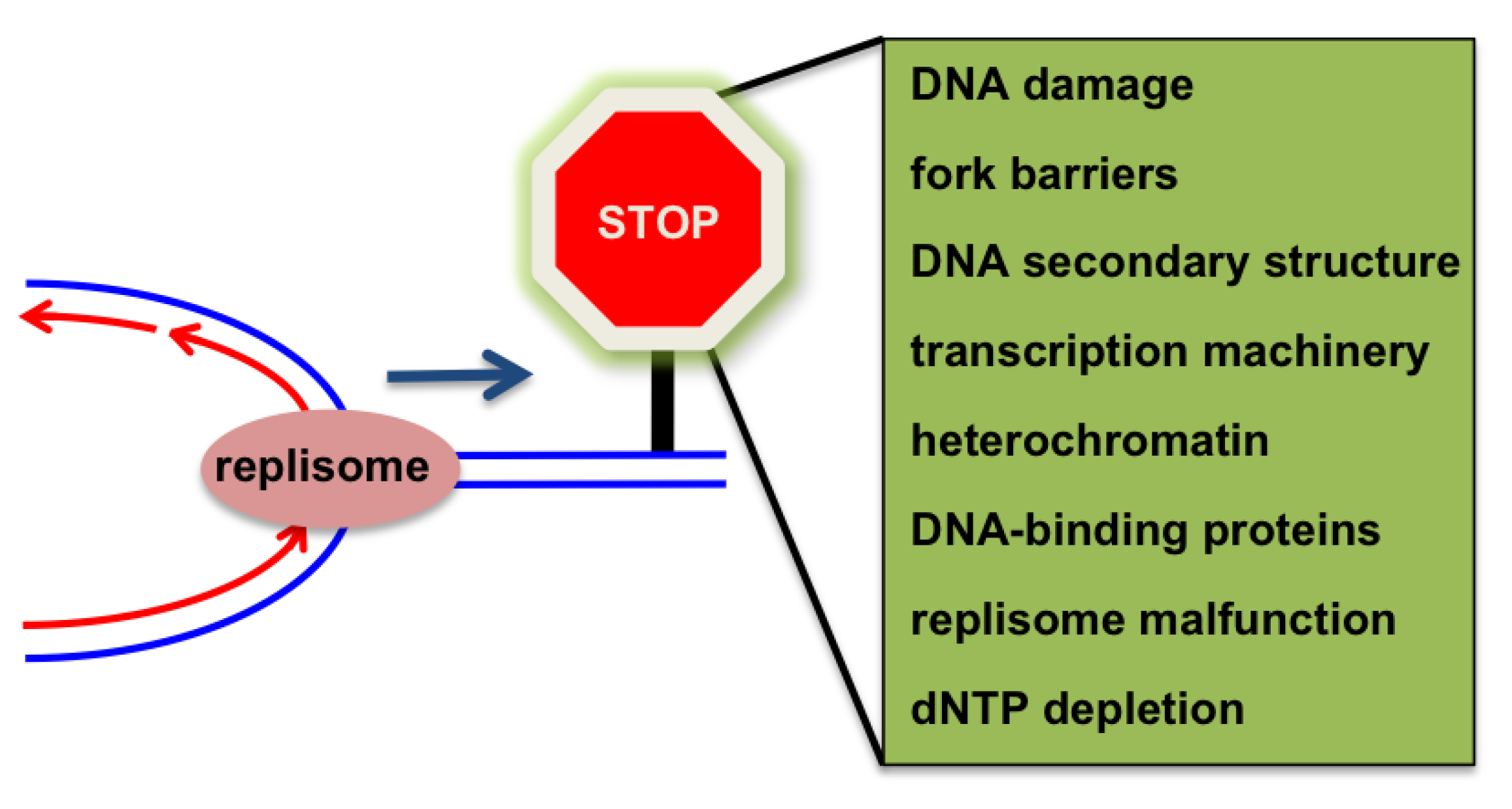
4. Challenges to Replication
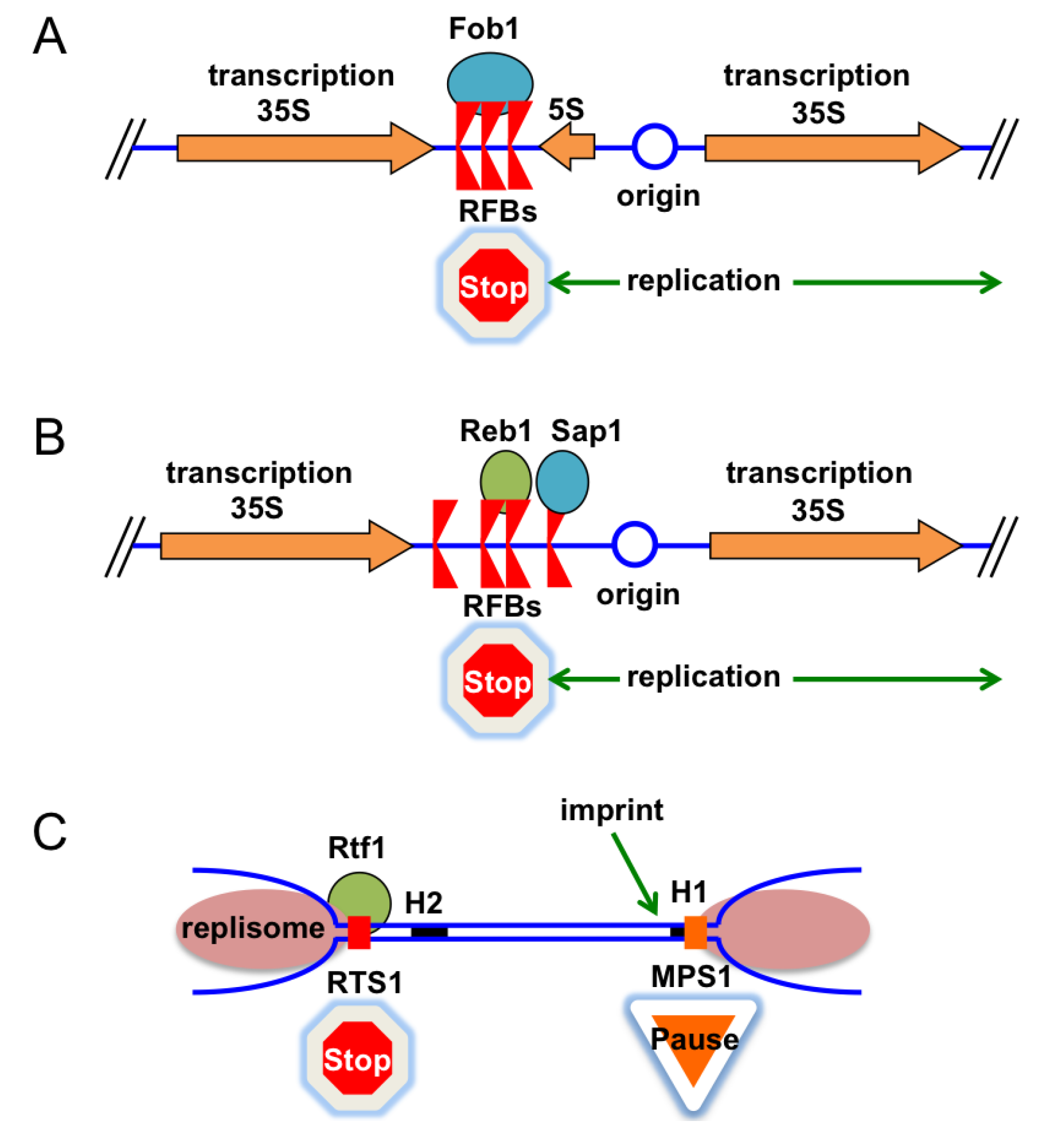
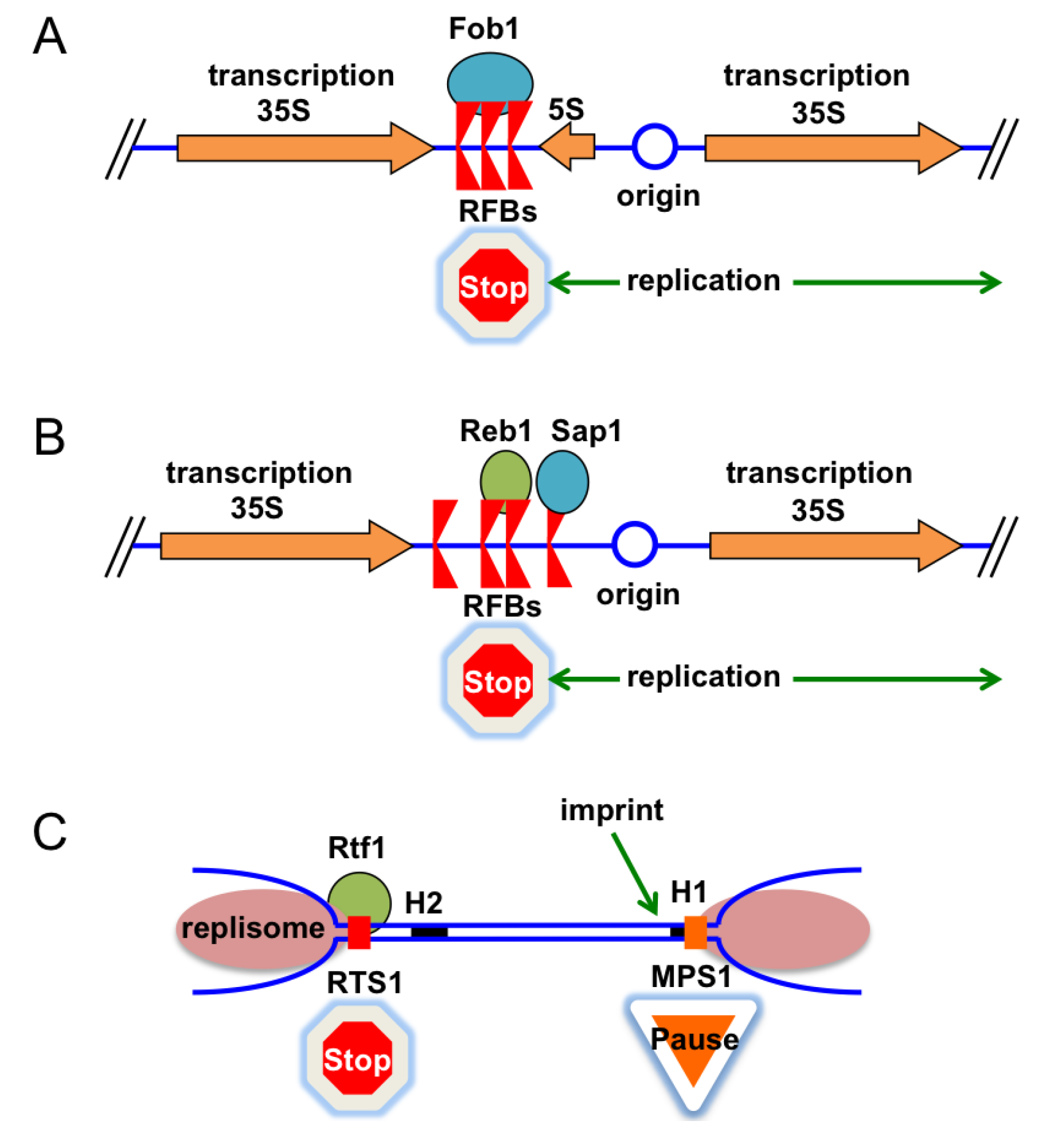
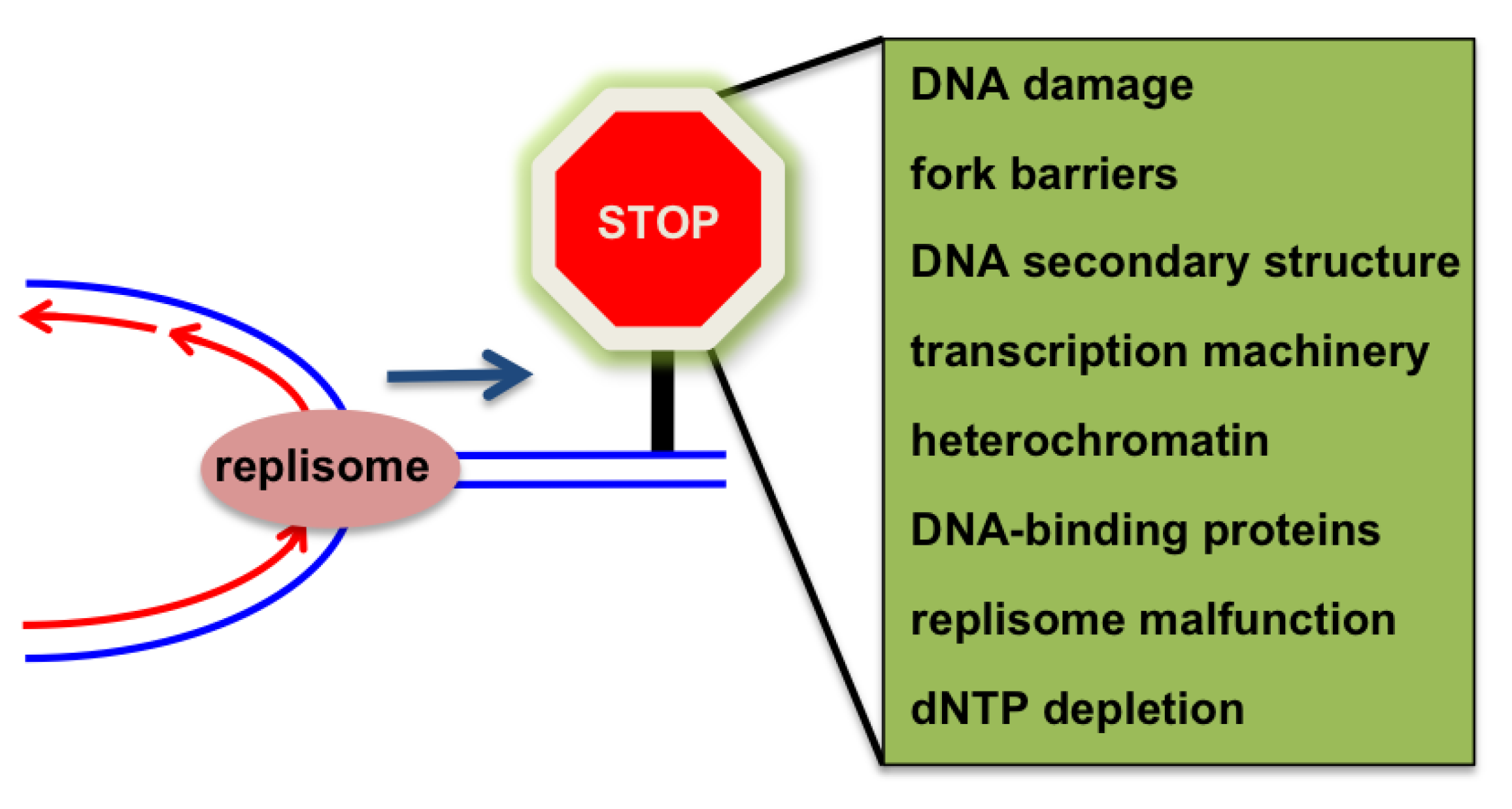
4.1. Replication Fork Barriers
4.2. Fork Barriers at rDNA Arrays in Eukaryotic Cells
4.3. Replication Termination at the Fission Yeast Mating-Type Locus
4.4. Replication Slow Zones
4.5. Fragile Sites
5. Conclusions and Closing Remarks
Acknowledgments
References
- Meselson, M.; Stahl, F.W. The replication of DNA in Escherichia coli. Proc. Natl. Acad. Sci. USA 1958, 44, 671–682. [Google Scholar] [CrossRef]
- Bessman, M.J.; Lehman, I.R.; Simms, E.S.; Kornberg, A. Enzymatic synthesis of deoxyribonucleic acid. II. General properties of the reaction. J. Biol. Chem. 1958, 233, 171–177. [Google Scholar]
- Lehman, I.R.; Bessman, M.J.; Simms, E.S.; Kornberg, A. Enzymatic synthesis of deoxyribonucleic acid. I. Preparation of substrates and partial purification of an enzyme from Escherichia coli. J. Biol. Chem. 1958, 233, 163–170. [Google Scholar]
- Okazaki, R.; Okazaki, T.; Sakabe, K.; Sugimoto, K.; Sugino, A. Mechanism of DNA chain growth. I. Possible discontinuity and unusual secondary structure of newly synthesized chains. Proc. Natl. Acad. Sci. USA 1968, 59, 598–605. [Google Scholar] [CrossRef]
- Wold, M.S.; Kelly, T. Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc. Natl. Acad. Sci. USA 1988, 85, 2523–2527. [Google Scholar] [CrossRef]
- Alani, E.; Thresher, R.; Griffith, J.D.; Kolodner, R.D. Characterization of DNA-binding and strand-exchange stimulation properties of y-RPA, a yeast single-strand-DNA-binding protein. J. Mol. Biol. 1992, 227, 54–71. [Google Scholar] [CrossRef]
- Siegal, G.; Turchi, J.J.; Myers, T.W.; Bambara, R.A. A 5' to 3' exonuclease functionally interacts with calf DNA polymerase ε. Proc. Natl. Acad. Sci. USA 1992, 89, 9377–9381. [Google Scholar]
- Goulian, M.; Richards, S.H.; Heard, C.J.; Bigsby, B.M. Discontinuous DNA synthesis by purified mammalian proteins. J. Biol. Chem. 1990, 265, 18461–18471. [Google Scholar]
- Waga, S.; Bauer, G.; Stillman, B. Reconstitution of complete SV40 DNA replication with purified replication factors. J. Biol. Chem. 1994, 269, 10923–10934. [Google Scholar]
- Budd, M.; Campbell, J.L. Temperature-sensitive mutations in the yeast DNA polymerase I gene. Proc. Natl. Acad. Sci. USA 1987, 84, 2838–2842. [Google Scholar] [CrossRef]
- Sitney, K.C.; Budd, M.E.; Campbell, J.L. DNA polymerase III, a second essential DNA polymerase, is encoded by the S. cerevisiae CDC2 gene. Cell 1989, 56, 599–605. [Google Scholar] [CrossRef]
- Boulet, A.; Simon, M.; Faye, G.; Bauer, G.A.; Burgers, P.M. Structure and function of the Saccharomyces cerevisiae CDC2 gene encoding the large subunit of DNA polymerase III. EMBO J. 1989, 8, 1849–1854. [Google Scholar]
- Morrison, A.; Araki, H.; Clark, A.B.; Hamatake, R.K.; Sugino, A. A third essential DNA polymerase in S. cerevisiae. Cell 1990, 62, 1143–1151. [Google Scholar] [CrossRef]
- Fisher, P.A.; Wang, T.S.; Korn, D. Enzymological characterization of DNA polymerase α. Basic catalytic properties processivity, and gap utilization of the homogeneous enzyme from human KB cells. J. Biol. Chem. 1979, 254, 6128–6137. [Google Scholar]
- Nethanel, T.; Zlotkin, T.; Kaufmann, G. Assembly of simian virus 40 Okazaki pieces from DNA primers is reversibly arrested by ATP depletion. J. Virol. 1992, 66, 6634–6640. [Google Scholar]
- Waga, S.; Stillman, B. Anatomy of a DNA replication fork revealed by reconstitution of SV40 DNA replication in vitro. Nature 1994, 369, 207–212. [Google Scholar] [CrossRef]
- Tsurimoto, T.; Stillman, B. Replication factors required for SV40 DNA replication in vitro. II. Switching of DNA polymerase α and δ during initiation of leading and lagging strand synthesis. J. Biol. Chem. 1991, 266, 1961–1968. [Google Scholar]
- Tsurimoto, T.; Melendy, T.; Stillman, B. Sequential initiation of lagging and leading strand synthesis by two different polymerase complexes at the SV40 DNA replication origin. Nature 1990, 346, 534–539. [Google Scholar] [CrossRef]
- Pursell, Z.F.; Isoz, I.; Lundström, E.-B.; Johansson, E.; Kunkel, T.A. Yeast DNA polymerase ε participates in leading-strand DNA replication. Science 2007, 317, 127–130. [Google Scholar]
- Nick McElhinny, S.A.; Gordenin, D.A.; Stith, C.M.; Burgers, P.M.; Kunkel, T.A. Division of labor at the eukaryotic replication fork. Mol. Cell 2008, 30, 137–144. [Google Scholar] [CrossRef]
- Larrea, A.A.; Lujan, S.A.; Nick McElhinny, S.A.; Mieczkowski, P.A.; Resnick, M.A.; Gordenin, D.A.; Kunkel, T.A. Genome-wide model for the normal eukaryotic DNA replication fork. Proc. Natl. Acad. Sci. USA 2010, 107, 17674–17679. [Google Scholar]
- Neuwald, A.F.; Aravind, L.; Spouge, J.L.; Koonin, E.V. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999, 9, 27–43. [Google Scholar]
- Labib, K.; Tercero, J.A.; Diffley, J.F.X. Uninterrupted MCM2-7 function required for DNA replication fork progression. Science 2000, 288, 1643–1647. [Google Scholar] [CrossRef]
- Pacek, M.; Walter, J.C. A requirement for MCM7 and Cdc45 in chromosome unwinding during eukaryotic DNA replication. EMBO J. 2004, 23, 3667–3676. [Google Scholar] [CrossRef]
- Aparicio, O.M.; Weinstein, D.M.; Bell, S.P. Components and Dynamics of DNA Replication Complexes in S. cerevisiae: Redistribution of MCM Proteins and Cdc45p during S Phase. Cell 1997, 91, 59–69. [Google Scholar] [CrossRef]
- Tye, B.-K. The MCM2-3-5 proteins: Are they replication licensing factors? Trends Cell Biol. 1994, 4, 160–166. [Google Scholar] [CrossRef]
- Chong, J.P.J.; Hayashi, M.K.; Simon, M.N.; Xu, R.-M.; Stillman, B. A double-hexamer archaeal minichromosome maintenance protein is an ATP-dependent DNA helicase. Proc. Natl. Acad. Sci. USA 2000, 97, 1530–1535. [Google Scholar]
- Ishimi, Y. A DNA helicase activity is associated with an MCM4, -6, and -7 protein complex. J. Biol. Chem. 1997, 272, 24508–24513. [Google Scholar] [CrossRef]
- Lee, J.K.; Hurwitz, J. Isolation and characterization of various complexes of the minichromosome maintenance proteins of Schizosaccharomyces pombe. J. Biol. Chem. 2000, 275, 18871–18878. [Google Scholar] [CrossRef]
- Nishitani, H.; Lygerou, Z. Control of DNA replication licensing in a cell cycle. Genes Cells 2002, 7, 523–534. [Google Scholar] [CrossRef]
- Lei, M.; Tye, B. Initiating DNA synthesis: From recruiting to activating the MCM complex. J. Cell Sci. 2001, 114, 1447–1454. [Google Scholar]
- Méchali, M. Eukaryotic DNA replication origins: Many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 2010, 11, 728–738. [Google Scholar] [CrossRef]
- Coleman, T.R.; Carpenter, P.B.; Dunphy, W.G. The Xenopus Cdc6 protein is essential for the initiation of a single round of DNA replication in cell-free extracts. Cell 1996, 87, 53–63. [Google Scholar] [CrossRef]
- Tanaka, T.; Knapp, D.; Nasmyth, K. Loading of an Mcm protein onto DNA replication origins is regulated by Cdc6p and CDKs. Cell 1997, 90, 649–660. [Google Scholar] [CrossRef]
- Ogawa, Y.; Takahashi, T.; Masukata, H. Association of fission yeast Orp1 and Mcm6 proteins with chromosomal replication origins. Mol. Cell. Biol. 1999, 19, 7228–7236. [Google Scholar]
- Maiorano, D.; Moreau, J.; Mechali, M. XCDT1 is required for the assembly of pre-replicative complexes in Xenopus laevis. Nature 2000, 404, 622–625. [Google Scholar]
- Nishitani, H.; Lygerou, Z.; Nishimoto, T.; Nurse, P. The Cdt1 protein is required to license DNA for replication in fission yeast. Nature 2000, 404, 625–628. [Google Scholar]
- Remus, D.; Beuron, F.; Tolun, G.; Griffith, J.D.; Morris, E.P.; Diffley, J.F. Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell 2009, 139, 719–730. [Google Scholar] [CrossRef]
- Evrin, C.; Clarke, P.; Zech, J.; Lurz, R.; Sun, J.; Uhle, S.; Li, H.; Stillman, B.; Speck, C. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl. Acad. Sci. USA 2009, 106, 20240–20245. [Google Scholar]
- Dahmann, C.; Diffley, J.F.; Nasmyth, K.A. S-phase-promoting cyclin-dependent kinases prevent re-replication by inhibiting the transition of replication origins to a pre-replicative state. Curr. Biol. 1995, 5, 1257–1269. [Google Scholar] [CrossRef]
- Mimura, S.; Takisawa, H. Xenopus Cdc45-dependent loading of DNA polymerase α onto chromatin under the control of S-phase Cdk. EMBO J. 1998, 17, 5699–5707. [Google Scholar] [CrossRef]
- Zou, L.; Stillman, B. Formation of a preinitiation complex by S-phase cyclin CDK-dependent loading of Cdc45p onto chromatin. Science 1998, 280, 593–596. [Google Scholar] [CrossRef]
- Nougarede, R.; Della Seta, F.; Zarzov, P.; Schwob, E. Hierarchy of S-phase-promoting factors: Yeast Dbf4-Cdc7 kinase requires prior S-phase cyclin-dependent kinase activation. Mol. Cell. Biol. 2000, 20, 3795–3806. [Google Scholar] [CrossRef]
- Sheu, Y.J.; Stillman, B. The Dbf4-Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature 2010, 463, 113–117. [Google Scholar]
- Zou, L.; Stillman, B. Assembly of a complex containing Cdc45p, replication protein A, and Mcm2p at replication origins controlled by S-phase cyclin-dependent kinases and Cdc7p-Dbf4p kinase. Mol. Cell. Biol. 2000, 20, 3086–3096. [Google Scholar] [CrossRef]
- Masai, H.; Taniyama, C.; Ogino, K.; Matsui, E.; Kakusho, N.; Matsumoto, S.; Kim, J.M.; Ishii, A.; Tanaka, T.; Kobayashi, T.; et al. Phosphorylation of MCM4 by Cdc7 kinase facilitates its interaction with Cdc45 on the chromatin. J. Biol. Chem. 2006, 281, 39249–39261. [Google Scholar]
- Muramatsu, S.; Hirai, K.; Tak, Y.-S.; Kamimura, Y.; Araki, H. CDK-dependent complex formation between replication proteins Dpb11, Sld2, Pol ε, and GINS in budding yeast. Genes Dev. 2010, 24, 602–612. [Google Scholar] [CrossRef]
- Handa, T.; Kanke, M.; Takahashi, T.S.; Nakagawa, T.; Masukata, H. DNA polymerization-independent functions of DNA polymerase epsilon in assembly and progression of the replisome in fission yeast. Mol. Biol. Cell 2012, 23, 3240–3253. [Google Scholar] [CrossRef]
- Saxena, S.; Dutta, A. Geminin-Cdt1 balance is critical for genetic stability. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2005, 569, 111–121. [Google Scholar] [CrossRef]
- Mendez, J.; Zou-Yang, X.H.; Kim, S.Y.; Hidaka, M.; Tansey, W.P.; Stillman, B. Human origin recognition complex large subunit is degraded by ubiquitin-mediated proteolysis after initiation of DNA replication. Mol. Cell 2002, 9, 481–491. [Google Scholar] [CrossRef]
- Diffley, J.F.; Cocker, J.H.; Dowell, S.J.; Rowley, A. Two steps in the assembly of complexes at yeast replication origins in vivo. Cell 1994, 78, 303–316. [Google Scholar] [CrossRef]
- Liang, C.; Stillman, B. Persistent initiation of DNA replication and chromatin-bound MCM proteins during the cell cycle in cdc6 mutants. Genes Dev. 1997, 11, 3375–3386. [Google Scholar] [CrossRef]
- Lee, K.Y.; Bang, S.W.; Yoon, S.W.; Lee, S.-H.; Yoon, J.-B.; Hwang, D.S. Phosphorylation of ORC2 protein dissociates origin recognition complex from chromatin and replication origins. J. Biol. Chem. 2012, 287, 11891–11898. [Google Scholar]
- Saha, P.; Chen, J.; Thome, K.C.; Lawlis, S.J.; Hou, Z.H.; Hendricks, M.; Parvin, J.D.; Dutta, A. Human CDC6/Cdc18 associates with Orc1 and cyclin-cdk and is selectively eliminated from the nucleus at the onset of S phase. Mol. Cell. Biol. 1998, 18, 2758–2767. [Google Scholar]
- Wohlschlegel, J.A.; Dwyer, B.T.; Dhar, S.K.; Cvetic, C.; Walter, J.C.; Dutta, A. Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science 2000, 290, 2309–2312. [Google Scholar]
- Hook, S.S.; Lin, J.J.; Dutta, A. Mechanisms to control rereplication and implications for cancer. Curr. Opin. Cell Biol. 2007, 19, 663–671. [Google Scholar] [CrossRef]
- O'Connell, B.C.; Harper, J.W. Ubiquitin proteasome system (UPS): What can chromatin do for you? Curr. Opin. Cell Biol. 2007, 19, 206–214. [Google Scholar] [CrossRef]
- Bravo, R.; Frank, R.; Blundell, P.A.; Macdonald-Bravo, H. Cyclin/PCNA is the auxiliary protein of DNA polymerase-δ. Nature 1987, 326, 515–517. [Google Scholar]
- Prelich, G.; Tan, C.K.; Kostura, M.; Mathews, M.B.; So, A.G.; Downey, K.M.; Stillman, B. Functional identity of proliferating cell nuclear antigen and a DNA polymerase-δ auxiliary protein. Nature 1987, 326, 517–520. [Google Scholar]
- Moldovan, G.L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef]
- Hoege, C.; Pfander, B.; Moldovan, G.-L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase η with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Yang, X.H.; Shiotani, B.; Classon, M.; Zou, L. Chk1 and Claspin potentiate PCNA ubiquitination. Genes Dev. 2008, 22, 1147–1152. [Google Scholar] [CrossRef]
- Branzei, D.; Seki, M.; Enomoto, T. Rad18/Rad5/Mms2-mediated polyubiquitination of PCNA is implicated in replication completion during replication stress. Genes Cells 2004, 9, 1031–1042. [Google Scholar] [CrossRef]
- Blastyak, A.; Pinter, L.; Unk, I.; Prakash, L.; Prakash, S.; Haracska, L. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol. Cell 2007, 28, 167–175. [Google Scholar] [CrossRef]
- Branzei, D.; Vanoli, F.; Foiani, M. SUMOylation regulates Rad18-mediated template switch. Nature 2008, 456, 915–920. [Google Scholar]
- Ulrich, H.D.; Vogel, S.; Davies, A.A. SUMO keeps a check on recombination during DNA replication. Cell Cycle 2005, 4, 1699–1702. [Google Scholar] [CrossRef]
- Leach, C.A.; Michael, W.M. Ubiquitin/SUMO modification of PCNA promotes replication fork progression in Xenopus laevis egg extracts. J. Cell Biol. 2005, 171, 947–954. [Google Scholar] [CrossRef]
- Pfander, B.; Moldovan, G.-L.; Sacher, M.; Hoege, C.; Jentsch, S. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 2005, 436, 428–433. [Google Scholar]
- Papouli, E.; Chen, S.; Davies, A.A.; Huttner, D.; Krejci, L.; Sung, P.; Ulrich, H.D. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol. Cell 2005, 19, 123–133. [Google Scholar] [CrossRef]
- Parnas, O.; Zipin-Roitman, A.; Pfander, B.; Liefshitz, B.; Mazor, Y.; Ben-Aroya, S.; Jentsch, S.; Kupiec, M. Elg1, an alternative subunit of the RFC clamp loader, preferentially interacts with SUMOylated PCNA. EMBO J. 2010, 29, 2611–2622. [Google Scholar] [CrossRef]
- Naryzhny, S.N.; Lee, H. The post-translational modifications of proliferating cell nuclear antigen: Acetylation, not phosphorylation, plays an important role in the regulation of its function. J. Biol. Chem. 2004, 279, 20194–20199. [Google Scholar] [CrossRef]
- Cai, J.; Yao, N.; Gibbs, E.; Finkelstein, J.; Phillips, B.; O'Donnell, M.; Hurwitz, J. ATP hydrolysis catalyzed by human replication factor C requires participation of multiple subunits. Proc. Natl. Acad. Sci. USA 1998, 95, 11607–11612. [Google Scholar]
- Tsurimoto, T.; Stillman, B. Replication factors required for SV40 DNA replication in vitro. I. DNA structure-specific recognition of a primer-template junction by eukaryotic DNA polymerases and their accessory proteins. J. Biol. Chem. 1991, 266, 1950–1960. [Google Scholar]
- Podust, L.M.; Podust, V.N.; Sogo, J.M.; Hubscher, U. Mammalian DNA polymerase auxiliary proteins: Analysis of replication factor C-catalyzed proliferating cell nuclear antigen loading onto circular double-stranded DNA. Mol. Cell. Biol. 1995, 15, 3072–3081. [Google Scholar]
- Zhang, G.; Gibbs, E.; Kelman, Z.; O'Donnell, M.; Hurwitz, J. Studies on the interactions between human replication factor C and human proliferating cell nuclear antigen. Proc. Natl. Acad. Sci. USA 1999, 96, 1869–1874. [Google Scholar]
- Yao, N.Y.; Johnson, A.; Bowman, G.D.; Kuriyan, J.; O'Donnell, M. Mechanism of proliferating cell nuclear antigen clamp opening by replication factor C. J. Biol. Chem. 2006, 281, 17528–17539. [Google Scholar]
- Skibbens, R.V.; Corson, L.B.; Koshland, D.; Hieter, P. Ctf7p is essential for sister chromatid cohesion and links mitotic chromosome structure to the DNA replication machinery. Genes Dev. 1999, 13, 307–319. [Google Scholar]
- Lengronne, A.; McIntyre, J.; Katou, Y.; Kanoh, Y.; Hopfner, K.P.; Shirahige, K.; Uhlmann, F. Establishment of sister chromatid cohesion at the S. cerevisiae replication fork. Mol. Cell 2006, 23, 787–799. [Google Scholar] [CrossRef]
- Ansbach, A.B.; Noguchi, C.; Klansek, I.W.; Heidlebaugh, M.; Nakamura, T.M.; Noguchi, E. RFCCtf18 and the Swi1-Swi3 complex function in separate and redundant pathways required for the stabilization of replication forks to facilitate sister chromatid cohesion in Schizosaccharomyces pombe. Mol. Biol. Cell 2008, 19, 595–607. [Google Scholar]
- Terret, M.E.; Sherwood, R.; Rahman, S.; Qin, J.; Jallepalli, P.V. Cohesin acetylation speeds the replication fork. Nature 2009, 462, 231–234. [Google Scholar]
- Bermudez, V.P.; Lindsey-Boltz, L.A.; Cesare, A.J.; Maniwa, Y.; Griffith, J.D.; Hurwitz, J.; Sancar, A. Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proc. Natl. Acad. Sci. USA 2003, 100, 1633–1638. [Google Scholar]
- Garg, P.; Stith, C.M.; Sabouri, N.; Johansson, E.; Burgers, P.M. Idling by DNA polymerase δ maintains a ligatable nick during lagging-strand DNA replication. Genes Dev. 2004, 18, 2764–2773. [Google Scholar] [CrossRef]
- Burgers, P.M.J. Polymerase dynamics at the eukaryotic DNA replication fork. J. Biol. Chem. 2009, 284, 4041–4045. [Google Scholar] [CrossRef]
- Jin, Y.H.; Ayyagari, R.; Resnick, M.A.; Gordenin, D.A.; Burgers, P.M.J. Okazaki fragment maturation in yeast. J. Biol. Chem. 2003, 278, 1626–1633. [Google Scholar]
- Budd, M.E.; Campbell, J.L. A yeast replicative helicase, Dna2 helicase, interacts with yeast FEN-1 nuclease in carrying out its essential function. Mol. Cell. Biol. 1997, 17, 2136–2142. [Google Scholar]
- Lee, K.H.; Kim, D.W.; Bae, S.H.; Kim, J.A.; Ryu, G.H.; Kwon, Y.N.; Kim, K.A.; Koo, H.S.; Seo, Y.S. The endonuclease activity of the yeast Dna2 enzyme is essential in vivo. Nucleic Acids Res. 2000, 28, 2873–2881. [Google Scholar] [CrossRef]
- Kao, H.-I.; Veeraraghavan, J.; Polaczek, P.; Campbell, J.L.; Bambara, R.A. On the roles of Saccharomyces cerevisiae Dna2p and flap endonuclease 1 in Okazaki fragment processing. J. Biol. Chem. 2004, 279, 15014–15024. [Google Scholar]
- Sogo, J.M.; Lopes, M.; Foiani, M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 2002, 297, 599–602. [Google Scholar] [CrossRef]
- Byun, T.S.; Pacek, M.; Yee, M.-C.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef]
- Sheu, Y.-J.; Stillman, B. Cdc7-Dbf4 Phosphorylates MCM Proteins via a Docking Site-Mediated Mechanism to Promote S Phase Progression. Mol. Cell 2006, 24, 101–113. [Google Scholar] [CrossRef]
- Matsumoto, S.; Shimmoto, M.; Kakusho, N.; Yokoyama, M.; Kanoh, Y.; Hayano, M.; Russell, P.; Masai, H. Hsk1 kinase and Cdc45 regulate replication stress-induced checkpoint responses in fission yeast. Cell Cycle 2010, 9, 4627–4637. [Google Scholar]
- Takayama, Y.; Kamimura, Y.; Okawa, M.; Muramatsu, S.; Sugino, A.; Araki, H. GINS, a novel multiprotein complex required for chromosomal DNA replication in budding yeast. Genes Dev. 2003, 17, 1153–1165. [Google Scholar] [CrossRef]
- Kubota, Y.; Takase, Y.; Komori, Y.; Hashimoto, Y.; Arata, T.; Kamimura, Y.; Araki, H.; Takisawa, H. A novel ring-like complex of Xenopus proteins essential for the initiation of DNA replication. Genes Dev. 2003, 17, 1141–1152. [Google Scholar] [CrossRef]
- Gambus, A.; Jones, R.C.; Sanchez-Diaz, A.; Kanemaki, M.; van Deursen, F.; Edmondson, R.D.; Labib, K. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat. Cell Biol. 2006, 8, 358–366. [Google Scholar] [CrossRef]
- Moyer, S.E.; Lewis, P.W.; Botchan, M.R. Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc. Natl. Acad. Sci. USA 2006, 103, 10236–10241. [Google Scholar]
- Im, J.S.; Ki, S.H.; Farina, A.; Jung, D.S.; Hurwitz, J.; Lee, J.K. Assembly of the Cdc45-Mcm2-7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 15628–15632. [Google Scholar]
- Ilves, I.; Petojevic, T.; Pesavento, J.J.; Botchan, M.R. Activation of the MCM2-7 helicase by association with Cdc45 and GINS proteins. Mol. Cell 2010, 37, 247–258. [Google Scholar] [CrossRef]
- Costa, A.; Ilves, I.; Tamberg, N.; Petojevic, T.; Nogales, E.; Botchan, M.R.; Berger, J.M. The structural basis for MCM2-7 helicase activation by GINS and Cdc45. Nat. Struct. Mol. Biol. 2011, 18, 471–477. [Google Scholar]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar]
- Bermudez, V.P.; Farina, A.; Tappin, I.; Hurwitz, J. Influence of the human cohesion establishment factor Ctf4/AND-1 on DNA replication. J. Biol. Chem. 2009, 285, 9493–9505. [Google Scholar]
- Gambus, A.; van Deursen, F.; Polychronopoulos, D.; Foltman, M.; Jones, R.C.; Edmondson, R.D.; Calzada, A.; Labib, K. A key role for Ctf4 in coupling the MCM2-7 helicase to DNA polymerase α within the eukaryotic replisome. EMBO J. 2009, 28, 2992–3004. [Google Scholar] [CrossRef]
- Zhu, W.; Ukomadu, C.; Jha, S.; Senga, T.; Dhar, S.K.; Wohlschlegel, J.A.; Nutt, L.K.; Kornbluth, S.; Dutta, A. Mcm10 and And-1/CTF4 recruit DNA polymerase α to chromatin for initiation of DNA replication. Genes Dev. 2007, 21, 2288–2299. [Google Scholar] [CrossRef]
- Lou, H.; Komata, M.; Katou, Y.; Guan, Z.; Reis, C.C.; Budd, M.; Shirahige, K.; Campbell, J.L. Mrc1 and DNA polymerase ε function together in linking DNA replication and the S phase checkpoint. Mol. Cell 2008, 32, 106–117. [Google Scholar] [CrossRef]
- Petermann, E.; Helleday, T.; Caldecott, K.W. Claspin promotes normal replication fork rates in human cells. Mol. Biol. Cell 2008, 19, 2373–2378. [Google Scholar] [CrossRef]
- Leman, A.R.; Noguchi, E. Local and global functions of Timeless and Tipin in replication fork protection. Cell Cycle 2012, 11, 3945–3955. [Google Scholar] [CrossRef]
- Tourriere, H.; Versini, G.; Cordon-Preciado, V.; Alabert, C.; Pasero, P. Mrc1 and Tof1 promote replication fork progression and recovery independently of Rad53. Mol. Cell 2005, 19, 699–706. [Google Scholar] [CrossRef]
- Unsal-Kacmaz, K.; Chastain, P.D.; Qu, P.-P.; Minoo, P.; Cordeiro-Stone, M.; Sancar, A.; Kaufmann, W.K. The human Tim/Tipin complex coordinates an intra-S checkpoint response to UV that slows replication fork displacement. Mol. Cell. Biol. 2007, 27, 3131–3142. [Google Scholar] [CrossRef]
- Brown, E.J.; Baltimore, D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000, 14, 397–402. [Google Scholar]
- De Klein, A.; Muijtjens, M.; van Os, R.; Verhoeven, Y.; Smit, B.; Carr, A.M.; Lehmann, A.R.; Hoeijmakers, J.H. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr. Biol. 2000, 10, 479–482. [Google Scholar] [CrossRef]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in checkpoint signaling. Science 2001, 294, 1713–1716. [Google Scholar] [CrossRef]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef]
- Dart, D.A.; Adams, K.E.; Akerman, I.; Lakin, N.D. Recruitment of the cell cycle checkpoint kinase ATR to chromatin during S-phase. J. Biol. Chem. 2004, 279, 16433–16440. [Google Scholar]
- Cha, R.S.; Kleckner, N. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 2002, 297, 602–606. [Google Scholar] [CrossRef]
- Ball, H.L.; Myers, J.S.; Cortez, D. ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol. Biol. Cell 2005, 16, 2372–2381. [Google Scholar] [CrossRef]
- Zou, L.; Cortez, D.; Elledge, S.J. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 2002, 16, 198–208. [Google Scholar] [CrossRef]
- Parrilla-Castellar, E.R.; Karnitz, L.M. Cut5 is required for the binding of Atr and DNA polymerase α to genotoxin-damaged chromatin. J. Biol. Chem. 2003, 278, 45507–45511. [Google Scholar] [CrossRef]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 Activates the ATR-ATRIP Complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef]
- Lee, J.; Kumagai, A.; Dunphy, W.G. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J. Biol. Chem. 2007, 282, 28036–28044. [Google Scholar]
- Yan, S.; Michael, W.M. TopBP1 and DNA polymerase-α directly recruit the 9-1-1 complex to stalled DNA replication forks. J. Cell Biol. 2009, 184, 793–804. [Google Scholar] [CrossRef]
- Kumagai, A.; Dunphy, W.G. Claspin, a Novel Protein Required for the Activation of Chk1 during a DNA Replication Checkpoint Response in Xenopus Egg Extracts. Mol. Cell 2000, 6, 839–849. [Google Scholar] [CrossRef]
- Chini, C.C.; Chen, J. Human claspin is required for replication checkpoint control. J. Biol. Chem. 2003, 278, 30057–30062. [Google Scholar] [CrossRef]
- Kumagai, A.; Kim, S.-M.; Dunphy, W.G. Claspin and the activated form of ATR-ATRIP collaborate in the activation of Chk1. J. Biol. Chem. 2004, 279, 49599–49608. [Google Scholar]
- Kumagai, A.; Dunphy, W.G. Repeated phosphopeptide motifs in Claspin mediate the regulated binding of Chk1. Nat. Cell Biol. 2003, 5, 161–165. [Google Scholar]
- Jin, J.; Shirogane, T.; Xu, L.; Nalepa, G.; Qin, J.; Elledge, S.J.; Harper, J.W. SCF-ßTRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 2003, 17, 3062–3074. [Google Scholar] [CrossRef]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 checkpoint pathway in mammals: Linkage of DNA damage to Cdk regulation through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar] [CrossRef]
- Peng, C.-Y.; Graves, P.R.; Thoma, R.S.; Wu, Z.; Shaw, A.S.; Piwnica-Worms, H. Mitotic and G2 checkpoint control: Regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 1997, 277, 1501–1505. [Google Scholar] [CrossRef]
- Kumagai, A.; Guo, Z.; Emami, K.H.; Wang, S.X.; Dunphy, W.G. The Xenopus Chk1 protein kinase mediates a caffeine-sensitive pathway of checkpoint control in cell-free extracts. J. Cell Biol. 1998, 142, 1559–1569. [Google Scholar] [CrossRef]
- Miao, H.; Seiler, J.A.; Burhans, W.C. Regulation of cellular and SV40 virus origins of replication by Chk1-dependent intrinsic and UVC radiation-induced checkpoints. J. Biol. Chem. 2003, 278, 4295–4304. [Google Scholar]
- Sorensen, C.S.; Syljuasen, R.G.; Lukas, J.; Bartek, J. ATR, Claspin and the Rad9-Rad1-Hus1 complex regulate Chk1 and Cdc25A in the absence of DNA damage. Cell Cycle 2004, 3, 941–945. [Google Scholar]
- Syljuasen, R.G.; Sorensen, C.S.; Hansen, L.T.; Fugger, K.; Lundin, C.; Johansson, F.; Helleday, T.; Sehested, M.; Lukas, J.; Bartek, J. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol. Cell. Biol. 2005, 25, 3553–3562. [Google Scholar]
- Kornberg, R.D. Chromatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871. [Google Scholar]
- Kornberg, R.D.; Thomas, J.O. Chromatin structure; oligomers of the histones. Science 1974, 184, 865–868. [Google Scholar]
- Benyajati, C.; Worcel, A. Isolation, characterization, and structure of the folded interphase genome of Drosophila melanogaste. Cell 1976, 9, 393–407. [Google Scholar] [CrossRef]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Reinberg, D.; Sims, R.J. de FACTo nucleosome dynamics. J. Biol. Chem. 2006, 281, 23297–23301. [Google Scholar]
- Wittmeyer, J.; Joss, L.; Formosa, T. Spt16 and Pob3 of Saccharomyces cerevisiae form an essential, abundant heterodimer that is nuclear, chromatin-associated, and copurifies with DNA polymerase α. Biochemistry 1999, 38, 8961–8971. [Google Scholar] [CrossRef]
- Wittmeyer, J.; Formosa, T. The Saccharomyces cerevisiae DNA polymerase alpha catalytic subunit interacts with Cdc68/Spt16 and with Pob3, a protein similar to an HMG1-like protein. Mol. Cell. Biol. 1997, 17, 4178–4190. [Google Scholar]
- Formosa, T.; Eriksson, P.; Wittmeyer, J.; Ginn, J.; Yu, Y.; Stillman, D.J. Spt16-Pob3 and the HMG protein Nhp6 combine to form the nucleosome-binding factor SPN. EMBO J. 2001, 20, 3506–3517. [Google Scholar] [CrossRef]
- Tan, B.C.; Chien, C.T.; Hirose, S.; Lee, S.C. Functional cooperation between FACT and MCM helicase facilitates initiation of chromatin DNA replication. EMBO J. 2006, 25, 3975–3985. [Google Scholar] [CrossRef]
- Tan, B.C.; Liu, H.; Lin, C.L.; Lee, S.C. Functional cooperation between FACT and MCM is coordinated with cell cycle and differential complex formation. J. Biomed. Sci. 2010, 17, 11. [Google Scholar] [CrossRef]
- Han, J.; Li, Q.; McCullough, L.; Kettelkamp, C.; Formosa, T.; Zhang, Z. Ubiquitylation of FACT by the cullin-E3 ligase Rtt101 connects FACT to DNA replication. Genes Dev. 2010, 24, 1485–1490. [Google Scholar] [CrossRef]
- Xin, H.; Takahata, S.; Blanksma, M.; McCullough, L.; Stillman, D.J.; Formosa, T. yFACT induces global accessibility of nucleosomal DNA without H2A-H2B displacement. Mol. Cell 2009, 35, 365–376. [Google Scholar] [CrossRef]
- Groth, A.; Corpet, A.; Cook, A.J.; Roche, D.; Bartek, J.; Lukas, J.; Almouzni, G. Regulation of replication fork progression through histone supply and demand. Science 2007, 318, 1928–1931. [Google Scholar] [CrossRef]
- Mello, J.A.; Sillje, H.H.; Roche, D.M.; Kirschner, D.B.; Nigg, E.A.; Almouzni, G. Human Asf1 and CAF-1 interact and synergize in a repair-coupled nucleosome assembly pathway. EMBO Rep. 2002, 3, 329–334. [Google Scholar] [CrossRef]
- Daganzo, S.M.; Erzberger, J.P.; Lam, W.M.; Skordalakes, E.; Zhang, R.; Franco, A.A.; Brill, S.J.; Adams, P.D.; Berger, J.M.; Kaufman, P.D. Structure and function of the conserved core of histone deposition protein Asf1. Curr. Biol. 2003, 13, 2148–2158. [Google Scholar] [CrossRef]
- Groth, A.; Ray-Gallet, D.; Quivy, J.P.; Lukas, J.; Bartek, J.; Almouzni, G. Human Asf1 regulates the flow of S phase histones during replicational stress. Mol. Cell 2005, 17, 301–311. [Google Scholar] [CrossRef]
- Stillman, B. Chromatin assembly during SV40 DNA replication in vitro. Cell 1986, 45, 555–565. [Google Scholar] [CrossRef]
- Smith, S.; Stillman, B. Purification and characterization of CAF-I, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell 1989, 58, 15–25. [Google Scholar] [CrossRef]
- Verreault, A.; Kaufman, P.D.; Kobayashi, R.; Stillman, B. Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell 1996, 87, 95–104. [Google Scholar]
- Shibahara, K.; Stillman, B. Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 1999, 96, 575–585. [Google Scholar] [CrossRef]
- Rolef Ben-Shahar, T.; Castillo, A.G.; Osborne, M.J.; Borden, K.L.; Kornblatt, J.; Verreault, A. Two fundamentally distinct PCNA interaction peptides contribute to chromatin assembly factor 1 function. Mol. Cell. Biol. 2009, 29, 6353–6365. [Google Scholar] [CrossRef]
- Huang, S.; Zhou, H.; Katzmann, D.; Hochstrasser, M.; Atanasova, E.; Zhang, Z. Rtt106p is a histone chaperone involved in heterochromatin-mediated silencing. Proc. Natl. Acad. Sci. USA 2005, 102, 13410–13415. [Google Scholar]
- Huang, S.; Zhou, H.; Tarara, J.; Zhang, Z. A novel role for histone chaperones CAF-1 and Rtt106p in heterochromatin silencing. EMBO J. 2007, 26, 2274–2283. [Google Scholar] [CrossRef]
- Ransom, M.; Dennehey, B.K.; Tyler, J.K. Chaperoning histones during DNA replication and repair. Cell 2010, 140, 183–195. [Google Scholar] [CrossRef]
- Ito, T.; Tyler, J.K.; Bulger, M.; Kobayashi, R.; Kadonaga, J.T. ATP-facilitated chromatin assembly with a nucleoplasmin-like protein from Drosophila melanogaster. J. Biol. Chem. 1996, 271, 25041–25048. [Google Scholar]
- Gasser, R.; Koller, T.; Sogo, J.M. The stability of nucleosomes at the replication fork. J. Mol. Biol. 1996, 258, 224–239. [Google Scholar] [CrossRef]
- Rothstein, R.; Michel, B.N.D.; Gangloff, S. Replication fork pausing and recombination or “gimme a break”. Genes Dev. 2000, 14, 1–10. [Google Scholar]
- Bastia, D.; Zzaman, S.; Krings, G.; Saxena, M.; Peng, X.; Greenberg, M.M. Replication termination mechanism as revealed by Tus-mediated polar arrest of a sliding helicase. Proc. Natl. Acad. Sci. USA 2008, 105, 12831–12836. [Google Scholar]
- Brewer, B.J.; Fangman, W.L. A replication fork barrier at the 3' end of yeast ribosomal RNA genes. Cell 1988, 55, 637–643. [Google Scholar] [CrossRef]
- Linskens, M.H.; Huberman, J.A. Organization of replication of ribosomal DNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 1988, 8, 4927–4935. [Google Scholar]
- Kobayashi, T.; Horiuchi, T. A yeast gene product, Fob1 protein, required for both replication fork blocking and recombinational hotspot activities. Genes Cells 1996, 1, 465–474. [Google Scholar]
- Kobayashi, T. The replication fork barrier site forms a unique structure with Fob1p and inhibits the replication fork. Mol. Cell. Biol. 2003, 23, 9178–9188. [Google Scholar] [CrossRef]
- Johzuka, K.; Horiuchi, T. Replication fork block protein, Fob1, acts as an rDNA region specific recombinator in S. cerevisiae. Genes Cells 2002, 7, 99–113. [Google Scholar] [CrossRef]
- Mohanty, B.K.; Bairwa, N.K.; Bastia, D. The Tof1p-Csm3p protein complex counteracts the Rrm3p helicase to control replication termination of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2006, 103, 897–902. [Google Scholar] [CrossRef]
- Ivessa, A.S.; Zhou, J.Q.; Zakian, V.A. The Saccharomyces Pif1p DNA helicase and the highly related Rrm3p have opposite effects on replication fork progression in ribosomal DNA. Cell 2000, 100, 479–489. [Google Scholar] [CrossRef]
- Ivessa, A.S.; Zhou, J.Q.; Schulz, V.P.; Monson, E.K.; Zakian, V.A. Saccharomyces Rrm3p, a 5' to 3' DNA helicase that promotes replication fork progression through telomeric and subtelomeric DNA. Genes Dev. 2002, 16, 1383–1396. [Google Scholar] [CrossRef]
- Ivessa, A.S.; Lenzmeier, B.A.; Bessler, J.B.; Goudsouzian, L.K.; Schnakenberg, S.L.; Zakian, V.A. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol. Cell 2003, 12, 1525–1536. [Google Scholar] [CrossRef]
- Torres, J.Z.; Schnakenberg, S.L.; Zakian, V.A. Saccharomyces cerevisiae Rrm3p DNA helicase promotes genome integrity by preventing replication fork stalling: Viability of rrm3 cells requires the intra-S-phase checkpoint and fork restart activities. Mol. Cell. Biol. 2004, 24, 3198–3212. [Google Scholar] [CrossRef]
- Torres, J.Z.; Bessler, J.B.; Zakian, V.A. Local chromatin structure at the ribosomal DNA causes replication fork pausing and genome instability in the absence of the S. cerevisiae DNA helicase Rrm3p. Genes Dev. 2004, 18, 498–503. [Google Scholar] [CrossRef]
- Schmidt, K.H.; Derry, K.L.; Kolodner, R.D. Saccharomyces cerevisiae RRM3, a 5' to 3' DNA helicase, physically interacts with proliferating cell nuclear antigen. J. Biol. Chem. 2002, 277, 45331–45337. [Google Scholar]
- Azvolinsky, A.; Dunaway, S.; Torres, J.Z.; Bessler, J.B.; Zakian, V.A. The S. cerevisiae Rrm3p DNA helicase moves with the replication fork and affects replication of all yeast chromosomes. Genes Dev. 2006, 20, 3104–3116. [Google Scholar] [CrossRef]
- Sanchez-Gorostiaga, A.; Lopez-Estrano, C.; Krimer, D.B.; Schvartzman, J.B.; Hernandez, P. Transcription termination factor reb1p causes two replication fork barriers at its cognate sites in fission yeast ribosomal DNA in vivo. Mol. Cell. Biol. 2004, 24, 398–406. [Google Scholar] [CrossRef]
- Krings, G.; Bastia, D. swi1- and swi3-dependent and independent replication fork arrest at the ribosomal DNA of Schizosaccharomyces pombe. Proc. Natl. Acad. Sci. USA 2004, 101, 14085–14090. [Google Scholar] [CrossRef]
- Krings, G.; Bastia, D. Sap1p binds to Ter1 at the ribosomal DNA of Schizosaccharomyces pombe and causes polar replication fork arrest. J. Biol. Chem. 2005, 280, 39135–39142. [Google Scholar] [CrossRef]
- Mejia-Ramirez, E.; Sanchez-Gorostiaga, A.; Krimer, D.B.; Schvartzman, J.B.; Hernandez, P. The mating type switch-activating protein Sap1 is required for replication fork arrest at the rRNA genes of fission yeast. Mol. Cell. Biol. 2005, 25, 8755–8761. [Google Scholar] [CrossRef]
- Little, R.D.; Platt, T.H.; Schildkraut, C.L. Initiation and termination of DNA replication in human rRNA genes. Mol. Cell. Biol. 1993, 13, 6600–6613. [Google Scholar]
- Evers, R.; Grummt, I. Molecular coevolution of mammalian ribosomal gene terminator sequences and the transcription termination factor TTF-I. Proc. Natl. Acad. Sci. USA 1995, 92, 5827–5831. [Google Scholar] [CrossRef]
- Kanei-Ishii, C.; Sarai, A.; Sawazaki, T.; Nakagoshi, H.; He, D.N.; Ogata, K.; Nishimura, Y.; Ishii, S. The tryptophan cluster: A hypothetical structure of the DNA-binding domain of the myb protooncogene product. J. Biol. Chem. 1990, 265, 19990–19995. [Google Scholar]
- Beach, D.H. Cell type switching by DNA transposition in fission yeast. Nature 1983, 305, 682–688. [Google Scholar]
- Beach, D.H.; Klar, A.J. Rearrangements of the transposable mating-type cassettes of fission yeast. EMBO J. 1984, 3, 603–610. [Google Scholar]
- Dalgaard, J.Z.; Klar, A.J. Orientation of DNA replication establishes mating-type switching pattern in S. pombe. Nature 1999, 400, 181–184. [Google Scholar]
- Arcangioli, B.; de Lahondes, R. Fission yeast switches mating type by a replication-recombination coupled process. EMBO J. 2000, 19, 1389–1396. [Google Scholar] [CrossRef]
- Dalgaard, J.Z.; Klar, A.J.S. A DNA replication-arrest site RTS1 regulates imprinting by determining the direction of replication at mat1 in S. pombe. Genes Dev. 2001, 15, 2060–2068. [Google Scholar] [CrossRef]
- Vengrova, S.; Codlin, S.; Dalgaard, J.Z. RTS1-an eukaryotic terminator of replication. Int. J. Biochem. Cell Biol. 2002, 34, 1031–1034. [Google Scholar] [CrossRef]
- Lambert, S.; Watson, A.; Sheedy, D.M.; Martin, B.; Carr, A.M. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell 2005, 121, 689–702. [Google Scholar] [CrossRef]
- Ahn, J.S.; Osman, F.; Whitby, M.C. Replication fork blockage by RTS1 at an ectopic site promotes recombination in fission yeast. EMBO J. 2005, 24, 2011–2023. [Google Scholar] [CrossRef]
- Codlin, S.; Dalgaard, J.Z. Complex mechanism of site-specific DNA replication termination in fission yeast. EMBO J. 2003, 22, 3431–3440. [Google Scholar] [CrossRef]
- Eydmann, T.; Sommariva, E.; Inagawa, T.; Mian, S.; Klar, A.J.S.; Dalgaard, J.Z. Rtf1-mediated eukaryotic site-specific replication termination. Genetics 2008, 180, 27–39. [Google Scholar] [CrossRef]
- Egel, R.; Beach, D.H.; Klar, A.J. Genes required for initiation and resolution steps of mating-type switching in fission yeast. Proc. Natl. Acad. Sci. USA 1984, 81, 3481–3485. [Google Scholar] [CrossRef]
- Dalgaard, J.Z.; Klar, A.J. swi1 and swi3 perform imprinting, pausing, and termination of DNA replication in S. pombe. Cell 2000, 102, 745–751. [Google Scholar] [CrossRef]
- Desany, B.A.; Alcasabas, A.A.; Bachant, J.B.; Elledge, S.J. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev. 1998, 12, 2956–2970. [Google Scholar] [CrossRef]
- Zhao, X.; Muller, E.G.; Rothstein, R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol. Cell 1998, 2, 329–340. [Google Scholar] [CrossRef]
- Hashash, N.; Johnson, A.L.; Cha, R.S. Regulation of fragile sites expression in budding yeast by MEC1, RRM3 and hydroxyurea. J. Cell Sci. 2010, 124, 181–185. [Google Scholar]
- Lopes, M.; Cotta-Ramusino, C.; Pellicioli, A.; Liberi, G.; Plevani, P.; Muzi-Falconi, M.; Newlon, C.S.; Foiani, M. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 2001, 412, 557–561. [Google Scholar] [CrossRef]
- Tercero, J.A.; Diffley, J.F.X. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 2001, 412, 553–557. [Google Scholar] [CrossRef]
- Magenis, R.E.; Hecht, F.; Lovrien, E.W. Heritable fragile site on chromosome 16: Probable localization of Haptoglobin locus in man. Science 1970, 170, 85–87. [Google Scholar]
- Harvey, J.; Judge, C.; Wiener, S. Familial X-linked mental retardation with an X chromosome abnormality. J. Med. Genet. 1977, 14, 46–50. [Google Scholar] [CrossRef]
- Sutherland, G.R. Fragile sites on human chromosomes: Demonstration of their dependence on the type of tissue culture medium. Science 1977, 197, 265–266. [Google Scholar]
- Sutherland, G.R. Heritable fragile sites on human chromosomes I. Factors affecting expression in lymphocyte culture. Am. J. Hum. Genet. 1979, 31, 125–135. [Google Scholar]
- Glover, T.W. FUdR induction of the X chromosome fragile site: Evidence for the mechanism of folic acid and thymidine inhibition. Am. J. Hum. Genet. 1981, 33, 234–242. [Google Scholar]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J.S.; Richards, S.; Verkerk, A.J.; Holden, J.J.; Fenwick, R.G., Jr.; Warren, S.T.; et al. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 1991, 67, 1047–1058. [Google Scholar] [CrossRef]
- Oberle, I.; Rousseau, F.; Heitz, D.; Kretz, C.; Devys, D.; Hanauer, A.; Boue, J.; Bertheas, M.F.; Mandel, J.L. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 1991, 252, 1097–1102. [Google Scholar]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef]
- Glover, T.W.; Berger, C.; Coyle, J.; Echo, B. DNA polymerase α inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 1984, 67, 136–142. [Google Scholar] [CrossRef]
- Sutherland, G.R.; Parslow, M.I.; Baker, E. New classes of common fragile sites induced by 5-azacytidine and bromodeoxyuridine. Hum. Genet. 1985, 69, 233–237. [Google Scholar] [CrossRef]
- LeBeau, M.M.; Rowley, J.D. Heritable fragile sites in cancer. Nature 1984, 308, 607–608. [Google Scholar]
- Gacy, A.M.; Goellner, G.; Juranic, N.; Macura, S.; McMurray, C.T. Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell 1995, 81, 533–540. [Google Scholar] [CrossRef]
- Hewett, D.R.; Handt, O.; Hobson, L.; Mangelsdorf, M.; Eyre, H.J.; Baker, E.; Sutherland, G.R.; Schuffenhauer, S.; Mao, J.I.; Richards, R.I. FRA10B structure reveals common elements in repeat expansion and chromosomal fragile site genesis. Mol. Cell 1998, 1, 773–781. [Google Scholar] [CrossRef]
- Zlotorynski, E.; Rahat, A.; Skaug, J.; Ben-Porat, N.; Ozeri, E.; Hershberg, R.; Levi, A.; Scherer, S.W.; Margalit, H.; Kerem, B. Molecular basis for expression of common and rare fragile sites. Mol. Cell. Biol. 2003, 23, 7143–7151. [Google Scholar] [CrossRef]
- Durkin, S.G.; Ragland, R.L.; Arlt, M.F.; Mulle, J.G.; Warren, S.T.; Glover, T.W. Replication stress induces tumor-like microdeletions in FHIT/FRA3B. Proc. Natl. Acad. Sci. USA 2008, 105, 246–251. [Google Scholar]
- Casper, A.M.; Nghiem, P.; Arlt, M.F.; Glover, T.W. ATR regulates fragile site stability. Cell 2002, 111, 779–789. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Leman, A.R.; Noguchi, E. The Replication Fork: Understanding the Eukaryotic Replication Machinery and the Challenges to Genome Duplication. Genes 2013, 4, 1-32. https://doi.org/10.3390/genes4010001
Leman AR, Noguchi E. The Replication Fork: Understanding the Eukaryotic Replication Machinery and the Challenges to Genome Duplication. Genes. 2013; 4(1):1-32. https://doi.org/10.3390/genes4010001
Chicago/Turabian StyleLeman, Adam R., and Eishi Noguchi. 2013. "The Replication Fork: Understanding the Eukaryotic Replication Machinery and the Challenges to Genome Duplication" Genes 4, no. 1: 1-32. https://doi.org/10.3390/genes4010001
APA StyleLeman, A. R., & Noguchi, E. (2013). The Replication Fork: Understanding the Eukaryotic Replication Machinery and the Challenges to Genome Duplication. Genes, 4(1), 1-32. https://doi.org/10.3390/genes4010001