Factors Behind Junk DNA in Bacteria


Abstract
:
1. Introduction
1.1. Genome Size and Junk DNA
1.2. Bacterial Endosymbionts as a Model
2. The Impact of Pseudogenes in Bacterial Genomes
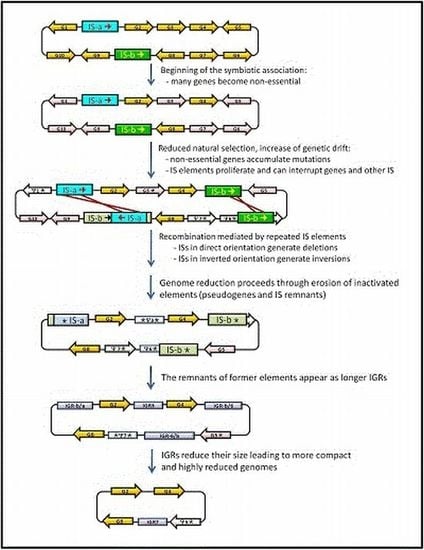
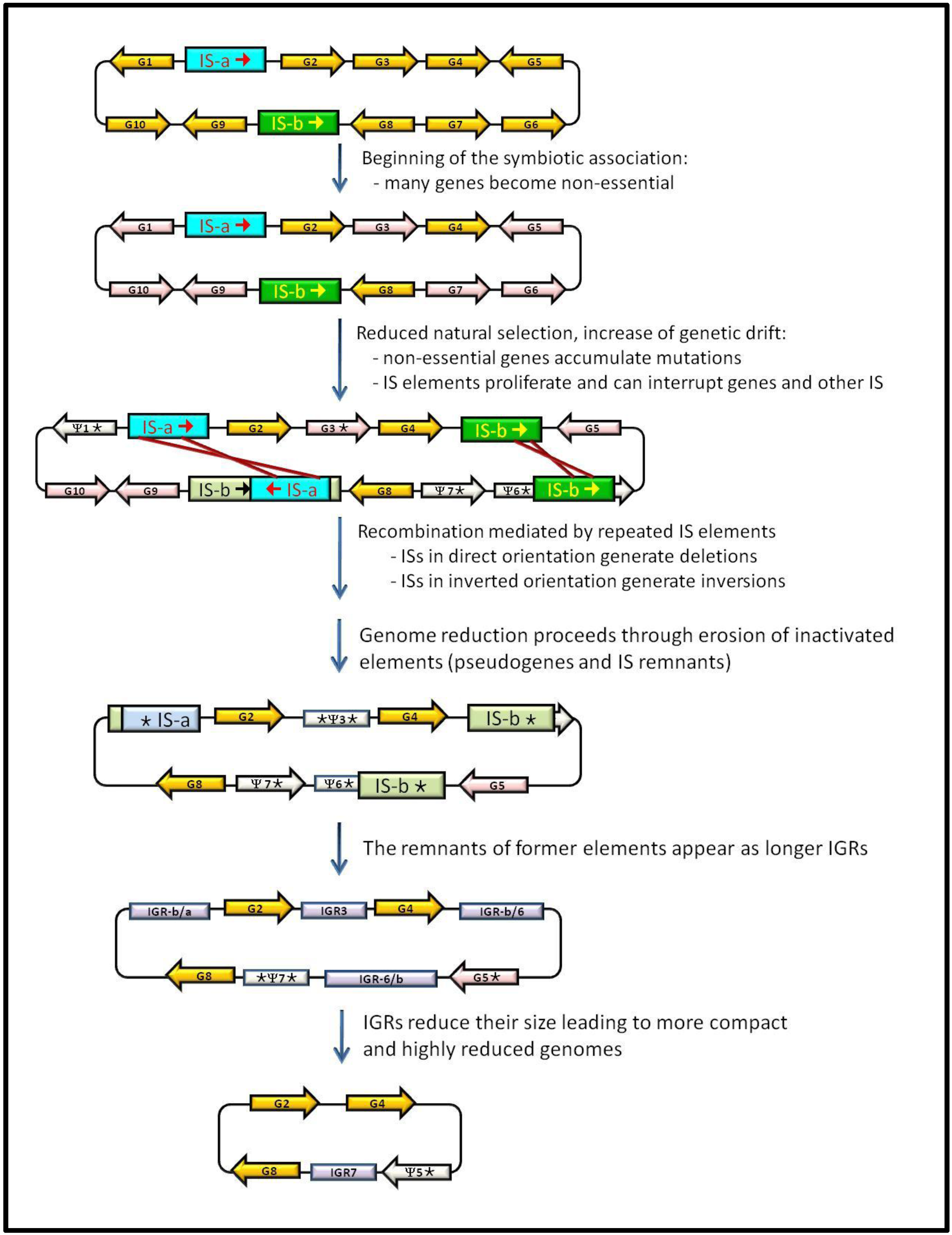
3. IS Elements Shaping Bacterial Genomes
4. Junk DNA as a Marker of the Symbiotic Integration Process
4.1. Serratia symbiotica, the Missing Link from Free-Living to Obligate Mutualism

{kind=link}
{kind=link}
| Species | Lifestyle | Genome Size (kb) | CDS | Pseudogenes | IGR Mean Size (bp) | % Coding Density | Presence of ISs (%) | Data Source |
|---|---|---|---|---|---|---|---|---|
| M. leprae TN | human parasite | 3,268 | 1614 | 1293 | ND | ND | Yes | [30,49] |
| M. tuberculosis H37Rv | human parasite | 4,411 | 4006 | 6* | ND | ND | 1.5 | [29,44,50] |
| S. symbiotica SAp | pea aphid facultative symbiont | 2,789 | 2098 | 550 | 204.3 | 60.9 | Yes | [9,12] |
| S. symbiotica SCc | cedar aphid facultative symbiont | 1,763 | 672 | 58 | 1672.01 | 38.7 | No | [12] |
| S. proteamaculans | free-living | 5,496 | 4942 | 12 | 165.67 | 87.1 | Yes | [12] |
| B. aphidicola BAp | pea aphid obligate endosymbiont | 656 | 574 | 1 | 126.9 | 86.7 | No | [12,51] |
| B. aphidicola BCc | cedar aphid obligate endosymbiont | 422 | 362 | 3 | 135.8 | 90.0 | No | [52] |
| S. glossinidius str. "morsitans" | tse-tse fly facultative symbiont | 4,293 | 2516 | 1501 | ND | 50.9 | 2.72 | [10,11] |
| W. pipientis wMel | D. melanogaster reproductive parasite | 1,268 | 1270 | 94 | ND | 80 | 7.7 | [44,53] |
| W. pipientis wRi | D. simulans reproductive parasite | 1,445 | 1150 | 114 | ND | ND | 10 | [54] |
| W. pipientis wPip | C. pipiens reproductive parasite | 1,482 | 1386 | 97 | ND | 82 | Yes | [55] |
| W. pipientis wBm | B. malayi obligate endosymbiont | 1,080 | 806 | 98 | ND | 67 | 5.4** | [56] |
| R. prowazekii str. Madrid E | human parasite | 1,112 | 835 | 12 | ND | 76 | 0.3 | [13] |
4.2. Wolbachia pipiensis, from Reproductive Parasite to Intracellular Mutualist
4.3. The Sodalis-like Group of Symbionts, at the Transition Point from Facultative to Obligate Symbionts
5. Concluding Remarks: Dynamics of Junk-DNA during the Evolutionary Reduction Process
Acknowledgments
References and Notes
- Gregory, T.R.; Nicol, J.A.; Tamm, H.; Kullman, B.; Kullman, K.; Leitch, I.J.; Murray, B.G.; Kapraun, D.F.; Greilhuber, J.; Bennett, M.D. Eukaryotic genome size databases. Nucleic Acids Res. 2007, 35, D332–D338. [Google Scholar]
- Gregory, T.R. Synergy between sequence and size in large-scale genomics. Nat. Rev. Genet. 2005, 6, 699–708. [Google Scholar] [CrossRef]
- Ohno, S. So much "Junk" DNA in our genome. Brookhaven Symp. Biol. 1972, 23, 366–370. [Google Scholar]
- Orgel, L.E.; Crick, F.H. Selfish DNA: The ultimate parasite. Nature 1980, 284, 604–607. [Google Scholar] [CrossRef]
- Delaye, L.; Gil, R.; Pereto, J.; Latorre, A.; Moya, A. Life with a few genes: A survey on naturally evolved reduced genomes. Open Evol. J. 2010, 4, 12–22. [Google Scholar] [CrossRef]
- Achaz, G.; Coissac, E.; Netter, P.; Rocha, E.P. Associations between inverted repeats and the structural evolution of bacterial genomes. Genetics 2003, 164, 1279–1289. [Google Scholar]
- Giovannoni, S.J.; Tripp, H.J.; Givan, S.; Podar, M.; Vergin, K.L.; Baptista, D.; Bibbs, L.; Eads, J.; Richardson, T.H.; Noordewier, M.; et al. Genome streamlining in a cosmopolitan oceanic bacterium. Science 2005, 309, 1242–1245. [Google Scholar]
- Cole, S.T.; Eiglmeier, K.; Parkhill, J.; James, K.D.; Thomson, N.R.; Wheeler, P.R.; Honore, N.; Garnier, T.; Churcher, C.; Harris, D.; et al. Massive gene decay in the leprosy bacillus. Nature 2001, 409, 1007–1011. [Google Scholar] [CrossRef]
- Burke, G.R.; Moran, N.A. Massive genomic decay in Serratia symbiotica, a recently evolved symbiont of aphids. Genome Biol. Evol. 2011, 3, 195–208. [Google Scholar] [CrossRef]
- Toh, H.; Weiss, B.L.; Perkin, S.A.; Yamashita, A.; Oshima, K.; Hattori, M.; Aksoy, S. Massive genome erosion and functional adaptations provide insights into the symbiotic lifestyle of Sodalis glossinidius in the tsetse host. Genome Res. 2006, 16, 149–156. [Google Scholar]
- Belda, E.; Moya, A.; Bentley, S.; Silva, F.J. Mobile genetic element proliferation and gene inactivation impact over the genome structure and metabolic capabilities of Sodalis glossinidius, the secondary endosymbiont of tsetse flies. BMC Genomics 2010, 11, 449. [Google Scholar] [CrossRef]
- Lamelas, A.; Gosalbes, M.J.; Manzano-Marin, A.; Pereto, J.; Moya, A.; Latorre, A. Serratia symbiotica from the aphid Cinara cedri: A missing link from facultative to obligate insect endosymbiont. PLoS Genet. 2011, 7, e1002357. [Google Scholar] [CrossRef]
- Andersson, S.G.; Zomorodipour, A.; Andersson, J.O.; Sicheritz-Ponten, T.; Alsmark, U.C.; Podowski, R.M.; Naslund, A.K.; Eriksson, A.S.; Winkler, H.H.; Kurland, C.G. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 1998, 396, 133–140. [Google Scholar] [CrossRef]
- Gil, R.; Belda, E.; Gosalbes, M.J.; Delaye, L.; Vallier, A.; Vincent-Monegat, C.; Heddi, A.; Silva, F.J.; Moya, A.; Latorre, A. Massive presence of insertion sequences in the genome of SOPE, the primary endosymbiont of the rice weevil Sitophilus oryzae. Int. Microbiol. 2008, 11, 41–48. [Google Scholar]
- Brussow, H.; Canchaya, C.; Hardt, W.D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef]
- Oliver, K.M.; Degnan, P.H.; Hunter, M.S.; Moran, N.A. Bacteriophages encode factors required for protection in a symbiotic mutualism. Science 2009, 325, 992–994. [Google Scholar] [CrossRef]
- Lima-Mendez, G.; Van Helden, J.; Toussaint, A.; Leplae, R. Prophinder: A computational tool for prophage prediction in prokaryotic genomes. Bioinformatics 2008, 24, 863–865. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Akhter, S.; Aziz, R.K.; Edwards, R.A. PhiSpy: A novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucleic Acids Res. 2012. [Google Scholar] [CrossRef]
- Canchaya, C.; Proux, C.; Fournous, G.; Bruttin, A.; Brussow, H. Prophage genomics. Microbiol. Mol. Biol. Rev. 2003, 67, 238–276. [Google Scholar] [CrossRef]
- Moya, A.; Pereto, J.; Gil, R.; Latorre, A. Learning how to live together: Genomic insights into prokaryote-animal symbioses. Nat. Rev. Genet. 2008, 9, 218–229. [Google Scholar] [CrossRef]
- Moran, N.A.; McCutcheon, J.P.; Nakabachi, A. Genomics and evolution of heritable bacterial symbionts. Annu. Rev. Genet. 2008, 42, 165–190. [Google Scholar] [CrossRef]
- Chain, P.S.; Carniel, E.; Larimer, F.W.; Lamerdin, J.; Stoutland, P.O.; Regala, W.M.; Georgescu, A.M.; Vergez, L.M.; Land, M.L.; Motin, V.L.; et al. Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia. pseudotuberculosis. Proc. Natl. Acad. Sci. USA 2004, 101, 13826–13831. [Google Scholar]
- Ogata, H.; Audic, S.; Renesto-Audiffren, P.; Fournier, P.E.; Barbe, V.; Samson, D.; Roux, V.; Cossart, P.; Weissenbach, J.; Claverie, J.M.; et al. Mechanisms of evolution in Rickettsia conorii and R. prowazekii. Science 2001, 293, 2093–2098. [Google Scholar] [CrossRef]
- Lerat, E.; Ochman, H. Recognizing the pseudogenes in bacterial genomes. Nucleic Acids Res. 2005, 33, 3125–3132. [Google Scholar] [CrossRef]
- Liu, Y.; Harrison, P.M.; Kunin, V.; Gerstein, M. Comprehensive analysis of pseudogenes in prokaryotes: Widespread gene decay and failure of putative horizontally transferred genes. Genome Biol. 2004, 5, R64. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Abby, S.; Daubin, V. Comparative genomics and the evolution of prokaryotes. Trends Microbiol. 2007, 15, 135–141. [Google Scholar] [CrossRef]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef]
- Gomez-Valero, L.; Rocha, E.P.; Latorre, A.; Silva, F.J. Reconstructing the ancestor of Mycobacterium leprae: The dynamics of gene loss and genome reduction. Genome Res. 2007, 17, 1178–1185. [Google Scholar] [CrossRef]
- Andersson, J.O.; Andersson, S.G. Pseudogenes, junk DNA, and the dynamics of Rickettsia genomes. Mol. Biol. Evol. 2001, 18, 829–839. [Google Scholar] [CrossRef]
- Perez-Brocal, V.; Latorre, A.; Gil, R.; Moya, A. Comparative analysis of two genomic regions among four strains of Buchnera aphidicola, primary endosymbiont of aphids. Gene 2005, 345, 73–80. [Google Scholar] [CrossRef]
- Dale, C.; Wang, B.; Moran, N.; Ochman, H. Loss of DNA recombinational repair enzymes in the initial stages of genome degeneration. Mol. Biol. Evol. 2003, 20, 1188–1194. [Google Scholar] [CrossRef]
- Gil, R.; Latorre, A.; Moya, A. Evolution of prokaryote-animal symbiosis from a genomics perspective. In (Endo) symbiotic Methanogenic Archaea; Hackstein, J.H.P., Ed.; Springer-Verlag: Berlin, Germany, 2010; pp. 207–233. [Google Scholar]
- Mahillon, J.; Chandler, M. Insertion sequences. Microbiol. Mol. Biol. Rev. 1998, 62, 725–774. [Google Scholar]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–36. [Google Scholar] [CrossRef]
- Nagy, Z.; Chandler, M. Regulation of transposition in bacteria. Res. Microbiol. 2004, 155, 387–398. [Google Scholar] [CrossRef]
- Wagner, A.; Lewis, C.; Bichsel, M. A survey of bacterial insertion sequences using IScan. Nucleic Acids Res. 2007, 35, 5284–5293. [Google Scholar] [CrossRef]
- Sawyer, S.A.; Dykhuizen, D.E.; DuBose, R.F.; Green, L.; Mutangadura-Mhlanga, T.; Wolczyk, D.F.; Hartl, D.L. Distribution and abundance of insertion sequences among natural isolates of Escherichia coli. Genetics 1987, 115, 51–63. [Google Scholar]
- Lawrence, J.G.; Ochman, H.; Hartl, D.L. The evolution of insertion sequences within enteric bacteria. Genetics 1992, 131, 9–20. [Google Scholar]
- Wagner, A. Periodic extinctions of transposable elements in bacterial lineages: Evidence from intragenomic variation in multiple genomes. Mol. Biol. Evol. 2006, 23, 723–733. [Google Scholar] [CrossRef]
- Touchon, M.; Rocha, E.P. Causes of insertion sequences abundance in prokaryotic genomes. Mol. Biol. Evol. 2007, 24, 969–981. [Google Scholar] [CrossRef]
- Newton, I.L.; Bordenstein, S.R. Correlations between bacterial ecology and mobile DNA. Curr. Microbiol. 62 2010, 198–208. [Google Scholar]
- Bordenstein, S.R.; Reznikoff, W.S. Mobile DNA in obligate intracellular bacteria. Nat. Rev. Microbiol. 2005, 3, 688–699. [Google Scholar] [CrossRef]
- Dale, C.; Moran, N.A. Molecular interactions between bacterial symbionts and their hosts. Cell. 2006, 126, 453–465. [Google Scholar] [CrossRef]
- Moran, N.A.; Plague, G.R. Genomic changes following host restriction in bacteria. Curr. Opin. Genet. Dev. 2004, 14, 627–633. [Google Scholar] [CrossRef]
- Schneider, D.; Lenski, R.E. Dynamics of insertion sequence elements during experimental evolution of bacteria. Res. Microbiol. 2004, 155, 319–327. [Google Scholar] [CrossRef]
- Latorre, A.; Gil, R.; Silva, F.J.; Moya, A. Chromosomal stasis versus plasmid plasticity in aphid endosymbiont Buchnera aphidicola. Heredity 2005, 95, 339–347. [Google Scholar] [CrossRef]
- Singh, P.; Cole, S.T. Mycobacterium leprae: Genes, pseudogenes and genetic diversity. Future Microbiol. 6 2010, 57–71. [Google Scholar]
- Camus, J.C.; Pryor, M.J.; Medigue, C.; Cole, S.T. Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv. Microbiol. 2002, 148, 2967–2973. [Google Scholar]
- Shigenobu, S.; Watanabe, H.; Hattori, M.; Sakaki, Y.; Ishikawa, H. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature 2000, 407, 81–86. [Google Scholar] [CrossRef]
- Perez-Brocal, V.; Gil, R.; Ramos, S.; Lamelas, A.; Postigo, M.; Michelena, J.M.; Silva, F.J.; Moya, A.; Latorre, A. A small microbial genome: The end of a long symbiotic relationship? Science 2006, 314, 312–313. [Google Scholar]
- Wu, M.; Sun, L.V.; Vamathevan, J.; Riegler, M.; Deboy, R.; Brownlie, J.C.; McGraw, E.A.; Martin, W.; Esser, C.; Ahmadinejad, N.; et al. Phylogenomics of the reproductive parasite Wolbachia. pipientis wMel: A streamlined genome overrun by mobile genetic elements. PLoS Biol. 2004, 2, E69. [Google Scholar] [CrossRef] [Green Version]
- Klasson, L.; Westberg, J.; Sapountzis, P.; Naslund, K.; Lutnaes, Y.; Darby, A.C.; Veneti, Z.; Chen, L.; Braig, H.R.; Garrett, R.; et al. The mosaic genome structure of the Wolbachia. wRi strain infecting Drosophila simulans. Proc. Natl. Acad. Sci. USA 2009, 106, 5725–5730. [Google Scholar]
- Klasson, L.; Walker, T.; Sebaihia, M.; Sanders, M.J.; Quail, M.A.; Lord, A.; Sanders, S.; Earl, J.; O'Neill, S.L.; Thomson, N.; et al. Genome evolution of Wolbachia strain wPip from the Culex pipiens group. Mol. Biol. Evol. 2008, 25, 1877–1887. [Google Scholar] [CrossRef]
- Foster, J.; Ganatra, M.; Kamal, I.; Ware, J.; Makarova, K.; Ivanova, N.; Bhattacharyya, A.; Kapatral, V.; Kumar, S.; Posfai, J.; et al. The Wolbachia genome of Brugia malayi: Endosymbiont evolution within a human pathogenic nematode. PLoS Biol. 2005, 3, e121. [Google Scholar] [CrossRef] [Green Version]
- Silva, F.J.; Latorre, A.; Moya, A. Genome size reduction through multiple events of gene disintegration in Buchnera APS. Trends Genet. 2001, 17, 615–618. [Google Scholar] [CrossRef]
- Lo, N.; Paraskevopoulos, C.; Bourtzis, K.; O'Neill, S.L.; Werren, J.H.; Bordenstein, S.R.; Bandi, C. Taxonomic status of the intracellular bacterium Wolbachia pipientis. Int. J. Syst. Evol. Microbiol. 2007, 57, 654–657. [Google Scholar] [CrossRef]
- Salzberg, S.L.; Puiu, D.; Sommer, D.D.; Nene, V.; Lee, N.H. Genome sequence of the Wolbachia endosymbiont of Culex quinquefasciatus JHB. J. Bacteriol. 2009, 191, 1725. [Google Scholar] [CrossRef]
- Heddi, A.; Charles, H.; Khatchadourian, C.; Bonnot, G.; Nardon, P. Molecular characterization of the principal symbiotic bacteria of the weevil Sitophilus oryzae: A peculiar G+C content of an endocytobiotic DNA. J. Mol. Evol. 1998, 47, 52–61. [Google Scholar] [CrossRef]
- Dale, C.; Plague, G.R.; Wang, B.; Ochman, H.; Moran, N.A. Type III secretion systems and the evolution of mutualistic endosymbiosis. Proc. Natl. Acad. Sci. USA 2002, 99, 12397–12402. [Google Scholar] [CrossRef]
- Lefevre, C.; Charles, H.; Vallier, A.; Delobel, B.; Farrell, B.; Heddi, A. Endosymbiont phylogenesis in the Dryophthoridae weevils: Evidence for bacterial replacement. Mol. Biol. Evol. 2004, 21, 965–973. [Google Scholar] [CrossRef]
- Conord, C.; Despres, L.; Vallier, A.; Balmand, S.; Miquel, C.; Zundel, S.; Lemperiere, G.; Heddi, A. Long-term evolutionary stability of bacterial endosymbiosis in Curculionoidea: Additional evidence of symbiont replacement in the Dryophthoridae family. Mol. Biol. Evol. 2008, 859–868. [Google Scholar]
- Koonin, E.V.; Makarova, K.S.; Aravind, L. Horizontal gene transfer in prokaryotes: Quantification and classification. Annu. Rev. Microbiol. 2001, 55, 709–742. [Google Scholar] [CrossRef]
- Lawrence, J.G.; Hendrix, R.W.; Casjens, S. Where are the pseudogenes in bacterial genomes? Trends Microbiol. 2001, 9, 535–540. [Google Scholar] [CrossRef]
- Ochman, H.; Davalos, L.M. The nature and dynamics of bacterial genomes. Science 2006, 311, 1730–1733. [Google Scholar] [CrossRef]
- Mira, A.; Ochman, H.; Moran, N.A. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001, 17, 589–596. [Google Scholar] [CrossRef]
- Andersson, J.O.; Andersson, S.G. Insights into the evolutionary process of genome degradation. Curr. Opin. Genet. Dev. 1999, 9, 664–671. [Google Scholar] [CrossRef]
- Petrov, D.A.; Hartl, D.L. Pseudogene evolution and natural selection for a compact genome. J. Hered. 2000, 91, 221–227. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gil, R.; Latorre, A. Factors Behind Junk DNA in Bacteria. Genes 2012, 3, 634-650. https://doi.org/10.3390/genes3040634
Gil R, Latorre A. Factors Behind Junk DNA in Bacteria. Genes. 2012; 3(4):634-650. https://doi.org/10.3390/genes3040634
Chicago/Turabian StyleGil, Rosario, and Amparo Latorre. 2012. "Factors Behind Junk DNA in Bacteria" Genes 3, no. 4: 634-650. https://doi.org/10.3390/genes3040634