Identifying and Characterizing Regulatory Sequences in the Human Genome with Chromatin Accessibility Assays


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
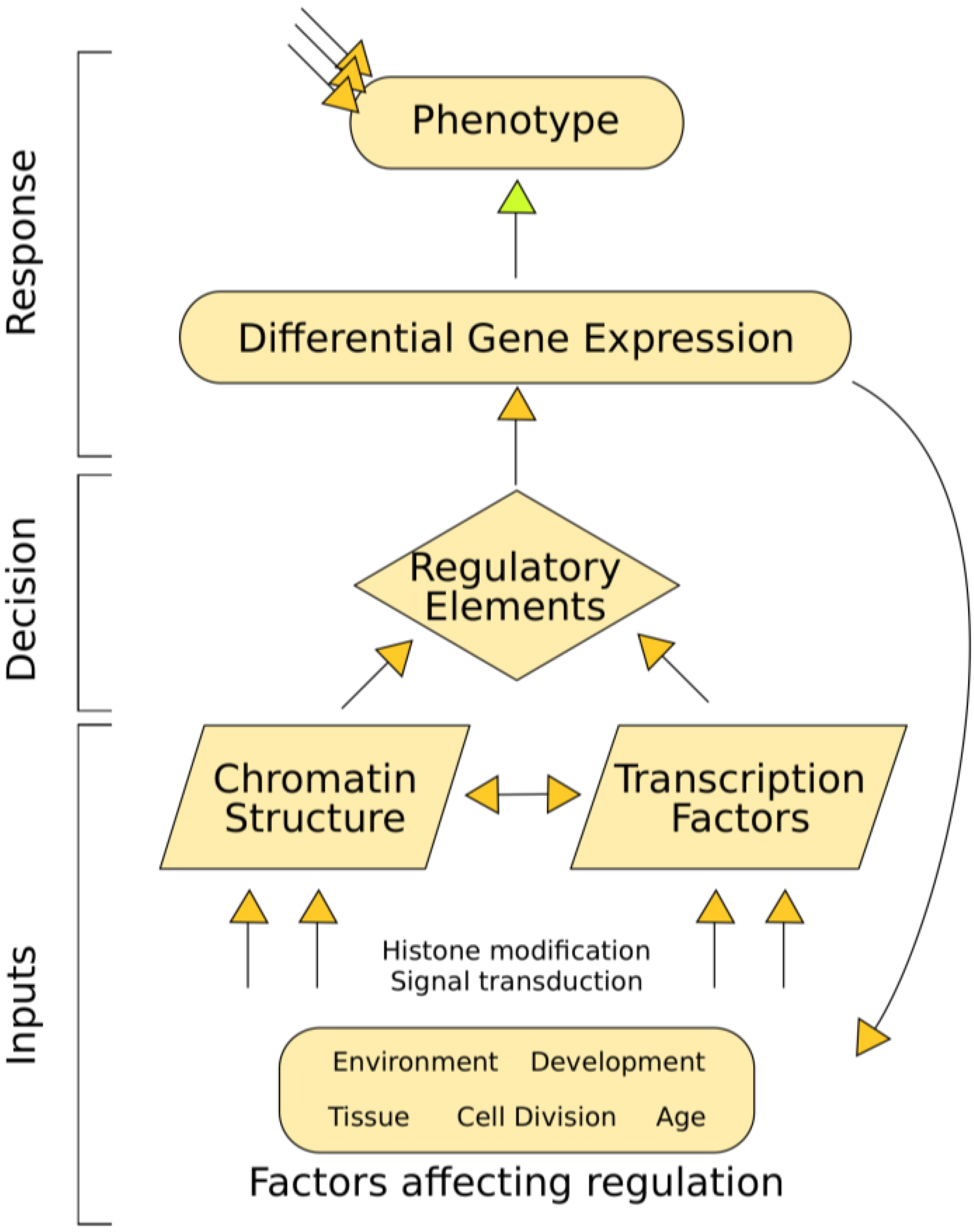
2. Background
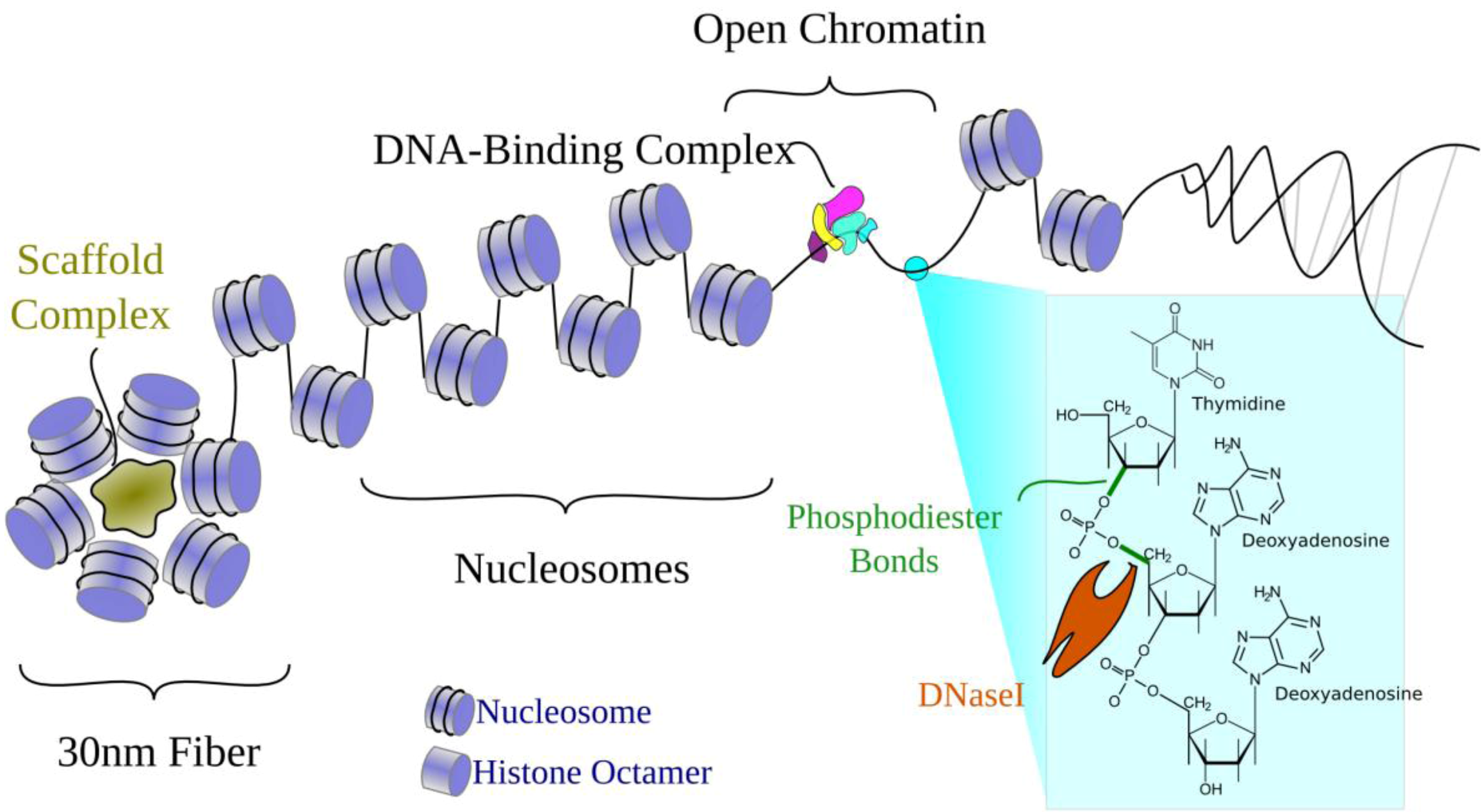
2.1. Chromatin Structure

2.2. Challenges to Studying Regulatory Elements
2.3. Open Chromatin and Regulatory Element Assays
2.3.1. DNaseI Hypersensitivity (DHS)
2.3.2. Formaldehyde-assisted Isolation of Regulatory Elements (FAIRE)
2.3.3. Chromatin Immunoprecipitation (ChIP)
2.3.4. Other Similar Assays
3. Identifying and Characterizing Regulatory Elements
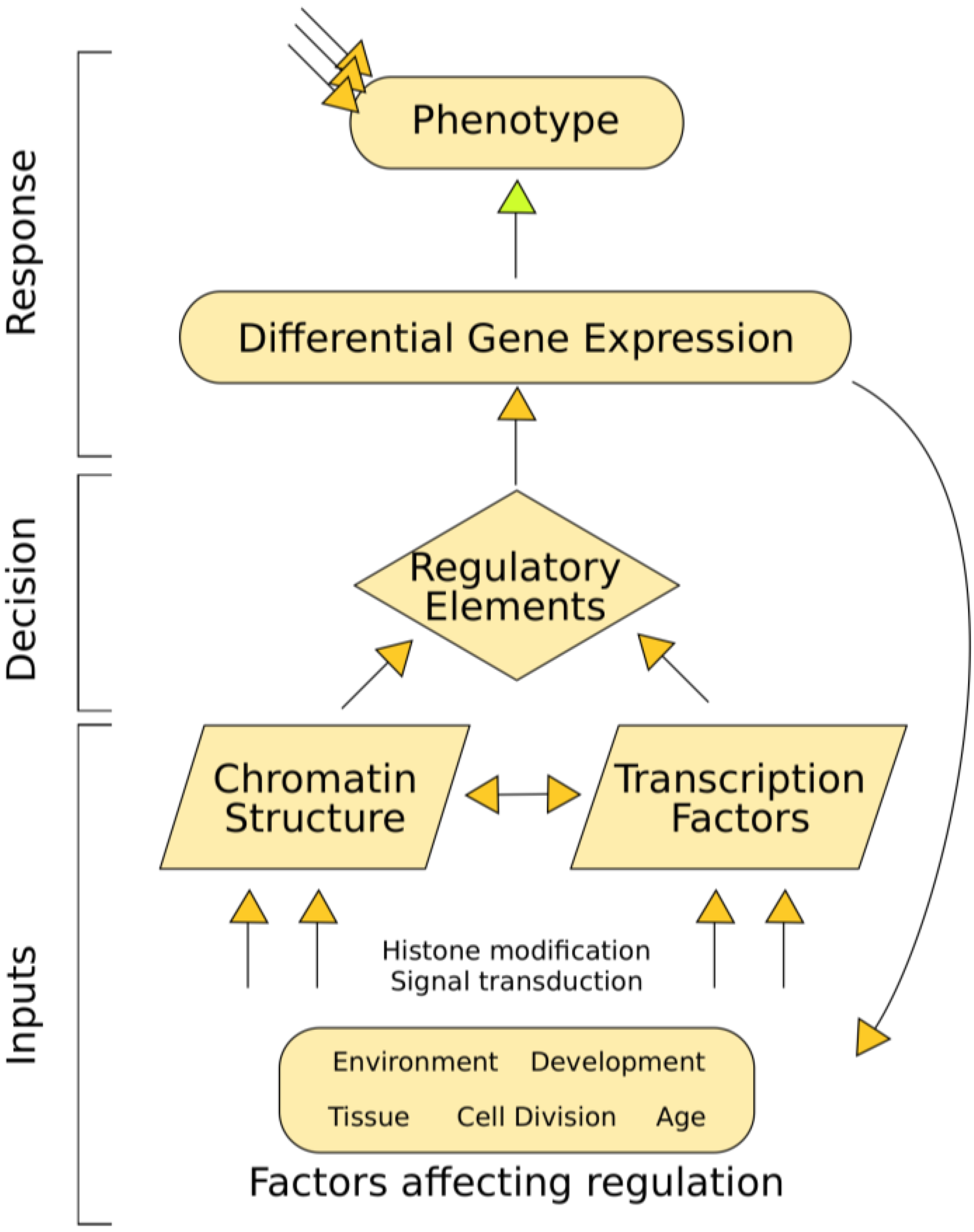
3.1. Open Chromatin Defines Regulatory Elements
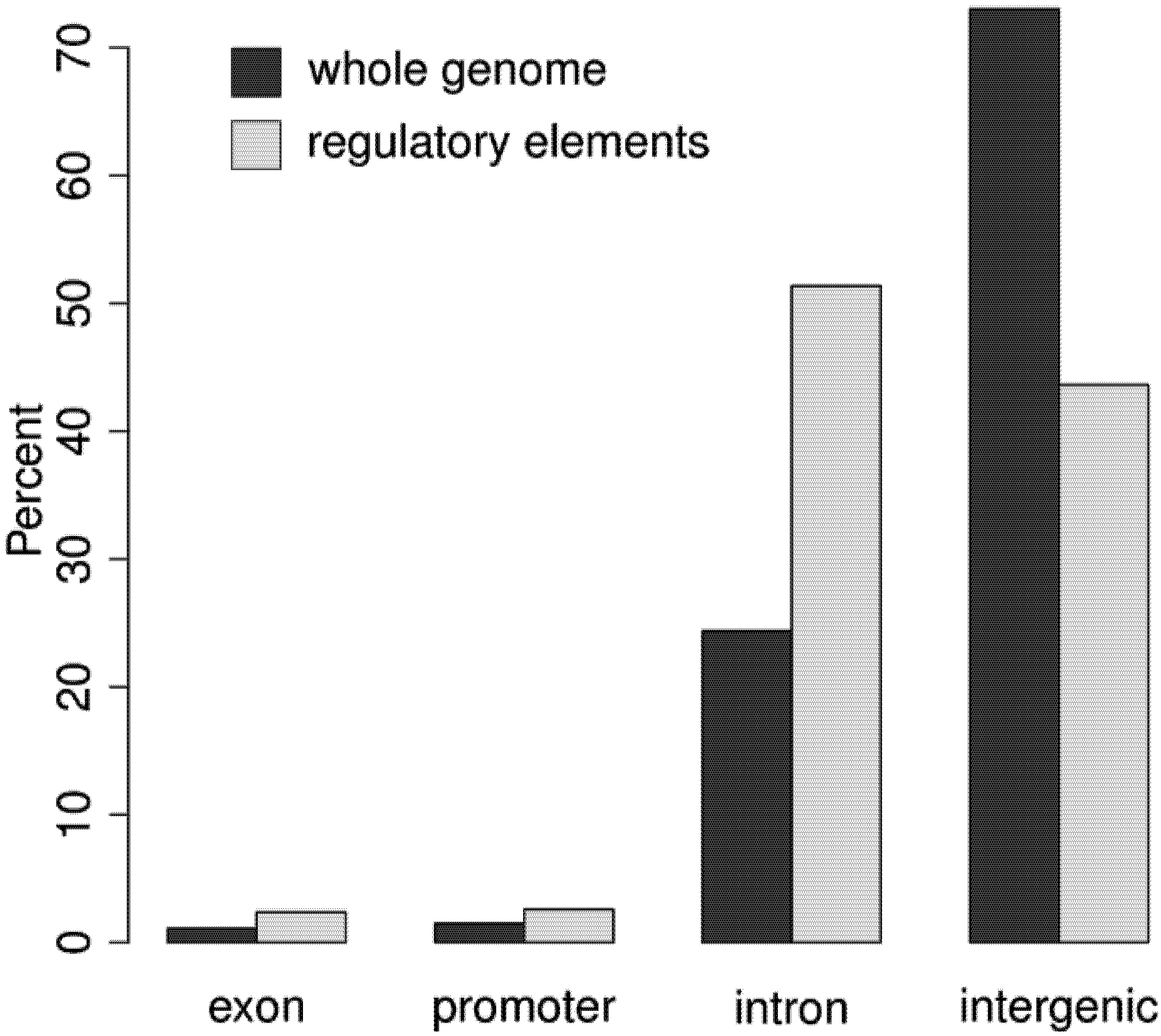
3.2. Regulatory Elements are Located in Promoter, Intergenic, and Intronic Regions
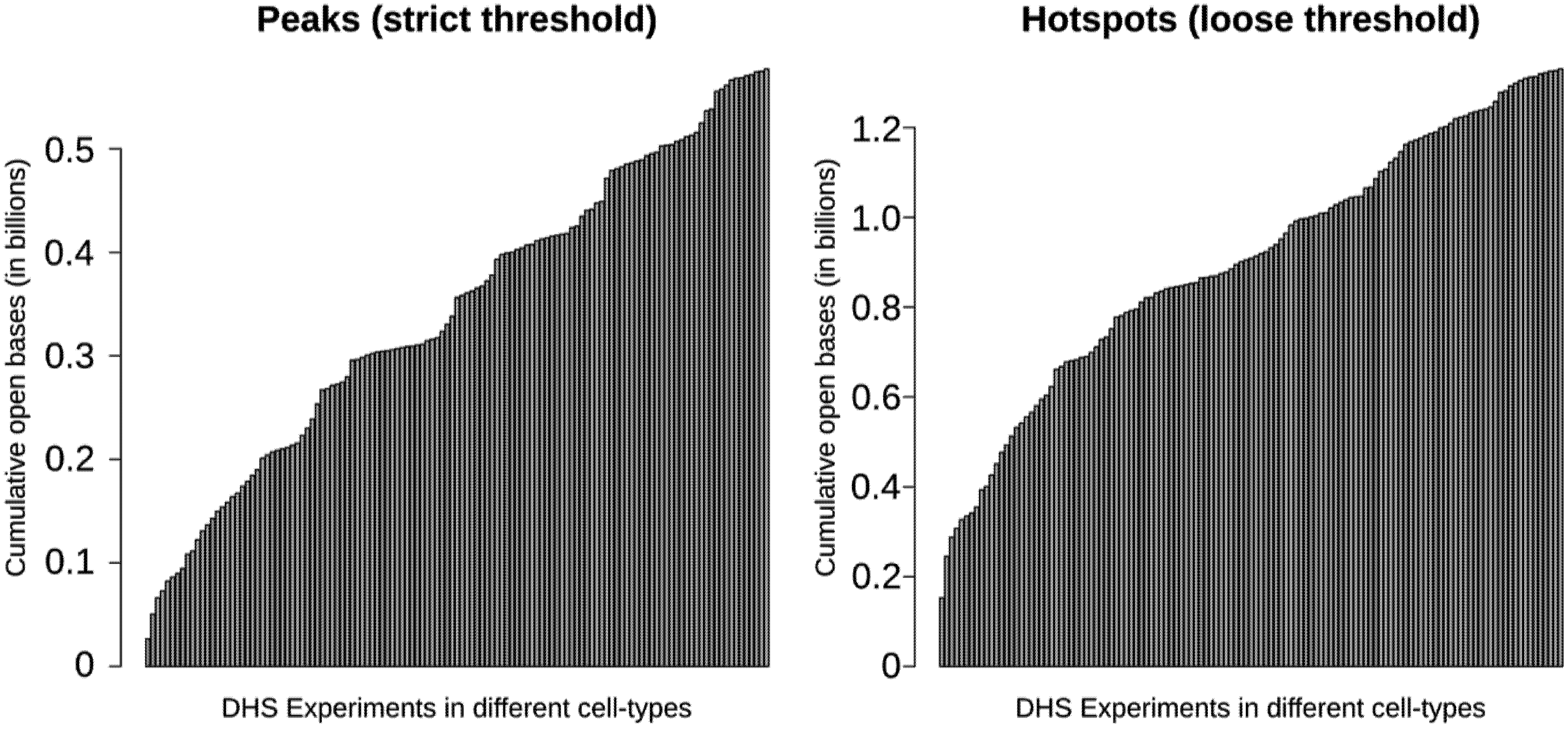
3.3. More than 30% of the Genome May be Regulatory
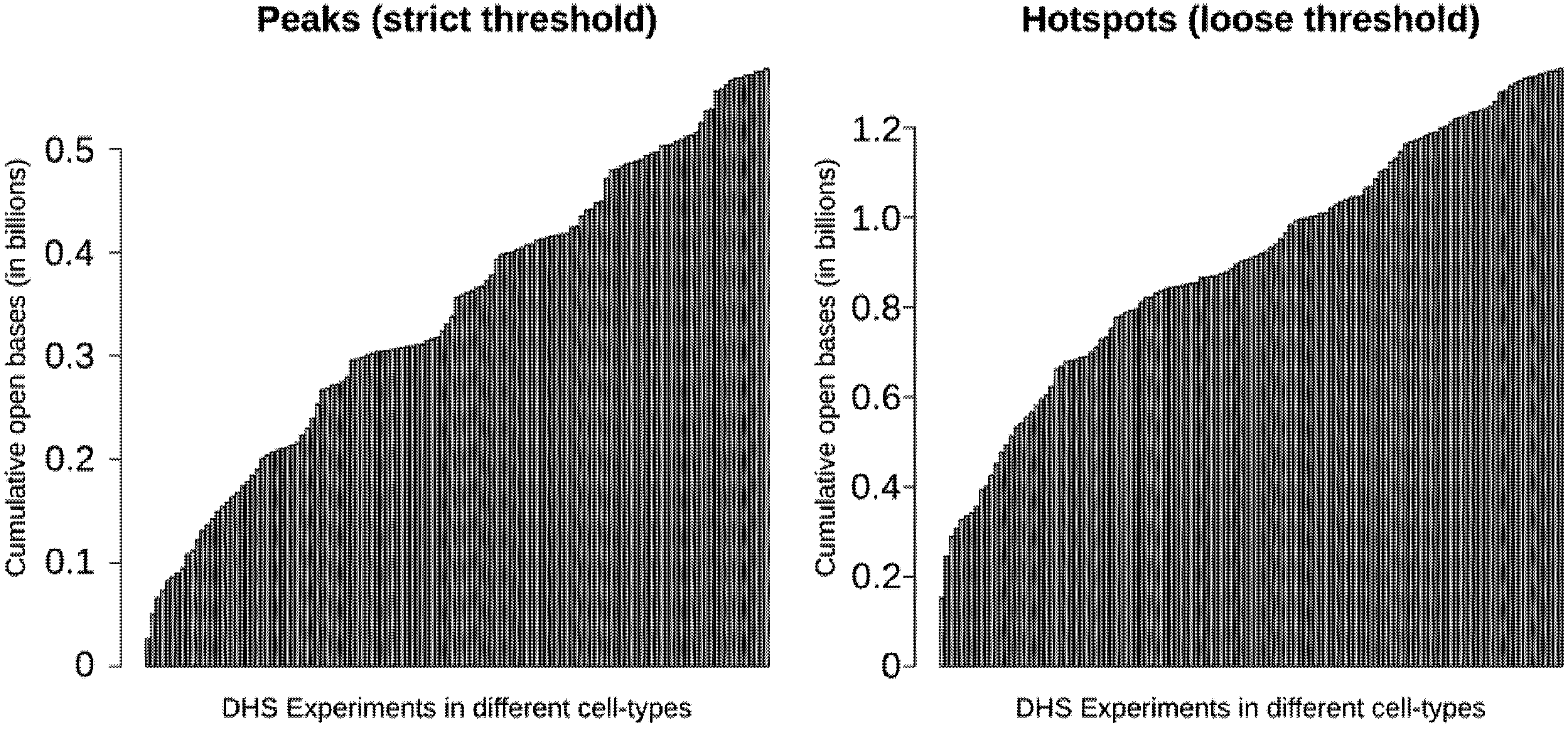
3.4. Most Regulatory Elements are Cell-Type-Specific

3.5. Transcription Factor Binding Affects Chromatin Structure
3.6. Chromatin Structure Affects Gene Expression
3.7. Regulatory Elements can be Classified by Factor, Function, or Cell-Type-Specificity
3.8. Perspectives
Acknowledgments
References and Notes
- Green, E.D.; Guyer, M.S. Charting a course for genomic medicine from base pairs to bedside. Nature 2011, 470, 204–213. [Google Scholar]
- Ohno, S. So much "junk" DNA in our genome. Brookhaven Symp Biol. 1972, 23, 366–370. [Google Scholar]
- Alexander, R.P.; Fang, G.; Rozowsky, J.; Snyder, M.; Gerstein, M.B. Annotating non-coding regions of the genome. Nat. Rev. Genet. 2010, 11, 559–571. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; Funke, R.; Gage, D.; Harris, K.; Heaford, A.; Howland, J.; Kann, L.; Lehoczky, J.; LeVine, R.; McEwan, P.; McKernan, K.; Meldrim, J.; Mesirov, J.P.; Miranda, C.; Morris, W.; Naylor, J.; Raymond, C.; Rosetti, M.; Santos, R.; Sheridan, A.; Sougnez, C.; Stange-Thomann, N.; Stojanovic, N.; Subramanian, A.; Wyman, D.; Rogers, J.; Sulston, J.; Ainscough, R.; Beck, S.; Bentley, D.; Burton, J.; Clee, C.; Carter, N.; Coulson, A.; Deadman, R.; Deloukas, P.; Dunham, A.; Dunham, I.; Durbin, R.; French, L.; Grafham, D.; Gregory, S.; Hubbard, T.; Humphray, S.; Hunt, A.; Jones, M.; Lloyd, C.; McMurray, A.; Matthews, L.; Mercer, S.; Milne, S.; Mullikin, J.C.; Mungall, A.; Plumb, R.; Ross, M.; Shownkeen, R.; Sims, S.; Waterston, R.H.; Wilson, R.K.; Hillier, L.W.; McPherson, J.D.; Marra, M.A.; Mardis, E.R.; Fulton, L.A.; Chinwalla, A.T.; Pepin, K.H.; Gish, W.R.; Chissoe, S.L.; Wendl, M.C.; Delehaunty, K.D.; Miner, T.L.; Delehaunty, A.; Kramer, J.B.; Cook, L.L.; Fulton, R.S.; Johnson, D.L.; Minx, P.J.; Clifton, S.W.; Hawkins, T.; Branscomb, E.; Predki, P.; Richardson, P.; Wenning, S.; Slezak, T.; Doggett, N.; Cheng, J.F.; Olsen, A.; Lucas, S.; Elkin, C.; Uberbacher, E.; Frazier, M.; Gibbs, R.A.; Muzny, D.M.; Scherer, S.E.; Bouck, J.B.; Sodergren, E.J.; Worley, K.C.; Rives, C.M.; Gorrell, J.H.; Metzker, M.L.; Naylor, S.L.; Kucherlapati, R.S.; Nelson, D.L.; Weinstock, G.M.; Sakaki, Y.; Fujiyama, A.; Hattori, M.; Yada, T.; Toyoda, A.; Itoh, T.; Kawagoe, C.; Watanabe, H.; Totoki, Y.; Taylor, T.; Weissenbach, J.; Heilig, R.; Saurin, W.; Artiguenave, F.; Brottier, P.; Bruls, T.; Pelletier, E.; Robert, C.; Wincker, P.; Smith, D.R.; Doucette-Stamm, L.; Rubenfield, M.; Weinstock, K.; Lee, H.M.; Dubois, J.; Rosenthal, A.; Platzer, M.; Nyakatura, G.; Taudien, S.; Rump, A.; Yang, H.; Yu, J.; Wang, J.; Huang, G.; Gu, J.; Hood, L.; Rowen, L.; Madan, A.; Qin, S.; Davis, R.W.; Federspiel, N.A.; Abola, A.P.; Proctor, M.J.; Myers, R.M.; Schmutz, J.; Dickson, M.; Grimwood, J.; Cox, D.R.; Olson, M.V.; Kaul, R.; Shimizu, N.; Kawasaki, K.; Minoshima, S.; Evans, G.A.; Athanasiou, M.; Schultz, R.; Roe, B.A.; Chen, F.; Pan, H.; Ramser, J.; Lehrach, H.; Reinhardt, R.; McCombie, W.R.; de la Bastide, M.; Dedhia, N.; Blöcker, H.; Hornischer, K.; Nordsiek, G.; Agarwala, R.; Aravind, L.; Bailey, J.A.; Bateman, A.; Batzoglou, S.; Birney, E.; Bork, P.; Brown, D.G.; Burge, C.B.; Cerutti, L.; Chen, H.C.; Church, D.; Clamp, M.; Copley, R.R.; Doerks, T.; Eddy, S.R.; Eichler, E.E.; Furey, T.S.; Galagan, J.; Gilbert, J.G.; Harmon, C.; Hayashizaki, Y.; Haussler, D.; Hermjakob, H.; Hokamp, K.; Jang, W.; Johnson, L.S.; Jones, T.A.; Kasif, S.; Kaspryzk, A.; Kennedy, S.; Kent, W.J.; Kitts, P.; Koonin, E.V.; Korf, I.; Kulp, D.; Lancet, D.; Lowe, T.M.; McLysaght, A.; Mikkelsen, T.; Moran, J.V.; Mulder, N.; Pollara, V.J.; Ponting, C.P.; Schuler, G.; Schultz, J.; Slater, G.; Smit, A.F.; Stupka, E.; Szustakowski, J.; Thierry-Mieg, D.; Thierry-Mieg, J.; Wagner, L.; Wallis, J.; Wheeler, R.; Williams, A.; Wolf, Y.I.; Wolfe, K.H.; Yang, S.P.; Yeh, R.F.; Collins, F.; Guyer, M.S.; Peterson, J.; Felsenfeld, A.; Wetterstrand, K.A.; Patrinos, A.; Morgan, M.J.; de Jong, P.; Catanese, J.J.; Osoegawa, K.; Shizuya, H.; Choi, S.; Chen, Y.J.; Szustakowki, J. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Schones, D.E.; Zhao, K. Genome-wide approaches to studying chromatin modifications. Nat. Rev. Genet. 2008, 9, 179–191. [Google Scholar] [CrossRef]
- Boyle, A.P.; Davis, S.; Shulha, H.P.; Meltzer, P.; Margulies, E.H.; Weng, Z.; Furey, T.S.; Crawford, G.E. High-resolution mapping and characterization of open chromatin across the genome. Cell 2008, 132, 311–322. [Google Scholar] [CrossRef]
- Song, L.; Crawford, G.E. DNase-seq: A high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. CSH Protoc. 2010, 2010. pdb.prot5384. [Google Scholar]
- Giresi, P.G.; Kim, J.; McDaniell, R.M.; Iyer, V.R.; Lieb, J.D. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007, 17, 877–885. [Google Scholar] [CrossRef]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-Wide Mapping of in Vivo Protein-DNA Interactions. Science 2007, 316, 1497–1502. [Google Scholar]
- Myers, R.M.; Stamatoyannopoulos, J.; Snyder, M.; Dunham, I.; Hardison, R.C.; Bernstein, B.E.; Gingeras, T.R.; Kent, W.J.; Birney, E.; Wold, B.; Crawford, G.E. A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 2011, 9, e1001046. [Google Scholar] [CrossRef] [Green Version]
- Elgin, S.C. Heterochromatin and gene regulation in Drosophila. Curr. Opin. Genet. Dev. 1996, 6, 193–202. [Google Scholar] [CrossRef]
- Wu, C.; Gilbert, W. Tissue-specific exposure of chromatin structure at the 5' terminus of the rat preproinsulin II gene. Proc. Natl. Acad. Sci. USA 1981, 78, 1577. [Google Scholar] [CrossRef]
- Weisbrod, S.; Weintraub, H. Isolation of a subclass of nuclear proteins responsible for conferring a DNase I-sensitive structure on globin chromatin. Proc. Natl. Acad. Sci. USA 1979, 76, 630–634. [Google Scholar] [CrossRef]
- Claverie, J.-M. Fewer genes, more noncoding RNA. Science 2005, 309, 1529–1530. [Google Scholar] [CrossRef]
- Lindblad-Toh, K.; Garber, M.; Zuk, O.; Lin, M.F.; Parker, B.J.; Washietl, S.; Kheradpour, P.; Ernst, J.; Jordan, G.; Mauceli, E.; Ward, L.D.; Lowe, C.B.; Holloway, A.K.; Clamp, M.; Gnerre, S.; Alföldi, J.; Beal, K.; Chang, J.; Clawson, H.; Cuff, J.; Di Palma, F.; Fitzgerald, S.; Flicek, P.; Guttman, M.; Hubisz, M.J.; Jaffe, D.B.; Jungreis, I.; Kent, W.J.; Kostka, D.; Lara, M.; Martins, A.L.; Massingham, T.; Moltke, I.; Raney, B.J.; Rasmussen, M.D.; Robinson, J.; Stark, A.; Vilella, A.J.; Wen, J.; Xie, X.; Zody, M.C.; Baldwin, J.; Bloom, T.; Chin, C.W.; Heiman, D.; Nicol, R.; Nusbaum, C.; Young, S.; Wilkinson, J.; Worley, K.C.; Kovar, C.L.; Muzny, D.M.; Gibbs, R.A.; Cree, A.; Dihn, H.H.; Fowler, G.; Jhangiani, S.; Joshi, V.; Lee, S.; Lewis, L.R.; Nazareth, L.V.; Okwuonu, G.; Santibanez, J.; Warren, W.C.; Mardis, E.R.; Weinstock, G.M.; Wilson, R.K.; Delehaunty, K.; Dooling, D.; Fronik, C.; Fulton, L.; Fulton, B.; Graves, T.; Minx, P.; Sodergren, E.; Birney, E.; Margulies, E.H.; Herrero, J.; Green, E.D.; Haussler, D.; Siepel, A.; Goldman, N.; Pollard, K.S.; Pedersen, J.S.; Lander, E.S.; Kellis, M. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 2011, 478, 476–482. [Google Scholar] [Green Version]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; Garg, K.; Sandstron, R.; Bates, D.; Canfield, T.K.; Diegel, M.; Dunn, D.; Ebrsol, A.K.; Frum, T.; Giste, E.; Harding, L.; Johnson, A.K.; Johnson, E.M.; Kutyavin, T.; Lajoie, B.; Lee, B.-K.; Lee, K.; London, D.; Lotakis, D.; Neph, S.; Fidencio, N.; Nguyen, E.D.; Reynolds, A.P.; Roach, V.; Safi, A.; Sanchez, M.E.; Sanyal, A.; Shafer, A.; Simon, J.M.; Song, L.; Vong, S.; Weaver, M.; Zhang, Z.; Zhang, Z.; Lenhard, B.; Tewari, M.; Hansen, R.S.; Navas, P.A.; Sunyaev, S.R.; Akey, J.M.; Sabo, P.J.; Kaul, R.; Iyer, V.R.; Lieb, J.D.; Furey, T.S.; Decker, J.; Crawford, G.E.; Stamatoyannopoulos, J.A. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar]
- Sandelin, A.; Wasserman, W.W.; Lenhard, B. ConSite: web-based prediction of regulatory elements using cross-species comparison. Nucleic Acids Res. 2004, 32, W249–W252. [Google Scholar] [CrossRef]
- Ewan Birney, J.A.S.; Anindya Dutta, R.G.; Thomas, R.G.; Elliott, H.M.; Zhiping Weng, M.S.; Emmanouil, T.D.; John, A.S.; Robert, E.T.; Michael, S.K.; Christopher, M.T.; Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigó, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; Kuehn, M.S.; Taylor, C.M.; Neph, S.; Koch, C.M.; Asthana, S.; Malhotra, A.; Adzhubei, I.; Greenbaum, J.A.; Andrews, R.M.; Flicek, P.; Boyle, P.J.; Cao, H.; Carter, N.P.; Clelland, G.K.; Davis, S.; Day, N.; Dhami, P.; Dillon, S.C.; Dorschner, M.O.; Fiegler, H.; Giresi, P.G.; Goldy, J.; Hawrylycz, M.; Haydock, A.; Humbert, R.; James, K.D.; Johnson, B.E.; Johnson, E.M.; Frum, T.T.; Rosenzweig, E.R.; Karnani, N.; Lee, K.; Lefebvre, G.C.; Navas, P.A.; Neri, F.; Parker, S.C.J.; Sabo, P.J.; Sandstrom, R.; Shafer, A.; Vetrie, D.; Weaver, M.; Wilcox, S.; Yu, M.; Collins, F.S.; Dekker, J.; Lieb, J.D.; Tullius, T.D.; Crawford, G.E.; Sunyaev, S.; Noble, W.S.; Dunham, I.; Denoeud, F.; Reymond, A.; Kapranov, P.; Rozowsky, J.; Zheng, D.; Castelo, R.; Frankish, A.; Harrow, J.; Ghosh, S.; Sandelin, A.; Hofacker, I.L.; Baertsch, R.; Keefe, D.; Dike, S.; Cheng, J.; Hirsch, H.A.; Sekinger, E.A.; Lagarde, J.; Abril, J.F.; Shahab, A.; Flamm, C.; Fried, C.; Hackermüller, J.; Hertel, J.; Lindemeyer, M.; Missal, K.; Tanzer, A.; Washietl, S.; Korbel, J.; Emanuelsson, O.; Pedersen, J.S.; Holroyd, N.; Taylor, R.; Swarbreck, D.; Matthews, N.; Dickson, M.C.; Thomas, D.J.; Weirauch, M.T.; Gilbert, J.; Drenkow, J.; Bell, I.; Zhao, X.; Srinivasan, K.G.; Sung, W.-K.; Ooi, H.S.; Chiu, K.P.; Foissac, S.; Alioto, T.; Brent, M.; Pachter, L.; Tress, M.L.; Valencia, A.; Choo, S.W.; Choo, C.Y.; Ucla, C.; Manzano, C.; Wyss, C.; Cheung, E.; Clark, T.G.; Brown, J.B.; Ganesh, M.; Patel, S.; Tammana, H.; Chrast, J.; Henrichsen, C.N.; Kai, C.; Kawai, J.; Nagalakshmi, U.; Wu, J.; Lian, Z.; Lian, J.; Newburger, P.; Zhang, X.X.; Bickel, P.; Mattick, J.S.; Carninci, P.; Hayashizaki, Y.; Weissman, S.; Hubbard, T.; Myers, R.M.; Rogers, J.; Stadler, P.F.; Lowe, T.M.; Wei, C.-L.; Ruan, Y.; Struhl, K.; Gerstein, M.; Antonarakis, S.E.; Fu, Y.; Green, E.D.; Karaöz, U.; Siepel, A.; Taylor, J.; Liefer, L.A.; Wetterstrand, K.A.; Good, P.J.; Feingold, E.A.; Guyer, M.S.; Cooper, G.M.; Asimenos, G.; Dewey, C.N.; Hou, M.; Nikolaev, S.; Montoya-Burgos, J.I.; Löytynoja, A.; Whelan, S.; Pardi, F.; Massingham, T.; Huang, H.; Zhang, N.R.; Holmes, I.; Mullikin, J.C.; Ureta-Vidal, A.; Paten, B.; Seringhaus, M.; Church, D.; Rosenbloom, K.; Kent, W.J.; Stone, E.A.; Batzoglou, S.; Goldman, N.; Hardison, R.C.; Haussler, D.; Miller, W.; Sidow, A.; Trinklein, N.D.; Zhang, Z.D.; Barrera, L.; Stuart, R.; King, D.C.; Ameur, A.; Enroth, S.; Bieda, M.C.; Kim, J.; Bhinge, A.A.; Jiang, N.; Liu, J.; Yao, F.; Vega, V.B.; Lee, C.W.H.; Ng, P.; Yang, A.; Moqtaderi, Z.; Zhu, Z.; Xu, X.; Squazzo, S.; Oberley, M.J.; Inman, D.; Singer, M.A.; Richmond, T.A.; Munn, K.J.; Rada-Iglesias, A.; Wallerman, O.; Komorowski, J.; Fowler, J.C.; Couttet, P.; Bruce, A.W.; Dovey, O.M.; Ellis, P.D.; Langford, C.F.; Nix, D.A.; Euskirchen, G.; Hartman, S.; Urban, A.E.; Kraus, P.; Van Calcar, S.; Heintzman, N.; Kim, T.H.; Wang, K.; Qu, C.; Hon, G.; Luna, R.; Glass, C.K.; Rosenfeld, M.G.; Aldred, S.F.; Cooper, S.J.; Halees, A.; Lin, J.M.; Shulha, H.P.; Xu, M.; Haidar, J.N.S.; Yu, Y.; Iyer, V.R.; Green, R.D.; Wadelius, C.; Farnham, P.J.; Ren, B.; Harte, R.A.; Hinrichs, A.S.; Trumbower, H.; Clawson, H.; Hillman-Jackson, J.; Zweig, A.S.; Smith, K.; Thakkapallayil, A.; Barber, G.; Kuhn, R.M.; Karolchik, D.; Armengol, L.; Bird, C.P.; de Bakker, P.I.W.; Kern, A.D.; Lopez-Bigas, N.; Martin, J.D.; Stranger, B.E.; Woodroffe, A.; Davydov, E.; Dimas, A.; Eyras, E.; Hallgrímsdóttir, I.B.; Huppert, J.; Zody, M.C.; Abecasis, G.R.; Estivill, X.; Bouffard, G.G.; Guan, X.; Hansen, N.F.; Idol, J.R.; Maduro, V.V.B.; Maskeri, B.; McDowell, J.C.; Park, M.; Thomas, P.J.; Young, A.C.; Blakesley, R.W.; Muzny, D.M.; Sodergren, E.; Wheeler, D.A.; Worley, K.C.; Jiang, H.; Weinstock, G.M.; Gibbs, R.A.; Graves, T.; Fulton, R.; Mardis, E.R.; Wilson, R.K.; Clamp, M.; Cuff, J.; Gnerre, S.; Jaffe, D.B.; Chang, J.L.; Lindblad-Toh, K.; Lander, E.S.; Koriabine, M.; Nefedov, M.; Osoegawa, K.; Yoshinaga, Y.; Zhu, B.; de Jong, P.J. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar]
- McGaughey, D.M.; Vinton, R.M.; Huynh, J.; Al-Saif, A.; Beer, M.A.; McCallion, A.S. Metrics of sequence constraint overlook regulatory sequences in an exhaustive analysis at phox2b. Genome Res. 2008, 18, 252–260. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Ren, B. Finding distal regulatory elements in the human genome. Curr. Opin. Genet. Dev. 2009, 19, 541–549. [Google Scholar] [CrossRef]
- Amano, T.; Sagai, T.; Tanabe, H.; Mizushina, Y.; Nakazawa, H.; Shiroishi, T. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev. Cell 2009, 16, 47–57. [Google Scholar] [CrossRef]
- Visser, M.; Kayser, M.; Palstra, R.J. HERC2 rs12913832 modulates human pigmentation by attenuating chromatin-loop formation between a long-range enhancer and the OCA2 promoter. Genome Res. 2012, 22, 446–455. [Google Scholar] [CrossRef]
- Spilianakis, C.G.; Lalioti, M.D.; Town, T.; Lee, G.R.; Flavell, R.A. Interchromosomal associations between alternatively expressed loci. Nature 2005, 435, 637–645. [Google Scholar] [CrossRef]
- Maston, G.A.; Evans, S.K.; Green, M.R. Transcriptional regulatory elements in the human genome. Annu. Rev. Genomics Hum. Genet. 2006, 7, 29–59. [Google Scholar] [CrossRef]
- Bulger, M.; Groudine, M. Functional and mechanistic diversity of distal transcription enhancers. Cell 2011, 144, 327–339. [Google Scholar] [CrossRef]
- Hou, C.; Zhao, H.; Tanimoto, K.; Dean, A. CTCF-dependent enhancer-blocking by alternative chromatin loop formation. Proc. Natl. Acad. Sci. USA 2008, 105, 20398–20403. [Google Scholar] [CrossRef]
- West, A.G.; Gaszner, M.; Felsenfeld, G. Insulators: many functions, many mechanisms. Gene. Dev. 2002, 16, 271–288. [Google Scholar] [CrossRef]
- Dean, A. On a chromosome far, far away: LCRs and gene expression. TRENDS Genet. 2006, 22, 38–45. [Google Scholar] [CrossRef]
- Simon, J.A.; Kingston, R.E. Mechanisms of polycomb gene silencing: Knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009, 10, 697–708. [Google Scholar]
- Carninci, P.; Sandelin, A.; Lenhard, B.; Katayama, S.; Shimokawa, K.; Ponjavic, J.; Semple, C.A.; Taylor, M.S.; Engstrom, P.G.; Frith, M.C.; Forrest, A.R.; Alkema, W.B.; Tan, S.L.; Plessy, C.; Kodzius, R.; Ravasi, T.; Kasukawa, T.; Fukuda, S.; Kanamori-Katayama, M.; Kitazume, Y.; Kawaji, H.; Kai, C.; Nakamura, M.; Konno, H.; Nakano, K.; Mottagui-Tabar, S.; Arner, P.; Chesi, A.; Gustincich, S.; Persichetti, F.; Suzuki, H.; Grimmond, S.M.; Wells, C.A.; Orlando, V.; Wahlestedt, C.; Liu, E.T.; Harbers, M.; Kawai, J.; Bajic, V.B.; Hume, D.A.; Hayashizaki, Y. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 2006, 38, 626–635. [Google Scholar]
- Lee, D.; Karchin, R.; Beer, M.A. Discriminative prediction of mammalian enhancers from DNA sequence. Genome Res. 2011, 21, 2167–2180. [Google Scholar] [CrossRef]
- Wu, C. The 5[prime] ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nature 1980, 286, 854–860. [Google Scholar] [CrossRef]
- Felsenfeld, G.; Groudine, M. Controlling the double helix. Nature 2003, 421, 448–453. [Google Scholar] [CrossRef]
- Gross, D.S.; Garrard, W.T. Nuclease Hypersensitive Sites in Chromatin. Annu. Rev. Biochem. 1988, 57, 159–197. [Google Scholar] [CrossRef]
- Song, L.; Zhang, Z.; Grasfeder, L.L.; Boyle, A.P.; Giresi, P.G.; Lee, B.-K.; Sheffield, N.C.; Gräf, S.; Huss, M.; Keefe, D.; Liu, Z.; London, D.; McDaniell, R.M.; Shibata, Y.; Showers, K.A.; Simon, J.M.; Vales, T.; Wang, T.; Winter, D.; Zhang, Z.; Clarke, N.D.; Birney, E.; Iyer, V.R.; Crawford, G.E.; Lieb, J.D.; Furey, T.S. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011, 21, 1757–1767. [Google Scholar] [CrossRef]
- Boyle, A.P.; Song, L.; Lee, B.-K.; London, D.; Keefe, D.; Birney, E.; Iyer, V.R.; Crawford, G.E.; Furey, T.S. High-resolution genome-wide in vivo footprinting of diverse transcription factors in human cells. Genome Res. 2011, 21, 456–464. [Google Scholar] [CrossRef]
- Pique-Regi, R.; Degner, J.F.; Pai, A.A.; Gaffney, D.J.; Gilad, Y.; Pritchard, J.K. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res. 2011, 21, 447–455. [Google Scholar] [CrossRef]
- Solomon, M.J.; Larsen, P.L.; Varshavsky, A. Mapping protein-DNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene. Cell 1988, 53, 937–947. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; Ching, K.A.; Antosiewicz-Bourget, J.E.; Liu, H.; Zhang, X.; Green, R.D.; Lobanenkov, V.V.; Stewart, R.; Thomson, J.A.; Crawford, G.E.; Kellis, M.; Ren, B. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [Green Version]
- Dahl, J.A.; Collas, P. A rapid micro chromatin immunoprecipitation assay (microChIP). Nature Protoc. 2008, 3, 1032–1045. [Google Scholar] [CrossRef]
- Statham, A.L.; Robinson, M.D.; Song, J.Z.; Coolen, M.W.; Stirzaker, C.; Clark, S.J. Bisulfite sequencing of chromatin immunoprecipitated DNA (BisChIP-seq) directly informs methylation status of histone-modified DNA. Genome Res. 2012, 22, 1120–1127. [Google Scholar] [CrossRef]
- Rhee, H.S.; Pugh, B.F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 2011, 147, 1408–1419. [Google Scholar] [CrossRef]
- Egelhofer, T.A.; Minoda, A.; Klugman, S.; Lee, K.; Kolasinska-Zwierz, P.; Alekseyenko, A.A.; Cheung, M.S.; Day, D.S.; Gadel, S.; Gorchakov, A.A.; Gu, T.; Kharchenko, P.V.; Kuan, S.; Latorre, I.; Linder-Basso, D.; Luu, Y.; Ngo, Q.; Perry, M.; Rechtsteiner, A.; Riddle, N.C.; Schwartz, Y.B.; Shanower, G.A.; Vielle, A.; Ahringer, J.; Elgin, S.C.; Kuroda, M.I.; Pirrotta, V.; Ren, B.; Strome, S.; Park, P.J.; Karpen, G.H.; Hawkins, R.D.; Lieb, J.D. An assessment of histone-modification antibody quality. Nat. Struct. Mol. Biol. 2011, 18, 91–93. [Google Scholar]
- Schones, D.E.; Cui, K.; Cuddapah, S.; Roh, T.-Y.Y.; Barski, A.; Wang, Z.; Wei, G.; Zhao, K. Dynamic regulation of nucleosome positioning in the human genome. Cell 2008, 132, 887–898. [Google Scholar] [CrossRef]
- Auerbach, R.K.; Euskirchen, G.; Rozowsky, J.; Lamarre-Vincent, N.; Moqtaderi, Z.; Lefrançois, P.; Struhl, K.; Gerstein, M.; Snyder, M. Mapping accessible chromatin regions using Sono-Seq. Proc. Natl. Acad. Sci. USA 2009, 106, 14926–14931. [Google Scholar]
- You, J.S.; Kelly, T.K.; De Carvalho, D.D.; Taberlay, P.C.; Liang, G.; Jones, P.A. OCT4 establishes and maintains nucleosome-depleted regions that provide additional layers of epigenetic regulation of its target genes. Proc. Natl. Acad. Sci USA 2011, 108, 14497–14502. [Google Scholar]
- Rosenbloom, K.R.; Dreszer, T.R.; Long, J.C.; Malladi, V.S.; Sloan, C.A.; Raney, B.J.; Cline, M.S.; Karolchik, D.; Barber, G.P.; Clawson, H.; Diekhans, M.; Fujita, P.A.; Goldman, M.; Gravell, R.C.; Harte, R.A.; Hinrichs, A.S.; Kirkup, V.M.; Kuhn, R.M.; Learned, K.; Maddren, M.; Meyer, L.R.; Pohl, A.; Rhead, B.; Wong, M.C.; Zweig, A.S.; Haussler, D.; Kent, W.J. ENCODE whole-genome data in the UCSC Genome Browser: update 2012. Nucleic Acids Res. 2012, 40, D912–D917. [Google Scholar]
- Park, P.J. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef]
- Collas, P. The current state of chromatin immunoprecipitation. Mol. Biotechnol. 2010, 45, 87–100. [Google Scholar] [CrossRef]
- Majewski, J.; Ott, J. Distribution and characterization of regulatory elements in the human genome. Genome Res. 2002, 12, 1827–1836. [Google Scholar] [CrossRef]
- Gaulton, K.J.; Nammo, T.; Pasquali, L.; Simon, J.M.; Giresi, P.G.; Fogarty, M.P.; Panhuis, T.M.; Mieczkowski, P.; Secchi, A.; Bosco, D.; Berney, T.; Montanya, E.; Mohlke, K.L.; Lieb, J.D.; Ferrer, J. A map of open chromatin in human pancreatic islets. Nat. Genet. 2010, 42, 255–259. [Google Scholar]
- Phillips, J.E.; Corces, V.G. CTCF: Master Weaver of the Genome. Cell 2009, 137, 1194–1211. [Google Scholar] [CrossRef]
- Hou, C.; Dale, R.; Dean, A. Cell type specificity of chromatin organization mediated by CTCF and cohesin. Proc. Natl. Acad. Sci. USA 2010, 107, 3651–3656. [Google Scholar] [CrossRef]
- Biddie, S.C.; John, S.; Sabo, P.J.; Thurman, R.E.; Johnson, T.A.; Schiltz, R.L.; Miranda, T.B.; Sung, M.-H.; Trump, S.; Lightman, S.L.; Vinson, C.; Stamatoyannopoulos, J.A.; Hager, G.L. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol. Cell 2011, 43, 145–155. [Google Scholar] [CrossRef]
- Shibata, Y.; Sheffield, N.C.; Fedrirgo, O.; Babbitt, C.C.; Wortham, M.; Tewari, A.K.; London, D.; Song, L.; Lee, B.-K.; Iyer, V.R.; Parker, S.C.; Margulies, E.H.; Wray, G.A.; Furey, T.S.; Crawford, G.E. Extensive evolutionary changes in regulatory element activity during human origins are associated with altered gene expression and positive selection. PLoS Genet. 2012, i 8, e1002789. [Google Scholar]
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.A.; van Berkum, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; Taatjes, D.J.; Dekker, J.; Young, R.A. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010, 467, 430–435. [Google Scholar] [Green Version]
- Degner, J.F.; Pai, A.A.; Pique-Regi, R.; Veyrieras, J.-B.; Gaffney, D.J.; Pickrell, J.K.; De Leon, S.; Michelini, K.; Lewellen, N.; Crawford, G.E.; Stephens, M.; Gilad, Y.; Pritchard, J.K. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature 2012, 482, 390–394. [Google Scholar] [CrossRef]
- Beer, M.A.; Tavazoie, S. Predicting gene expression from sequence. Cell 2004, 117, 185–198. [Google Scholar] [CrossRef]
- Yuan, Y.; Guo, L.; Shen, L.; Liu, J.S. Predicting gene expression from sequence: a reexamination. PLoS Comput. Biol. 2007, 3, e243. [Google Scholar] [CrossRef]
- Segal, E.; Raveh-Sadka, T.; Schroeder, M.; Unnerstall, U.; Gaul, U. Predicting expression patterns from regulatory sequence in Drosophila segmentation. Nature 2008, 451, 535–540. [Google Scholar] [CrossRef]
- Natarajan, A.; Yardimci, G.G.; Sheffield, N.C.; Crawford, G.E.; Ohler, U. Predicting Cell-Type Specific Gene Expression from Regions of Open Chromatin. Genome Res. 2012, 22, 1711–1722. [Google Scholar] [CrossRef]
- Pérez-Lluch, S.; Blanco, E.; Carbonell, A.; Raha, D.; Snyder, M.; Serras, F.; Corominas, M. Genome-wide chromatin occupancy analysis reveals a role for ASH2 in transcriptional pausing. Nucleic Acids Res. 2011, 39, 4628–4639. [Google Scholar] [CrossRef]
- Reddy, T.E.; Pauli, F.; Sprouse, R.O.; Neff, N.F.; Newberry, K.M.; Garabedian, M.J.; Myers, R.M. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009, 19, 2163–2171. [Google Scholar] [CrossRef]
- Wontakal, S.N.; Guo, X.; Will, B.; Shi, M.; Raha, D.; Mahajan, M.C.; Weissman, S.; Snyder, M.; Steidl, U.; Zheng, D.; Skoultchi, A.I. A Large Gene Network in Immature Erythroid Cells Is Controlled by the Myeloid and B Cell Transcriptional Regulator PU. 1. PLoS Genet. 2011, 7, 15. [Google Scholar]
- Wu, J.Q.; Seay, M.; Schulz, V.P.; Hariharan, M.; Tuck, D.; Lian, J.; Du, J.; Shi, M.; Ye, Z.; Gerstein, M.; Snyder, M.P.; Weissman, S. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS Genet. 2012, 8, e1002565. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Tatonetti, N.P.; Landt, S.G.; Yang, X.; Slifer, T.; Altman, R.B.; Snyder, M. Cooperative transcription factor associations discovered using regulatory variation. Proc. Natl. Acad. Sci. USA 2011, 108, 13353–13358. [Google Scholar]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010, 28, 817–825. [Google Scholar] [CrossRef] [Green Version]
- Pennacchio, L.A.; Ahituv, N.; Moses, A.M.; Prabhakar, S.; Nobrega, M.A.; Shoukry, M.; Minovitsky, S.; Dubchak, I.; Holt, A.; Lewis, K.D.; Plajzer-Frick, I.; Akiyama, J.; De Val, S.; Afzal, V.; Black, B.L.; Couronne, O.; Eisen, M.B.; Visel, A.; Rubin, E.M. In vivo enhancer analysis of human conserved non-coding sequences. Nature 2006, 444, 499–502. [Google Scholar]
- Birney, E.; Lieb, J.D.; Furey, T.S.; Crawford, G.E.; Iyer, V.R. Allele-specific and heritable chromatin signatures in humans. Hum. Mol. Genet. 2010, 19, R204–R209. [Google Scholar] [CrossRef]
- Wei, G.; Zhao, K. 3C-based methods to detect long-range chromatin interactions. Front. Biol. 2011, 6, 76–81. [Google Scholar] [CrossRef]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef]
- Fullwood, M.J.; Liu, M.H.; Pan, Y.F.; Liu, J.; Xu, H.; Mohamed, Y.B.; Orlov, Y.L.; Velkov, S.; Ho, A.; Mei, P.H.; Chew, E.G.Y.; Huang, P.Y.H.; Welboren, W.-J.; Han, Y.; Ooi, H.S.; Ariyaratne, P.N.; Vega, V.B.; Luo, Y.; Tan, P.Y.; Choy, P.Y.; Wansa, K.D.S.A.; Zhao, B.; Lim, K.S.; Leow, S.C.; Yow, J.S.; Joseph, R.; Li, H.; Desai, K.V.; Thomsen, J.S.; Lee, Y.K.; Karuturi, R.K.M.; Herve, T.; Bourque, G.; Stunnenberg, H.G.; Ruan, X.; Cacheux-Rataboul, V.; Sung, W.-K.; Liu, E.T.; Wei, C.-L.; Cheung, E.; Ruan, Y. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009, 462, 58–64. [Google Scholar]
- Sexton, T.; Bantignies, F.; Cavalli, G. Genomic interactions: chromatin loops and gene meeting points in transcriptional regulation. Semin. Cell Dev. Biol. 2009, 20, 849–855. [Google Scholar] [CrossRef]
- Fraser, P.; Bickmore, W. Nuclear organization of the genome and the potential for gene regulation. Nature 2007, 447, 413–417. [Google Scholar] [CrossRef]
- Dekker, J. The three 'C' s of chromosome conformation capture: controls, controls, controls. Nat. Methods 2006, 3, 17–21. [Google Scholar] [CrossRef]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.D.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; Ku, M.; Durham, T.; Kellis, M.; Bernstein, B.E. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011, 473, 43–49. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sheffield, N.C.; Furey, T.S. Identifying and Characterizing Regulatory Sequences in the Human Genome with Chromatin Accessibility Assays. Genes 2012, 3, 651-670. https://doi.org/10.3390/genes3040651
Sheffield NC, Furey TS. Identifying and Characterizing Regulatory Sequences in the Human Genome with Chromatin Accessibility Assays. Genes. 2012; 3(4):651-670. https://doi.org/10.3390/genes3040651
Chicago/Turabian StyleSheffield, Nathan C., and Terrence S. Furey. 2012. "Identifying and Characterizing Regulatory Sequences in the Human Genome with Chromatin Accessibility Assays" Genes 3, no. 4: 651-670. https://doi.org/10.3390/genes3040651