Comparative Genomics of Aeschynomene Symbionts: Insights into the Ecological Lifestyle of Nod-Independent Photosynthetic Bradyrhizobia

Abstract
:1. Introduction
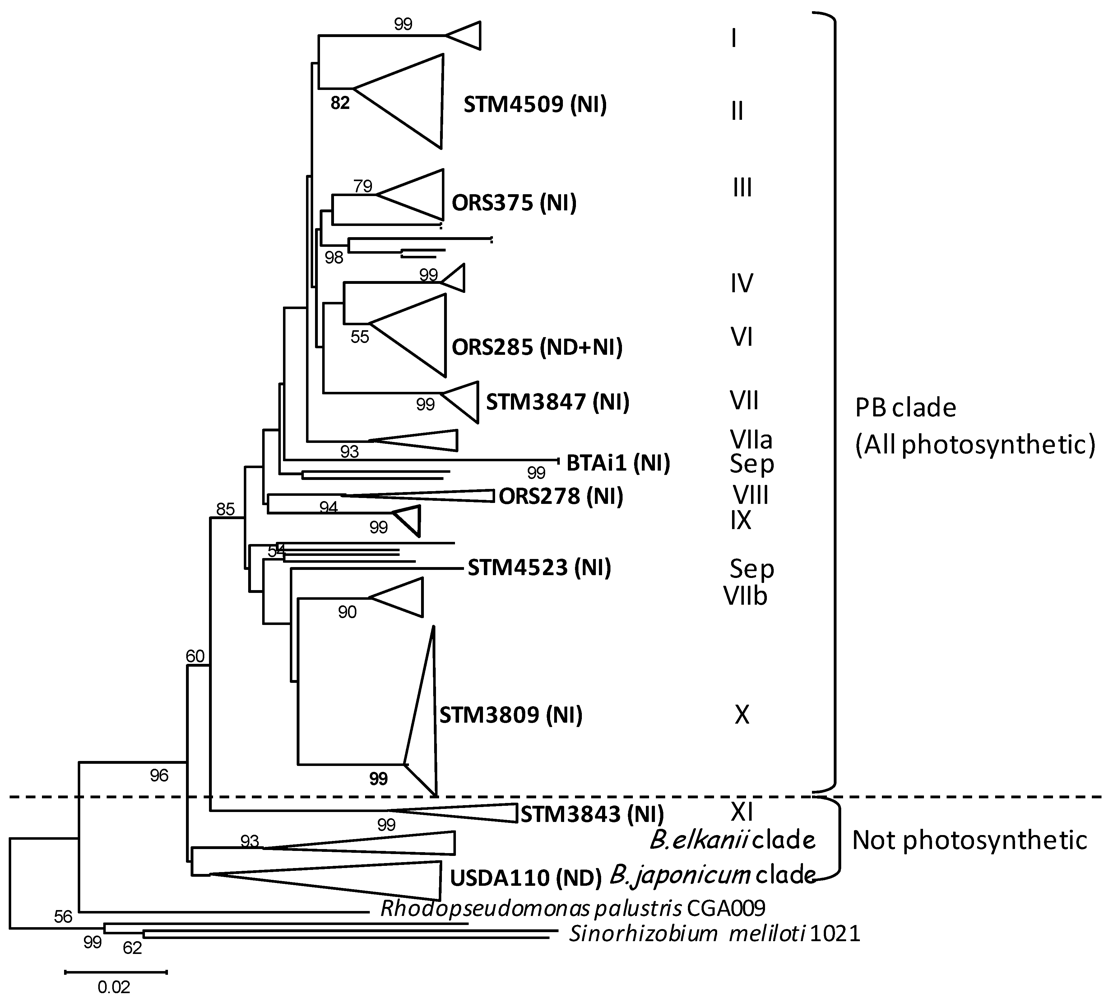
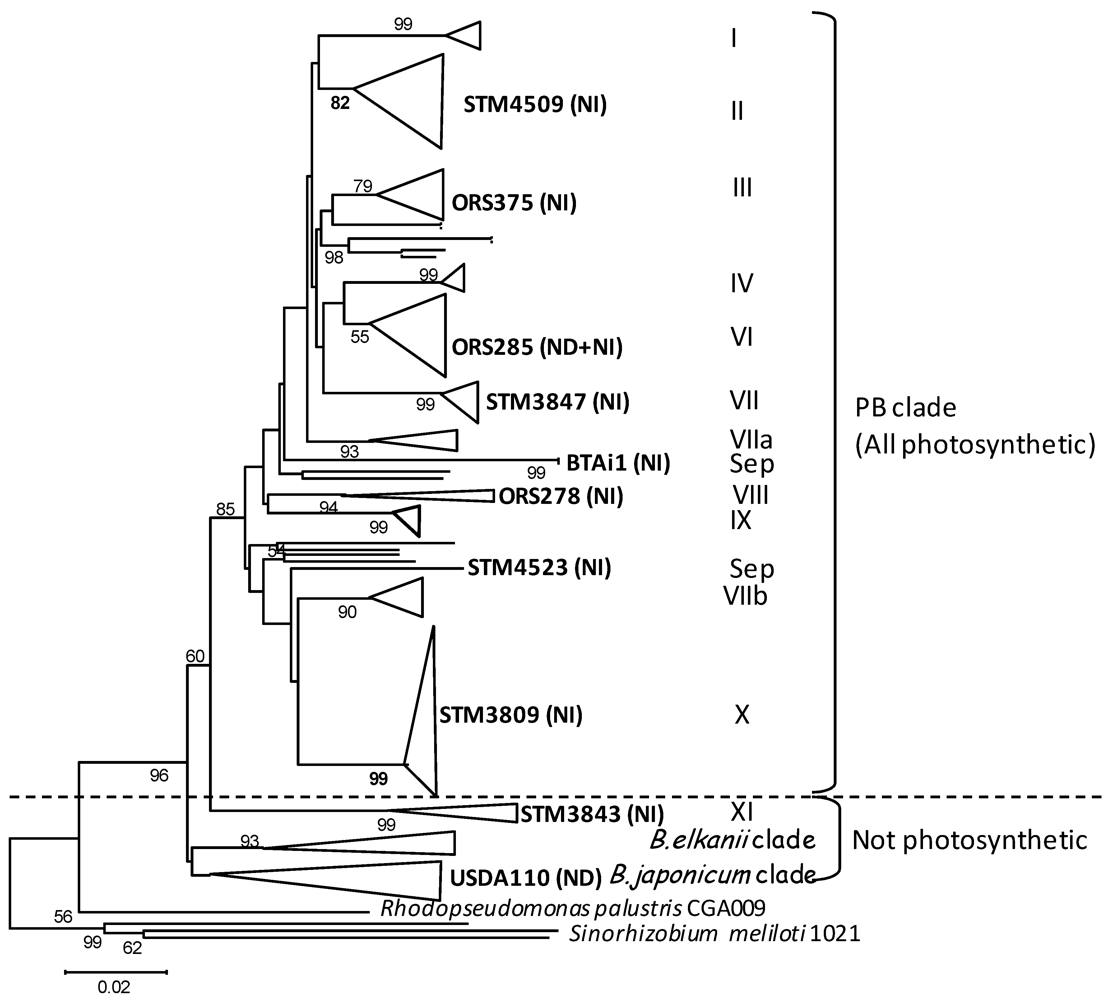
2. Results and Discussion
2.1. Genomic Features of Bradyrhizobial Genomes

{kind=link}
{kind=link}
{kind=link}
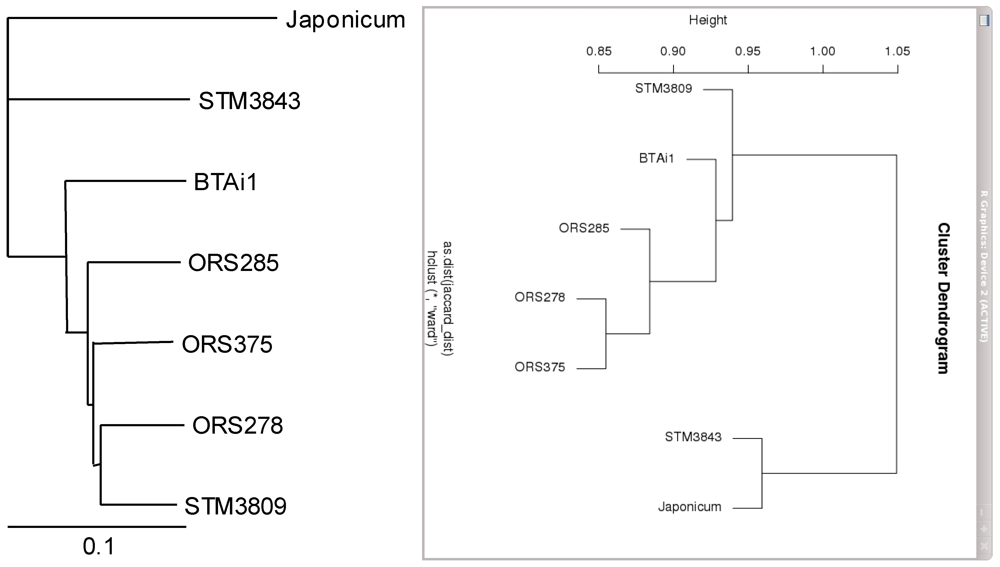{kind=link}
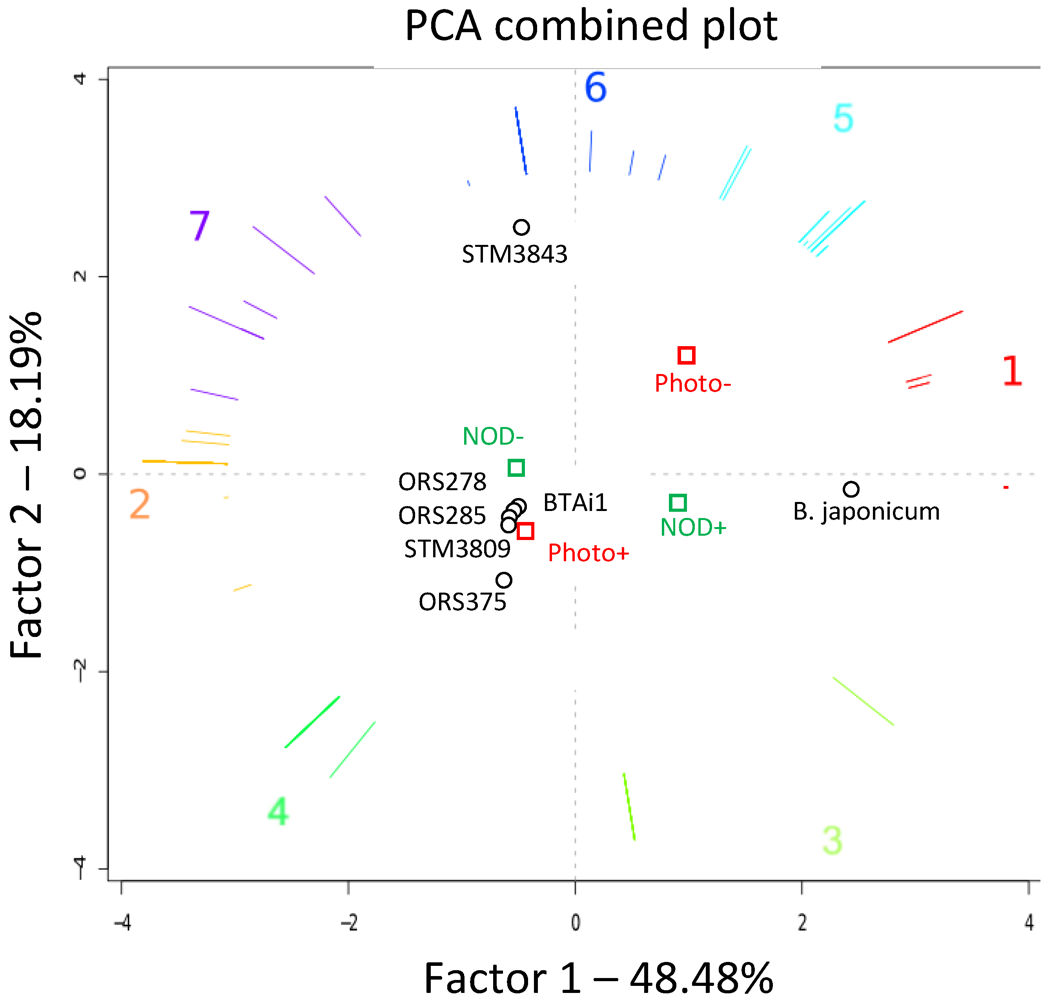{kind=link}
{kind=link}
| Strain/Replicon | Group * | CI Group | Geographical Origin | ND/NI | Sequencing Status | Nb Contigs | Size (bp) | Accession Number |
|---|---|---|---|---|---|---|---|---|
| Photosynthetic Bradyrhizobium clade | ||||||||
| ORS278 | VIII | 3 | Senegal | NI | Complete | 1 | 7456587 | NC_009445 |
| BTAi1 Chr | Sep | 3 | USA | NI | Complete | 1 | 8264689 | NC_009485 |
| BTAi1 Pl | Complete | 1 | 228826 | NC_009475 | ||||
| ORS375 | III | 3 | Senegal | NI | 454 + Solexa | 497 | 7909110 | CAFI01000001-497 & |
| STM3809 | X | 3 | F. Guiana | NI | 454 + Solexa | 803 | 7391986 | CAFJ01000001-803 & |
| ORS285 | VI | 2 | Senegal | Both % | 454 (30X) | 301 | 7632258 | CAFH01000001-301 & |
| STM3847 | VII | 3 | F. Guiana | NI | Solexa 20X | 152475 | 10121035 § | ERP000868 $ |
| STM4509 | II | 3 | Mexico | NI | Solexa 20X | 190139 | 9206235 § | ERP000868 $ |
| STM4523 | Sep | 3 | Mexico | NI | Solexa 20X | 158371 | 8610503 § | ERP000868 $ |
| Non-photosynthetic strain | ||||||||
| STM3843 | XI | 3 | F. Guiana | NI | 454 + Solexa | 350 | 8469730 | CAFK01000001-350 & |
| B. japonicum | ||||||||
| USDA110 | Sep | 1 | USA | ND | Complete | 1 | 9105828 | NC_004463 |
| Strain | GC% | CDS N | CDS L | IGR (bp) | PCD (%) | rRNA | tRNA | MscRNA | NRR (%) |
|---|---|---|---|---|---|---|---|---|---|
| ORS278 | 65.51 | 6748 | 952.1 | 180.23 | 85.50 | 6 | 50 | 10 | 8.76 |
| BTAi1 chr | 64.92 | 7466 | 959.42 | 176.72 | 85.59 | 6 | 52 | 12 | 9.33 |
| BTAi1 pl | 60.71 | 257 | 800.2 | 186.02 | 79.42 | - | - | - | 4.75 |
| ORS285 | 65.23 | 6848 | 955.92 | 184.32 | 85.41 | 4 | 49 | 11 | 10.45 |
| ORS375 | 65.49 | 7348 | 921.28 | 182.92 | 84.99 | 3 | 52 | 11 | 10.22 |
| STM3809 | 66.18 | 7142 | 879.79 | 182.97 | 84.19 | 3 | 47 | 13 | 9.42 |
| STM3843 | 63.3 | 8399 | 878.57 | 159.54 | 86.30 | 3 | 48 | 10 | 5.10 |
| BjUSDA110 | 64.06 | 9648 | 862.74 | 135.17 | 88.98 | 3 | 50 | 3 | 9.53 |
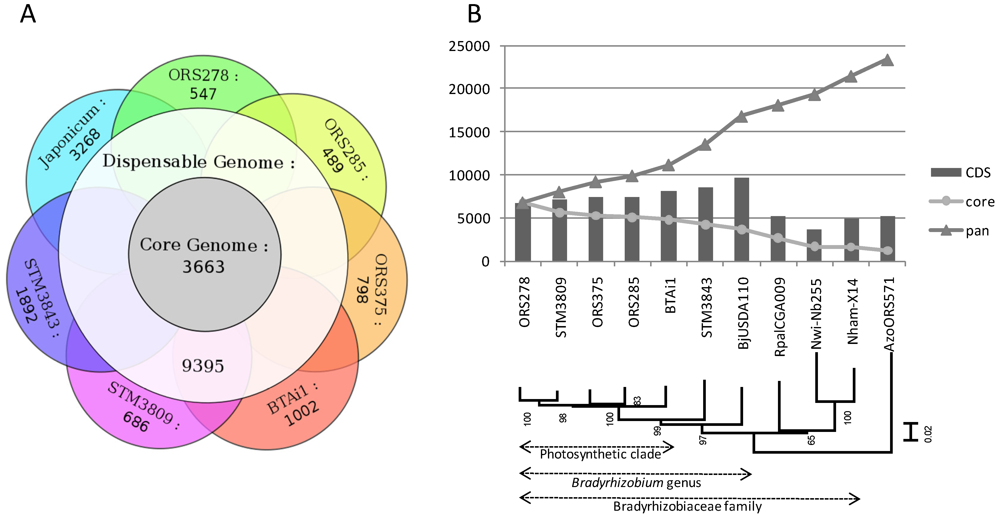
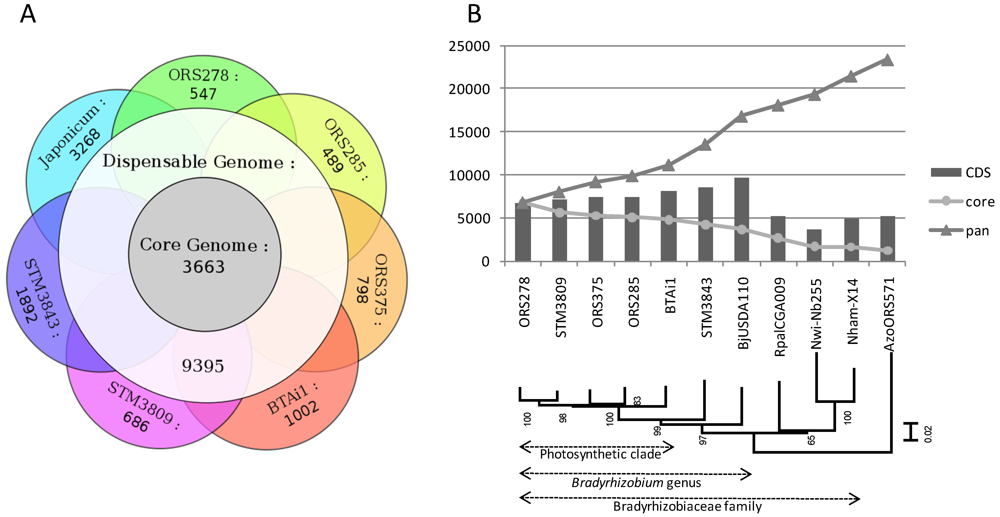
2.2. Core, Dispensable and Strain-Specific Genes
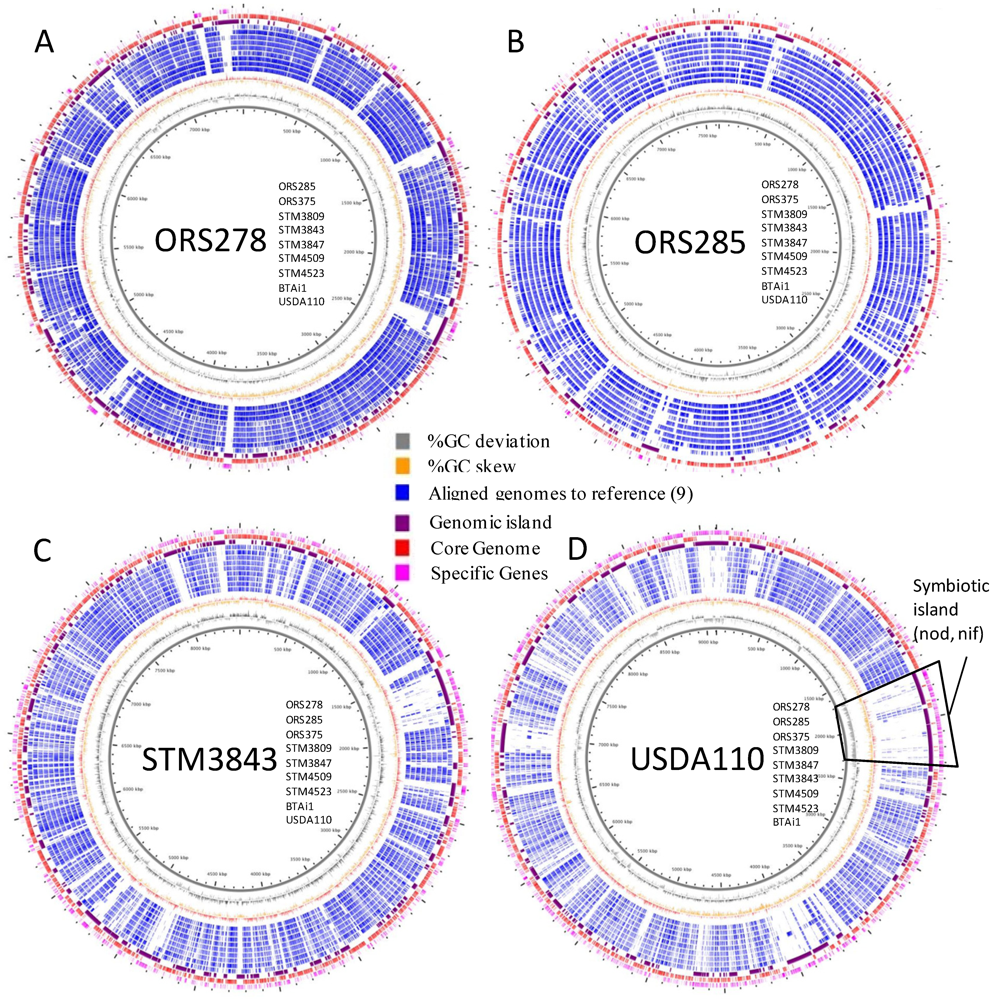
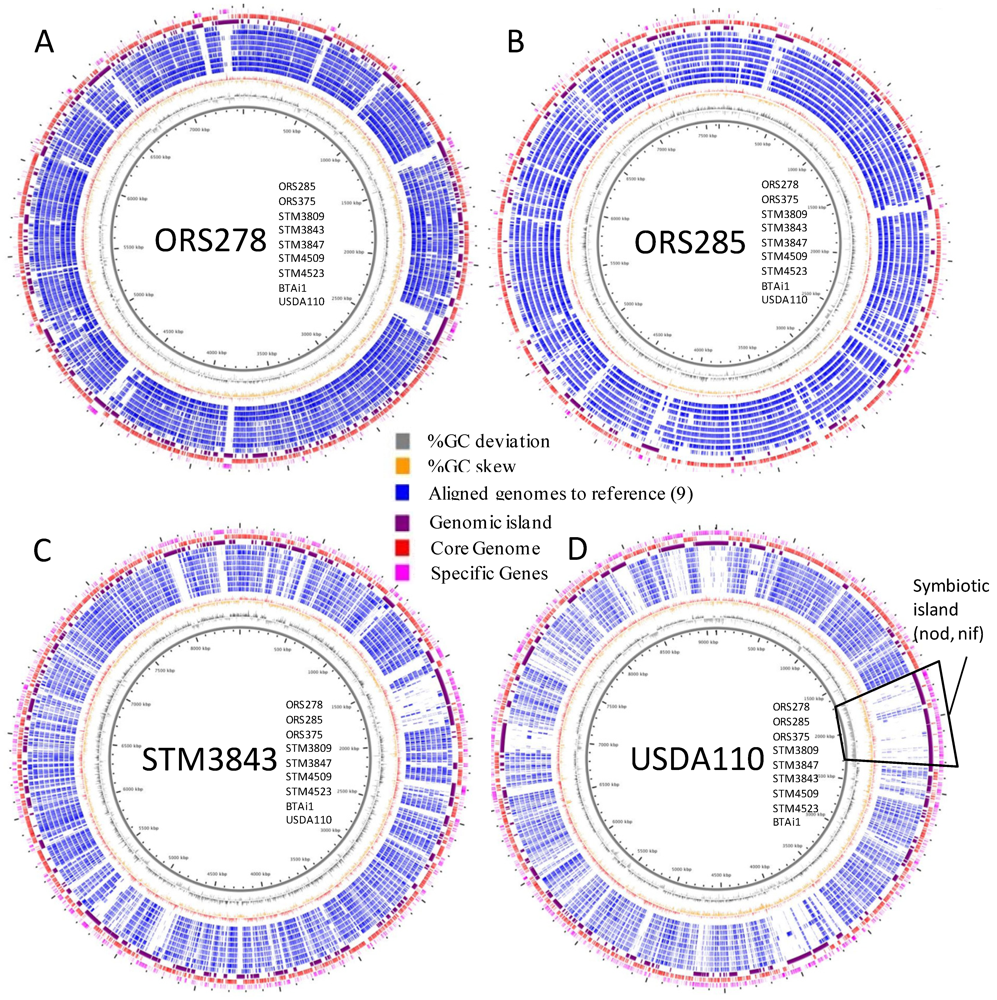
2.3. Whole Genome Comparative Analyses and Distribution of Genomic Islands

2.4. Metabolic Network Comparison of Strains
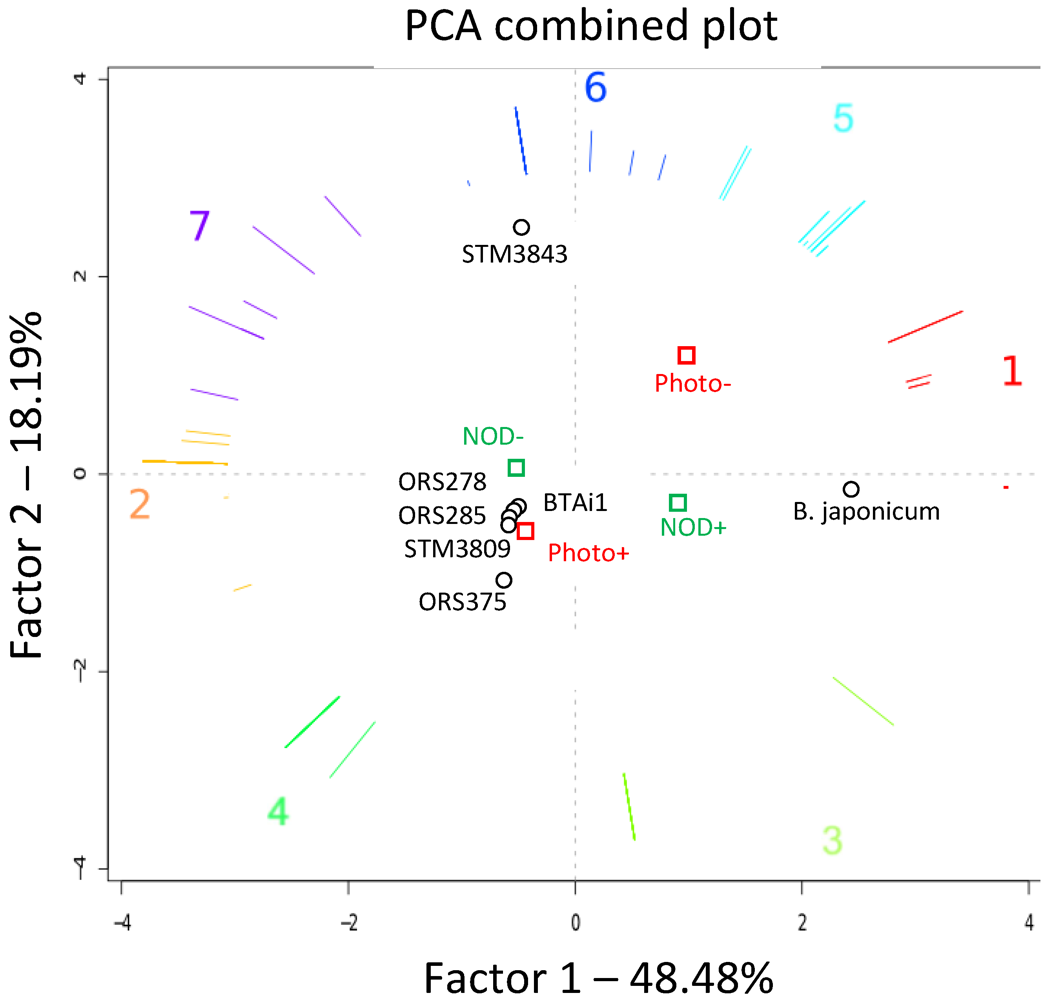
2.5. Functional Exploration of Genomes: Focus on Photosynthetic and Symbiotic Abilities

2.5.1. Photosynthetic Versus Non-Photosynthetic Strains
2.5.2. Focus On Nod-Dependent (ND) and Nod-Independent (NI) Symbiotic Strains
- (i)
- Regulatory genes of which many are two-component regulatory proteins (9 genes with sensor/histidine kinase domains), diguanylate cyclase (BRADO0188, BRADO1402, BRADO6510), circadian clock proteins KaiB and KaiC involved in circadian rythms (BRADO1478 and 1479);
- (ii)
- Catabolic enzymes: catabolic pathways for protocatechuate (BRADO2235 to 2343, BRADO2378-2379) and vanillate degradation (BRADO1851), two compounds derived from lignin monomers [30] (lignin being an abundant constituent of plant cell walls); NUDIX hydrolases that hydrolyse a wide range of organic pyrophosphates and reflect the metabolic complexity and adaptability of a given organism [31]; various catabolic enzymes especially in Taurine degradation;
- (iii)
- N-acetyl-glucosamine modifying enzymes as UDP-N-Acetylglucosamine 1-carboxyvinyltransferase (BRADO6639), with some proteins containing TPR repeats (BRADO4235, BRADO7123);
- (iv)
- Polysaccharide & glycan biosynthesis with 2 specific NI gene regions: BRADO4794-4814 with enzymes involved in wall techoic acid biosynthesis, an anionic polymer that decorate the peptidoglycan layers of many Gram-positive organisms; and BRADO5154-5204 located on a GI in all PB strains;
- (v)
- Biosynthesis of various molecules including carotenoids (BRADO0659), Indole-3-acetic acid (IAA) (BRADO7016), antibiotics (BRADO1346), cobalamin (B12 vitamin, BRADO4898-4917), and a type 3 polyketide synthase (BRADO0151);
- (vi)
- Nitrogen fixation as nifV (BRADO5390), encoding an homocitrate synthase responsible for ex planta N2 fixation ability [32], as well as several nitrogen-fixation related genes (BRADO5414, 5416, 5418, 5424) clustered in an operon conserved among all NI strains as well as in Azorhizobium caulinodans ORS571, but not in rhizobia as B. japonicum USDA110, S. melioti 1021 or M. loti MAFF303099 (that do not fix nitrogen in the free-living state). Interestingly, this gene cluster is up-regulated in A. caulinodans ORS571 bacteroids on Sesbania rostrata stem nodules, indicating a probable specific role in symbiotic nitrogen fixation in these symbionts;
- (vii)
- Carbon monoxide oxidation (BRADO6025-6032), that catalyzes the reversible conversion between carbon dioxide (CO2) and carbon monoxide (CO), and found in many marine bacterial genomes;
- (viii)
- Chemotaxis: several methyl-accepting chemotaxis proteins (receptor/sensory transducer);
- (ix)
- LuxI-LuxR quorum sensing with BRADO0941 involved in the biosynthesis of a cinnamoyl-HSL (Homo Serine Lactone), an unusual aryl-HSL [33];
- (x)
- Transport: General secretion pathway protein (BRADO6338-type II, BRADO6341, 6344), HlyD-family protein (BRADO7122).
| CDS labels | Gene | Product |
|---|---|---|
| Regulation | ||
| Two component regulatory systems | ||
| BRADO3895 | putative two-component system, response regulator receiver | |
| BRADO4688 | putative two-component system sensor protein with Hpt domain | |
| BRADO4689 | putative sensor histidine kinase with a response regulator receiver domain | |
| BRADO5320 | putative sensor histidine kinase (PAS & response regulator receiver) | |
| BRADO6874 | putative two component system, regulator receiver (CheY-like protein) | |
| BRADO7009 | putative response regulator receiver (CheY-like protein) | |
| BRADO7010 | putative sensor histidine kinase (receiver & phosphotransferase domains) | |
| BRADO5682 | putative two component sensor histidine kinase | |
| BRADO1407 | putative Two-component system histidine kinase | |
| BRADO5100 | Putative Two-component sensor histidine kinase | |
| Transcriptional Regulators | ||
| BRADO3272 | putative transcriptional regulatory protein, TetR/AcrR family | |
| BRADO3678 | putative transcriptional regulatory protein, TetR family | |
| BRADO5987 | putative transcriptional regulator, TetR family | |
| Cyclases | ||
| BRADO0188 | putative diguanylate cyclase (GGDEF) domain | |
| BRADO1402 | putative diguanylate cyclase (GGDEF) | |
| BRADO2821 | putative Adenylate cyclase with a CHAD domain | |
| BRADO6510 | putative diguanylate cyclase with GGDEF and EAL domains | |
| Circadian clock operon | ||
| BRADO1478 | kaiC | circadian clock protein kinase kaiC |
| BRADO1479 | kaiB | circadian clock protein |
| BRADO1480 | putative signal transduction histidine kinase with PAS/PAC domains | |
| BRADO1481 | putative response regulator receiver (CheY-like protein) | |
| Catabolism | ||
| Plant wall degradation | ||
| BRADO1851 | vanB | vanillate O -demethylase oxidoreductase (Vanillate degradation) |
| BRADO2335-2343 | protochatechuate transport and degradation (ligJABC) | |
| BRADO2379 | protocatechuate 4,5-dioxygenase (4,5-PCD), alpha chain | |
| BRADO2380 | 2,3-dihydroxyphenylpropionate 1,2-dioxygenase | |
| BRADO2382 | putative transcriptional regulator, PadR-like family | |
| BRADO4665 | hpcB | homoprotocatechuate 2,3-dioxygenase |
| Various Catabolism/Detoxification | ||
| BRADO0718 | putative intradiol ring-cleavage dioxygenase | |
| BRADO1842 | putative amine oxidase | |
| BRADO1843 | putative ATPase, AAA family | |
| BRADO1844 | putative Glutathione S-transferase | |
| BRADO1848 | putative thiosulfate sulfurtransferase with Rhodanese-like domain | |
| BRADO1849 | conserved HP; putative oxidoreductase | |
| BRADO4030 | putative TauD/TfdA family dioxygenase (Taurine catabolism) | |
| BRADO4031 | putative dioxygenase; putative taurine dioxygenase (Taurine catabolism) | |
| NUDIX hydrolase | ||
| BRADO3028 | putative MutT/nudix family protein | |
| BRADO3892 | putative NUDIX-like hydrolase (modular protein) | |
| BRADO4664 | putative NUDIX hydrolase | |
| Biosynthesis and modification enzymes | ||
| N -AcetylGlucosamine modifying enzymes | ||
| BRADO3973 | putative UDP- N -Acetylgucosamine-Peptide- N -Acetylglucosaminyl transferase subunit-related | |
| BRADO4235 | putative O -GlcNAc transferase related protein (TRP repeats) | |
| BRADO6639 | UDP- N -acetylglucosamine 1-carboxyvinyltransferase | |
| BRADO7123 | putative O -GlcNAc transferase related protein (TRP repeats) | |
| Polysaccharide biosynthesis & glycans | ||
| BRADO4794 | putative D-3-phosphoglycerate dehydrogenase | |
| BRADO4795 | putative carbohydrate kinase (xylulose/erythritol kinase, lyx/eryA-like) | |
| BRADO4796-4800 | putative sugar ABC transporter (permease + ATP-binding) | |
| BRADO4801 | putative transcription regulator (EryD-like) | |
| BRADO4802 | putative carbohydrate kinase | |
| BRADO4803 | putative hydrolase (HAD superfamily) | |
| BRADO4804 | putative aldolase/epimerase (AraD-like) | |
| BRADO4807 | putative 3-oxoacyl-acyl-carrier-protein reductase | |
| BRADO4814 | putative N -acetyl-mannosamine transferase (teich | |
| BRADO5154 | putative Capsular polysaccharide biosynthesis protein | |
| BRADO5156 | putative Asparagine synthetase | |
| BRADO5164 | putative bifunctional enzyme (sugar kinase/cytidylyltransferase) | |
| BRADO5165 | putative sugar-phosphate nucleotidyl transferase | |
| BRADO5167 | putative Phosphoheptose isomerase | |
| BRADO5168 | putative dTDP-glucose 4,6-dehydratase | |
| BRADO5173 | putative aminoglycoside phosphotransferase family protein | |
| BRADO5197 | putative Glycosyl transferase, group 1 | |
| BRADO5204 | phosphoheptose isomerase (Sedoheptulose 7-phosphate isomerase) | |
| BRADO6336 | putative NiFe-hydrogenase/urease accessory HupE/UreJ family protein | |
| Others | ||
| BRADO0151 | putative Type III polyketide synthase; putative chalcone synthase | |
| BRADO0860 | putative asparagine synthetase (glutamine-hydrolyzing) | |
| BRADO1346 | putative Phenazine biosynthesis protein | |
| BRADO1560 | putative aldolase | |
| BRADO4898 | putative cobalt-precorrin-6A synthase, cbiD (Vitamin B12 biosynthesis) | |
| BRADO4900 | putative CobE protein (Vitamin B12 biosynthesis) | |
| BRADO4906 | putative precorrin-3B synthase (cobG) (Vitamin B12 biosynthesis) | |
| BRADO4910 | putative HupE/UreJ protein | |
| BRADO4916 | putative cobalamin synthase (CobS family) (Vitamin B12 biosynthesis) | |
| BRADO4917 | alpha-ribazole phosphatase (anaerobic pathway cobalamin biosynthesis) | |
| BRADO7016 | indole-3-pyruvate decarboxylase (IAA biosynthesis) | |
| Nitrogen fixation | ||
| BRADO5389 | cysE | serine acetyltransferase |
| BRADO5390 | nifV | homocitrate synthase |
| BRADO5396 | putative transcriptional regulatory protein (protein cheY homolog) | |
| BRADO5403 | putative carboxymethylenebutenolidase(DLH) | |
| BRADO5410 | putative Ferredoxin, 2Fe-2S | |
| BRADO5411 | putative Aminotransferase, DegT/DnrJ/EryC1/StrS family | |
| BRADO5414 | conserved HP (NifZ domain) | |
| BRADO5416 | LRV protein FeS4 cluster in N -terminus | |
| BRADO5417 | conserved HP; TPR repeat | |
| BRADO5418 | putative iron-sulfur cluster assembly protein (SufA-like) | |
| BRADO5424 | putative nifU protein (C-terminal fragment) | |
| Carbon monoxide fixation | ||
| BRADO6025 | putative Sensor histidine kinase (HWE family) with a GAF domain | |
| BRADO6026 | coxF | carbon monoxide dehydrogenase, coxF accessory protein |
| BRADO6027 | coxE | carbon monoxide dehydrogenase, coxE accessory protein |
| BRADO6029 | coxL | carbon monoxide dehydrogenase large chain |
| BRADO6031 | coxM | carbon monoxide dehydrogenase medium chain (CO-DH M) |
| BRADO6032 | coxC | carbon monoxide dehydrogenase, coxC signalling protein |
| Photosynthesis & Carotenoid | ||
| BRADO0659-0662 | carotenoid synthesis | |
| BRADO2007 | putative heme oxygenase | |
| BRADO2008 | putative bacteriophytochrome | |
| Quorum sensing | ||
| BRADO0941 | autoinducer (acylhomoserine lactone) synthase | |
| Chemotaxis | ||
| BRADO2926 | putative Methyl-accepting chemotaxis protein | |
| BRADO0678 | putative methyl-accepting chemotaxis protein | |
| BRADO3031 | putative methyl-accepting chemotaxis receptor/sensory transducer | |
| BRADO4514 | putative bacterial chemotaxis sensory transducer | |
| BRADO5051 | conserved HP-cheL | |
| Transport | ||
| Transporters | ||
| BRADO0677 | putative TRAP-dicarboxylate transporter (DctP subunit) | |
| Secretion | ||
| BRADO6338-6344 | general secretion pathway proteins (GspI, GspL) | |
| BRADO7122 | putative HlyD-family secretion protein | |
3. Experimental Section
3.1. Bacterial Strains Maintenance and Preparation of Genomic DNA
3.2. Sequencing and Assembly of 7 Bradyrhizobium Genomes
3.3. Automatic and Expert Annotation of Bradyrhizobial Genomes
3.4. Whole Genome Alignments
3.5. GC% Deviation
3.6. Core-Genome and Pangenome
3.7. Phylogenomics
3.8. Search for Genomic Islands (GI)
3.9. Metabolic Comparisons
4. Conclusions
Acknowledgments
References
- Gyaneshwar, P.; Hirsch, A.M.; Moulin, L.; Chen, W.M.; Elliott, G.N.; Bontemps, C.; Estrada de Los Santos, P.; Gross, E.; dos Reis, F.B., Jr.; Sprent, J.; et al. Legume-nodulating betaproteobacteria: Diversity, host range and future prospects. Mol. Plant Microbe Interact. 2011, 24, 1276–1288. [Google Scholar]
- Nzoue, A.; Miche, L.; Klonowska, A.; Laguerre, G.; de Lajudie, P.; Moulin, L. Multilocus sequence analysis of bradyrhizobia isolated from Aeschynomene species in Senegal. Syst. Appl. Microbiol. 2009, 32, 400–412. [Google Scholar] [CrossRef]
- Rivas, R.; Martens, M.; de Lajudie, P.; Willems, A. Multilocus sequence analysis of the genus Bradyrhizobium. Syst. Appl. Microbiol. 2009, 32, 101–110. [Google Scholar] [CrossRef]
- James, E.K.; de Fatima Loureiro, M.; Pott, A.; Pott, V.J.; Martins, C.M.; Franco, A.A.; Sprent, J.I. Flooding-tolerant legumes symbioses from the Brazilian Pantanal. New Phytol. 2001, 150, 723–738. [Google Scholar]
- Boivin, C.; Ndoye, I.; Lortet, G.; Ndiaye, A.; de Lajudie, P.; Dreyfus, B. The sesbania root symbionts Sinorhizobium saheli and S. teranga bv. sesbaniae can form stem nodules on Sesbania rostrata, although they are less adapted to stem nodulation than Azorhizobium caulinodans. Appl. Environ. Microbiol. 1997, 63, 1040–1047. [Google Scholar]
- Bonaldi, K.; Gargani, D.; Prin, Y.; Fardoux, J.; Gully, D.; Nouwen, N.; Goormachtig, S.; Giraud, E. Nodulation of Aeschynomene afraspera and Aeschynomene indica by photosynthetic Bradyrhizobium sp. strain ORS285: The Nod-dependent versus the Nod-independent symbiotic interaction. Mol. Plant Microbe Interact. 2011, 24, 1359–1371. [Google Scholar] [CrossRef]
- Capoen, W.; Goormachtig, S.; Holsters, M. Water-tolerant legume nodulation. J. Exp. Bot. 2010, 61, 1251–1255. [Google Scholar] [CrossRef]
- D'Haeze, W.; Mergaert, P.; Prome, J.C.; Holsters, M. Nod factor requirements for efficient stem and root nodulation of the tropical legume Sesbania rostrata. J. Biol. Chem. 2000, 275, 15676–15684. [Google Scholar]
- Evans, W.R.; Fleischman, D.E.; Calvert, H.E.; Pyati, P.V.; Alter, G.M.; Rao, N.S. Bacteriochlorophyll and photosynthetic reaction centers in Rhizobium strain BTAi 1. Appl. Environ. Microbiol. 1990, 56, 3445–3449. [Google Scholar]
- Giraud, E.; Hannibal, L.; Fardoux, J.; Vermeglio, A.; Dreyfus, B. Effect of Bradyrhizobium photosynthesis on stem nodulation of Aeschynomene sensitiva. Proc. Natl. Acad. Sci. USA 2000, 97, 14795–14800. [Google Scholar]
- Giraud, E.; Fleischman, D. Nitrogen-fixing symbiosis between photosynthetic bacteria and legumes. Photosynth. Res. 2004, 82, 115–130. [Google Scholar]
- Bonaldi, K.; Gourion, B.; Fardoux, J.; Hannibal, L.; Cartieaux, F.; Boursot, M.; Vallenet, D.; Chaintreuil, C.; Prin, Y.; Nouwen, N.; et al. Large-scale transposon mutagenesis of photosynthetic Bradyrhizobium sp. strain ORS278 reveals new genetic loci putatively important for nod-independent symbiosis with Aeschynomene indica. Mol. Plant Microbe Interact. 2011, 23, 760–770. [Google Scholar]
- Giraud, E.; Moulin, L.; Vallenet, D.; Barbe, V.; Cytryn, E.; Avarre, J.C.; Jaubert, M.; Simon, D.; Cartieaux, F.; Prin, Y.; et al. Legumes symbioses: Absence of Nod genes in photosynthetic bradyrhizobia. Science 2007, 316, 1307–1312. [Google Scholar]
- Molouba, F.; Lorquin, J.; Willems, A.; Hoste, B.; Giraud, E.; Dreyfus, B.; Gillis, M.; de Lajudie, P.; Masson-Boivin, C. Photosynthetic bradyrhizobia from Aeschynomene spp. are specific to stem-nodulated species and form a separate 16S ribosomal DNA restriction fragment length polymorphism group. Appl. Environ. Microbiol. 1999, 65, 3084–3094. [Google Scholar]
- Alazard, D. Stem and root nodulation in Aeschynomene spp. Appl. Environ. Microbiol. 1985, 50, 732–734. [Google Scholar]
- Miche, L.; Moulin, L.; Chaintreuil, C.; Contreras-Jimenez, J.L.; Munive-Hernandez, J.A.; del Carmen Villegas-Hernandez, M.; Crozier, F.; Bena, G. Diversity analyses of Aeschynomene symbionts in Tropical Africa and Central America reveal that nod-independent stem nodulation is not restricted to photosynthetic bradyrhizobia. Environ. Microbiol. 2010, 12, 2152–2164. [Google Scholar]
- Kaneko, T.; Nakamura, Y.; Sato, S.; Minamisawa, K.; Uchiumi, T.; Sasamoto, S.; Watanabe, A.; Idesawa, K.; Iriguchi, M.; Kawashima, K.; et al. Complete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110. DNA Res. 2002, 9, 189–197. [Google Scholar] [CrossRef]
- European Nucleotide Archive. Available online: http://www.ebi.ac.uk/ena/data/view/ERP000868/ (accessed on 8 November 2011).
- Cytryn, E.J.; Jitacksorn, S.; Giraud, E.; Sadowsky, M.J. Insights learned from pBTAi1, a 229-kb accessory plasmid from Bradyrhizobium sp. strain BTAi1 and prevalence of accessory plasmids in other Bradyrhizobium sp. strains. ISME J. 2008, 2, 158–170. [Google Scholar] [CrossRef]
- Gil, R.; Silva, F.J.; Pereto, J.; Moya, A. Determination of the core of a minimal bacterial gene set. Microbiol. Mol. Biol. Rev. 2004, 68, 518–537. [Google Scholar]
- Damerval, T.; Castets, A.M.; Guglielmi, G.; Houmard, J.; Tandeau de Marsac, N. Occurrence and distribution of gas vesicle genes among cyanobacteria. J. Bacteriol. 1989, 171, 1445–1452. [Google Scholar]
- Buts, L.; Lah, J.; Dao-Thi, M.H.; Wyns, L.; Loris, R. Toxin-antitoxin modules as bacterial metabolic stress managers. Trends Biochem. Sci. 2005, 30, 672–679. [Google Scholar]
- Oda, Y.; Larimer, F.W.; Chain, P.S.; Malfatti, S.; Shin, M.V.; Vergez, L.M.; Hauser, L.; Land, M.L.; Braatsch, S.; Beatty, J.T.; et al. Multiple genome sequences reveal adaptations of a phototrophic bacterium to sediment microenvironments. Proc. Natl. Acad. Sci. USA 2008, 105, 18543–18548. [Google Scholar]
- Giraud, E.; Fardoux, J.; Fourrier, N.; Hannibal, L.; Genty, B.; Bouyer, P.; Dreyfus, B.; Vermeglio, A. Bacteriophytochrome controls photosystem synthesis in anoxygenic bacteria. Nature 2002, 417, 202–205. [Google Scholar]
- Jaubert, M.; Zappa, S.; Fardoux, J.; Adriano, J.M.; Hannibal, L.; Elsen, S.; Lavergne, J.; Vermeglio, A.; Giraud, E.; Pignol, D. Light and redox control of photosynthesis gene expression in Bradyrhizobium: Dual roles of two PpsR. J. Biol. Chem. 2004, 279, 44407–44416. [Google Scholar]
- Jaubert, M.; Vuillet, L.; Hannibal, L.; Adriano, J.M.; Fardoux, J.; Bouyer, P.; Bonaldi, K.; Fleischman, D.; Giraud, E.; Vermeglio, A. Control of peripheral light-harvesting complex synthesis by a bacteriophytochrome in the aerobic photosynthetic bacterium Bradyrhizobium strain BTAi1. J. Bacteriol. 2008, 190, 5824–5831. [Google Scholar]
- Jaubert, M.; Lavergne, J.; Fardoux, J.; Hannibal, L.; Vuillet, L.; Adriano, J.M.; Bouyer, P.; Pignol, D.; Giraud, E.; Vermeglio, A. A singular bacteriophytochrome acquired by lateral gene transfer. J. Biol. Chem. 2007, 282, 7320–7328. [Google Scholar]
- Lepo, J.E.; Hanus, F.J.; Evans, H.J. Chemoautotrophic growth of hydrogen-uptake-positive strains of Rhizobium japonicum. J. Bacteriol. 1980, 141, 664–670. [Google Scholar]
- Gourion, B.; Delmotte, N.; Bonaldi, K.; Nouwen, N.; Vorholt, J.A.; Giraud, E. Bacterial RuBisCO is required for efficient Bradyrhizobium/Aeschynomene symbiosis. PLoS ONE 2011, 6, e21900. [Google Scholar]
- Sudtachat, N.; Ito, N.; Itakura, M.; Masuda, S.; Eda, S.; Mitsui, H.; Kawaharada, Y.; Minamisawa, K. Aerobic vanillate degradation and C1 compound metabolism in Bradyrhizobium japonicum. Appl. Environ. Microbiol. 2009, 75, 5012–5017. [Google Scholar]
- McLennan, A.G. The Nudix hydrolase superfamily. Cell. Mol. Life Sci. 2006, 63, 123–143. [Google Scholar]
- Hakoyama, T.; Niimi, K.; Watanabe, H.; Tabata, R.; Matsubara, J.; Sato, S.; Nakamura, Y.; Tabata, S.; Jichun, L.; Matsumoto, T.; et al. Host plant genome overcomes the lack of a bacterial gene for symbiotic nitrogen fixation. Nature 2009, 462, 514–517. [Google Scholar]
- Ahlgren, N.A.; Harwood, C.S.; Schaefer, A.L.; Giraud, E.; Greenberg, E.P. Aryl-homoserine lactone quorum sensing in stem-nodulating photosynthetic bradyrhizobia. Proc. Natl. Acad. Sci. USA 2011, 108, 7183–7188. [Google Scholar]
- Giraud, E. LSTM, Montpellier, France. Unpublished work, 2011.
- Downie, J.A. The roles of extracellular proteins, polysaccharides and signals in the interactions of rhizobia with legume roots. FEMS Microbiol. Rev. 2010, 34, 150–170. [Google Scholar]
- D'Haeze, W.; Holsters, M. Nod factor structures, responses, and perception during initiation of nodule development. Glycobiology 2002, 12, 79–105. [Google Scholar] [CrossRef]
- Maillet, F.; Poinsot, V.; Andre, O.; Puech-Pages, V.; Haouy, A.; Gueunier, M.; Cromer, L.; Giraudet, D.; Formey, D.; Niebel, A.; et al. Fungal lipochitooligosaccharide symbiotic signals in arbuscular mycorrhiza. Nature 2011, 469, 58–63. [Google Scholar]
- Hamel, L.P.; Beaudoin, N. Chitooligosaccharide sensing and downstream signaling: Contrasted outcomes in pathogenic and beneficial plant-microbe interactions. Planta 2010, 232, 787–806. [Google Scholar]
- Kambara, K.; Ardissone, S.; Kobayashi, H.; Saad, M.M.; Schumpp, O.; Broughton, W.J.; Deakin, W.J. Rhizobia utilize pathogen-like effector proteins during symbiosis. Mol. Microbiol. 2009, 71, 92–106. [Google Scholar]
- Krause, A.; Doerfel, A.; Gottfert, M. Mutational and transcriptional analysis of the type III secretion system of Bradyrhizobium japonicum. Mol. Plant Microbe Interact. 2002, 15, 1228–1235. [Google Scholar] [CrossRef]
- Renier, A.; Maillet, F.; Fardoux, J.; Poinsot, V.; Giraud, E.; Nouwen, N. Photosynthetic Bradyrhizobium ORS285 synthesizes 2-O-methylfucosylated lipochitooligosaccharides for nod gene dependent interaction with Aeschynomene plants. Mol. Plant Microbe Interact. 2011, 24, 1440–1447. [Google Scholar] [CrossRef]
- Hueck, C.J. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol. Mol. Biol. Rev. 1998, 62, 379–433. [Google Scholar]
- Wenzel, M.; Friedrich, L.; Gottfert, M.; Zehner, S. The type III-secreted protein NopE1 affects symbiosis and exhibits a calcium-dependent autocleavage activity. Mol. Plant Microbe Interact. 2010, 23, 124–129. [Google Scholar]
- Vincent, J.M. A Manual for the Pratical Study of Root-Nodule Bacteria. In International Biological Programme Handbook; Blackwell Scientific Publications: Oxford, UK, 1970; Volume 15. [Google Scholar]
- Joint Genome Institute. Available online: http://my.jgi.doe.gov/general/ (accessed on 8 November 2011).
- Jeck, W.R.; Reinhardt, J.A.; Baltrus, D.A.; Hickenbotham, M.T.; Magrini, V.; Mardis, E.R.; Dangl, J.L.; Jones, C.D. Extending assembly of short DNA sequences to handle error. Bioinformatics 2007, 23, 2942–2944. [Google Scholar]
- EMBL Nucleotide Sequence Database (EBI). Available online: http://www.ebi.ac.uk/genomes/ (accessed on 8 November 2011).
- MAGE. MicroScope Annotation and Comparative Genomics Platform. Available online: http://www.genoscope.cns.fr/agc/microscope/ (accessed on 8 November 2011).
- Vallenet, D.; Labarre, L.; Rouy, Z.; Barbe, V.; Bocs, S.; Cruveiller, S.; Lajus, A.; Pascal, G.; Scarpelli, C.; Medigue, C. MaGe: A microbial genome annotation system supported by synteny results. Nucleic Acids Res. 2006, 34, 53–65. [Google Scholar]
- Bocs, S.; Cruveiller, S.; Vallenet, D.; Nuel, G.; Medigue, C. AMIGene: Annotation of microbial genes. Nucleic Acids Res. 2003, 31, 3723–3726. [Google Scholar]
- Vallenet, D.; Engelen, S.; Mornico, D.; Cruveiller, S.; Fleury, L.; Lajus, A.; Rouy, Z.; Roche, D.; Salvignol, G.; Scarpelli, C.; et al. MicroScope: A platform for microbial genome annotation and comparative genomics. Database (Oxford) 2009, 2009, ap021. [Google Scholar]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar]
- O'Brien, K.P.; Remm, M.; Sonnhammer, E.L. Inparanoid: A comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 2005, 33, D476–D480. [Google Scholar]
- Overbeek, R.; Fonstein, M.; D'Souza, M.; Pusch, G.D.; Maltsev, N. The use of gene clusters to infer functional coupling. Proc. Natl. Acad. Sci. USA 1999, 96, 2896–2901. [Google Scholar]
- Mavromatis, K.; Chu, K.; Ivanova, N.; Hooper, S.D.; Markowitz, V.M.; Kyrpides, N.C. Gene context analysis in the Integrated Microbial Genomes (IMG) data management system. PLoS ONE 2009, 4, e7979. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K. BLAST+: Architecture and applications. BMC Bioinformatics 2009, 10, 421. [Google Scholar] [CrossRef]
- Sarkar, I.N.; Egan, M.G.; Coruzzi, G.; Lee, E.K.; DeSalle, R. Automated simultaneous analysis phylogenetics (ASAP): An enabling tool for phlyogenomics. BMC Bioinformatics 2008, 9, 103. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony method. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar]
- Roche, D. LABGeM, CEA Genoscope, Evry, France. Unpublished work, 2011.
- Karp, P.D.; Paley, S.; Romero, P. The pathway tools software. Bioinformatics 2002, 18, S225–S232. [Google Scholar]
- Caspi, R.; Foerster, H.; Fulcher, C.A.; Kaipa, P.; Krummenacker, M.; Latendresse, M.; Paley, S.; Rhee, S.Y.; Shearer, A.G.; Tissier, C.; et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2008, 36, D623–D631. [Google Scholar]
- Microcyc. Available online: https://www.genoscope.cns.fr/agc/microscope/metabolism/microcyc.php/ (accessed on 8 November 2011).
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mornico, D.; Miché, L.; Béna, G.; Nouwen, N.; Verméglio, A.; Vallenet, D.; Smith, A.A.T.; Giraud, E.; Médigue, C.; Moulin, L. Comparative Genomics of Aeschynomene Symbionts: Insights into the Ecological Lifestyle of Nod-Independent Photosynthetic Bradyrhizobia. Genes 2012, 3, 35-61. https://doi.org/10.3390/genes3010035
Mornico D, Miché L, Béna G, Nouwen N, Verméglio A, Vallenet D, Smith AAT, Giraud E, Médigue C, Moulin L. Comparative Genomics of Aeschynomene Symbionts: Insights into the Ecological Lifestyle of Nod-Independent Photosynthetic Bradyrhizobia. Genes. 2012; 3(1):35-61. https://doi.org/10.3390/genes3010035
Chicago/Turabian StyleMornico, Damien, Lucie Miché, Gilles Béna, Nico Nouwen, André Verméglio, David Vallenet, Alexander A.T. Smith, Eric Giraud, Claudine Médigue, and Lionel Moulin. 2012. "Comparative Genomics of Aeschynomene Symbionts: Insights into the Ecological Lifestyle of Nod-Independent Photosynthetic Bradyrhizobia" Genes 3, no. 1: 35-61. https://doi.org/10.3390/genes3010035
APA StyleMornico, D., Miché, L., Béna, G., Nouwen, N., Verméglio, A., Vallenet, D., Smith, A. A. T., Giraud, E., Médigue, C., & Moulin, L. (2012). Comparative Genomics of Aeschynomene Symbionts: Insights into the Ecological Lifestyle of Nod-Independent Photosynthetic Bradyrhizobia. Genes, 3(1), 35-61. https://doi.org/10.3390/genes3010035