
A Bidirectional Non-Coding RNA Promoter Mediates Long-Range Gene Expression Regulation

 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
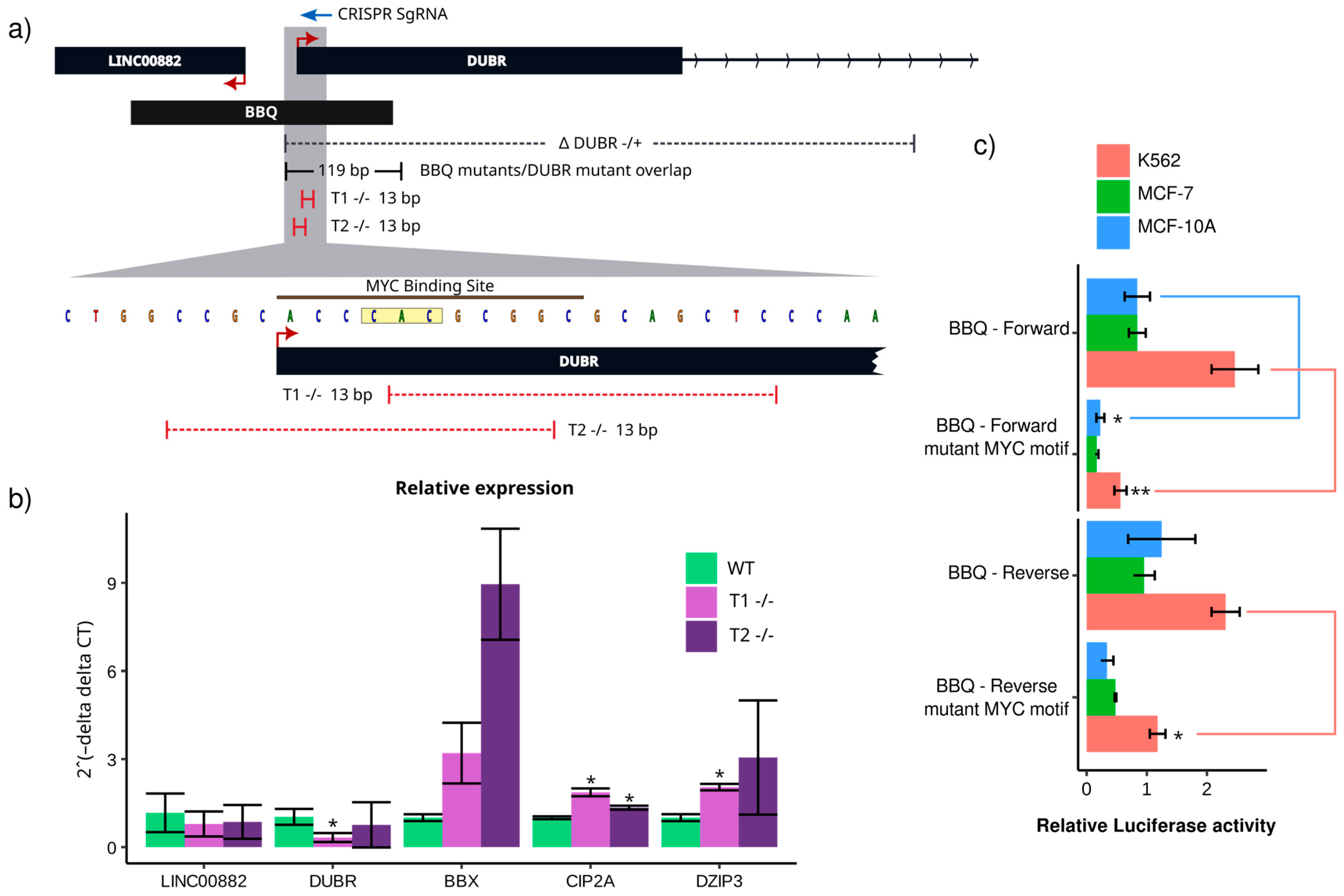{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Luciferase Reporter Assay
2.3. Lentiviral Production and Cell Infection
2.4. CRISPR-Cas9 Plasmid Construction
2.5. CRISPR-Cas9 Assay
2.6. Cell Proliferation Assays
2.7. RNA Isolation and RT-qPCR
2.8. RNA Sequencing (RNA-Seq) and Data Analysis
2.9. Site-Directed Mutagenesis
2.10. Chromatin Immunoprecipitation (ChIP)
2.11. Chromosome Conformation Capture Assay (4C-Seq) and Data Analysis
2.12. Retrieval of Public Data Utilized in This Work
3. Results
3.1. LINC00882-DUBR Intergenic Region Behaves In Vitro as a Bidirectional Promoter
3.2. BBQ Monoallelic Deletion Affects Cell Proliferation
3.3. BBQ Deletion Induces Transcriptional Changes Independently of LINC00882 or DUBR Expression


3.4. Canonical DUBR Transcription Start Site Affects Distal Gene Expression
3.5. BBQ Physically Interacts with Differentially Expressed Genes
3.6. BBQ Acts as a Quencher of BBX Expression and Modulates Regulatory Chromatin Interactions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andersson, R.; Sandelin, A.; Danko, C.G. A Unified Architecture of Transcriptional Regulatory Elements. Trends Genet. 2015, 31, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kellis, M. ChromHMM: Automating Chromatin-State Discovery and Characterization. Nature Methods 2012, 9, 215. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.M.; Buske, O.J.; Wang, J.; Weng, Z.; Bilmes, J.A.; Noble, W.S. Unsupervised Pattern Discovery in Human Chromatin Structure through Genomic Segmentation. Nat. Methods 2012, 9, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Core, L.J.; Martins, A.L.; Danko, C.G.; Waters, C.T.; Siepel, A.; Lis, J.T. Analysis of Nascent RNA Identifies a Unified Architecture of Initiation Regions at Mammalian Promoters and Enhancers. Nat. Genet. 2014, 46, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-K.; Shiekhattar, R. Architectural and Functional Commonalities between Enhancers and Promoters. Cell 2015, 162, 948–959. [Google Scholar] [CrossRef] [PubMed]
- Zabidi, M.A.; Stark, A. Regulatory Enhancer–Core-Promoter Communication via Transcription Factors and Cofactors. Trends Genet. 2016, 32, 801–814. [Google Scholar] [CrossRef]
- Nguyen, T.A.; Jones, R.D.; Snavely, A.R.; Pfenning, A.R.; Kirchner, R.; Hemberg, M.; Gray, J.M. High-Throughput Functional Comparison of Promoter and Enhancer Activities. Genome Res. 2016, 26, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Haines, J.E.; Munson, G.; Chen, J.; Perez, E.M.; Kane, M.; McDonel, P.E.; Guttman, M.; Lander, E.S. Neighborhood Regulation by lncRNA Promoters, Transcription, and Splicing. BioRxiv 2016, 050948. [Google Scholar] [CrossRef]
- Latos, P.A.; Pauler, F.M.; Koerner, M.V.; Şenergin, H.B.; Hudson, Q.J.; Stocsits, R.R.; Allhoff, W.; Stricker, S.H.; Klement, R.M.; Warczok, K.E.; et al. Airn Transcriptional Overlap, but Not Its lncRNA Products, Induces Imprinted Igf2r Silencing. Science 2012, 338, 1469–1472. [Google Scholar] [CrossRef]
- Paralkar, V.R.; Mishra, T.; Luan, J.; Yao, Y.; Kossenkov, A.V.; Anderson, S.M.; Dunagin, M.; Pimkin, M.; Gore, M.; Sun, D.; et al. Lineage and Species-Specific Long Noncoding RNAs during Erythro-Megakaryocytic Development. Blood 2014, 123, 1927–1937. [Google Scholar] [CrossRef]
- Yin, Y.; Yan, P.; Lu, J.; Song, G.; Zhu, Y.; Li, Z.; Zhao, Y.; Shen, B.; Huang, X.; Zhu, H.; et al. Opposing Roles for the lncRNA Haunt and Its Genomic Locus in Regulating HOXA Gene Activation during Embryonic Stem Cell Differentiation. Cell Stem Cell 2015, 16, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Sandelin, A. Determinants of Enhancer and Promoter Activities of Regulatory Elements. Nat. Rev. Genet. 2020, 21, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Mikhaylichenko, O.; Bondarenko, V.; Harnett, D.; Schor, I.E.; Males, M.; Viales, R.R.; Furlong, E.E.M. The Degree of Enhancer or Promoter Activity Is Reflected by the Levels and Directionality of eRNA Transcription. Genes Dev. 2018, 32, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Adachi, N.; Lieber, M.R. Bidirectional Gene Organization. Cell 2002, 109, 807–809. [Google Scholar] [CrossRef] [PubMed]
- Duttke, S.H.C.; Lacadie, S.A.; Ibrahim, M.M.; Glass, C.K.; Corcoran, D.L.; Benner, C.; Heinz, S.; Kadonaga, J.T.; Ohler, U. Perspectives on Unidirectional versus Divergent Transcription. Mol. Cell 2015, 60, 348–349. [Google Scholar] [CrossRef] [PubMed]
- Trinklein, N.D.; Aldred, S.F.; Hartman, S.J.; Schroeder, D.I.; Otillar, R.P.; Myers, R.M. An Abundance of Bidirectional Promoters in the Human Genome. Genome Res. 2004, 14, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.Q.; Elnitski, L.L. Diversity of Core Promoter Elements Comprising Human Bidirectional Promoters. BMC Genom. 2008, 9, S3. [Google Scholar] [CrossRef] [PubMed]
- Hon, C.-C.; Ramilowski, J.A.; Harshbarger, J.; Bertin, N.; Rackham, O.J.L.; Gough, J.; Denisenko, E.; Schmeier, S.; Poulsen, T.M.; Severin, J.; et al. An Atlas of Human Long Non-Coding RNAs with Accurate 5′ Ends. Nature 2017, 543, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.B.; Johnston, R.L.; Inostroza-Ponta, M.; Fox, A.H.; Fortini, E.; Moscato, P.; Dinger, M.E.; Mattick, J.S. Genome-Wide Analysis of Long Noncoding RNA Stability. Genome Res. 2012, 22, 885–898. [Google Scholar] [CrossRef]
- Luo, S.; Lu, J.Y.; Liu, L.; Yin, Y.; Chen, C.; Han, X.; Wu, B.; Xu, R.; Liu, W.; Yan, P.; et al. Divergent lncRNAs Regulate Gene Expression and Lineage Differentiation in Pluripotent Cells. Cell Stem Cell 2016, 18, 637–652. [Google Scholar] [CrossRef]
- Yang, M.Q.; Koehly, L.M.; Elnitski, L.L. Comprehensive Annotation of Bidirectional Promoters Identifies Co-Regulation among Breast and Ovarian Cancer Genes. PLoS Comput. Biol. 2007, 3, e72. [Google Scholar] [CrossRef]
- Wei, W.; Pelechano, V.; Järvelin, A.I.; Steinmetz, L.M. Functional Consequences of Bidirectional Promoters. Trends Genet. 2011, 27, 267–276. [Google Scholar] [CrossRef]
- Zhu, L.; Huang, F.; Wan, T.; Xu, H.; Zhao, Q. Overexpression of Long Noncoding RNA LINC00882 Is Associated with Poor Prognosis in Hepatocellular Carcinoma. OncoTargets Ther. 2018, 11, 5209–5217. [Google Scholar] [CrossRef]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Wakano, C.; Byun, J.S.; Di, L.-J.; Gardner, K. The Dual Lives of Bidirectional Promoters. Biochim. Et Biophys. Acta (BBA) Gene Regul. Mech. 2012, 1819, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Schagat, T.; Paguio, A.; Kopish, K. Normalizing Genetic Reporter Assays: Approaches and Considerations for Increasing Consistency and Statistical Significance. Cell Notes 2007, 17, 9–12. [Google Scholar]
- Villanueva, R.A.M.; Chen, Z.J. Ggplot2: Elegant Graphics for Data Analysis (2nd Ed.). Meas. Interdiscip. Res. Perspect. 2019, 17, 160–167. [Google Scholar] [CrossRef]
- Concordet, J.-P.; Haeussler, M. CRISPOR: Intuitive Guide Selection for CRISPR/Cas9 Genome Editing Experiments and Screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing Real-Time PCR Data by the Comparative CT Method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Sandelin, A.; Alkema, W.; Engström, P.; Wasserman, W.W.; Lenhard, B. JASPAR: An Open-access Database for Eukaryotic Transcription Factor Binding Profiles. Nucleic Acids Res. 2004, 32, D91–D94. [Google Scholar] [CrossRef] [PubMed]
- Franke, M.; De la Calle-Mustienes, E.; Neto, A.; Almuedo-Castillo, M.; Irastorza-Azcarate, I.; Acemel, R.D.; Tena, J.J.; Santos-Pereira, J.M.; Gómez-Skarmeta, J.L. CTCF Knockout in Zebrafish Induces Alterations in Regulatory Landscapes and Developmental Gene Expression. Nat. Commun. 2021, 12, 5415. [Google Scholar] [CrossRef]
- Schmidl, C.; Rendeiro, A.F.; Sheffield, N.C.; Bock, C. ChIPmentation: Fast, Robust, Low-Input ChIP-Seq for Histones and Transcription Factors. Nat. Methods 2015, 12, 963–965. [Google Scholar] [CrossRef] [PubMed]
- Krijger, P.H.L.; Geeven, G.; Bianchi, V.; Hilvering, C.R.E.; de Laat, W. 4C-Seq from Beginning to End: A Detailed Protocol for Sample Preparation and Data Analysis. Methods 2020, 170, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Otsu, N. A Threshold Selection Method from Gray-Level Histograms. IEEE Trans. Syst. Man Cybern. 1979, 9, 62–66. [Google Scholar] [CrossRef]
- The ENCODE Project Consortium. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Noguchi, S.; Arakawa, T.; Fukuda, S.; Furuno, M.; Hasegawa, A.; Hori, F.; Ishikawa-Kato, S.; Kaida, K.; Kaiho, A.; Kanamori-Katayama, M.; et al. FANTOM5 CAGE Profiles of Human and Mouse Samples. Sci. Data 2017, 4, 170112. [Google Scholar] [CrossRef]
- Karolchik, D.; Hinrichs, A.S.; Furey, T.S.; Roskin, K.M.; Sugnet, C.W.; Haussler, D.; Kent, W.J. The UCSC Table Browser Data Retrieval Tool. Nucleic Acids Res. 2004, 32, D493–D496. [Google Scholar] [CrossRef]
- Wang, H.; Fan, Z.; Shliaha, P.V.; Miele, M.; Hendrickson, R.C.; Jiang, X.; Helin, K. H3K4me3 Regulates RNA Polymerase II Promoter-Proximal Pause-Release. Nature 2023, 615, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, M.M.; Oguchi, A.; Suzuki, A.; Murakawa, Y. Cap Analysis of Gene Expression (CAGE) and Noncoding Regulatory Elements. Semin. Immunopathol. 2022, 44, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Li, W.; Liu, J.; Chen, C.-H.; Liao, Q.; Xu, P.; Xu, H.; Xiao, T.; Cao, Z.; Peng, J.; et al. Genome-Scale Deletion Screening of Human Long Non-Coding RNAs Using a Paired-Guide RNA CRISPR–Cas9 Library. Nat. Biotechnol. 2016, 34, 1279–1286. [Google Scholar] [CrossRef]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef]
- Javierre, B.M.; Burren, O.S.; Wilder, S.P.; Kreuzhuber, R.; Hill, S.M.; Sewitz, S.; Cairns, J.; Wingett, S.W.; Várnai, C.; Thiecke, M.J.; et al. Lineage-Specific Genome Architecture Links Enhancers and Non-Coding Disease Variants to Target Gene Promoters. Cell 2016, 167, 1369–1384.e19. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Hitz, B.C.; Gabdank, I.; Hilton, J.A.; Kagda, M.S.; Lam, B.; Myers, Z.; Sud, P.; Jou, J.; Lin, K.; et al. New Developments on the Encyclopedia of DNA Elements (ENCODE) Data Portal. Nucleic Acids Res. 2020, 48, D882–D889. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.M.; Ernst, J.; Wilder, S.P.; Kundaje, A.; Harris, R.S.; Libbrecht, M.; Giardine, B.; Ellenbogen, P.M.; Bilmes, J.A.; Birney, E.; et al. Integrative Annotation of Chromatin Elements from ENCODE Data. Nucleic Acids Res. 2013, 41, 827–841. [Google Scholar] [CrossRef]
- Koyanagi, K.O.; Hagiwara, M.; Itoh, T.; Gojobori, T.; Imanishi, T. Comparative Genomics of Bidirectional Gene Pairs and Its Implications for the Evolution of a Transcriptional Regulation System. Gene 2005, 353, 169–176. [Google Scholar] [CrossRef]
- Liu, B.; Chen, J.; Shen, B. Genome-Wide Analysis of the Transcription Factor Binding Preference of Human Bi-Directional Promoters and Functional Annotation of Related Gene Pairs. BMC Syst. Biol. 2011, 5, S2. [Google Scholar] [CrossRef] [PubMed]
- Ørom, U.A.; Derrien, T.; Beringer, M.; Gumireddy, K.; Gardini, A.; Bussotti, G.; Lai, F.; Zytnicki, M.; Notredame, C.; Huang, Q.; et al. Long Noncoding RNAs with Enhancer-like Function in Human Cells. Cell 2010, 143, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Young, R.S.; Kumar, Y.; Bickmore, W.A.; Taylor, M.S. Bidirectional Transcription Initiation Marks Accessible Chromatin and Is Not Specific to Enhancers. Genome Biol. 2017, 18, 242. [Google Scholar] [CrossRef] [PubMed]
- Franke, M.; Ibrahim, D.M.; Andrey, G.; Schwarzer, W.; Heinrich, V.; Schöpflin, R.; Kraft, K.; Kempfer, R.; Jerković, I.; Chan, W.-L.; et al. Formation of New Chromatin Domains Determines Pathogenicity of Genomic Duplications. Nature 2016, 538, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Furlan-Magaril, M.; Ando-Kuri, M.; Arzate-Mejía, R.G.; Morf, J.; Cairns, J.; Román-Figueroa, A.; Tenorio-Hernández, L.; Poot-Hernández, A.C.; Andrews, S.; Várnai, C.; et al. The Global and Promoter-Centric 3D Genome Organization Temporally Resolved during a Circadian Cycle. Genome Biol. 2021, 22, 162. [Google Scholar] [CrossRef]
- Li, G.; Ruan, X.; Auerbach, R.K.; Sandhu, K.S.; Zheng, M.; Wang, P.; Poh, H.M.; Goh, Y.; Lim, J.; Zhang, J.; et al. Extensive Promoter-Centered Chromatin Interactions Provide a Topological Basis for Transcription Regulation. Cell 2012, 148, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Shao, J.; Mitra, J.; Xiong, F.; D’Antonio, M.; Wang, R.; Garcia-Bassets, I.; Ma, Q.; Zhu, X.; Lee, J.-H.; et al. Enhancer Release and Retargeting Activates Disease-Susceptibility Genes. Nature 2021, 595, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-Y.; Chang, W.-S.W.; Lai, Y.-K.; Wu, C.-W. C-Myc Regulates the Coordinated Transcription of Brain Disease-Related PDCD10–SERPINI1 Bidirectional Gene Pair. Mol. Cell. Neurosci. 2009, 42, 23–32. [Google Scholar] [CrossRef]
- See, Y.X.; Chen, K.; Fullwood, M.J. MYC Overexpression Leads to Increased Chromatin Interactions at Super-Enhancers and MYC Binding Sites. Genome Res. 2022, 32, 629–642. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peralta-Alvarez, C.A.; Núñez-Martínez, H.N.; Cerecedo-Castillo, Á.J.; Poot-Hernández, A.C.; Tapia-Urzúa, G.; Garza-Manero, S.; Guerrero, G.; Recillas-Targa, F. A Bidirectional Non-Coding RNA Promoter Mediates Long-Range Gene Expression Regulation. Genes 2024, 15, 549. https://doi.org/10.3390/genes15050549
Peralta-Alvarez CA, Núñez-Martínez HN, Cerecedo-Castillo ÁJ, Poot-Hernández AC, Tapia-Urzúa G, Garza-Manero S, Guerrero G, Recillas-Targa F. A Bidirectional Non-Coding RNA Promoter Mediates Long-Range Gene Expression Regulation. Genes. 2024; 15(5):549. https://doi.org/10.3390/genes15050549
Chicago/Turabian StylePeralta-Alvarez, Carlos Alberto, Hober Nelson Núñez-Martínez, Ángel Josué Cerecedo-Castillo, Augusto César Poot-Hernández, Gustavo Tapia-Urzúa, Sylvia Garza-Manero, Georgina Guerrero, and Félix Recillas-Targa. 2024. "A Bidirectional Non-Coding RNA Promoter Mediates Long-Range Gene Expression Regulation" Genes 15, no. 5: 549. https://doi.org/10.3390/genes15050549
APA StylePeralta-Alvarez, C. A., Núñez-Martínez, H. N., Cerecedo-Castillo, Á. J., Poot-Hernández, A. C., Tapia-Urzúa, G., Garza-Manero, S., Guerrero, G., & Recillas-Targa, F. (2024). A Bidirectional Non-Coding RNA Promoter Mediates Long-Range Gene Expression Regulation. Genes, 15(5), 549. https://doi.org/10.3390/genes15050549